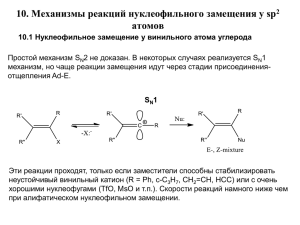
реакции мономолекулярного нуклеофильного замещения
advertisement

Органические галогениды. Механизмы реакций нуклеофильного замещения и элиминирования Алкилгалогениды Типичные реакции c участием галогена: SN Органические галогениды Арилгалогениды Типичные реакции c участием галогена: SN E Механизмы: Механизмы: SN1, SN2 E1, E2 Ареновый Ариновый Типовые реакции алифатического нуклеофильного замещения с участием алкилгалогенидов Название Гидролиз алкилгалогенидов Алкилирование алкилгалогенидами Алкилирование аминов Реакции с участием ацетиленовой группы Реакция Механизм реакции SN1 ( реакции мономолекулярного нуклеофильного замещения) (substitution nucleophilic monomolecular) Стадия 1: Ионизация субстрата, с образованием карбокатиона (медленная стадия): Стадия 2: Нуклеофильная атака карбкатиона (быстрая стадия): Стадия 1: R−X → R+ + X− Стадия 2:R+ + Y− → R−Y Скорость реакции = k × [RX] CH3 Br CH3 H C2H5 D(+)-2-Бромбутан Молекула хиральна BrH C2H5 Карбкатион плоский Ахирален 4 Реакции SN1 характерны для субстратов, образующих относительно стабильные карбокатионы: Alk3C-X ArCH2-X CH2=CH-CH2X, X= Hal, OCOR, OSO2Ar, OSO2CF3 Механизм реакции SN2 реакции бимолекулярного нуклеофильного замещения (substitution nucleophilic bimolecular) R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Скорость реакции = k [RX] [Y] В реакциях SN2 оптически активных субстратов происходит «обращение конфигурации» и образуется продукт противоположной конфигурации. Т.е. (+) субстрат дает (-)-продукт и наоборот. Одна стадия: Атака нуклеофила и отщепление уходящей группы происходят синхронно, карбокатион не образуется Реакции SN2 характерны для субстратов с первичными алкильными группами AlkCH2-X и особенно для метильных производных CH3-X, т.к. в этих случаях минимальны стерические препятствия в переходных состояниях при атаке нуклеофилами: R=H Кроме того, для AlkCH2-X и CH3-X невыгоден SN1 маршрут реакции, т.к. нестабильные карбокатионы AlkCH2+ и особенно CH3+ R= Alk Влияния растворителей на протекание органических реакций Межмолекулярные взаимодействия Свойства атомов, молекул, ионов в растворах сильно отличаются от свойств в твердом состоянии или газовой фазе ! Растворитель может менять направление реакции, а скорости одной и той же реакции в в различных растворителях могут отличаться в миллионы раз. Основная причина - СОЛЬВАТАЦИЯ G влияиние сольватации на свободные энергии Сольватация снижает свободную энергию молекул и ионов (DG solv) за счет выделения энергии при межмолекулярном взаимодействии A+B C Модель сольватации s s s s M s Как правило, сольватация возрастает с увеличением полярности растворителя. Полярность измеряют через диэлектрическую проницаемость (e) и дипольный момент (m) s s s сольватная оболочка Сольватация – выделение энергии DGsolv = DGрастворения - DGкрист.решетки Сольватное число: число мол-л ра-теля, приходящееся на один ион, которые остаются связанными с ионом настольно долго, что могут двигаться вместе с ионом Диэлектрическая проницаемость (e) и дипольный момент (m) растворителей Ра-тель e m При 25оС вода 78.5 1.8 этиленгликоль 37.7 2.0 метанол 32.6 1.65 этанол 24.3 1.7 AcOH 6.2 1.5 пропанол 20.1 1.7 MeNO2 38.6 3.1 MeCN 37.5 3.5 ДМСО 48.9 3.9 сульфолан 44.0 4.8 Трет-бутанол 12.2 1.7 ДМФА 36.7 3.8 ацетон 20.7 2.7 PhNO2 34.8 4.0 PhCN 25.2 3.9 дихлорэтан 10.4 1.75 G влияиние сольватации на свободные энергии В реальных процессах сольватированы все участники реакции. Вопрос в том, кто из них сольватирован в большей степени ? Ответ может дать качественная теория влияния растворителей (теория ХьюзаИнгольда). A+B C Постулат: энергия сольватации выше для ионов, чем для нейтральных молекул; энергия сольватации возрастает с уменьшением размеров молекул и ионов. Реакции, при которых в переходном состоянии возникают заряды или уже имеющиеся в реагентах заряды концентрируются в меньшем объеме, ускоряются с повышением полярности растворителя. Если ионные заряды реагентов нейтрализуются в переходном состоянии или распределяются в большем объеме, то такие реакции замедляются с увеличением полярности растворителя. Влияние увеличения полярности растворителей на скорости SN реакций Механизм Реагенты Переходное состояние Изменение заряда в переходном состоянии Изменение скорости с возрастанием полярности а) SN1 R-X Rd+……Xd- Разделение зарядов увеличение б) SN1 R-X+ Rd+……Xd+ Распределение заряда в большем объеме уменьшение в) SN2 Y: + R-X Y d-…….R…..X d- Разделение зарядов увеличение г) SN2 Y:- + R-X Y d-…….R…..X d- Распределение заряда в большем объеме уменьшение д) SN2 Y: + R-X+ Y d+…….R…..X d+ Распределение заряда в большем объеме уменьшение е) SN2 Y:- + R-X+ Y d-…….R…..X d+ Нейтрализация зарядов уменьшение Сравнительный фактор Кинетика SN 1 SN 2 k × [RX] Стереохимический результат рацемизация k × [RX] × [Y] инверсия Влияние структуры субстрата на скорость реакции CH3-Х реакция не идёт очень хорошо R-CH2-X реакция не идёт хорошо R2CH-X реакция идёт реакция идёт R3C-X очень хорошо реакция не идёт R-CH=CH-CH2-X реакция идёт хорошо С6H5-CH2-X реакция идёт хорошо R-CO-CH2-X реакция не идёт отлично R-O-CH2-X отлично хорошо R2N-CH2-X отлично хорошо 1,2-Элиминирование (Механизмы Е1 и Е2) 1 Если XY: 2 HHal дегидрогалогенирование Н Нейтральная молекула расщепление ониевых солей HOH дегидратация Уходящая группа Механизм Е1 (мономолекулярное элиминирование) Скорость реакции = k [RX] Лимитирующая стадия – образование карбокатиона Возможны перегруппировки карбокатиона Механизм Е2 (бимолекулярное элиминирование) Скорость реакции = k [RX] [В] Стереоэлектронные ограничения Е2: Связи, разрывающиеся в активированном комплексе Е2 должны быть в транс-положении по отношению друг к другу • Реакции замещения и элиминирования являются конкурирующими (SN1/E1 и SN2/E2), поскольку протекают через одно и то же переходное состояние «Хорошие» и «плохие» уходящие группы X – уходящие группы. Зависят ли скорости этих реакций от природы X ? Уходящая группа отщепляется тем легче, чем стабильней она как свободная молекула или ион. Обычно это находится в обратной зависимости от основности, поэтому наилучшие уходящие группы – это самые слабые основания (сильная кислот – слабое основание). Хорошие уходящие группы называют «нуклеофугами» Хорошие уходящие группы Плохие уходящие группы (Нуклеофуги) Сильные основания Слабые основания R-Hal Hal- R-Hal Hal- Hal = I-, Br- Hal = F- R-O+H2 H2O (Сила кислот: HI > HBr > HCl > HF) R-OH HO- R'-OCOR RCOOR-N+R3' NR3' R-NH2 NH2R-H H:- - гидрид ион, самая плохая уходящая группа. Очень неустойчив и в свободном виде не существует. Какая кислота отвечает этому основанию ? Лучшие из уходящих групп (хорошие нуклеофуги): CH3SO2O- (OMs), CF3SO2O- (TfO), p-CH3C6H4SO2O- (TsO), ClO4-, N2 k2 >> k1 Реакции нуклеофильного замещения в ароматическом ряду Типичные реакции нуклеофильного замещения арилгалогенидов Получение фенолов Получение ароматических простых эфиров Получение замещенных аминов Получение ароматических сульфидов X= F, Cl, Br, I X≠H Механизмы нуклеофильного ароматического замещения Реакции нуклеофильного ароматического замещения ускоряются электроноакцепторными заместителями ! SNAr механизм Промежуточный комплекс (комплекс Мейзенгеймера) (стабилизируется электроноакцепторными группами) В каких положениях должна находиться нитро-группа (о-, м-, п-), чтобы стабилизировать промежуточный комплекс и ускорять SNAr реакции ? Ариновый (бензиновый) механизм Как доказать ? Этот механизм характерен для галиодаренов, не имеющих электроноакцепторных заместителей