1 К заседанию Бюро ОМБН РАМН «25» сентября 2012 г
advertisement

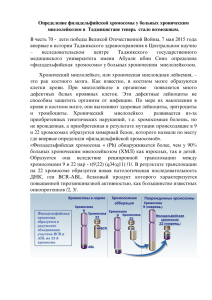
1 К заседанию Бюро ОМБН РАМН «25» сентября 2012 г. СПРАВКА к научному докладу д.м.н.С.И.Куцева на тему: «Геномные основы таргетной терапии миелопролиферативных заболеваний» Миелопролиферативные заболевания – группа клональных онкогематологических заболеваний, развивающихся вследствие появления мутаций в стволовых кроветворных клетках, но характеризующихся избыточной пролиферацией преимущественно миелоидного ростка кроветворения с сохранением способности опухолевых клеток к дифференцировке. К этой группе болезней относят хронический миелолейкоз, эссенциальную тромбоцитемию, истинную полицитемию, хронический идиопатический миелофиброз, хронический эозинофильный лейкоз, хронический нейтрофильный лейкоз. Геномные исследования одного из этих заболеваний – хронического миелолейкоза, привели к созданию первого препарата таргетного действия - иматиниба (STI571), применение которого для лечения хронического миелолейкоза стало революционным прорывом в области целенаправленной противоопухолевой терапии. Впервые концепция тотального неспецифического уничтожения активно пролиферирующих клеток с помощью цитостатических препаратов была заменена парадигмой целенаправленной элиминации опухолевых клеток, имеющих идентичную клональную природу. Хронический миелолейкоз (ХМЛ) характеризуется появлением в стволовой кроветворной клетке специфического цитогенетического маркера – филадельфийской (Ph) хромосомы. Ph-хромосома образуется в результате реципрокной транслокации t(9;22)(q34;q11.2), сопровождающейся появлением химерного гена BCR-ABL. Экспрессия гена BCR-ABL приводит к появлению тирозинкиназы с патологически высокой функциональной активностью, играющей ключевую роль в патогенезе ХМЛ. На момент установления диагноза хронического миелоидного лейкоза 95-100% клеток костного мозга являются Ph-позитивными при цитогенетическом исследовании, а уровень экспрессии гена BCR-ABL достигает 100% относительно контрольного гена ABL. Иматиниб селективно ингибирует BCR-ABL тирозинкиназу, встраиваясь в АТФсвязывающий карман киназного домена белка BCR-ABL и удерживая его таким образом в неактивном состоянии. Клинические испытания иматиниба показали необыкновенно высокую эффективность этого препарата в терапии ХМЛ. Так в исследовании IRIS показано, что к пяти годам полный гематологический ответ на терапию иматинибом достигается у 98% больных ХМЛ, большой цитогенетический ответ – у 92%, полный цитогенетический ответ – у 87%; 5-летняя выживаемость без прогрессии заболевания составила 93%. К настоящему моменту прошли клинические испытания и зарегистрированы в США, Европе и Российской Федерации более эффективные ингибиторы тирозинкиназ II поколения – нилотиниб и дазатиниб. Тем не менее, несмотря на столь впечатляющие результаты, существует проблема резистентности к терапии иматинибом. К восьми годам терапии иматинибом количество больных ХМЛ с отсутствием ответа или рецидивом ХМЛ достигает 35-45%. Появление новых препаратов ингибиторов тирозинкиназ второго поколения, нилотиниба и дазатиниба, и их использование в качестве препаратов первой линии терапии ХМЛ повысило эффективность лечения. Тем не менее, по предварительным данным проблема резистентности сохраняется и при использовании этих новейших препаратов приблизительно в 20-25% случаев ХМЛ. Понятие резистентности к иматинибу включает по меньшей мере две дефиниции. Первичная резистентность (или рефрактерность) – это отсутствие или недостаточный ответ на лечение. Вторичная, или приобретенная, резистентность определяется как потеря достигнутого ответа и прогрессирование заболевания до фазы акселерации и бластного криза. Механизмы резистентности к терапии ХМЛ ингибиторами тирозинкиназ различны 2 и включают в себя как связанные с BCR-ABL тирозинкиназой, так и не связанные. К первой группе, относят амплификацию гена BCR-ABL и мутации гена BCR-ABL. Амплификация гена BCR-ABL обусловливает повышенную экспрессию белка BCR-ABL и, как следствие, резистентность к проводимой терапии. Впервые было установлено, что обнаружение даже небольшого клона (2-7%) лейкозных клеток с амплификацией гена BCR-ABL на фоне терапии иматинибом является неблагоприятным прогностическим фактором для достижения цитогенетического и молекулярного ответа на проводимое лечение и предполагает изменение тактики терапии ХМЛ. Анализ амплификации гена BCR-ABL у пациентов с первичной или вторичной резистентностью к терапии иматинибом и другими ингибиторами тирозинкиназ рекомендован для включения в обязательный мониторинг эффективности терапии ХМЛ. Некоторые точечные мутации в химерном гене BCR-ABL приводят к замене одной из аминокислот в белке BCR-ABL тирозинкиназы и изменению конформации сайта связывания иматиниба, что может приводить к неэффективности проводимой терапии. Впервые проведенный анализ мутаций гена BCR-ABL в когорте первично резистентных пациентов с ХМЛ позволил обнаружить миссенс-мутации в 20% случаях как причину формировании первичной резистентности к терапии ингибиторами тирозинкиназ. Большая часть этих мутаций (63,6%) локализованы в участке гена BCR-ABL, кодирующем P-петлю BCR-ABL-тирозикиназы, ответственную за связывание с иматинибом. Учитывая различную чувствительность мутантных форм BCR-ABL тирозиникназы к разным ингибиторам тирозиникназ, результаты мутационного анализа гена BCR-ABL позволили использовать дифференцированный подход к назначению ингибиторов тирозинкиназ второго и третьего поколения. Выполненный сравнительный анализ влияния амплификации гена BCR-ABL и мутаций гена BCR-ABL на развитие резистентности к терапии ХМЛ впервые показал, что сочетание этих двух механизмов резистентности встречается всего лишь в 3% случаев. Нестабильность генома опухолевых клеток, обусловленная повышенной активностью BCR-ABL тирозинкиназы и высоким уровнем свободных радикалов кислорода, предполагает стохастический характер появления амплификации или мутации гена BCRABL с последующей селекцией клонов опухолевых клеток. Вторую группу механизмов резистентности к терапии ХМЛ ингибиторами тирозинкиназ составляют BCR-ABL независимые механизмы: клональная цитогенетическая эволюция (появление дополнительных хромосомных аберраций в Phпозитивных и Ph-негативных клетках и связанная с этим активация альтернативных, помимо BCR-ABL опосредованных, сигнальных путей), нарушения фармакокинетики препаратов группы ингибиторов тирозинкиназ. В результате проведенных исследований было показано, что появление дополнительных хромосомных аберраций в Ph-позитивных клетках костного мозга статистически достоверно снижает выживаемость пациентов с ХМЛ, находящихся на терапии иматинибом. Тем не менее, клональная эволюция в Ph-позитивных клетках у некоторых пациентов является транзиторным фактом. На фоне продолжающейся терапии иматинибом в высоких дозах или в результате перехода на терапию ингибиторами тирозинкиназ второго поколения, клон опухолевых клеток с признаками цитогенетической эволюции может исчезать. В тоже время, появление дополнительных хромосомных аберраций в Ph-негативных клетках у пациентов с частичным или полным цитогенетическим ответом не влияет на безрецидивную и общую выживаемость больных с ХМЛ. Проведенные фарамкокинетические исследования показали, что концентрация иматиниба в плазме коррелирует с достижением цитогенетического и молекулярного ответов на терапию. Терапевтически эффективная концентрация иматиниба в плазме и костном мозге стабилизирует достигнутый ответ и снижает риск развития рецидива. Исследования показали зависимость концентрации иматиниба в плазме от активности 3 ряда ферментов цитохрома P-450, которые играют важную роль в деградации фармакологических препаратов, снижая их активность и увеличивая их элиминацию. Результаты фармакогенетических исследований подтверждают эти данные и могут быть использованы в перспективе в индивидуальном подборе дозы иматиниба для терапии первичных больных ХМЛ. Эффективность терапии ХМЛ ингибиторами тирозинкиназ во многом зависит от своевременного цитогенетического и молекулярного мониторинга, раннего выявления и выяснения причин первичной и вторичной резистентности, своевременной коррекции терапии ХМЛ. Однако, до настоящего времени причины резистентности к терапии ингибиторами тирозинкиназ и прогрессия хронической фазы вплоть до бластного криза у ряда пациентов неизвестны. Более того, у пациентов достигших по данным количественной ОТ-ПЦР в режиме реального времени полного молекулярного ответа (транскрипт BCR-ABL не определяется) более чувствительные методы ПЦР анализа (5-6 log) выявляют небольшое количество опухолевых клеток. По современным представлениям полная эрадикация опухолевых клеток при терапии ингибиторами тирозинкиназ невозможна. В костном мозге сохраняются BCRABL-позитивные лейкозные стволовые клетки, нечувствительные к ингибиторам тирозинкиназ, которые могут являться источником для рецидива опухоли. В настоящее время основной областью исследований в терапии ХМЛ является изучение профиля мутаций и экспрессии различных генов лейкозных стволовых клеток, а также межклеточные взаимоотношения стволовых клеток и клеток стромы в костномозговых “нишах” с целью поиска дополнительных мишеней для таргетной терапии и достижения полной эрадикации опухолевых клеток. Классические Ph-негативные хронические миелопролиферативные заболевания включают в себя истинную полицитемию (ИП), эссенциальную тромбоцитемию (ЭТ) и идиопатический миелофиброз (ИМ), объединенных сходством клинической картины, морфологических проявлений и, как выяснилось в последние годы, генетических изменений. В частности, мутация V617F в псевдокиназном домене гена JAK2 обнаруживается более чем в 90% случаев ИП, в 70% случаев ЭТ и 50% случаев ИМ. Данная мутация приводит к аутоингибированию янус тирозинкиназы 2 в гемопоэтических клетках и, как следствие, активации сигнальных путей, регулирующих пролиферацию гемпопоэтических клеток. В ряде случаев JAK2V617F – негативных миелопролиферативных заболеваний обнаруживаются мутации в экзоне 12 гена JAK2, с тем же эффектом аутоингибирования янус киназы 2. В 5-10% случаев ЭТ и ИМ обнаруживаются мутации W515L/K гена тромбопоэтинового рецептора MPL, которая приводит к активации регуляторного пути JAK2-STAT, как и мутация V617F в гене JAK2. Проведенные исследования показали, что соотношение клона клеток с мутацией JAK2 V617F относительно количества клеток, несущих ген JAK2wt дикого типа, различно в группе Ph-негативных хронических миелопролиферативных заболеваний. Более того, тяжесть течения заболевания может зависеть от величины клона клеток с мутацией JAK2 V617F . В клиническую практику терапии Ph-негативных хронических миелопролиферативных заболеваний внедряются препараты таргетного действия ингибиторы янус тирозинкиназы 1/2. Клинические исследования первого из них – руксолитиниба показали достижимость клинической ремиссии (например, редукцию размеров селезенки), улучшение качества жизни и значительное увеличение выживаемости больных с ИМ. Тем не менее, соотношение аллелей JAK2 V617F/ JAK2wt при терапии руксолитинибом JAK V617F – позитивных миелопролиферативных заболеваний не меняется, что свидетельствует об отсутствии процесса эрадикации опухолевого клона. Данное обстоятельство объясняется тем, что мутации в генах JAK2 и MPL являются вторичными, имеют определенное значение для диагностики и 4 мониторинга эффективности терапии и небольшое значение в качестве цели для таргетной терапии Разработка эффективных подходов к терапии миелопролиферативных заболеваний основана на поиске ведущих (“drive”) мутаций и подборе соответствующих препаратов таргетного действия. Внедрение в научные исследования современных технологий секвенирования нового поколения, позволяющих анализировать геном, экзом, транскриптом опухолевых клеток и клеток микроокружения, позволит расширить и дополнить наши знания о патогенезе миелопролиферативных заболеваний, идентифицировать новые молекулярные мишени и разработать новые таргетные препараты для терапии миелопролиферативных и других онкогематологических заболеваний.