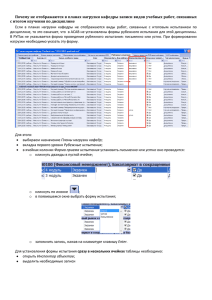
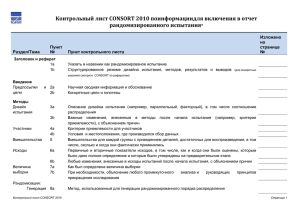
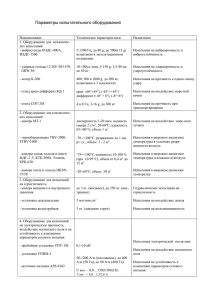
Document 3683657
advertisement

ГОСУДАРСТВЕННЫЙ СТАНДАРТ РЕСПУБЛИКИ КАЗАХСТАН НАДЛЕЖАЩАЯ КЛИНИЧЕСКАЯ ПРАКТИКА. ОСНОВНЫЕ ПОЛОЖЕНИЯ СТ РК 1616-2006 Издание официальное Комитет по техническому регулированию и метрологии Министерства индустрии и торговли Республики Казахстан (Госстандарт) Астана Предисловие 1 РАЗРАБОТАН Республиканским государственным предприятием «Национальный центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» 2 ВНЕСЕН Комитетом фармации Министерства здравоохранения Республики Казахстан 3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ приказом Комитета по техническому регулированию и метрологии Министерства индустрии и торговля Республики Казахстан от 29 декабря 2006 года №557. 4 В настоящем стандарте учтены основные нормативные положения следующих международных документов: ICH GCP - Руководство по Надлежащей клинической практике (Guideline for Good Clinical Practice) Международной конференции по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH), которое, разработано с учетом действующих требований надлежащей клинической практики Европейского Союза, Соединенных Штатов Америки и Японии, а также Австралии, Канады и Всемирной организации здравоохранения (ВОЗ) 5 В настоящем стандарте реализованы нормы законов Республики Казахстан от 09.11.04 г. №603-II «О техническом регулировании», от 11.07.1997 г. №151-I «О языках в Республике Казахстан», от 13.01.01 г. №522-II «О лекарственных средствах», от 04.06.03 г. №430 «О системе здравоохранения» от 07.07.06 г. №170 «Об охране здоровья граждан». 6 СРОК ПЕРВОЙ ПРОВЕРКИ 2011 год ПЕРИОДИЧНОСТЬ ПРОВЕРКИ 5 лет 7 ВВЕДЕН ВПЕРВЫЕ Настоящий стандарт не может быть полностью или частично воспроизведен, тиражирован и распространен в качестве официального издания без разрешения Комитета по техническому регулированию и метрологии Министерства индустрии и торговли Республики Казахстан Введение Надлежащая клиническая практика (Good Clinical Practice (GCP)) - стандарт клинических исследований/испытаний с участием человека в качестве испытуемого, охватывающий планирование, проведение, завершение, проверку, анализ результатов, составление отчетов и ведение документации. Данный стандарт обеспечивает научную значимость исследований/испытаний, их этическую приемлемость и полную документированность клинических характеристик изучаемого лекарственного средства, доверие к полученным результатам, а также их признаваемость уполномоченными органами других стран. Соблюдение данного стандарта служит гарантией безопасности, благополучия, защищенности испытуемых, согласованности с принципами, заложенными Хельсинкской декларацией Всемирной Медицинской Ассоциацией, а также достоверности данных клинического исследования/испытания. Настоящий стандарт построен на основе ICH GCP - Руководство по Надлежащей клинической практике (Guideline for Good Clinical Practice) Международной конференции по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH), которое, разработано с учетом действующих требований надлежащей клинической практики Европейского Союза, Соединенных Штатов Америки и Японии, а также Австралии, Канады и Всемирной организации здравоохранения (ВОЗ) [1]. ГОСУДАРСТВЕННЫЙ СТАНДАРТ РЕСПУБЛИКИ КАЗАХСТАН НАДЛЕЖАЩАЯ КЛИНИЧЕСКАЯ ПРАКТИКА. ОСНОВНЫЕ ПОЛОЖЕНИЯ Дата введения 2008.01.01 1 Область применения Настоящий стандарт необходимо соблюдать при проведении клинических исследований/испытаний, данные которых запланировано представлять в уполномоченные органы. Принципы, установленные настоящим стандартом, применимы и к иным клиническим исследованиям/испытаниям, которые могут оказать влияние на безопасность и благополучие человека, выступающего в качестве испытуемого. 2 Нормативные ссылки В данном стандарте использованы ссылки на следующие нормативные документы: СТ РК 1.2-2002 «Государственная система стандартизации. Порядок разработки государственных стандартов». СТ РК 1.5-2004 «Общие требования к построению, изложению, оформлению и содержанию стандартов». 3 Термины и определения В настоящем стандарте применены следующие термины с соответствующими определениями, используемые в области применения данного стандарта: 3.1 Аудит: Систематическая, независимая и документированная проверка документации и деятельности, вовлеченных в проведении клинического исследования/испытания сторон, которая проводится для подтверждения факта осуществления этой деятельности, а также для оценки соответствия процедур сбора, обработки и представления данных, требованиям протокола исследования/испытания, стандартных операционных процедур и настоящего Стандарта. 3.2 Биодоступность: Скорость и степень, с которой активная субстанция или ее активная часть абсорбируется из лекарственной формы и становится доступной в месте ее предполагаемого действия. 3.3 Биоэквивалентность: Сравнительная характеристика двух лекарственных средств при одинаковых условиях, которая подтверждает их фармацевтическую и биологическую эквивалентность в отношении эффективности и безопасности после использования в одинаковых молярных дозах. 3.4 Благополучие испытуемых: Физическое и психическое состояние здоровья испытуемых, позволяющее им участвовать в клиническом исследовании/испытании. 3.5 Брошюра исследователя: Документ, содержащий реферативное изложение результатов доклинического (неклинического) и клинического изучения исследуемого лекарственного препарата, значимых для его исследования/испытания на человеке. 3.6 Воспроизведенное лекарственное средство (генерик): Лекарственное средство, идентичное оригинальному лекарственному средству по составу активных субстанций, показателям качества, безопасности, эффективности и поступившее в обращение после истечения срока действия охранных документов на оригинальное лекарственное средство. 3.7 Договор: Документ, содержащий письменное, датированное, подписанное соглашение между двумя или более сторонами, определяющее договоренности, касающиеся распределения объема работ, прав, обязанностей, и при необходимости, финансовые вопросов. 3.8 Доклиническое (неклиническое) исследование/испытание: Биомедицинские исследования/испытания лекарственного средства in vitro и на различных биологических объектах, для выявления их физических и химических свойств, механизма действия, фармакодинамических и фармакокинетических свойств, токсичности, влияния на репродуктивность и концерогенность, не включающие в себя эксперименты на человеке. 3.9 Документальный след: Документация, позволяющая полностью восстановить ход событий. 3.10 Документация: Записи в любой форме (на бумажных, электронных, магнитных или оптических носителях, сканограммы, рентгеновские снимки, электрокардиограммы и др.), описывающие либо регистрирующие методы, организацию и/или результаты клинического исследования/испытания, факторы влияющие на исследование/испытание и принятые меры. 3.11 Законный представитель: Физическое или юридическое лицо, имеющее в соответствии с действующим законодательством право давать согласие на участие в клиническом исследовании/испытании от имени потенциального испытуемого. 3.12 Идентификационный код испытуемого: Уникальный код, присваиваемый исследователем индивидуально каждому испытуемому для обеспечения конфиденциальности его личных данных и используемый вместо имени испытуемого в отчетах и/или других данных, относящихся к исследованию/испытанию. 3.13 Индивидуальная регистрационная форма: ИРФ - Печатный и электронный документ, предназначенный для внесения в него информации по каждому испытуемому, предусмотренный протоколом клинического исследования/испытания. 3.14 Инспекция: Официальная проверка с целью оценки полученных данных и качества проведения клинического исследования/испытания, материальной базы и документов, относящихся к клиническому исследованию/испытанию, уполномоченным органом в области здравоохранения с привлечением специалистов экспертной организации обеспечивающей безопасность, эффективность и качество лекарственных средств. 3.15 Испытуемый: Пациент или здоровый человек добровольно принимающий участие в клиническом исследовании/испытании лекарственного препарата. 3.16 Информация для испытуемого: Документ, описывающий общие цели исследования/испытания, риск и пользу участия в нем, а также процедуры, в которых потребуется участие испытуемого. 3.17 Информированное согласие: Документально оформленное добровольное согласие испытуемого на участие в клиническом исследовании/испытании после ознакомления со всеми необходимыми документами, подписанное и датированное испытуемым. 3.18 Исследователь: Физическое лицо, непосредственно проводящее и отвечающее за порядок проведения клинического исследования/испытания лекарственного средства на клинической базе. 3.19 Ответственный исследователь: Руководитель коллектива клинической базы. 3.20 Исследователь-координатор: Исследователь, отвечающий за координацию деятельности исследователей, участвующих в многоцентровом клиническом исследовании/испытании. 3.21 Исследуемое лекарственное средство: Лекарственная форма активного вещества или плацебо, изучаемая или используемая для контроля в клиническом исследовании/испытании. 3.22 Клиническая база: Медицинская организация, определенная уполномоченным органом на основании рекомендации экспертной организации обеспечивающей безопасность, эффективность и качество лекарственных средств, для проведения клинических исследований/испытаний лекарственного средства. 3.23 Клиническое исследование/испытание: Исследование/испытание с участием человека в качестве испытуемого, проводимое для выявления или подтверждения клинических эффектов, фармакологических свойств и/или выявления всех побочных реакций исследуемого лекарственного средства, в целях установления его безопасности и эффективности. 3.24 Контрактная исследовательская организация: Физическое или юридическое лицо, которое в рамках договора со спонсором выполняет одну или несколько его функций, связанных с проведением клинического исследования/испытания. 3.25 Контроль качества: Методы и меры, являющиеся частью системы обеспечения качества и используемые для проверки качества выполняемых в рамках исследования/испытания процедур. 3.26 Комиссия по вопросам этики: Независимый орган, образованный при исследовательском центре (клинической базе), в состав которого входят специалисты в области здравоохранения, науки, представители общественных организаций, осуществляющий защиту прав, безопасности и благополучия испытуемых и исследователей, этическую и нравственно-правовую оценку материалов клинического исследования/испытания. 3.27 Республиканская комиссия по вопросам этики: Независимый орган, образованный при уполномоченном органе, в состав которого входят ведущие специалисты уполномоченного органа, науки, представители общественных организаций, осуществляющий защиту прав, безопасности и благополучия испытуемых и исследователей, этическую и нравственно-правовую оценку материалов клинического исследования/испытания в случаях возникновения спорных вопросов на всех этапах клинического исследования/испытания, а также после их завершения. 3.28 Координационный комитет: Орган, который может быть организован спонсором для координации проведения многоцентрового клинического исследования/испытания. 3.29 Конфиденциальность: Сохранение в тайне от неуполномоченных лиц информации, принадлежащей спонсору, или информации, позволяющей идентифицировать испытуемого. 3.30 Многоцентровое клиническое исследование/испытание: Клиническое исследование/испытание, осуществляемое в нескольких клинических базах по единому протоколу. 3.31 Монитор: Лицо, назначаемое спонсором, контролирующее проведение клинического исследования/испытания в соответствии с протоколом. 3.32 Мониторинг: Процедура контроля за ходом клинического исследования/испытания и обеспечения его проведения, сбора данных и представления результатов исследования/испытания согласно протоколу, стандартным операционным процедурам и настоящего стандарта. 3.33 Надлежащая клиническая практика: Стандарт планирования, организации, проведения, мониторинга, аудита, документирования, анализа и представления результатов клинических исследований/испытаний, являющийся гарантией достоверности и точности полученных данных, обеспечивающий защиту прав, здоровья и конфиденциальности испытуемых. 3.34 Нежелательная побочная реакция: Все отрицательные или непредвиденные реакции организма, связанные с введением любой дозы лекарственного средства, в случаях, когда не может быть исключена причинно-следственная связь между этой реакцией и применением лекарственного средства. 3.35 Нежелательное побочное явление: Любое неблагоприятное изменение в состоянии здоровья испытуемого, независимо от причинно-следственной связи с применением исследуемого лекарственного средства. 3.36 Совет по мониторингу исследования/испытания: Независимый орган, который может быть образован по инициативе спонсора для периодического рассмотрения хода клинического исследования/испытания, данных по безопасности и/или основных параметров эффективности, а также для выработки рекомендаций спонсору о целесообразности продолжения, прекращения исследования/испытания или внесения в него изменений. 3.37 Незаинтересованный свидетель: Физическое лицо, непричастное к проведению клинического исследования/испытания, на которое не могут оказать давление участники клинического исследования/испытания и которое в случае, если испытуемый или его законный представитель не умеет или не может читать, присутствует во время получения информированного согласия, зачитывает текст информированного согласия и любые другие предоставляемые испытуемому письменные материалы. 3.38 Непредвиденная побочная реакция: Реакция организма испытуемого, которая не соответствует имеющейся информации о побочных реакциях исследуемого лекарственного средства. 3.39 Обеспечение качества: Совокупность систематических и планомерных действий, обеспечивающих соответствие проведения исследования/испытания, сбора, регистрации и предоставления данных настоящему стандарту и нормативным правовым актам Республики Казахстан. 3.40 Основные документы: Документы, в совокупности или по отдельности позволяющие оценить ход клинического исследования/испытания и качество полученных данных. 3.41 Отчет монитора: Письменный документ монитора спонсору после каждого посещения исследовательского центра (клинической базы) и/или контакта с исследователями в соответствии со стандартными операционными процедурами (СОП) спонсора. 3.42 Отчет об аудите: Письменное заключение аудитора о результатах аудита, предоставляемое спонсору. 3.43 Отчет о клиническом исследовании/испытании: Предоставленные в письменной форме результаты клинического исследования/испытания и их анализ, в соответствии с требованиями настоящего стандарта. 3.44 Первичная документация: Исходные документы, данные и записи (истории болезни, амбулаторные карты, лабораторные записи, заметки, дневники исследуемых, вопросники, журналы выдачи медикаментов, записи автоматических устройств, микрофиши, фотонегативы, микропленки или магнитные носители, рентгеновские снимки, любые записи, относящиеся к пациенту, в том числе хранящиеся в аптеке, лабораториях и отделениях инструментальной диагностики и др.), используемые в клиническом исследовании/испытании. 3.45 Первичные данные: Информация, содержащаяся в первичной документации, описывающая результаты клинических наблюдений, обследований и другой деятельности, позволяющая воссоздать картину клинического исследования/испытания и оценить ее. 3.46 Побочное действие: Нежелательное действие лекарственного средства, обусловленное его фармакологическими свойствами, возникающее при применении в рекомендуемых дозах. 3.47 Побочная реакция: Неблагоприятные или непредвиденные реакции на исследуемое лекарственное средство, связанные с введением его в любой дозе. 3.48 Побочное явление: Неблагоприятное проявление у испытуемого, независимо от наличия причинно-следственной связи с применением исследуемого лекарственного средства. 3.49 Поправка к протоколу: Внесение изменений в протокол клинического исследования/испытания. 3.50 Препарат сравнения: Зарегистрированное лекарственное средство или плацебо, используемые в качестве контроля в клиническом исследовании/испытании. 3.51 Промежуточный отчет о клиническом исследовании/испытании: Документ о промежуточных результатах и оценка клинического исследования/испытания, основанная на анализе данных. 3.52 Протокол: Документ, содержащий основные задачи, методологию, процедуры, статистические аспекты, организацию клинического исследования/испытания, ранее полученные данные относительно исследуемого лекарственного средства, обоснования исследования/испытания и его последующие поправки. 3.53 Рандомизация: Процесс отнесения участников исследования/испытания к основным и контрольным группам случайным образом, позволяющий свести к минимуму систематическую ошибку. 3.54 Резюме исследователя: Документ, подтверждающий образование, профессиональную подготовку и опыт исследователя. 3.55 Серьезная побочная реакция/явление: Неблагоприятное клиническое проявление, независимо от дозы лекарственного средства, представляющее угрозу для жизни испытуемого, приводящее к смерти, стойкой или выраженной нетрудоспособности/инвалидности, госпитализации/продления срока госпитализации, врожденным аномалиям/порокам развития. 3.56 Серьезное побочное действие: Клинически доказанная и документально оформленная серьезная побочная реакция/явление. 3.57 Слепой метод: Неосведомленность одной или нескольких участников клинического исследования/испытания о назначенном испытуемому лечении. Слепой метод дифференцируется на простой слепой метод и двойной слепой метод. 3.58 Простой слепой метод: Неосведомленность испытуемого о назначенном ему лечении. 3.59 Двойной слепой метод: Неосведомленность испытуемого, исследователей, мониторов и лиц, выполняющих статистическую обработку данных о назначенном испытуемому лечении. 3.60 Cоисследователь: Член исследовательского коллектива (интерн, ординатор, научный сотрудник), имеющий соответствующую подготовку, назначенный исследователем и осуществляющий под его контролем определенные этапы клинического исследования/испытания. 3.61 Спонсор: Физическое или юридическое лицо, являющееся инициатором клинического исследования/испытания и несущее ответственность за его организацию и/или финансирование. 3.62 Cпонсор-исследователь: Физическое лицо, которое самостоятельно или совместно с другими лицами инициирует и проводит клиническое исследование/испытание. Обязанности спонсораисследователя включают в себя обязанности как спонсора, так и исследователя. 3.63 Стандартная операционная процедура; СОП: Инструкция, содержащая описание четкой последовательности по выполнению функций или работ. 3.64 Уязвимые испытуемые: Испытуемые, страдающие неизлечимыми заболеваниями, находящиеся в неотложном состоянии, проживающие в домах по уходу, нищие, безработные, представители национальных меньшинств, бездомные, кочевники, беженцы, несовершеннолетние и лица, находящиеся под опекой или попечительством, а также лица, неспособные дать согласие. 4 Принципы надлежащей клинической практики Клиническое исследование/испытание должно проводиться в соответствии с этическими принципами, заложенными Хельсинкской декларацией Всемирной Медицинской Ассоциации [1], отраженными в настоящем стандарте и законодательстве Республики Казахстан. 4.1 Проведению клинического исследования/испытания предшествует тщательная оценка возможного риска в сравнении с ожидаемой пользой для испытуемого и общества. 4.2 Права, безопасность и благополучие испытуемого имеют первостепенное значение и должны превалировать над интересами науки и общества. 4.3 Информация (доклиническая и клиническая) о исследуемом лекарственном средстве должна быть достаточной для обоснования предполагаемого клинического исследования/испытания. 4.4 Клиническое исследование/испытание должно быть научно обосновано, четко и подробно описано в протоколе исследования/испытания. 4.5 Клиническое исследование/испытание должно проводиться в соответствии с протоколом, одобренным комиссией по вопросам этики. 4.6 Ответственность за состояние здоровья испытуемого и оказываемую ему медицинскую помощь возлагается на исследователя. 4.7 Исследователь должен иметь соответствующие образование, профессиональную подготовку, квалификацию и опыт для проведения клинического исследования/испытания. 4.8 Добровольное информированное согласие должно быть получено у каждого испытуемого до включения его в исследования/испытание. 4.9 Вся полученная в клиническом исследования/испытании информация документируется и хранится соответствующим образом. 4.10 Конфиденциальность записей, позволяющих идентифицировать испытуемых, обеспечивается с соблюдением права на частную жизнь. Защита конфиденциальности гарантируется в соответствии с законодательством Республики Казахстан. 4.11 Производство и хранение исследуемого лекарственного средства, обращение с ним необходимо осуществлять в соответствии с правилами надлежащей производственной практики (GMP). Исследуемые лекарственные средства должны применяться в соответствии с утвержденным протоколом. 4.13 Для обеспечения качества каждого исследования/испытания должны быть внедрены соответствующие системы качества и операционные процедуры. 5 Комиссия по вопросам этики 5.1 Комиссия по вопросам этики (далее - Комиссия) создается на клинической базе. Комиссия осуществляет свою деятельность в соответствии с Положением о комиссии, стандартными операционными процедурами, соблюдает требования настоящего стандарта и законодательства Республики Казахстан. Состав комиссии и Положение о комиссии утверждаются первым руководителем клинической базы. 5.2 Комиссия по вопросам этики защищает права, безопасность и здоровье испытуемых, при этом особое внимание уделяется уязвимым испытуемым. 5.3 В Комиссию представляются следующие документы: а) протокол исследования/испытания и последующие поправки к нему; б) письменную форму информированного согласия испытуемого и последующие его редакции; в) документированное подтверждение материалов привлечения испытуемых к участию в исследования/испытании; г) материалы, предоставляемые испытуемому; д) брошюру исследователя; е) известную информацию, касающуюся безопасности исследуемого лекарственного средства; ж) информацию о методах и порядках выплат, компенсаций испытуемым; з) текущую версию научной биографии исследователя и/или другие материалы, подтверждающие его квалификацию; и) другие документы, необходимые комиссии по вопросам этики. 5.4 Комиссия рассматривает вопрос о проведении клинического исследования/испытания, его сроках и документально оформляет свое решение относительно рассмотренных документов при вынесении следующих решений в виде: а) одобрения и/или рекомендаций; б) требования о внесении изменений, необходимых для получения одобрения; в) отказа; г) отмены/приостановления данных ранее решений. 5.5 Комиссия оценивает соответствие квалификации исследователя на основании его научной биографии и/или другой соответствующей документации. 5.6 Комиссия рассматривает каждое текущее исследование/испытание с периодичностью, адекватной риску для испытуемого, но не реже одного раза в год. 5.7 Комиссия вправе потребовать, чтобы испытуемым помимо информации, указанной в 7.8.1, были предоставлены дополнительные сведения об исследовании/испытании, если эта информация позволит существенно повысить степень защиты прав, безопасности и/или благополучия испытуемых. 5.8 Комиссия, в случаях, когда согласие на участие испытуемого в исследовании/испытании, дает его законный представитель, требует, чтобы предоставленный протокол и/или другая документация соответствовали этическим нормам и законодательству Республики Казахстан требованиям для таких исследований/испытаний. 5.9 В случаях, когда протокол предусматривает невозможность получения согласия испытуемого или его законного представителя на участие в исследовании/испытании до момента включения испытуемого в исследование/испытание, комиссия по вопросам этики требует соответствия, предоставленного протокола и/или другой документации этическим нормам и требованиям законодательства Республики Казахстан для такого типа исследований/испытаний (например, при неотложных состояниях). 5.10 Комиссия рассматривает размер и порядок выплат испытуемому. Размер выплат испытуемому определяется до начала исследования/испытания и не зависит от того, завершил испытуемый исследование/испытание полностью или нет. 5.11 Комиссия требует, чтобы информация, касающаяся выплат испытуемому, включая методы, суммы и график выплат, была отражена в письменной форме информированного согласия. 5.12 Состав комиссии формируется из специалистов, обладающих необходимым опытом и квалификацией, представителей науки, общественных организаций, независящих от клинической базы и спонсора, для оценки научных, медицинских и этических аспектов предлагаемого исследования/испытания. В состав комиссии входит не менее пяти человек. 5.13 Комиссия ведет список своих членов с указанием их квалификации. 5.14 Комиссия документирует свою работу, ведет протоколы заседаний, соблюдает требования настоящего стандарта и законодательства Республики Казахстан. 5.15 Комиссия принимает решения на заседаниях при наличии кворума, установленного ее стандартными операционными процедурами. 5.16 Право голосовать имеют только те члены Комиссии, которые участвуют в рассмотрении документации и обсуждении. 5.17 Исследователь предоставляет информацию по любым аспектам исследования/испытания, но не участвует в прениях или в голосовании Комиссии. 5.18 Комиссия вправе обращаться за помощью к другим независимым экспертам, не являющихся ее членами. 5.19 В случаях возникновения спорных вопросов, комиссия обязана обращаться в Республиканскую комиссию по вопросам этики. 5.20 Комиссия разрабатывает, документально оформляет: а) Положение о комиссии; б) состав Комиссии (фамилии и квалификацию членов) и учредившую её клиническую базу; в) план проведения заседаний, оповещения членов Комиссии о предстоящих заседаниях, а также организацию заседаний; г) порядок первичного и последующего рассмотрения документации по исследованию/испытанию; д) периодичность последующего рассмотрения документации по исследованию/испытанию; е) порядок ускоренного рассмотрения и вынесения решения относительно незначительных изменений в ходе исследований/испытаний, ранее рассмотренных комиссией; ж) недопустимость включения испытуемого в исследование/испытание до того, как комиссия выдаст письменное заключение и/или рекомендации на проведение исследования/испытания; з) недопустимость отклонений от протокола или его изменений без предварительного заключения и/или рекомендации, либо соответствующих поправок комиссии, кроме тех случаев, когда необходимо устранить непосредственную угрозу испытуемому или когда изменения касаются только административных или материально-технических аспектов исследования/испытания (например, замена монитора, изменение номера телефона и др.); и) обязанность исследователя незамедлительно сообщать комиссии: 1) об отклонениях от протокола или изменениях протокола, произведенных для устранения непосредственной угрозы испытуемому; 2) об изменениях, увеличивающих риск для испытуемых и/или существенно влияющих на проведение исследования/испытания; 3) обо всех непредвиденных серьезных побочных реакциях на лекарственный препарат; 4) о новых данных, которые могут свидетельствовать о возрастании риска для испытуемого или неблагоприятно повлиять на ход исследования/испытания; к) обязанность комиссии в письменном виде сообщать исследователю: 1) о своих заключениях и обоснованиях относительно исследования/испытания; 2) о порядке обжалования его заключений и/или рекомендаций. 5.21 Комиссия хранит все документы, включая свои стандартные операционные процедуры, списки членов с указанием рода деятельности и места работы, представленные на рассмотрение документы, протоколы заседаний и переписку в течение трех лет после завершения исследования/испытания и предоставляет их по запросу уполномоченных органов, Республиканской комиссии по вопросам этики, исследователя, спонсора. 6 Республиканская комиссия по вопросам этики 6.1 Республиканская комиссия по вопросам этики (далее - Республиканская комиссия) создается при уполномоченном органе. Республиканская комиссия осуществляет свою деятельность в соответствии с Положением о Республиканской комиссии, стандартными операционными процедурами, соблюдает требования настоящего стандарта и законодательства Республики Казахстан. Состав Республиканской комиссии и Положение о Республиканской комиссии утверждается руководителем уполномоченного органа. 6.2 Республиканская комиссия по вопросам этики защищает права, безопасность и здоровье испытуемых, при этом особое внимание уделяет уязвимым испытуемым. 6.3 Республиканская комиссия рассматривает спорные вопросы, возникшие до начала, в ходе проведения или после завершения клинического исследования/испытания и документально оформляет свое решение относительно рассмотренных документов. 6.4 Республиканская комиссия вправе потребовать от комиссии по вопросам этики, любую информацию относительно клинического исследования/испытания, дополнительные сведения об исследовании/испытании, если, по мнению национальной комиссии, эта информация позволит существенно повысить степень защиты прав, безопасности и/или благополучия испытуемых. 6.5 Состав Республиканской комиссии формируется из ведущих специалистов уполномоченного органа, представителей науки, общественных организаций для оценки научных, медицинских и этических аспектов исследования/испытания и утверждается. 6.6 Республиканская комиссия ведет список своих членов с указанием их квалификации. 6.7 Республиканская комиссия осуществляет свою деятельность в соответствии со стандартными операционными процедурами, документирует свою работу, ведет протоколы заседаний, соблюдает требования настоящего стандарта и законодательства Республики Казахстан. 6.8 Республиканская комиссия принимает решения на заседаниях при наличии кворума, установленного его письменными процедурами. 6.9 Право голосовать имеют только те члены Республиканской комиссии, которые участвуют в рассмотрении документации и обсуждении. 6.10 Республиканская комиссия вправе обращаться за помощью к другим независимым экспертам, не являющимися его членами. 6.11 Республиканская комиссия разрабатывает, документально оформляет: а) состав Республиканской комиссии (фамилии и квалификацию членов) и учредивший его орган; б) порядок назначения заседаний, оповещения его членов о предстоящих заседаниях, а также организацию заседаний; в) порядок рассмотрения документации по возникшим спорным вопросам относительно исследования/испытания; г) недопустимость включения испытуемого в исследование/испытание до того, как Республиканская комиссия по вопросам этики выдаст письменное заключение и/или рекомендации по возникшим спорным вопросам на проведение исследования/испытания; д) обязанность Республиканской комиссии по вопросам этики в письменном виде сообщать уполномоченным органам, комиссиям по вопросам этики, исследователю, спонсору: 1) о своих заключениях и обоснованиях касающихся исследования/испытания; 2) о порядке обжалования его заключений и/или рекомендаций. 6.12 Республиканская комиссия по вопросам этики хранит все документы, включая свои процедуры, списки членов с указанием рода деятельности и места работы, представленные на рассмотрение документы, протоколы заседаний и переписку в течение трех лет, после завершения рассмотрения спорных вопросов и предоставляет их по запросу уполномоченных органов, комиссий по вопросам этики, исследователя, спонсора. 7 Исследователь 7.1 Квалификация и обязательства исследователя 7.1.1 Исследователь должен иметь образование, профессиональную подготовку и опыт, позволяющие ему принять на себя ответственность за надлежащее проведение клинического исследования/испытания. Квалификация исследователя должна быть подтверждена его научной биографией (curriculum vitae) и/или другими документами, которые могут быть запрошены спонсором, комиссией и/или национальной комиссией по вопросам этики и/или уполномоченным органом. 7.1.2 Исследователь должен детально знать применение исследуемого лекарственного средства в соответствии с протоколом, текущей редакцией брошюры исследователя, информацией о лекарственном средстве и другими источниками, предоставляемыми спонсором. 7.1.3 Исследователь должен знать и соблюдать правила настоящего стандарта и требования законодательства Республики Казахстан, относительно проведения клинических испытаний. 7.1.4 Исследователь не препятствует мониторингу и аудиту со стороны спонсора, а также инспекциям уполномоченного органа. 7.1.5 Исследователь ведет список лиц, обладающих необходимой квалификацией, которые по его поручению осуществляют определенную деятельность в рамках исследования/испытания. 7.1.6 Исследователь обязан продемонстрировать (например, на основании ретроспективных данных) возможность набора в течение оговоренного периода требуемого количества подходящих испытуемых. 7.1.7 Исследователь должен иметь достаточное количество времени, чтобы надлежащим образом провести и завершить исследование/испытание в течение оговоренного периода. 7.1.8 Исследователь должен располагать достаточным количеством квалифицированных сотрудников и материальных ресурсов (помещения, оборудование) в период исследования/испытания, для того чтобы провести это исследование/испытание надлежащим и безопасным образом. 7.1.9 Исследователь несет ответственность, чтобы все занятые в клиническом исследовании/испытании сотрудники были хорошо знакомы с протоколом и исследуемым лекарственным средством, а также со своими функциями и обязанностями. 7.2 Оказание медицинской помощи испытуемым 7.2.1 Исследователь или соисследователь, являющийся врачом, несет ответственность за все принимаемые в рамках исследования/испытания решения медицинского характера. 7.2.2 Исследователь обеспечивает оказание испытуемому необходимой медицинской помощи в случае любых связанных с исследованием/испытанием нежелательных явлений, включая клинически значимые изменения лабораторных показателей во время и по завершении участия испытуемого в исследовании/испытании. Исследователь обязан информировать испытуемого о ставших известными исследователю интеркуррентных заболеваниях, требующих медицинской помощи. 7.2.3 Исследователь сообщает лечащему врачу, если таковой имеется, об участии испытуемого в исследовании/испытании при условии, что испытуемый не возражает против информирования лечащего врача. 7.2.4 Исследователь должен попытаться установить причины побудившие испытуемого досрочно прекратить участие в исследовании/испытании, проявляя при этом полное уважение к правам испытуемого (испытуемый имеет право не сообщать о причинах). 7.3 Контакты с комиссией по вопросам этики 7.3.1 Перед началом исследования/испытания, исследователь обязан получить письменное, датированное одобрение комиссии по вопросам этики протокола исследования/испытания, письменной формы информированного согласия и ее последующих редакций, мер, направленных на привлечение испытуемых к участию в исследовании/испытании (например, рекламных объявлений), и любых других письменных материалов, которые необходимо предоставить испытуемым. 7.3.2 Вместе с другими документами исследователь предоставляет комиссии последнюю редакцию брошюры исследователя. Если брошюру дополняют в ходе исследования/испытания, исследователь обязан предоставить комиссии новую редакцию брошюры исследователя. 7.3.3 В течение исследования/испытания исследователь предоставляет комиссии по вопросам этики все подлежащие рассмотрению документы. 7.4 Соблюдение протокола 7.4.1 Исследователь проводит исследование/испытание в соответствии с протоколом, согласованным со спонсором, одобренным комиссией по вопросам этики и утвержденным уполномоченным органом. Документом, подтверждающим договоренность исследователя и спонсора, служит подписанный протокол и договор. 7.4.2 Исследователь не вправе отклоняться от протокола или вносить в него изменения без согласия спонсора и документально оформленного одобрения поправки к протоколу комиссией по вопросам этики, кроме тех случаев, когда изменения касаются только административных или технических аспектов исследования/испытания (например, замена монитора, изменение номера телефона). 7.4.3 Исследователь документально оформляет любые отклонения от утвержденного протокола. 7.4.4 Исследователь, при возникновении непосредственной угрозы испытуемым, вправе отклоняться от протокола или вносить в него изменения, для их немедленного устранения без предварительного одобрения комиссией по вопросам этики. Описание отклонений или изменений с указанием их причин и предлагаемые поправки к протоколу в кратчайшие сроки направляются исследователем в: а) комиссию по вопросам этики для рассмотрения и принятия решения; б) спонсору для согласования с ним; в) уполномоченному органу. 7.5 Исследуемое лекарственное средство 7.5.1 Ответственность за учет исследуемых лекарственных средств на клинической базе несет исследователь. 7.5.2 Исследователь вправе передать некоторые или все обязанности по учету исследуемых лекарственных средств на клинической базе аптечному работнику или иному лицу, подконтрольному исследователю. 7.5.3 Исследователь и/или аптечный работник или иное лицо, уполномоченное исследователем, относительно исследуемого лекарственного средства ведут учет: а) поставок исследуемых лекарственных средств в исследовательский центр; б) фактического количества исследуемых лекарственных средств; в) использования исследуемых лекарственных средств испытуемыми; г) возврата исследуемых лекарственных средств спонсору; д) иного распоряжения неиспользованными лекарственными средствами. 7.5.4 Записи по учету включают в себя даты, количество, номера партий/серий, срок годности, уникальные коды исследуемых лекарственных средств. Исследователь ведет записи, подтверждающие получение испытуемыми исследуемых лекарственных средств в дозах, предусмотренных протоколом, в количествах, согласующихся с общим количеством исследуемых лекарственных средств, полученным от спонсора. 7.5.5 Исследуемые лекарственные средства хранятся в соответствии с инструкциями спонсора и требованиями нормативных правовых актов [7]. 7.5.6 Исследователь обеспечивает использование исследуемых лекарственных средств в соответствии с утвержденным протоколом. 7.5.7 Исследователь или уполномоченное исследователем лицо обязаны объяснить каждому испытуемому правила применения исследуемых лекарственных средств и через определенные интервалы времени (в зависимости от исследования/испытания) контролировать соблюдение этих инструкций каждым испытуемым. 7.6 Рандомизация и раскрытие кода Исследователь соблюдает предусмотренную в исследования/испытании методику рандомизации, если таковая имеется, и обеспечивает раскрытие кода только в соответствии с протоколом. Если исследования/испытание проводится слепым методом, исследователь незамедлительно документально оформляет и объясняет спонсору любое преждевременное раскрытие кода исследуемых лекарственных средств (например, случайное раскрытие кода, раскрытие кода в связи с серьезным нежелательным явлением и др.). 7.7 Информированное согласие испытуемых 7.7.1 При получении и документальном оформлении информированного согласия исследователь соблюдает требования законодательства Республики Казахстан, настоящего стандарта и этических принципов. До начала исследования/испытания исследователь обязан получить одобрение комиссии по вопросам этики относительно письменной формы информированного согласия и любых других письменных материалов, предоставляемых испытуемым. 7.7.2 Письменная форма информиро¬ван¬ного согласия и любые другие письменные материалы, предоставляемые испытуемому, дополняются и/или исправляются по мере появления новой важной информации, которая может оказаться существенной для его согласия. Любая дополненная и/или исправленная письменная форма информированного согласия и любые другие дополнен¬ные и/или исправленные письменные материалы, предоставляемые испытуемым, передаются для одобрения в комиссию по вопросам этики до их использования в исследовании/испытании. Испытуемому или его законному представителю своевременно предоставляется для ознакомления новая информация, способная повлиять на желание испытуемого продолжать участие в исследовании/испытании. Факт ознакомления их с этой информацией оформляется документально. 7.7.3 Ни исследователь, ни другие занятые в исследовании/испытании лица не вправе принуждать испытуемого или использовать иные некорректные методы воздействия с целью склонить его к участию либо продолжению участия в исследовании/испытании. 7.7.4 Ни устная, ни письменная информация, касающаяся исследования/испытания, включая письменную форму информированного согласия, не должны содержать формулировок, прямо или косвенно склоняющих испытуемого или его законного представителя отказаться от законных прав, а также формулировок, прямо или косвенно освобождающих исследователя, организацию, спонсора или их представителей от ответственности за халатность. 7.7.5 Исследователь или уполномоченное им лицо информируют испытуемого или его законного представителя обо всех аспектах исследования/испытания, включая информационные материалы, одобренные комиссией по вопросам этики. 7.7.6 В устную и письменную информацию об исследовании/испытании, включая письменную форму информированного согласия, включается минимальное количество специальных терминов. Они должны быть понятны испытуемому или его законному представителю и незаинтересованному свидетелю. 7.7.7 Перед получением информированного согласия исследователь или назначенное им лицо предоставляют испытуемому или его законному представителю достаточное количество времени и возможность для получения более подробной информации об исследовании/испытании и принятия решения об участии или отказе от участия в нем. Испытуемый или его законный представитель должны получить исчерпывающие ответы на все вопросы об исследовании/испытании. 7.7.8 До начала участия в исследовании/испытании испытуемый или его законный представитель, а также лицо, проводившее разъяснительную беседу, подписывают и собственноручно датируют письменную форму информированного согласия. 7.7.9 Если испытуемый или его законный представитель не способны читать, то при проведении разъяснительной беседы необходимо обязательное присутствие незаинтересованного свидетеля. После того как испытуемому или его законному представителю разъяснили письменную форму информированного согласия и другие письменные материалы и испытуемый или его законный представитель дали устное согласие на участие испытуемого в исследовании/испытании и, если способны, подписали и собственноручно датировали письменную форму информированного согласия, свидетель подписывает их и датирует. Подписывая форму согласия, свидетель подтверждает, что информация, содержащаяся в форме согласия и всех других письменных материалах, была точно разъяснена и понята испытуемому или его законному представителю, и что согласие на участие в исследовании/испытании дано испытуемым или его законным представителем добровольно. 7.7.10 Как в ходе разъяснительной беседы, так и в письменной форме информированного согласия и других письменных материалах, предоставляемых испытуемым, разъясняется следующее: а) исследование/испытание носит экспериментальный характер; б) цель исследования/испытания; в) варианты лечения в процессе исследования/испытания и вероятность случайного распределения в одну из групп лечения; г) процедуры исследования/испытания, включая все инвазивные процедуры; д) обязанности испытуемого; е) аспекты исследования/испытания, которые носят экспериментальный характер; ж) ожидаемый риск или неудобства для испытуемого, а также, в соответствующих случаях, для эмбриона, плода или грудного ребенка; з) ожидаемые выгода и/или польза. Если пользы с медицинской точки зрения не предполагается, то испытуемый должен быть поставлен об этом в известность; и) иные, помимо предусмотренных в исследовании/испытании, методы лечения, которые могут быть назначены испытуемому, а также их потенциальные риск и/или польза; к) компенсация и/или лечение, доступные испытуемому в случае причинения вреда его здоровью в результате участия в исследовании/испытании; л) планируемые выплаты испытуемому за его участие в исследовании/испытании, если таковые предусмотрены; м) планируемые расходы испытуемого, если таковые ожидаются, связанные с его участием в исследовании/испытании; н) участие испытуемого в исследовании/испытании является добровольным, и он вправе отказаться от участия или выйти из исследования/испытания в любой момент без каких-либо санкций для себя или потери предусмотренных выгод; о) мониторы, аудиторы, комиссия по вопросам этики и уполномоченный орган, в пределах своей компетенции, имеют прямой доступ к оригинальным медицинским записям испытуемого для проверки процедур и/или данных клинического исследования/испытания, не нарушая при этом конфиденциальности данных испытуемого. Испытуемый или его законный представитель, подписывая письменную форму информированного согласия, дают разрешение на такой доступ; п) записи, идентифицирующие испытуемого, сохраняются в тайне и могут быть раскрыты только в той мере, в какой это допускается законодательством. При публикации результатов исследования/испытания конфиденциальность данных испытуемого строго сохраняется; р) испытуемый или его законный представитель должны быть своевременно ознакомлены с новой информацией, способной повлиять на желание испытуемого продолжать участие в исследовании/испытании; с) адреса и телефоны лиц, к которым можно обратиться для получения дополнительной информации о ходе исследования/испытания, правах испытуемых и в случае причинения вреда здоровью испытуемого; т) возможные обстоятельства и/или причины, по которым участие испытуемого в исследовании/испытании может быть прекращено; у) предполагаемая длительность участия испытуемого в исследовании/испытании; ф) количество испытуемых, которое предполагается включить в исследование/испытание. 7.7.11 Перед включением в исследование/испытание испытуемому или его законному представителю предоставляются подписанный и датированный экземпляр формы информированного согласия, и другие информационные материалы. Во время участия испытуемого в исследовании/испытании он или его законный представитель должны получить подписанные и датированные экземпляры всех последующих редакций формы информированного согласия и копии всех поправок к другим письменным материалам, предоставляемым испытуемым. 7.7.12 Если в клиническом исследовании/испытании участвуют испытуемые на основании согласия их законных представителей (например, несовершеннолетние, пациенты с выраженным слабоумием), то испытуемого информируют об исследовании/испытании в соответствии с его способностью понять эту информацию, и, если испытуемый в состоянии, он должен подписать и собственноручно датировать письменную форму информированного согласия. 7.7.13 Кроме случаев, описанных в 6.7.14, в нелечебное исследовании/испытание, могут быть включены испытуемые, которые лично дают свое согласие, подписывают и датируют письменную форму информированного согласия. 7.7.14 В нелечебное исследовании/испытание могут быть включены испытуемые с согласия их законных представителей при соблюдении следующих положений: а) цели исследования/испытания требуют включения испытуемых, состояние которых не позволяет им дать свое согласие лично; б) ожидаемый риск для испытуемых незначителен; в) вредное воздействие на здоровье испытуемых сведено к минимуму и незначительно; г) исследование/испытание не запрещено законодательством Республики Казахстан; д) для включения таких испытуемых в исследование/испытание, должно быть получено специальное одобрение комиссии по вопросам этики. Подобные исследования/испытания (за исключением обоснованных случаев) проводятся с участием пациентов, имеющих заболевание, для лечения которого предназначен исследуемый продукт. Испытуемые в таких исследованиях/испытаниях находятся под особо тщательным наблюдением, и их участие прекращается, если есть основания полагать, что они испытывают чрезмерный дискомфорт. 7.7.15 При неотложных состояниях, когда до включения в исследование/испытание невозможно получить согласие самого испытуемого, оно запрашивается у его законного представителя, если таковой присутствует. Если предварительное согласие самого испытуемого невозможно и отсутствует его законный представитель, то для включения испытуемого в исследование/испытание предпринимаются меры предусмотренные протоколом, одобренные комиссией по вопросам этики, направленные на защиту прав, безопасности и благополучия испытуемого, а также обеспечивающие соответствия требованиям законодательства Республики Казахстан. Испытуемый или его законный представитель должны быть в кратчайшие сроки поставлены в известность об исследовании/испытании, и у них запрашивается согласие на продолжение участия в исследовании/испытании. 7.8 Записи и отчеты 7.8.1 Исследователь обеспечивает правильность, полноту, разборчивость и своевременность предоставления спонсору данных, содержащихся в индивидуальной регистрационной форме и во всех требуемых отчетах. 7.8.2 Данные в индивидуальной регистрационной форме должны соответствовать первичной документации, из которой они перенесены, имеющиеся расхождения должны быть объяснены. 7.8.3 Любые изменения или исправления в индивидуальной регистрационной форме подписываются, датируются, объясняются (при необходимости). В них не допускается скрытие первоначальной записи (т.е. должен быть сохранен «документальный след»). Это относится как к письменным, так и к электронным изменениям или исправлениям. Спонсор предоставляет исследователям и/или их уполномоченным представителям инструкцию о порядке оформления таких исправлений. Исследователь должен получить от спонсора письменные процедуры, предусматривающие, изменения или исправления в индивидуальной регистрационной форме, вносимые его уполномоченными представителями, документально оформляются, являются необходимыми и одобряются исследователем. Исследователь хранит записи об изменениях и исправлениях. 7.8.4 Исследователь ведет документацию по исследованию/испытанию согласно разделу 10 требованиям настоящего стандарта. Исследователь обязан принимать меры, предотвращающие случайное или преждевременное уничтожение этих документов. 7.8.5 Основные документы хранятся не менее двух лет после утверждения последней заявки на регистрацию препарата в Республике Казахстан и до тех пор, пока ни одна из заявок не будет находиться на рассмотрении, и не будет планироваться новых заявок, или не менее двух лет после официального прекращения клинической разработки исследуемого продукта. Эти документы храниться более длительный срок в случае, если это предусмотрено требованиями законодательства Республики Казахстан или договором со спонсором. Ответственностью спонсора является информирование исследователя об истечении срока хранения документации. 7.8.6 Финансовые аспекты исследования/испытания отражаются в договоре между спонсором и исследователем. 7.8.7 По запросу монитора, аудитора, комиссии по вопросам этики или уполномоченного органа исследователь обеспечивает прямой доступ ко всем записям, относящимся к исследованию/испытанию. 7.9 Отчеты о ходе исследования/испытания 7.9.1 Исследователь представляет комиссии по вопросам этики краткие письменные отчеты о ходе исследования/испытания ежегодно или чаще, если этого требует комиссия по вопросам этики. 7.9.2 Исследователь незамедлительно предоставляет письменные отчеты спонсору, комиссии по вопросам этики и, при необходимости администрации клинической базы о любых изменениях, существенно влияющих на проведение исследования/испытания и/или увеличивающих риск для испытуемых. 7.10 Отчетность по безопасности 7.10.1 Обо всех серьезных нежелательных явлениях в течение 24 часов сообщается спонсору, за исключением тех серьезных нежелательных явлениях, которые в протоколе или в другом документе (например, в брошюре исследователя) определены, как не требующие немедленного сообщения. После первичного немедленного сообщения в кратчайшие сроки составляется подробный письменный отчет. Первичный и последующие отчеты идентифицируют испытуемых исследования/испытания по присвоенным им уникальным кодам, а не по их именам, персональным идентификационным номерам и/или адресам. Исследователь соблюдает требования нормативных правовых актов [8], регламентирующие предоставление отчетов о непредвиденных серьезных нежелательных реакциях уполномоченному органу и комиссии по вопросам этики. 7.10.2 Обо всех нежелательных явлениях и/или отклонениях лабораторных показателей от нормы, определенных протоколом как критические для оценки безопасности, обязательно сообщается спонсору в соответствии с требованиями к отчетам и в сроки, определенные спонсором в протоколе. 7.10.3 При сообщениях о смерти испытуемого исследователь обязан по запросу спонсора и комиссии по вопросам этики предоставить любую дополнительную информацию (например, протокол вскрытия и посмертный эпикриз). 7.11 Досрочное прекращение или приостановление исследования/испытания 7.11.1 Если по какой-либо причине исследование/испытание досрочно прекращено или приостановлено, исследователь незамедлительно информирует испытуемых, обеспечивает им соответствующее лечение и наблюдение и информирует уполномоченный орган. Кроме того: а) в случаях, когда исследователь досрочно прекращает или приостанавливает исследование/испытание без предварительного согласия спонсора, он обязан сообщить об этом администрации клинической базы и незамедлительно проинформировать спонсора и комиссию по вопросам этики, предоставляя им подробное письменное объяснение причины приостановки или прекращения исследования/испытания; б) в случаях, когда спонсор прекращает или приостанавливает исследование/испытание, исследователь незамедлительно сообщает об этом администрации клинической базы и предоставляет в комиссию по вопросам этики подробное письменное объяснение причины приостановки или прекращения исследования/испытания; в) в случаях, когда комиссия по вопросам этики окончательно или временно отзывает одобрение на проведение исследования/испытания, исследователь сообщает об администрации клинической базы и незамедлительно информирует об этом спонсора, предоставляя ему подробное письменное объяснение причины приостановки или прекращения исследования/испытания. 7.11.2 По завершении исследования/испытания, исследователь сообщает об этом администрацию клинической базы, предоставляет отчет об итогах исследования/испытания в комиссию по вопросам этики и в уполномоченный орган (по требованию). 8 Спонсор 8.1 Обеспечение и контроль качества 8.1.1 Спонсор обеспечивает систему мер по качеству исследования/испытания и его контролю, несет ответственность за их соблюдение с целью гарантии проведения исследования/испытания, получения, регистрации и предоставления данных в соответствии с протоколом исследования/испытания, требованиями настоящего стандарта и законодательства Республики Казахстан, руководствуясь стандартными операционными процедурами (СОП). 8.1.2 Спонсор несет ответственность за получение от всех вовлеченных в клиническое исследование/испытание сторон письменного согласия на предоставление прямого доступа во все участвующие в исследовании/испытании клинические базы, ко всем первичным документам и отчетам, с целью проведения их мониторинга и аудита, а также инспекции уполномоченными органами. 8.1.3 Контроль качества осуществляется на всех этапах работы с целью обеспечения достоверности данных исследования/испытания и правильности обработки. 8.1.4 Договоры между спонсором и организацией или любой другой участвующей в исследовании/испытании стороной составляются в письменной форме, как часть протокола или в качестве самостоятельных документов. 8.2 Контрактная исследовательская организация 8.2.1 Спонсор вправе полностью или частично передавать обязанности и функции, связанные с проведением исследования/испытания, контрактной исследовательской организации. Контрактная исследовательская организация осуществляет меры по обеспечению и контролю качества. Ответственность за качество и полноту полученных в ходе исследования/испытания данных несет спонсор. 8.2.2 Передача контрактной исследовательской организации любых связанных с исследованием/испытанием обязанностей и функций оформляется документально. 8.2.3 Все связанные с исследованием/испытанием обязанности и функции, не переданные контрактной исследовательской организации, остаются в компетенции спонсора. 8.2.4 Контрактная исследовательская организация принимает на себя ответственность за проведение исследования/испытания, согласно принятым обязанностям и функциям, оформленным документально. 8.3 Медицинская квалификация 8.3.1 Спонсор назначает персонал, обладающий соответствующей медицинской квалификацией, для решения вопросов медицинского характера, возникающих в ходе исследования/испытания. При необходимости для этой цели могут быть привлечены внешние консультанты. 8.3.2 Спонсор назначает квалифицированных специалистов (врачей, клинических фармакологов, биостатистиков), на всех этапах исследования/испытания - от разработки протокола, индивидуальной регистрационной формы и плана статистического анализа до подготовки промежуточного и итогового отчетов. 8.4 Дизайн исследования/испытания Дизайн исследования/испытания составляется в соответствии с разделом 8 настоящего стандарта. 8.5 Менеджмент исследования/испытания, работа с данными и ведение документации 8.5.1 Спонсор привлекает специалистов, обладающих соответствующей квалификацией для осуществления контроля за проведением исследования/испытания, сбора, верификации и обработки данных, а также подготовки отчетов об исследовании/испытании. 8.5.2 Спонсор при необходимости создает совет по мониторингу исследования/испытания для рассмотрения хода клинического исследования/испытания, данных по безопасности и эффективности и выработки рекомендаций спонсору о целесообразности продолжения, прекращения исследования/испытания или внесения в него изменений. Совет по мониторингу данных ведет письменные записи всех своих заседаний и руководствуется письменными операционными процедурами. 8.5.3 При использовании компьютерных систем для работы с данными исследования/испытания и/или систем удаленного ввода данных спонсор обязан: а) обеспечивать и документально оформлять соответствие систем компьютерной обработки данных к полноте, точности и надежности данных, а также стабильности достижения требуемого результата (т.е. валидацию); б) иметь СОП по использованию этих систем; в) обеспечивать работу систем, позволяющих изменять данные и при этом вносимые изменения документируются, а ранее введенные данные не удаляются (документальный след); г) иметь систему защиты, предотвращаю¬щую несанкционированный доступ к данным; д) иметь список лиц, наделенных полномочиями вносить изменения в данные; е) обеспечивать адекватное резервное копирование данных; ж) сохранять маскировку в исследованиях/испытаниях, проводимых слепым методом (т.е. сохранять маскировку при вводе и обработке данных). 8.5.4 В случаях, если в процессе обработки данные подвергаются изменениям, необходимо проводить сравнения исходных данных с обработанными. 8.5.5 Спонсор использует индивидуальный идентификационный код испытуемого, позволяющий идентифицировать все данные по каждому испытуемому. 8.5.6 Спонсор или другие владельцы данных хранят все основные документы по исследованию/испытанию согласно списку «Основные документы клинического исследования/испытания» (раздел 11 настоящего стандарта). 8.5.7 Спонсор хранит все относящиеся к нему основные документы по исследованию/испытанию в соответствии с нормативными требованиями тех стран, в которых препарат зарегистрирован и/или планируется подача заявки на его регистрацию. 8.5.8 В случаях если спонсор прекращает клиническую разработку исследуемого лекарственного средства (по одному или всем показаниям, путям введения, лекарственным формам), он обязан хранить документы по исследованию/испытанию в течение двух лет с момента официального прекращения разработки. 8.5.9 В случаях, если спонсор прекращает клиническую разработку лекарственного средства, он обязан сообщить об этом участникам исследования/испытания и уполномоченному органу. 8.5.10 Любая передача прав собственности на данные о лекарственном средстве должна быть доведена до сведения уполномоченного органа. 8.5.11 Относящиеся к спонсору основные документы хранятся не менее двух лет после утверждения последней заявки на регистрацию препарата в Республике Казахстан. 8.5.12 Спонсор обязан в письменной форме информировать исследователей/организации о необходимости обязательного хранения связанных с исследованием/испытанием записей, а также письменно известить их, как только необходимость в дальнейшем хранении отпадет. 8.6 Выбор исследователя 8.6.1 Спонсор самостоятельно выбирает исследователей и организации, проводящие исследования/испытания. Каждый исследователь должен иметь квалификацию, опыт и возможности для полноценного проведения исследования/испытания. В случаях если в многоцентровых исследованиях/испытаниях необходимо организовать координационный комитет и/или выбрать координаторов из числа исследователей, то это является обязанностью спонсора. 8.6.2 До подписания договора с исследователем/организацией на проведение исследования/испытания спонсор обязан предоставить исследователю /организации протокол и брошюру исследователя в текущей редакции и дать исследователю/организации достаточное время для ознакомления с протоколом и предоставленной информацией. 8.6.3 Спонсор должен получить согласие исследователя/организации: а) проводить исследование/испытание в соответствии с правилами настоящего стандарта и законодательством Республики Казахстан, а также с протоколом, согласованным со спонсором и одобренным комиссией по вопросам этики; б) соблюдать процедуры сбора и представления данных по ходу исследования/испытания; в) на проведение мониторинга, аудита и инспектирование; г) хранить основные документы, связанные с исследованием/испытанием, до тех пор, пока спонсор не сообщит исследователю/организации, что эти документы больше не требуются. Спонсор и исследователь/организация должны подписать протокол или иной документ, подтверждающий это согласие. 8.7 Распределение обязанностей До начала исследования/испытания спонсор устанавливает и распределяет все связанные с исследованием/испытанием обязанности и функции. 8.8 Компенсации испытуемым и исследователем 8.8.1 Спонсор обеспечивает страхование или гарантирует юридическую и финансовую поддержку исследователя/организации в случае предъявления им исков, связанных с исследованием/испытанием, за исключением тех из них, которые возникли в результате халатности и/или неосторожности со стороны исследователя или членов исследовательского коллектива. 8.8.2 Правила и процедуры спонсора обязывают его предусматривать возмещение расходов на лечение испытуемых в случае причинения вреда их здоровью в ходе исследования/испытания, в соответствии с действующими законодательством Республики Казахстан. 8.8.3 Спонсор обеспечивает выплату испытуемым, размер, порядок и способ выплат должны соответствовать требованиям законодательства Республики Казахстан. 8.9 Финансирование Финансовые аспекты исследования/испытания должны быть документально оформлены в виде договора между спонсором и исследователем/организацией. 8.10 Подача заявки в уполномоченный орган 8.10.1 До начала клинического исследования/испытания спонсор обязан подать в уполномоченный орган заявку и необходимые документы для их рассмотрения, принятия и/или получения разрешения для начала исследования/испытания. 8.10.2 Уполномоченный орган после рассмотрения заявки и необходимых документов утверждает протокол клинического исследования/испытания, на основании рекомендаций экспертной организации обеспечивающей безопасность, эффективность и качество лекарственных средств. 8.10.3 Утверждение уполномоченным органом протокола клинического исследования/испытания служит разрешением для начала исследования/испытания. 8.11 Получение одобрения комиссии по вопросам этики 8.11.1 Спонсор получает от исследователя: а) наименование и адрес комиссии по вопросам этики, проводящей экспертизу исследования/испытания; б) справку-подтверждение от комиссии по вопроса этики о том, что она основана и действует согласно требованиям настоящего стандарта и соответствующего законодательства; в) документально оформленное одобрение комиссии по вопроса этики, копию текущей версии протокола, письменной формы информированного согласия и любых других предоставляемых испытуемым письменных материалов, а также описание действий по привлечению испытуемых к участию в исследовании/испытании, документов, относящихся к предусмотренным для испытуемых выплатам и компенсациям, и любых иных документов, которые могли быть затребованы комиссией по вопроса этики. 8.11.2 В случаях если комиссия по вопроса этики одобряет внесение изменений в какие-либо аспекты исследования/испытания (например в протокол, письменную форму информированного согласия или любые иные предоставляемые испытуемым письменные материалы) и/или другие процедуры, исследователь обязан предоставить спонсору копии всех измененных документов и дату полученного одобрения комиссии по вопроса этики. 8.11.3 Спонсор должен получить от исследователя документацию и даты любых повторных одобрений комиссии по вопросам этики, а также заключения об отзыве или приостановке ранее выданного одобрения. 8.12 Информация об исследуемом препарате 8.12.1 При планировании исследований/испытаний спонсор обязан располагать достаточным объемом результатов доклинических/неклинических и/или клинических исследований/испытаний, свидетельствующих о безопасности и эффективности исследуемого препарата с указанием путей введения, доз и длительности применения популяцией. 8.12.2 Спонсор обязан обновлять брошюру исследователя по мере получения новой информации. Спонсор обеспечивает исследователей информацией о наличии у исследуемых препаратов (включая активные препараты сравнения и плацебо) характеристик, соответствующих стадии разработки, производству лекарственных средств в соответствии с правилами GMP, кодировке и маркировке таким образом, чтобы обеспечить «слепоту исследования/испытания». Маркировка должна соответствовать требованиям нормативных правовых актов [4]. 8.12.3 Спонсор указывает температуру, условия и срок хранения для исследуемых препаратов, растворители и процедуры для их разведения и восстановления, а также, если таковые предусмотрены, устройства для введения лекарственного средства. Спонсор информирует об этих требованиях всех участников исследования/испытания. 8.12.4 Упаковка исследуемого лекарственного средства должна защищать его от загрязнения и предотвращать от порчи при транспортировке и хранении. 8.12.5 Для исследований/испытаний, в которых используется слепой метод, система кодирования исследуемого препарата включает в себя механизм быстрой идентификации препарата в случае неотложных состояний и в то же время, не допускающий возможности раскрытия кода. 8.12.6 В случаях изменения лекарственной формы и/или технологии производства исследуемого препарата или препарата сравнения в ходе исследования/испытания, должны быть получены результаты дополнительных исследований/испытаний данной лекарственной формы лекарственного средства (например, стабильности, растворимости, биодоступности), необходимые для оценки того, способны ли данные изменения существенно повлиять на фармакокинетический профиль лекарственного средства. 8.13 Поставка исследуемых лекарственных препаратов 8.13.1 Спонсор отвечает за поставку исследователю исследуемых препаратов. 8.13.2 Спонсор поставляет исследуемый препарат исследователю в соответствии со своими стандартными операционными процедурами, после получения всей требуемой документации. Письменные процедуры спонсора включают в себя инструкции для исследователя/организации по правилам хранения исследуемого препарата и обращения с ним, а также по ведению соответствующей документации. Процедуры содержат описание безопасного получения исследуемого препарата, обращения с ним, его хранение и выдачу, изъятие неиспользованного исследуемого препарата у испытуемых и возврат его спонсору (либо иные способы уничтожения препарата, установленные спонсором и соответствующие нормативным требованиям) [9]. 8.13.3 Спонсор: а) обеспечивает своевременную поставку исследуемого лекарственного средства исследователям; б) документально оформляет процедуру доставки, приема, выдачи, возврата и уничтожения исследуемого лекарственного средства; в) имеет систему возврата исследуемого лекарственного средства и документирования подобного изъятия (например, отзыва бракованного лекарственного средства, возврата лекарственного средства после окончания исследования/испытания или по истечении срока годности); г) имеет систему уничтожения неиспользованных исследуемых лекарственных средств и документирования подобного уничтожения. д) принимает меры по обеспечению стабильности исследуемых лекарственных средств на протяжении всего периода использования; е) имеет достаточное количество исследуемого лекарственного средства, чтобы подтвердить, в случае необходимости, его соответствие спецификациям; ж) ведет учет анализов и характеристик образцов лекарственного средства из партии. В зависимости от стабильности образцы сохраняют либо до окончания анализа данных по исследованию/испытанию, либо в течение срока, определенного требованиями нормативно-технической документацией. 8.14 Доступ к записям 8.14.1 Спонсор предусматривает в протоколе или ином письменном соглашении обязанность исследователя/организации обеспечить прямой доступ к первичным данным/документации для целей мониторинга, аудита, экспертизы национальной комиссии и/или комиссии по вопросам этики , а также инспекции со стороны уполномоченных органов. 8.14.2 Спонсор должен убедиться в том, что каждый испытуемый дал письменное согласие на прямой доступ к его оригинальным медицинским записям для целей мониторинга, аудита, экспертизы национальной комиссии и/или комиссии по вопросам этики, а также инспекции со стороны уполномоченных органов. 8.15 Информация о безопасности 8.15.1 Спонсор отвечает за постоянную оценку безопасности исследуемых препаратов в течение всего клинического исследования/испытания. 8.15.2 Спонсор незамедлительно уведомляет всех участников исследования/испытания и уполномоченный орган о полученных данных, которые могут неблагоприятно отразиться на безопасности испытуемых, повлиять на проведение исследования/испытания или изменить одобрение комиссии по вопросам этики на продолжение исследования/испытания. 8.16 Информация о нежелательных реакциях 8.16.1 Спонсор в короткий срок сообщает всем участникам исследования/испытания, комиссию по вопросам этики и уполномоченному органу обо всех нежелательных реакциях, которые одновременно являются серьезными и непредвиденными. Отчеты о побочных реакциях должны соответствовать требованиям нормативных правовых актов [10] и настоящего стандарта. 8.16.2 Спонсор представляет на рассмотрение уполномоченному органу все новые данные и периодические отчеты по безопасности исследуемого лекарственного средства в соответствии с требованиями нормативных правовых актов [10]. 8.17 Мониторинг 8.17.1 Целью мониторинга клинического исследования/испытания является: а) оценка обеспечения защиты прав и благополучия испытуемых; б) проверка представленных данных на предмет точности, полноты и подтверждения первичной документацией; в) контроль за проведением исследования/испытания в соответствии с утвержденным протоколом и поправками к нему, требованиями настоящего стандарта и нормативных правовых актов [10]. 8.17.2 Выбор мониторов и их квалификация: а) мониторы назначаются спонсором; б) мониторы должны иметь соответствующую подготовку, а также обладать научными и/или клиническими знаниями, необходимыми для проведения надлежащего мониторинга исследования/испытания. Квалификация мониторов подтверждается документально; в) мониторы детально изучают исследуемые препараты, протокол, письменную форму информированного согласия и все другие предоставляемые испытуемым письменные материалы, стандартные операционные процедуры спонсора, требования настоящего стандарта и законодательства Республики Казахстан. 8.17.3 Спонсор обеспечивает надлежащий мониторинг исследований/испытаний. Спонсор определяет необходимые объем и содержание мониторинга исходя из целей, задач, методологии, сложности, маскировки, объема и оцениваемых параметров исследования/испытания. С целью мониторинга организуются посещения клинических баз до, во время и после окончания исследования/испытания. В исключительных случаях спонсор может решить, что мониторинг в клинических базах в сочетании с тренингом и проведением совещаний исследователей, предоставлением им подробного письменного руководства может гарантированно обеспечить надлежащее проведение исследования/испытания в соответствии с настоящим стандартом. Выбор данных для проверки может быть основан на статистических методах. 8.17.4 Монитор, в соответствии с требованиями спонсора, обеспечивает надлежащее проведение и документальное оформление исследования/испытания. С этой целью монитор: а) выступает в качестве связующего звена между спонсором и исследователем; б) проверяет на протяжении всего исследования/испытания квалификацию и возможности исследователя и его персонала, а также помещения для исследований/испытаний, включая лаборатории, оборудование на предмет соответствия их, требованиям для проведения клинических исследований/испытаний качественным и безопасным образом; в) по отношению к исследуемым препаратам монитор проверяет: 1) приемлемость сроков и условий хранения препарата и достаточность его количества до конца исследования/испытания; 1) назначение исследуемого лекарственного средства в соответствии с критериями отбора испытуемых и в дозах, установленных протоколом; 2) предоставление испытуемым инструкции по применению, обращению, хранению и возврату исследуемого лекарственного средства; 3) документацию на получение, применение и возврат исследуемых препаратов на клинической базе; 4) осуществление уничтожения (либо иное распоряжение) неиспользованных исследуемых препаратов в клинической базе в соответствии с требованиями нормативных правовых актов[9] и по согласованию со спонсором; г) следит за соблюдением исследователем утвержденного протокола и всех утвержденных поправок к нему, если таковые имеются; д) проверяет получение письменного информированного согласия каждого испытуемого до начала участия в исследовании/испытании; е) следит за получением исследователем брошюры для исследователя в текущей редакции, всех других документов и материалов, необходимых для проведения исследования/испытания надлежащим образом и в соответствии с требованиями законодательства Республики Казахстан; ж) проверяет информированность исследователя и его сотрудников о проводимом исследовании/испытании. з) проверяет выполнение обязанностей исследователя и его сотрудников в соответствии с протоколом и всеми другими письменными соглашениями между спонсором и исследователем/организацией и отсутствие передачи своих функций неуполномоченным лицам; и) проверяет соблюдение исследователем критериев отбора при включении испытуемых; к) сообщает о соблюдении сроков набора испытуемых в исследование/испытание; л) проверяет правильность, полноту и своевременность регистрации данных в первичных и других относящихся к исследованию/испытанию документах, а также порядок их ведения; м) проверяет правильность, полноту, своевременность, дату и идентификацию исследования/испытания и предоставление всех требуемых отчетов, уведомлений, запросов, заявок и других документов и содержание точной и подробной информации в них; н) проверяет правильность и полноту данных в индивидуальной регистрационной форме, первичных документах и других записях путем сопоставления их между собой. С особым вниманием монитор проверяет: 1) правильность внесения требуемых протоколом данных в индивидуальной регистрационной форме и их соответствие данным в первичной документации; 2) документальное оформление любых изменений дозы и/или терапии для каждого испытуемого; 3) регистрацию в индивидуальной регистрационной форме нежелательных явлений, сопутствующих лечений и интеркуррентных заболеваний, в соответствии с требованиями протокола; 4) регистрацию сведений о нарушении режима исследований/испытаний (пропущенных испытуемым визитах, обследованиях и т.д.); 5) регистрацию и объяснение всех случаев исключения и выбывания испытуемых; о) сообщает исследователю о любых, допущенных в индивидуальной регистрационной форме ошибках, пропусках и неразборчивых записях. Монитор следит за тем, чтобы соответствующие исправления, добавления или вычеркивания были сделаны, датированы, объяснены (если необходимо) и подписаны самим исследователем либо уполномоченным на подписание за него изменений в индивидуальной регистрационной форме членом исследовательского коллектива. Данные полномочия должны быть закреплены документально; п) проверяет своевременность сообщения исследователем о нежелательных явлениях, согласно требованиям настоящего стандарта, протокола, комиссии по вопросам этики, спонсора и уполномоченного органа; р) проверяет ведение исследователем основных документов; с) сообщает исследователю о допущенных отклонениях от протокола, СОП, требований настоящего стандарта и законодательства Республики Казахстан и предпринимает необходимые действия с целью предотвращения повторения установленных отклонений. 8.17.5 Монитор соблюдает письменные СОП спонсора, а также процедуры, специально определенные спонсором для мониторинга конкретного исследования/испытания. 8.17.6 Отчет монитора: а) после каждого визита в исследовательский центр или связанного с исследованием/испытанием контакта монитор представляет спонсору письменный отчет; б) отчеты включают в себя дату, наименование центра, имя монитора, имя исследователя или иного лица, с которым состоялся контакт; в) отчеты содержат краткое описание объектов проверки, сообщение монитора о существенных данных/фактах, отклонениях и недостатках, выводы, описание действий, предпринятых либо планируемых и/или рекомендуемых для обеспечения соблюдения требований протокола, настоящего стандарта и уполномоченного органа; г) проверка отчета и последующие действия по нему должны быть документально оформлены уполномоченным представителем спонсора. 8.18 Аудит При проведении аудита в пределах мер, направленных на обеспечение качества исследования/испытания, спонсор учитывает: 8.18.1 Цель аудита Целью спонсорского аудита является оценка соответствия проведения исследования/испытания протоколу, СОП, требованиям настоящего стандарта и законодательства правовым актам. Аудит является самостоятельной процедурой, не связанной с мониторингом и контролем качества. 8.18.2 Выбор и квалификацию аудиторов: а) Для проведения аудита спонсор назначает лиц, независимых от клинических исследований/испытаний. б) Спонсор обязан удостовериться в том, что аудиторы обладают достаточной квалификацией, подготовкой и опытом для проведения аудита надлежащим образом. Квалификация аудитора подтверждается документально. 8.18.3 Процедуру аудита: а) спонсор должен убедиться, что аудит клинических исследований/испытаний программ проводится в соответствии с письменными процедурами спонсора, определяющими объект аудита, методы проведения аудита, частоту аудитов, а также форму и содержание отчетов об аудите; б) разработанный спонсором план аудита и процедуры аудита определяются значимостью данного исследования/испытания, количеством испытуемых, участвующих в исследовании/испытании, типом и сложностью исследования/испытания, степенью риска для испытуемых, а также любыми выявленными проблемами; в) замечания и выводы аудита оформляются документально; г) уполномоченный орган запрашивает отчеты об аудите, в случаях, когда имеются свидетельства серьезного несоответствия требованиям настоящего стандарта, или в случае судебных разбирательств; д) в случаях определенных законодательством или подзаконными актами, спонсор представляет заинтересованной стороне свидетельство о проведенном аудите. 8.19 Несоблюдение требований 8.19.1 При несоблюдении исследователем требований протокола, СОП, требований настоящего стандарта и/или законодательства Республики Казахстан, спонсор безотлагательно принимает меры, направленные на обеспечение их соблюдения. 8.19.2 При обнаружении в ходе мониторинга или аудита серьезных и/или повторяющихся случаев несоблюдения исследователем требований протокола, СОП, настоящего стандарта и/или законодательства Республики Казахстан, спонсор отстраняет исследователя от участия в исследовании/испытании. Об этом спонсор безотлагательно уведомляет уполномоченный орган, с указанием причины отстранения. 8.20 Досрочное прекращение или приостановка исследования/испытания Если исследование/испытание досрочно прекращено или приостановлено, спонсор незамедлительно сообщает об этом исследователям, комиссия по вопросам этики и уполномоченному органу, с указанием причины. 8.21 Отчет о клиническом исследовании/испытании Независимо от того, было ли исследование/испытание завершено по протоколу или прекращено досрочно, спонсор предоставляет в уполномоченный орган отчеты о клиническом исследовании/испытании, в соответствии с требованиями нормативных правовых актов [10]. Спонсор обеспечивает соответствие отчетов требованиям настоящего стандарта. 8.22 Многоцентровые исследования/испытания При многоцентровых исследованиях/испытаниях спонсор обеспечивает следующее: 8.22.1 Проведение исследования/испытания в строгом соответствии с протоколом, одобренным комиссией по вопросам этики и утвержденным уполномоченным органом. 8.22.2 Разработку индивидуальной регистрационной формы таким образом, чтобы собрать все требуемые данные из всех клинических баз, участвующих в многоцентровом исследовании/испытании. Тем исследователям, которые собирают дополнительные данные, должны быть также предоставлены дополнительные индивидуальные регистрационные формы, разработанные для сбора дополнительных данных. 8.22.3 Документальное оформление обязанностей исследователей-координаторов и других исследователей до начала исследования/испытания. 8.22.4 Предоставление инструкции по соблюдению протокола, единых стандартов оценки клинических и лабораторных данных, заполнению индивидуальной регистрационной формы всем исследователям. 8.22.5 Возможность связи между исследователями. 9 Протокол клинического исследования/испытания и поправки к протоколу Содержание протокола исследования/испытания имеет указанную ниже структуру. Информация, имеющая отношение только к одной клинической базе, представляется на отдельных страницах протокола или содержится в отдельном соглашении, а часть приведенной ниже информации может также содержаться в других документах, ссылки на которые имеются в протоколе, в брошюре исследователя. 9.1 Общие сведения 9.1.1 Название протокола, идентификационный номер протокола и дата. Любая поправка должна иметь номер и дату. 9.1.2 Наименование/имя и адрес спонсора и монитора (если они различаются). 9.1.3 Фамилия, имя и должность лиц, уполномоченных от имени спонсора подписывать протокол и поправки к нему. 9.1.4 Фамилия, имя, должность, адрес и номер телефона медицинского эксперта назначенного спонсором по данному исследованию/испытанию. 9.1.5 Фамилия, имя и должность исследователей, отвечающих за проведение исследования/испытания, а также адреса и номера телефонов клинических баз. 9.1.6 Фамилия, имя, должность, адрес и номер телефона квалифицированного врача, отвечающего за принятие всех решений медицинского характера (если данное лицо не является исследователем). 9.1.7 Наименования и адреса клинических лабораторий и других медицинских и/или технических служб и/или организаций, вовлеченных в исследование/испытание. 9.2 Обоснование исследования/испытания 9.2.1 Название и описание исследуемых продуктов. 9.2.2 Сводное изложение результатов доклинических и клинических исследований/испытаний имеющих клиническую значимость. 9.2.3 Краткое описание известных и потенциальных рисков и пользы для испытуемых, если таковые имеются. 9.2.4 Описание и обоснование способа введения, дозировки, режима дозирования и курса лечения. 9.2.5 Указание на то, что исследование/испытание будет проводиться в соответствии с протоколом, требованиями настоящего стандарта и законодательства Республики Казахстан. 9.2.6 Описание исследуемой популяции. 9.2.7 Ссылки на литературные источники и данные, существенные для исследования/испытания и представляющие собой обоснование данного исследования/испытания. 9.3 Подробное описание целей и задач исследования/испытания 9.4 Дизайн исследования/испытания 9.4.1 Указание основных и дополнительных (при наличии) исследуемых параметров, которые будут оцениваться в ходе исследования/испытания. 9.4.2 Описание типа/дизайна проводимого исследования/испытания (например, двойное слепое, плацебо-контролируемое, параллельное) и графическую схему дизайна исследования/испытания, процедур и этапов исследования/испытания. 9.4.3 Описание мер, направленных на минимизацию/исключение субъективности, в том числе рандомизации и слепого метода/маскировки. 9.4.4 Описание используемого в исследовании/испытании лекарственного средства, с указанием дозировки, схемы применения, лекарственной формы, упаковки и маркировки. 9.4.5 Ожидаемая продолжительность участия испытуемых в исследовании/испытании, описание последовательности и продолжительности всех периодов исследования/испытания, включая период последующего наблюдения, если таковой предусмотрен. 9.4.6 Описание "правил остановки" или "критериев исключения" для отдельных испытуемых, этапов исследования/испытания или исследования/испытания в целом. 9.4.7 Процедуры учета исследуемых лекарственных средств, включая, при наличии, плацебо и препараты сравнения. 9.4.8 Хранение рандомизационных кодов и процедуры их раскрытия. 9.4.9 Перечень всех данных, регистрируемых непосредственно в индивидуальной регистрационной форме (т.е. без предварительной записи в письменном или электронном виде) и рассматриваемых в качестве первичных данных. 9.5 Включение и исключение испытуемых 9.5.1 Критерии включения испытуемых в исследование/испытание. 9.5.2 Критерии невключения испытуемых в исследование/испытание. 9.5.3 Критерии исключения испытуемых (основания прекращения применения исследуемого лекарственного средства) и процедуры, определяющие: а) Когда и как испытуемых исключать из исследования/испытания. б) Какие данные, и в какие сроки должны быть собраны по исключенным пациентам. в) Способ замены выбывших испытуемых. г) Последующее наблюдение за испытуемыми, исключенными из исследования/испытания. 9.6 Лечение испытуемых 9.6.1 Для каждой группы испытуемых предоставляются сведения о всех применяемых в ходе исследования/испытания и лечения лекарственных средствах. Сведения должны содержать информацию о названии всех лекарственных средствах, их дозировки, частоту приема, пути/способы введения, а также продолжительность лечения, включая периоды последующего наблюдения. 9.6.2 Лекарственные средства и способы лечения, применение которых разрешено протоколом (включая неотложную терапию) или не разрешено использование их до и/или во время исследования/испытания. 9.6.3 Методы контроля за соблюдением испытуемыми режима лечения. 9.7 Оценка эффективности 9.7.1 Перечень параметров эффективности. 9.7.2 Методы и сроки оценки, регистрации и анализа параметров эффективности. 9.8 Оценка безопасности 9.8.1 Перечень параметров безопасности. 9.8.2 Методы и сроки оценки, регистрации и анализа параметров безопасности. 9.8.3 Требования к отчетности, процедурам регистрации и сообщениям о нежелательных явлениях и интеркуррентных заболеваниях. 9.8.4 Метод и продолжительность наблюдения за испытуемыми после возникновения нежелательных явлений. 9.9 Статистика 9.9.1 Описание статистических методов, которые предполагается использовать, включая сроки каждого планируемого промежуточного анализа. 9.9.2 Планируемое количество испытуемых. При многоцентровых исследованиях/испытаниях планируемое количество испытуемых определяется в каждой клинической базе. Обоснование размера выборки или вычисления для обоснования статистической мощности и клинической значимости исследования/испытания. 9.9.3 Применяемый уровень значимости. 9.9.4 Критерии прекращения исследования/испытания. 9.9.5 Процедуры учета отсутствующих, не подлежащих анализу, сомнительных и фальсифицированных данных. 9.9.6 Процедуры сообщения о любых отклонениях от первоначального плана статистической обработки (все отклонения от первоначального плана статистической обработки должны быть описаны и обоснованы в протоколе и/или итоговом отчете об исследовании/испытании). 9.9.7 Категории испытуемых, данные которых включаются в статистический анализ, (например, все рандомизированные испытуемые, все испытуемые, получившие хотя бы одну дозу исследуемого лекарственного средства или все испытуемые, соответствующие критериям отбора, данные которых пригодны для включения в анализ). 9.10 Прямой доступ к первичным данным/документации Спонсор предусматривает в протоколе или ином письменном соглашении обязанность исследователей не препятствовать прямому доступу к первичным данным/документации для проведения связанных с исследованием/испытанием мониторинга, аудита, этической экспертизы, а также инспекции со стороны уполномоченного органа. 9.11 Контроль качества и обеспечение качества 9.12 Описание этических аспектов исследования/испытания 9.13 Сбор данных и ведение записей 9.14 Указание о финансировании и страховании, если они не представлены в отдельном договоре 9.15 Указание о политике в отношении публикаций, если она не описана в отдельном договоре 9.16 Приложения 10 Брошюра исследователя 10.1 Введение Брошюра исследователя представляет собой сводное изложение клинических и доклинических данных по исследуемому лекарственному средству, которые имеют значение для его изучения с участием человека в качестве испытуемого. Брошюра исследователя предназначена для предоставления исследователям и другим участникам исследования/испытания, информации, способствующей пониманию и соблюдению положений протокола, таких как доза, частота/периодичность доз, способы введения и процедуры мониторинга безопасности. Брошюра исследователя обеспечивает понимание аспектов оказания медицинской помощи испытуемым в ходе исследования/испытания. Информация должна быть представлена в краткой, простой, объективной, взвешенной и лишенной рекламного оттенка форме, позволяющей клиницисту или потенциальному исследователю понять ее и сформировать свою собственную объективную оценку целесообразности предлагаемого исследования/испытания исходя из соотношения риска и пользы. В редактировании брошюры исследователя должен принимать участие медицинский эксперт. Содержание брошюры исследователя должно быть согласовано с профильными специалистами тех областей, где были получены описываемые данные. Минимальный объем информации, который содержится в брошюре исследователя, а также структура его изложения определяются настоящим стандартом. Характер и объем доступной информации изменяются в зависимости от стадии разработки исследуемого лекарственного средства. В случае, если исследуемое лекарственное средство находится на рынке и большинству практикующих врачей хорошо известны его фармакологические свойства, брошюра исследователя может быть менее подробной. По решению уполномоченного органа, вместо брошюры исследователя могут быть использованы материалы, содержащие основную информацию о лекарственном средстве: листок-вкладыш или информация на этикетке. Альтернативный вариант должен содержит актуальную, всестороннюю и подробную информацию обо всех характеристиках исследуемого лекарственного средства, которые важны для исследователя. Если зарегистрированное лекарственное средство исследуется по новому показанию к применению, брошюра исследователя должна быть составлена с учетом нового применения. Брошюру исследователя следует пересматривать не реже одного раза в год и, при необходимости, дополнять в соответствии с письменными процедурами спонсора. В зависимости от стадии разработки продукта и по мере поступления новой значимой информации брошюру исследователя можно дополнять и более часто. В соответствии с требованиями настоящего стандарта новая информация может быть настолько важна, что до ее включения в новую редакцию брошюры исследователя с ней необходимо ознакомить исследователей, комиссию по вопросам этики и/или уполномоченный орган. Спонсор отвечает за предоставление исследователям текущей редакции брошюры исследователя. Исследователь несет ответственность за ее предоставление в комиссию по вопросам этики. Если спонсором исследования/испытания является исследователь, он обязан рассмотреть возможность получения брошюры от производителя лекарственного средства. Если исследуемое лекарственное средство предоставляется самим спонсором-исследователем, он доводит необходимую информацию до сведения занятого в исследовании/испытании персонала. В случаях, когда составление брошюры исследователя неосуществимо, в качестве альтернативы спонсор-исследователь представляет необходимую информацию в протоколе в разделе обоснования исследования/испытания. 10.2 Общие положения Брошюра исследователя включает в себя: 10.2.1 Титульный лист На титульном листе указываются информация о спонсоре (название и адрес), идентификаторы каждого исследуемого лекарственного средства (номер исследования/испытания, код, химическое, международное непатентованное названия, торговое/патентованное название исследуемого лекарственного препарата, если это согласуется с желанием спонсора и не противоречит действующему законодательству Республики Казахстан и дата редакции брошюры исследователя). Рекомендуется указывать номер текущего издания брошюры исследователя, а также номер и дату предыдущего издания. В приложении 1 к настоящему стандарту приведен пример титульного листа. 10.2.2 Указание на конфиденциальность Уведомление для исследователей и получателей документа о том, что брошюра исследователя должна рассматриваться, как конфиденциальная информация, предназначенная исключительно для ознакомления и использования коллективом исследователей, комиссией/комитетом по вопросам этики и уполномоченным органом. 10.3 Содержание брошюры исследователя Брошюра исследователя имеет следующее содержание: 10.3.1 Содержание. Пример содержания приведен в приложении 2 к настоящему стандарту. 10.3.2 Резюме Приводится информация о физических, химических, фармацевтических свойствах исследуемого лекарственного средства, а также данные о его токсикологии, фармакокинетики, фармакодинамики и клинической активности в контексте соответствующей стадии клинического изучения. 10.3.3 Введение В кратком вводном разделе указываются химическое, международное непатентованное названия (а также торговое/патентованное название, если зарегистрированы) исследуемого лекарственного средства, все активные вещества. Также указываются фармакологическая группа, к которой относится исследуемое лекарственное средство, и место, на которое он в ней претендует (например, преимущества, препарат выбора). Приводятся доказательства в пользу дальнейшего изучения исследуемого лекарственного средства, а также его потенциальные показания к профилактическому, терапевтическому или диагностическому применению. В этом разделе должен быть сформулирован общий подход к оценке исследуемого лекарственного средства. 10.3.4 Физические, химические и фармацевтические свойства и состав лекарственной формы Представляются описание компонентов (активного и вспомогательных веществ) исследуемого лекарственного средства (включая химические и/или структурные формулы), краткая справка об их основных физических, химических и фармацевтических свойствах. С целью обеспечения и соблюдения, адекватных мер предосторожности в ходе клинического исследования/испытания указывается состав лекарственной формы, включая вспомогательные вещества, и приводится обоснование использования данной лекарственной формы для клинического изучения. Также предоставляются инструкции по хранению и использованию лекарственной формы. Следует указывать любое структурное сходство с другими известными лекарственными средствами. 10.3.5 Доклинические исследования/испытания Введение В вводной части в краткой форме приводятся результаты всех доклинических исследований/испытаний фармакодинамических и фармакокинетических свойств, токсичности, канцерогенности, мутагенности, тератогенности и эмбриотоксичности исследуемого лекарственного средства. Описываются использованные методы изучения и полученные результаты с обсуждением их потенциальной клинической значимости в контексте предполагаемых лечебных и возможных неблагоприятных или неожидаемых реакций у человека. Далее в этом разделе приводятся следующие данные: Использованные в исследованиях/испытаниях виды животных; Количество и пол животных в каждой группе; Условия содержания и рацион кормления животных; Единицы измерения дозы (например, миллиграмм/килограмм (мг/кг и др.); Кратность введения; Путь введения; Длительность курса введения; Продолжительность последующего наблюдения после окончания введения исследуемого лекарственного средства. Для большей наглядности данные следует, представлять в виде таблиц/списков. Последующие разделы должны содержать обсуждение наиболее важных результатов исследований/испытаний, их экстраполяцию на человека, а также необходимость их подтверждения в клинических исследованиях/испытаниях. Должны быть представлены данные сравнения результатов исследований/испытаний на одном и том же виде животных при использовании как эффективных, так и нетоксических доз исследуемого лекарственного средства (т.е. определить терапевтический индекс). Следует указать, как эти данные соотносятся с дозировками, планируемыми для исследования/испытания на людях. Во всех случаях, при проведении сравнений рекомендуется указывать концентрации исследуемого лекарственного средства в крови/ткани, а не дозировки, выраженные в мг/кг); а) Фармакодинамика Данный раздел включает в себя описание фармакодинамических свойств исследуемого лекарственного средства и, его активных метаболитов по результатам исследований/испытаний на животных. Описывается следующее: изучение специфической активности и механизма действия (эффективность при экспериментальной патологии, лиганд-рецепторное взаимодействие, специфичность действия); характер, выраженность фармакологических эффектов, скорость их развития, продолжительность, обратимость и дозозависимость эффектов, терапевтический индекс; тесты, направленные на оценку безопасности (специальные эксперименты для изучения фармакологических действий, выходящих за рамки планируемых клинических показаний). В процессе изучения фармакодинамики исследуемого лекарственного средства устанавливают не только его специфическую, но и возможные побочные действия, связанные с фармакологической активностью. Действие исследуемого лекарственного средства на здоровый и больной организм может различаться, поэтому изучение его фармакодинамических свойств должны проводиться на моделях соответствующих патологических состояний). б) Фармакокинетика В этом разделе описываются особенности всасывания, местная и системная биодоступность, связь с белками, липопротеидами плазмы крови, распределения с указанием объема распределения, органотропность, пути метаболизма (биотрасформация, конъюгация) и пути выведения исследуемого лекарственного средства. Необходимо указать биодуступность (абсолютную, относительную), максимальную концентрацию исследуемого лекарственного средства в плазме крови Сmax и время, в течение которой она достигается Tmax, влияние на активность микросомальных ферментов печени, периоды полувыведения из плазмы и тканей. Особенности изменения фармакокинетики (линейная и нелинейная) при повышении доз исследуемого лекарственного средства. Следует провести корреляцию между фармакокинетическими параметрами и результатами фармакодинамических и токсикологических экспериментальных исследований/испытаний. В этом же разделе могут быть описаны результаты экспериментального исследования/испытания лекарственного взаимодействия исследуемого лекарственного средства с другими препаратам). в) Токсикология В данном разделе описываются токсические свойства исследуемого лекарственного средства, изученные на разных видах животных. При токсикологических исследованиях/испытаниях на экспериментальных животных должны быть установлены характер и выраженность возможного повреждающего воздействия исследуемого лекарственного средства. Токсикологические исследования/испытания проводятся в двух направлениях: на изучение общетоксического действия и изучение специфических видов токсичности (канцерогенность, мутагенность, эмбриотоксичность, тератогенность, иммунореактивность, в том числе аллергенность). Данные исследования/испытания общетоксического действия позволяют определить: переносимые и токсические дозы; наиболее чувствительные к исследуемому лекарственному средству органы и системы организма, характер и степень патологических изменений в них, а также определить обратимость вызываемых повреждений; зависимость токсических действий от дозы и длительности применения исследуемого лекарственного средства. Изучение общетоксического действия исследуемого лекарственного средства подразделяется на два этапа: изучение «острой» токсичности при однократном или дробном введении через короткие интервалы в течение суток; изучение «хронической» токсичности при длительном введении (продолжительность введения определяется предполагаемым курсом клинического применения). Данные исследования/испытания токсических свойств исследуемого лекарственного средства описывается в следующей последовательности: токсичность при однократном введении; токсичность при многократном введении; канцерогенность; влияние на иммунореактивность (местно-раздражающее и аллергизирующее действие); репродуктивная токсичность (эмбриотоксичность, фетотоксичность); генотоксичность (мутагенность и тератогенность). В каждом случае следует описать характер, частоту, выраженность, степень тяжести, дозозависимость, скорость развития, продолжительность и обратимость токсических действий. Следует указать переносимые дозы, минимальные и максимальные токсические дозы, а также летальные. Если лекарственная форма содержит вспомогательные вещества, неразрешенные для применения в медицинской практике, то каждое из этих веществ исследуют отдельно. При комбинации нескольких фармакологически активных веществ в одной лекарственной форме, изучают токсичность комбинации в целом и каждого ингредиента в отдельности, если он не был ранее разрешен для применения в медицинской практике). 10.3.6 Клинические исследования/испытания Введение В этом разделе подробно описываются результаты изучений исследуемого лекарственного средства на людях, включая данные по фармакодинамике, фармакокинетике, дозозависимости действий, эффективности и безопасности. Необходимо кратко описать каждое завершенное клиническое исследование/испытание по данному исследуемому лекарственному средству. Указывается информация об исследуемом лекарственном средстве, полученная как в результате клинических исследованияй/испытаний, так и на основе обобщенного пострегистрационного применения препарата). а) Фармакокинетика исследуемого лекарственного средства у человека Данный раздел должен содержать следующую информацию, относящуюся к фармакокинетике и предоставляется по следующим разделам: всасывание, связывание с белками плазмы, распределение, метаболизм и выведение; биодоступность (абсолютная, если это возможно, и/или относительная) с использованием определенной лекарственной формы в качестве сравнения; фармакокинетика у различных групп испытуемых (в зависимости от пола, возраста или нарушений функций органов); взаимодействия с другими лекарственными средствами и пищей; другие данные по фармакокинетике (например, результаты проведенных в рамках клинических исследований/испытаний фармакокинетических исследований/испытаний на различных группах). б) Безопасность и эффективность Представляется информация по безопасности, фармакодинамике, эффективности и дозозависимости действий исследуемого лекарственного средства (и его метаболитов, если это изучалось), информация, полученная в ходе проведенных ранее клинических исследований/испытаний (с участием здоровых добровольцев и/или пациентов). Приводится интерпретация этих данных. Если часть клинических исследований/испытаний уже завершена, требуется представить обобщенные отчеты всех исследований/испытаний по эффективности и безопасности исследуемого лекарственного средства по отдельным показаниям у различных групп испытуемых. Также предоставляются сводные таблицы побочных реакций по всем клиническим исследованиям/испытаниям и для всех изученных показаний. Необходимо указать существенные различия в характере и частоте встречаемости побочных реакций, как между различными показаниями, так и между различными группами испытуемых. В брошюре исследователя описываются возможные риски и ожидаемые нежелательные реакции, которые основываются на накопленном опыте применения, как самого исследуемого лекарственного средства, так и сходных с ним препаратов. Требуется описать меры предосторожности и рекомендуемые методы обследования, которые следует использовать при применении лекарственного средства с исследовательскими целями. в) пострегистрационный опыт применения В данном разделе указываются страны, в которых исследуемое лекарственное средство уже имеется в продаже или был зарегистрирован. Вся информация, полученная в ходе пострегистрационного применения исследуемого лекарственного средства, представляется в обобщенном виде с указанием лекарственной формы, дозировки, пути введения, нежелательных побочных реакций и их исход. Должны быть указаны страны, в которых заявителю было отказано в регистрации исследуемого лекарственного средства или он был изъят из обращения). 10.3.7 Заключение и рекомендации для исследователя В этом разделе представляется обсуждение доклинических, клинических данных и обобщается информация, полученная из разных источников по различным свойствам исследуемого лекарственного средства. Таким образом, исследователю предоставляются наиболее информативная интерпретация имеющихся данных, а также выводы о значимости этой информации для последующих клинических исследований/испытаний. Освещаются опубликованные работы по сходным продуктам, если таковые имеются. Это позволит исследователю быть готовым к нежелательным реакциям или другим проблемам, которые могут возникнуть в ходе клинических исследований/испытаний. Основная цель данного раздела заключается в том, чтобы помочь исследователю получить четкое представление о возможных рисках и нежелательных реакциях, а также о специальных тестах, методах наблюдения и мерах предосторожности, которые могут понадобиться в ходе клинического исследования/испытания. Это представление основывается на доступной информации о физических, химических, фармацевтических, фармакологических, токсикологических и клинических свойствах исследуемого лекарственного средства. Клиническому исследователю также предоставляются инструкции по диагностике и лечению возможных передозировок и нежелательных реакций, которые основаны на предыдущем клиническом опыте и фармакологических свойствах исследуемого лекарственного средства. Приложение А (справочное) Пример оформления Титульного листа НАЗВАНИЕ и АДРЕС СПОНСОРА Исследуемый лекарственный препарат Номер исследования/испытания: Код исследуемого лекарственного препарата: Химическое название: Международное непатентованное название: Патентованное/торговое название (если это согласуется с желанием спонсора и не противоречит действующему законодательству Республики Казахстан) БРОШЮРА ИССЛЕДОВАТЕЛЯ Номер текущего издания: Дата издания: Вводится взамен: _____________________ номер предыдущего издания Дата издания: Приложение Б (справочное) Пример оформления Содержания брошюры исследователя Гриф конфиденциальности (необязательно) Подписи (необязательно) А. 1 Содержание А. 2 Резюме А. 3 Введение А. 4 Физические, химические, фармацевтические свойства и состав лекарственной формы А. 5 Доклинические исследования/испытания: А. 5.1 Фармакодинамика А. 5.2 Фармакокинетика А. 5.3 Токсикология А. 6 Клинические исследования/испытания: А. 6.1 Фармакокинетика исследуемого лекарственного средства у человека А. 6.2 Безопасность и эффективность А. 6.3 Пострегистрационный опыт применения А. 7 Заключение и рекомендации для исследователя Ссылки на: Публикации Отчеты Данные ссылки должны быть приведены в конце каждой главы Приложения (при наличии) 11. Основные документы клинического исследования/испытания 11.1 Введение Основными документами клинического исследования/испытания являются те, которые вместе или по отдельности позволяют оценить проведение исследования/испытания и качество полученных данных. Они служат доказательством соблюдения исследователем, спонсором и монитором требований настоящего стандарта и законодательства Республики Казахстан. Основные документы необходимы для выполнения ряда важных задач. Своевременное оформление основных документов в файлы исследователя способствует успешному выполнению своих функций исследователем, спонсором и монитором. Документы являются объектом независимого аудита со стороны спонсора и инспекции уполномоченного органа. Ниже приведен минимальный перечень основных документов. Документы делятся на три группы в зависимости от стадии клинического исследования/испытания: а) до начала клинической фазы исследования/испытания; б) в ходе клинической фазы исследования/испытания; в) после завершения или досрочного прекращения исследования/испытания. В каждом документе указываются файлы хранения: в файлах исследователя и/или спонсора. Допускается объединение некоторых документов при условии, что отдельные элементы легко идентифицируются. Файлы исследования/испытания формируются в начале исследования/испытания на клинической базе и в офисе спонсора. Исследование/испытание можно считать завершенным только после того, как монитор проверит файлы исследователя и спонсора и подтвердит наличие всех необходимых документов. Любой документ, перечисленный в настоящем стандарте может быть затребован спонсором для проведения аудита и уполномоченным органом для проведения инспекции. 11.2 В период планирования клинического исследования/испытания На стадии планирования исследования/испытания до его формального начала создаются документы, согласно Таблице 1. Таблица 1 Наименование документа Обоснование наличия документа в файле Брошюра исследователя Подписанный протокол и поправки к нему (если таковые имеются), образец индивидуальной регистрационной карты (ИРФ) Документально закрепить факт передачи исследователю необходимой и актуальной научной информации об исследуемом продукте Документально закрепить факт утверждения спонсором и исследователем протокола/поправок и ИРФ Отметка о наличии в файлах ИсследоСпонсор ватель + + + + Информация, предоставляемая испытуемому: - Форма информированного согласия (включая все необходимые разъяснительные материалы) - Документально оформить факт получения информированного согласия + + Документально подтвердить, что испытуемым будет предоставлена соответствующая (с точки зрения содержания и доступности для понимания) письменная информация, помогающая им дать согласие на основе полной информированности + + + + + + + - Любая другая письменная информация - - Рекламные объявления для привлечения испытуемых в исследование/ испытание (если используются) Страховое обязательство (если требуется) Подписанное соглашение вовлеченных сторон, например: - между исследователем и спонсором - между исследователем и контрактной Документально подтвердить, что меры по привлечению испытуемых адекватны, и отсутствует элемент принуждения Документально закрепить финансовое соглашение по исследованию/испытанию между спонсором и исследователем/организацией Документально подтвердить, что в случае причинения ущерба, связанного с исследованием/испытанием, испытуемым будет доступна компенсация Документально закрепить соглашения: исследовательской организацией - между спонсором и контрактной исследовательской организацией + + + + (если требуется) + Датированное и документально оформленное одобрение Республиканской комиссии и/или комиссии по вопросам этики следующих документов: - протокола и любых поправок Документально подтвердить, что данное исследование/испытание было рассмотрено и одобрено Республиканская комиссия и/или комиссией по вопросам этики. Указывают номер версии и дату документа + + + - ИРФ (если требуется) - формы информированного согласия - любой другой письменной информации, предоставляемой испытуемым; - рекламных объявлений для привлечения испытуемых в исследование/испытание (если используются) - информации о компенсации испытуемым (при наличии) - любых иных /одобренных документов Состав национальной комиссии и/или комиссии по вопросам этики Утверждение протокола уполномоченным органом Curriculum vitae и/или другие документы, подтверждающие квалификацию исследователей и соисследователей Документально закрепить соответствие состава национальной комиссии и/или комиссии по вопросам этики + + (если требуется) Документально подтвердить разрешение уполномоченного органа получено до начала исследования/испытания Документально подтвердить квалификацию и пригодность для проведения исследования/испытания и/или осуществления медицинского наблюдения за + + + + Границы нормальных показателей для клинических/ медицинских/лабораторных/технических процедур и/или тестов предусмотренных протоколом Медицинские/лабораторные/технические процедуры/тесты сертификация или испытуемыми Документально закрепить границы нормальных показателей для лабораторных тестов + + Документально подтвердить пригодность оборудования для проведения требуемых тестов и обеспечения надежности результатов + (если требуется) + Документально подтвердить соблюдение соответствующих требований к маркировке исследуемого препарата и пригодность инструкций для испытуемых Документально закрепить инструкции для обеспечения надлежащего хранения, упаковки, распределения и утилизации исследуемых продуктов и расходных материалов. Документально закрепить даты и способ поставки, номера серий исследуемых продуктов и расходных материалов. Позволяет отследить серию продукта, контролировать условия поставки и вести учет Документально подтвердить подлинность, чистоту и количественное содержание активного вещества (дозировку) исследуемых продуктов Документально закрепить процедуру экстренной идентификации маскированного исследуемого продукта без нарушения маскировки для остальных испытуемых Документально закрепить метод рандомизации испытуемых - + + + + + - + + + (третья сторона, если требуется) + Документально подтвердить приемлемость исследовательского центра - + (третья сторона, если требуется) + аккредитация или внутренний и/или внешний контроль качества или другие методы подтверждения (где требуется) Образцы этикеток на упаковках исследуемых продуктов Инструкция по обращению с исследуемыми продуктами и расходными материалами (если не включено в протокол или Брошюру исследователя) Учет поставок исследуемых продуктов и расходных материалов Сертификаты анализов поставленных исследуемых продуктов Процедуры раскрытия кода для исследований/ испытаний, проводимых слепым методом Рандомизационный список Отчет монитора о предварительном визите для данного исследования/испытания Документально подтвердить факт ознакомления исследователя и занятого в исследовании/испытании персонала с процедурами исследования/испытания Отчет монитора о стартовом визите + + 11.3 В период клинической фазы исследования/испытания В подтверждение того, что вся необходимая новая информация документально оформляется по мере ее поступления, в дополнение к вышеперечисленным документам, имеющимся в файле, по ходу исследования/испытания необходимо добавлять документы, указанные в Таблице 2. Таблица 2 Обновленные исследователя версии Брошюры Любое изменение: - протокола/поправок и ИРФ - формы информированного согласия - любой информации, испытуемым - рекламных привлечения исследование/ Документально закрепить факт своевременного сообщения исследователю необходимой информации по мере ее поступления Документально закрепить изменения данных документов, произведенные во время исследования/испытания + + + + Документально подтвердить факт рассмотрения и утверждения/одобрения национальной комиссии и/или комиссии по вопросам этики поправок и/или новых редакций. Указывается редакция и дата документа + + другой письменной предоставляемой объявлений испытуемых для в испытание (если используются) Датированное и документально оформленное утверждение/одобрение национальной комиссии и/или комиссии по вопросам этики следующих документов: - поправок к протоколу - новых редакций: - формы согласия информированного - предоставляемых материалов - рекламных испытуемым объявлений для привлечения исследование/ испытуемых в испытание (если используется) - других утвержденных/одобренных документов - результатов периодического рассмотрения документации по исследование/ испытанию (где требуется) Где требуется, разрешение/одобрение/уведомление уполномоченных органов для: - поправок к протоколу и других документов Научную биографию (curriculum vitae) новых исследователей и/или соисследователей Изменения границ нормальных показателей для предусмотренных протоколом медицинских/лабораторных/технических процедур/тестов Изменения в медицинских/лабораторных/технических процедурах/тестах: - сертификация Документально закрепить соответствие нормативным требованиям Документально закрепить нормальный диапазон значений тестов, измененных в ходе исследования/испытания Документально подтвердить, что тесты продолжают отвечать требованиям в течение периода исследования/испытания + (если требуется) + + + + + + (если требуется) + + + - + - + + + - или аккредитация или внутренний контроль качества - - и/или внешний или другие методы подтверждения (где требуется) Документация по поставкам исследуемых продуктов и расходных материалов Сертификаты анализа новых серий исследуемых продуктов Отчеты мониторов о визитах Существенные исследования/испытания переговоры/переписка визитов): для (помимо - переписка - записи встреч - записи телефонных переговоров Документально закрепить визиты мониторов и их результаты Документально закрепить любые соглашения либо существенные переговоры, касающиеся вопросов проведения исследования/испытания, его административных аспектов, нарушений протокола, отчетности по нежелательным явлениям Подписанные информированного согласия формы Первичная документация Заполненные, датированные и подписанные индивидуальные регистрационные карты (ИРК) Документирование исправлений в ИРК Уведомление спонсора исследователем о серьезных нежелательных явлениях и соответствующие отчеты Уведомление спонсором и/или исследователем (что применимо) уполномоченных органов и Республиканскую комиссию/комиссию по вопросам этики о непредвиденных серьезных нежелательных реакциях и о другой информации по безопасности. Сообщение спонсором исследователю информации по безопасности Промежуточные или годовые отчеты, предоставляемые Республиканской комиссией/комиссией по вопросам этики и уполномоченным органам Журнал скрининга испытуемых Документально подтвердить, что согласие каждого испытуемого получено в соответствии с GCP и протоколом до начала участия в исследовании/испытании. Кроме того, документально закрепить разрешение на прямой доступ Документально подтвердить факт существования испытуемого и достоверность собранных данных. Включить исходные документы, относящиеся к исследованию/испытанию, лечению и анамнезу испытуемого Документально оформить подтверждение исследователем или уполномоченными сотрудниками исследователя зарегистрированных данных Документально закрепить все изменения/дополнения или исправления, сделанные в ИРК после записи первоначальных данных Уведомление спонсора исследователем о серьезных нежелательных явлениях и соответствующие отчеты согласно Уведомление спонсором и/или исследователем (что применимо) уполномоченных органов и Республиканскую комиссию/комиссию по вопросам этики о непредвиденных серьезных нежелательных реакциях и о другой информации по безопасности Сообщение спонсором исследователю информации по безопасности Промежуточные или годовые отчеты, предоставляемые Республиканской комиссией/комиссией по вопросам этики уполномоченным органам Документально закрепить + + + + + (копия) + (оригинал) + (копия) + (оригинал) + + + + + + + + (если требуется) + + (если идентификацию испытуемых, прошедших перед исследованием/испытании Список идентификационных испытуемых Журнал регистрации испытуемых кодов включения Учет исследуемого продукта исследовательском центре в Лист образцов подписей Учет хранящихся образцов биологических жидкостей/тканей (если имеются) требуется) скрининг Документально подтвердить, что исследователь/организация хранит конфиденциальный список имен всех испытуемых, которым при включении в исследование/испытание были присвоены идентификационные коды. Позволяет исследователю/организации идентифицировать любого испытуемого Документально закрепить хронологическую последовательность включения испытуемых по идентификационным кодам Документально закрепить использование исследуемого продукта в соответствии с протоколом Документально закрепить образцы подписей и инициалов всех лиц, уполномоченных вносить данные и/или исправления в ИРК Документально закрепить местонахождение и идентификацию хранящихся образцов в случае необходимости проведения повторных анализов + - + - + + + + + + 11.4 После завершения или прекращения исследования/испытания После завершения или прекращения исследования/испытания все документы, перечисленные в 11.2 и 11.3 должны содержаться в файле исследования/испытания вместе с документами указанными в Таблице 3. Таблица 3 Учет исследуемого лекарственного препарата на клинической базе Документально закрепить использование исследуемого продукта в соответствии с протоколом. Документально закрепить результаты окончательного подсчета количества исследуемого + + Документация уничтожению продукта по исследуемого Итоговый идентификационных испытуемых Сертификат аудита имеется) Отчет монитора завершающем визите список кодов (если о Документация по распределению испытуемых по группам и раскрытию кодов Итоговый отчет исследователя, предоставляемый в Республиканскую комиссию и/или комиссию по вопросам этики (если требуется) и уполномоченным органам (где применимо) Отчет о клиническом исследовании/испытании продукта, полученного исследовательским центром, выданного испытуемым, возвращенного испытуемыми и возвращенного спонсору Документально закрепить факт уничтожения неиспользованных исследуемых продуктов спонсором или в исследовательском центре Сделать возможной идентификацию всех включенных в исследование/испытание испытуемых в случае необходимости их последующего наблюдения. Список должен храниться с соблюдением требований конфиденциальности в течение согласованного срока Документально закрепить факт проведения аудита Документально закрепить окончание всех мероприятий исследования/испытания, необходимых для завершающего визита, и наличие копий основных документов в соответствующих файлах Возвращается спонсору для документального закрепления имевших место случаев вскрытия кодов Документально закрепить завершение исследования/испытания Документально закрепить результаты исследования/испытания и их интерпретацию + (если уничтожен на клинической базе) + + + + - + - + + + + (если необходимо) + - Приложение В (справочное) Библиография [1] ICH GCP. Guideline for Good Clinical Practice. International Руководство по Надлежащей клинической практике. Международной конференции по гармонизации технических требований к регистрации фармацевтических продуктов, предназначенных для применения человеком, которое, разработано с учетом действующих требований надлежащей клинической практики Европейского Союза, Соединенных Штатов Америки и Японии, а также Австралии, Канады и Всемирной организации здравоохранения (ВОЗ) Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH [2] Закон Республики Казахстан «О техническом регулировании» от 09.11.04 г. № 603-II [3] Закон Республики Казахстан «О языках в Республике Казахстан» от 11.07.1997 г. №151-I [4] Закон Республики Казахстан «О лекарственных средствах» от 13.01.04 г. №522-II [5] Закон Республики Казахстан от 04.06.03 «О системе здравоохранения» г. №430 [6] Закон Республики Казахстан «Об охране здоровья граждан» от 07.07.06 г. №170 [7] Приказ Комитета здравоохранения МЗ образования и спорта от 10.05.1999 г. №238 [8] Приказ МЗ РК от 14.02.2005 года №52 [9] Постановление Правительства «Инструкция по хранению различных групп лекарственных средств и изделий медицинского назначения в аптечных организациях, фармацевтических предприятиях и лечебнопрофилактических учреждениях» и «Инструкция о порядке хранения и обращения с лекарственными средствами, обладающими огнеопасными и взрывоопасными свойствами в аптечных организациях и фармацевтических предприятиях» «Об утверждении Инструкции по проведению мониторинга побочных действий лекарственных средств в Республике Казахстан» «Об утверждении порядка по уничтожению или дальнейшей переработке продукции и товаров в случае Республики Казахстан признания их от 29.12.1995 г. №1894 [10] Приказ МЗ РК от 14.02.2005 г. №53 «Об утверждении Инструкции по проведению клинических исследований и (или) испытаний фармакологических и лекарственных средств в Республике Казахстан» __________________________________________________________________ УДК 615.03.004.14:001.32 МКС 11.220.01 Ключевые слова: государственный стандарт, клиническая база, клинические исследования/испытания, испытуемый, исследователь, исследуемое лекарственное средство, побочная реакция, спонсор, уполномоченный орган, комиссия по вопросам этики, комитет по вопросам этики __________________________________________________________________