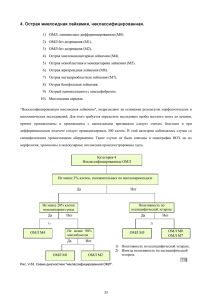
Немировченко Валентина Святославовна
advertisement

Федеральное государственное бюджетное учреждение «Российский онкологический научный центр имени Н.Н.Блохина» Российской академии медицинских наук На правах рукописи Немир овченко Валентина Свято сл авовна РОЛЬ ВАЛЬПРОЕВОЙ И ПОЛНОСТЬЮ ТРАНС-РЕТИНОЕВОЙ КИСЛОТ В ЛЕЧЕНИИ ДЕТЕЙ С ОСТРЫМИ МИЕЛОИДНЫМИ ЛЕЙКОЗАМИ 14.01.12 – Онкология ДИССЕРТАЦИЯ на соискание ученой степени кандидата медицинских наук Научный руководитель: Заведующий гемобластозов отделением НИИ химиотерапии ДОГ им.Н.Н.Блохина РАМН, д.м.н. Попа А.В. Москва, 2014 РОНЦ ОГЛАВЛЕНИЕ Введение Актуальность……………………………………………………………....стр. 3 Цель работы……………………………………………………….....стр. 4 Задачи исследования..………………………………………........... стр. 4 Научная новизна……………………………………………............ стр. 4 Практическая ценность……………………………………………....стр. 4 Глава 1. Обзор литературы……………………………………..…….....стр. 5 Глава 2. Характеристика пациентов, методов исследования и лечения…………………………………………………………………. стр. 38 Лечение…………………………………………………………..…...стр.43 Протокол ОМЛ НИИ ДО 2002…………………………………...стр. 44 Протокол ОМЛ НИИ ДОГ 2007…………………………………стр. 48 Статистическая обработка данных……………………………….стр. 53 Глава 3. Результаты лечения детей, получавших терапию согласно протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 ……….…стр. 54 Частота достижений ремиссий……………………………………...стр. 54 Результаты выживаемости……………………………………..…....стр.58 Переносимость терапии……………………………………………..стр. 85 Инфекционные осложнения………………………………………….стр. 85 Глава 4. Роль высокодозной химиотерапии и трансплантации гемопоэтических стволовых клеток в лечении детей с ОМЛ…….….стр. 90 Заключение………………………………………………………..…….…. стр. 95 Выводы……………………………………………………………..……... стр. 102 Список литературы…………………………………………………....… стр. 104 3 Введение АКТУАЛЬНОСТЬ. Результаты лечения детей, больных острыми миелоидными лейкозами (ОМЛ), за последние 20 лет улучшились, но их выживаемость ниже, чем больных острым лимфобластным лейкозом. Большинство неудач в лечении детей с ОМЛ связано с рефрактерностью опухолевых клеток к проводимой химиотерапии и возникновением рецидивов, что требует разработки и применение новых методов и препаратов. Острые миелоидные лейкозы – это гетерогенная группа острых лейкозов, основной характеристикой которой является клональная экспансия миелоидными бластами периферической крови, костного мозга или других тканей. Изучение хромосомных аномалий, ведущих к повреждению нормальной экспрессии генов при острых миелоидных лейкозах, позволило выявить, что изменения обнаруживаются как на генетическом, так и на эпигенетическом уровне, т.е. на уровне регуляции считывания генетической информации с участием белковых и нуклеотидных структур, присутствующих в клетке. Тот факт, что эпигенетические аномалии, в отличие от генетических мутаций, потенциально обратимы и могут быть восстановлены, делает данное направление терапии достаточно перспективным. Эпигенетические механизмы, которые изменяют структуру хроматина можно разделить на основные категории: метилирование ДНК, ковалентная модификация гистонов, нековалентные механизмы. Взаимодействие этих изменений создает "эпигенетический пейзаж", который регулирует способность генома проявляться в различных типах клеток, их стадий развития и различных заболеваний, в том числе опухолевых. Применение направленной терапии, веществ, модифицирующих действие гистондеацетилазы, ДНК-метилтрансферазы, синтетических РНК делает возможным изменение цикла жизни злокачественной клетки, возврат её к нормальной дифференцировке и естественному апоптозу. 4 Вальпроевая кислота (ВК) и полностью транс-ретиноевая кислота (ATRA) представляют собой препаратами с эпигенетическим эффектом: действуя в сочетании они, репрессирующего не изменяя структуру комплекса, ДНК, приводят деацетилирующего к гистоны разрушению регуляторных элементов генов, ответственных за дифференцировку клеток. Мы полагаем, что добавление эпигенетического лечения к химиотерапии позволит улучшить результаты лечения группы больных острыми миелоидными лейкозами. В протоколе ОМЛ НИИ ДО 2002 применялось только химиотерапия, в протоколе ОМЛ НИИ ДОГ 2007 – сочетание химиопрепаратов с эпигенетическими. ЦЕЛЬ РАБОТЫ: Повысить частоту полных ремиссий и выживаемость детей, больных острым миелоидным лейкозом с помощью сочетания эпигенетического лечения и химиотерапии. ЗАДАЧИ ИССЛЕДОВАНИЯ: 1. Оценить частоту ремиссий в результате терапии индукции у детей с ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007. 2. Сравнить выживаемость больных с ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007. 3. Определить токсичность вальпроевой кислоты и полностью транс-ретиноевой кислоты в протоколе ОМЛ НИИ ДОГ 2007. 4. Сравнить эффективность поддерживающей терапии с эпигенетическим лечением и без него у больных со стандартным риском ОМЛ. НАУЧНАЯ НОВИЗНА: Впервые в России применен протокол для лечения детей, больных ОМЛ, основанный на сочетании химиопрепаратов с лекарствами, влияющими на активность генов, участвующих в дифференцировке и пролиферации бластов. ПРАКТИЧЕСКАЯ ЗНАЧИМОСТЬ: На основании проведенного исследования, будут даны рекомендации по лечению детей, больных острыми миелоидными лейкозами с химиотерапии. использованием комбинации эпигенетического лечения и 5 ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ Острый лейкоз – злокачественное новообразование, характеризующееся клоновым распространением различными по дифференцировке миелоидными или лимфоидными предшественниками [10]. Он представляет собой наиболее распространенный вид опухоли в детском и подростковом возрасте, на который приходится 32% всех опухолей диагностируемых у детей в возрасте до 15 лет и 26% – до 20 лет [98]. За последние несколько десятилетий достигнуты хорошие результаты в лечении детей, больных острым лейкозом, благодаря стратификации по группам риска, оптимизации химиотерапии, расширение показаний к трансплантации гемопоэтических стволовых клеток и улучшению сопроводительной терапии. Применяя современные протоколы лечения детей бессобытийная выживаемость (БСВ) при остром лимфобластном лейкозе (ОЛЛ) достигает 80% [87, 80, 81]. Подобный успех не достигнут при острых миелоидных лейкозах. Улучшение результатов лечения ОМЛ, вероятно, зависит от поиска новых терапевтических стратегий, для чего необходимо: 1) дальнейшее уточнение специфических подклассов, которые требуют различных по интенсивности режимов лечения; 2) более глубокое изучение биологии лейкозной стволовой клетки, для полной эрадикации патологического клона, а не только для сокращения числа и объема опухолевых клеток; 3) исследование генетических и эпигенетических изменений, играющих важную роль в патогенезе, для определения потенциальных мишеней и создания новых препаратов [20, 67]. Важность понимания биологии опухолевой клетки отражает современное определение острого миелоидного лейкоза, который является генетически гетерогенным заболеванием, характеризующимся соматическими проявлениями генетических и эпигенетических перестроек в гемоэпоэтических клетках предшественниках, что нарушает нормальные механизмы самообновления, пролиферации и дифференцировки [33]. 6 Длительное время (с 1976г.) использовалась морфоцитохимическая классификация. Наиболее распространенной морфоцитохимической классификацией ОМЛ до сих пор остается предложенная франко-американобританской (FAB) группой. Принцип FAB-классификации основан на морфоцитохимических критериях и заключается в установлении линейной принадлежности лейкоза (гранулоцитарные, монобластные, эритробластные, мегакариобластные) и уровня дифференцировки клеток в пределах гранулоцитарных лейкозов. FAB-классификация[4]: М0 – ОМЛ с минимальной дифференцировкой; М1 – миелобластный лейкоз без созревания; М2 – миелобластный лейкоз с созреванием; М3 – промиелоцитарный лейкоз; М4 – миеломонобластный лейкоз; М5 - монобластный лейкоз, М5a – острый монобластный лейкоз недифференцированный M5b – острый моноцитарный лейкоз с признаками дифференцировки М6 - эритролейкоз; М7 - мегакариобластный лейкоз. Важным дополнением к FAB-классификации является иммунофенотипирование опухолевых клеток костного мозга, посредством которого можно определить антигены на поверхности бластов. В некоторых случаях именно определение поверхностных антигенов являются диагностически значимыми для установления диагноз. Это особенно важно для диагностирования острого мегакариоцитарного (FAB-М7) варианта, так как морфоцитохимически очень тяжело отличить клетки FAB-М7 от FAB-L2 морфологического варианта острого лимфобластного лейкоза. М0: Острый недифференцированный лейкоз В признаки. бластных В клетках единичных отсутствуют бластных специфические клетках морфологические определяется активность 7 миелопероксидазы и липидов (менее 3%), ШИК-позитивное вещество диффузно распределено в бластных клетках. М1: Острый миелобластный лейкоз без признаков созревания Бластные клетки не имеют специфические морфологические признаки. В единичных клетках выявляется пероксидазная активность (более 3%). М2: Острый миелобластный лейкоз с признаками созревания. При FAB-М2 ОМЛ в бластных клетках определяется зернистость (более 10% бластных клеток), в единичных бластах могут встречаться палочеки Ауэра. Бластные клетки пероксидазоположительные, содержат небольшое количество неспецифической эстеразы, ШИК-позитивное вещество в диффузной форме. М3: Острый промиелоцитарный лейкоз (ОПЛ) ОПЛ морфологически характеризуется крупными бластными клетками, часто с неправильной формой ядра, наличием крупной зернистости и палочек Ауэра в цитоплазме, которые могут располагаться пучками. Иногда встречаются нетипичные гипогранулярные случаи FAB-М3 варианта. В бластных клетках содержится значительное количество липидов и умеренное количество неспецифической эстеразы. ШИК-позитивное вещество в диффузной форме. Дети с диагнозом ОПЛ, были исключены из исследования, так как получали лечение по протоколу APL-2008. М4: Острый миеломонобластный лейкоз (ОММЛ) При FAB-М4 ОМЛ опухолевый пул клеток с неправильной и округлой формой ядра. В цитоплазме встречается зернистость и палочки Ауэра. От 20% до 100% бластных клеток содержат миелопероксидазу. В 20% - 100% бластов выявляется высокая активность неспецифической эстеразы, подавляемая фторидом натрия. Кроме того, выделяют вариант FAB-М4 с присутствием большого числа эозинофилов (>5%) с аномальной грануляцией – М4Ео. М5: Острый монобластный лейкоз Острый монобластный лейкоз отличается от FAB-М4 (миеломонобластного) присутствием более 80% клеток моноцитоидной линии. Различают две формы FAB-М5: 8 М5а - более 80% опухолевых клеток относятся к незрелым монобластам, которые характеризуются признаков, содержат отсутствием большое специфических количество морфологических неспецифической эстеразы, ингибируемой фторидом натрия, а активность миелопероксидазы выявляется в небольшом количестве. М5b - сочетание монобластов, промоноцитов или моноцитов составляет более 80% опухолевых клеток. Бластные клетки содержат ядра моноцитоидной формы. Неспецифическая эстераза в большом количестве, подавляется фторидом натрия. Миелопероксидаза в отдельных клетках в небольшом количестве. М6: Острый эритроцитарный лейкоз Диагноз острого эритроцитарного лейкоза – FAB-М6 устанавливается в случае определения более 50% эритробластов в костном мозге. Если присутствие опухолевых эритробластов колеблется в пределах 30% только от неэритроидных клеток, то такое состояние костного мозга необходимо рассматривать как миелодиспластический синдром (МДС). М7: Острый мегакариоцитарный лейкоз. Опухолевые клетки при FAB-М7 лейкозе полиморфны с округлыми ядрами и нежным сетчатым ядерным хроматином. Кроме того, бласты могут иметь овальную в виде «ручного зеркала» или круглую форму с выростами цитоплазмы вокруг голых ядер. Обычно активность миелопероксидазы отсутствует, также негативна реакция с суданом черным, в то же время они могут быть ШИК-позитивны. Эти признаки часто способствуют ошибкам, возникающим при диагностике, когда диагностируется не FAB-М7 лейкоз, а FABL2 морфологический вариант ОЛЛ. Для подтверждения FAB-М7 варианта ОМЛ обязательно необходимо иммунофенотипирование бластных клеток костного мозга: экспрессия поверхностных антигенов CD41, CD42 или CD61 подтверждает диагноз FAB-М7 лейкоза [4]. Всемирной Организации Здравоохранения (ВОЗ) в 2001 году была предложена новая классификация, которая характеризует ОМЛ не только в соответствии с морфоцитохимическими особенностями бластных клеток, но и 9 с генотипом, иммунофенотипом. В 2008 году, в связи с появлением новых данных, была пересмотрена классификации опухолей гемопоэтической и лимфоидной тканей ВОЗ. Согласно четвертому изданию этой классификации ОМЛ делится на следующие группы [100]: ОМЛ с повторяющимися генетическими аномалиями: ОМЛ с t(8;21)(q22;q22); RUNX1-RUNX1T1 ОМЛ с inv(16)(p13.1q22) или t(16;16)(p13.1;q22); CBFB-MYH11 Острый промиелоцитарный лейкоз t(15;17)(q22;q12); PML-RARA ОМЛ с t(9;11)(p22;q23); MLLT3-MLL ОМЛ с t(6;9)(p23;q34); DEK-NUP214 ОМЛ с inv(3)(q21q26.2) или t(3;3)(q21;q26.2); RPN1-EVI1 ОМЛ (мегакариоцитарный) с t (1;22)(p13;q13); RBM15- MKL1 ОМЛ с мутацией NPM1 ОМЛ с мутацией CEBPA ОМЛ, связанный с миелодиспластическими изменениями Миелоидные опухоли, связанные с лечением Миелоидная саркома Миелопролиферативный синдром, связанный с синдромом Дауна - транзиторный анормальный миелопоэз - миелоидный лейкоз, ассоциированный с синдромом Дауна Острый лейкоз из плазмацитоидных дендритных клеток ОМЛ с нормальным кариотипом: ОМЛ с минимальной дифференцировкой (FAB-M0) ОМЛ без созревания (FAB-M1) ОМЛ с созреванием (FAB-M2) Острый лейкоз миеломоноцитарный (FAB-M4) Острый монобластный и моноцитарный лейкоз (FAB-M5a, M5b) Острый лейкоз эритроидный (FAB-M6) Острый лейкоз мегакариобластный (FAB-M7) 10 Острый лейкоз базофильный Острый панмиелофиброз. Классификации ВОЗ основана на наличии основных генетических изменениях, поскольку они, как правило, связаны с характерными клиникоморфологическими особенностями и могут служить специфическими диагностическими и прогностическими маркерами. Группа с повторяющимися генетическими аномалиями была расширена, так как наличие определенных аберраций ведет к образованию химерного гена. Группа ОМЛ с миелодисплазией была переименована в ОМЛ связанный с миелодиспластическими изменениями, для того что бы подчеркнуть биологическую значимость миелодиспластического синдрома, в развитии данного вида лейкоза[10, 23, 118, 122]. Но даже при хорошей генетической диагностике в 60-80% случаев выявить хромосомные аномалии не удается, имеется нормальный кариотип. Повышение доступности секвенирования генома обещает раскрыть новые генетические повреждения, но остается открытым вопрос о том, какие изменения первичны, а какие вторичны и связаны с опухолевой прогрессией [70]. На основании результатов многих исследований, больные ОМЛ стратифицируются по группам риска. Выделение групп риска в различных исследовательских группах схожа (Таблица 1). Пациенты стратифицируются либо на две: низкого (стандартного) и высокого (AIEOP LAM, AML-BFM, NOPHOAML), или, как и в нашем исследовании (ОМЛ НИИ ДОГ), на три – с группой промежуточного (среднего) риска (MRC/DCOG AML, JPLSG AML, St Jude AML, COG AAML). В основе деления на группы риска лежит наличие хромосомных аномалий: благоприятных, таких как t(8;21) и inv(16)/t(16;16), для группы низкого (стандартного) риска, и неблагоприятных перестроек – t(6;9), t(9;11), t(9;22), del (7q-), del(5q-), -7, -5,-3, более 3 хромосомных аномалий – для группы высокого риска, а также ответ на 15 день от начала индукции ремиссии. К промежуточному (среднему) риску, относят пациентов, которые не вошли ни в стандартную, ни в группу высокого риска [6, 8, 83, 109]. 11 Применяя современную химиотерапию, у 90% детей с ОМЛ удается достичь ремиссии [59, 86, 114, 109,110], но в тоже время, общая выживаемость остается низкой 45-53% [60, 59, 98,110, 109, 112, 113, 114]. Лечение ОМЛ состоит из индукции и постиндуктивной химиотерапии. Несмотря на усиление терапии и применения новых химиотерапевтических препаратов, частота рецидивов составляет 35%, наибольшая часть которых развивается в первые два года после лечения. Современные европейские исследования, в том числе AML-BFM, MRCAML, NOPHO-AML, включая протокол НИИ ДО 2002 дали аналогичные оценки бессобытийной выживаемости (от 41% до 56%) наиболее частой причиной неудач в этих исследованиях был ранний рецидив, что требует разработки эффективных методов позволяющих преодолеть лекарственную резистентность опухолевых клеток [6, 10, 49, 58, 59, 60, 64, 83, 112, 114]. В последние годы основным направлением в терапии является преодоление резистентности опухолевых клеток, путем интенсификации терапии за счет увеличения доз, количества курсов химиопрепаратов. В исследовании MRC AML 12 доказано отсутствие улучшения выживаемости больных при увеличении числа курсов химиотерапии (общая выживаемость при проведении 4 или 5 курсов была одинакова и составила 74%, бессобытийная – 62% и 63% соответственно, риск рецидива в обеих группах – 36%)[114]. 12 Таблица 1 Стратификация больных ОМЛ по группам риска Исследовательская Группы риска Группа низкого риска Группа стандартно группа риска AIEOP LAM [109] Группы низкого и t(8;21), inv(16)/t(16;16), высокого риска и ремиссия после 1 курса химиотерапии AML-BFM [8] Группы низкого и FAB-M1/M2 с высокого риска палочками Ауэра, FABM4Eo+, t(8;21), inv(16), количество бластов на 15 день менее 5% при отсутствии FLT3/ITD St Jude AML [8] Группы низкого, t(8;21), inv(16), t(16;16), Все остальные стандартного и хороший ответ на высокого лечение Группа высокого риска Все остальные Все остальные t(6;9), t(8;16), t(16;21), -7, -5, 5q-, исключая FLT3-ITD с негативным MRD после 1го индукционного курса, FAB-M0 или M6, FAB-M7 безt t(1;22), ОМЛ после лечения, вторичный ОМЛ, пациенты с плохим ответом на лечение (MRD более 5% на 22 день, и/или MRD более 0,1% после 2х курсов индукции 13 Продолжение таблицы 1 Группы низкого и Меньше 15% бластов высокого риска после 1го курса и ремиссия после 2го курса, или t(8;21), inv(16), t(16;16), t(9;11) и ремиссия после 2го курса химиотерапии COG AAML0531 [8] Группы низкого, t(8;21), inv(16), t(16;16) стандартного и высокого 11q23 аномалия, t(9;11), более 15% бластов на 15 день, или неполная ремиссия после 2х курсов химиотерапии NOPHO-AML 200425 [83] Все остальные -7, -5, -5q; ответ по костному мозгу M-3 (более 15% бластов) после 1го курса, исключаю всех больных с низким риском по цитогенетики -7, -5q, t(9;22), t(6;9) ELAM02 [53] Группы низкого, стандартного и высокого t(8;21) Все остальные JPLSG AML [8] Группы низкого, стандартного и высокого t(8;21), inv(16), t(16;16) Все остальные -7, -5q, t(9;22), t(16;21), FLT3/ITD, но с полным ответом после 1 курса химиотерапии MRC/DCOG AML [8] Группы низкого, стандартного и высокого t(8;21), inv(16)/t(16;16), независимо от ответа после 1го курса химиотерапии Все остальные Более 15% бластов после 1го курса химиотерапии, или неблагоприятный кариотип [-5, -7, del(5q), abn(3q), t(9;22), комплексный кариотип] Продолжение таблицы 1 ОМЛ НИИ ДОГ [6] Группы стандартного, среднего и высокого 14 t(8;21), inv(16) или FAB-М1, М2, М4 с t(16;16), без экспрессии нормальным В-клеточных антигенов кариотипом или с утратой половой хромосомы или с вовлечением 11q23 исключая t(10;11) или +8 или аномалия длинного плеча хромосомы 3; экспрессия Вклеточных или эритроидных маркеров. FAB-М0, М5, М6, М7, ОМЛ с мультилинейной дисплазией, FABМ1, М2, М4 в сочетании с t(6;9), t(9;11), t(9;22), del (7q-), del(5q-), -7, -5,-3, кольцевидной хромосомой, более 3 хромосомными аномалиями, более 25% бластов в миелограмме на 15 день от начала индукции ремиссии у больных со стандартным и средним риском. 15 Группа BFM отметила улучшение общей и безрецидивной выживаемость при применении митоксантрона у больных ОМЛ с t(8;21) [93]. С другой стороны, часть исследований показало увеличение токсичности, как острой (включающей сильную миелосупрессию с инфекциями в нейтропении, мукозит, острую кардиотоксичность), так и поздней (поздняя кардиотоксичность, в том числе сердечная недостаточность, аритмии, нейрокогнитивные дисфункции), а также развитие вторичных злокачественных новообразований при увеличении доз химиопрепаратов [21, 27]. Все это указывает на невозможность безграничной интенсификации химиотерапии. Другим направлением развития лечения детей с ОМЛ было расширение показаний к проведению трансплантации гемопоэтических стволовых клеток (ТГСК). При сравнении групп детей, получивших аллогенную ТГСК во время первой ремиссии с группой больных, получивших только химиотерапию (ХТ), получены достаточно схожие данные в разных исследовательских группах (Таблица 2). Вопрос о проведении ТГСК у детей, больных ОМЛ в первую ремиссию остается достаточно спорным. В 2002 году проведение ТГСК было рекомендовано в Европе у всех больных, исключая группу низкого риска [29]. В общем, частота рецидивов после аллогеной ТГСК значительно ниже – 18%, чем после химиотерапии, но при этом токсичность, полученная в результате ТГСК значительно выше (7-17% против 1-8% при химиотерапии) [7, 22, 64, 73, 78, 84]. Кроме того, вероятность достижения второй ремиссии после аллогеной ТГСК зачастую ниже, чем после химиотерапии. Еще одна из проблем ТГСК – это поиск донора, известно, что очень малый процент больных имеет родственного совместимого донора, а поиск в регистре потенциальных доноров костного мозга не всегда позволяет найти соответствующего. В связи с выраженной токсичностью от ТГСК, результаты общей выживаемости, в сравнении с группой детей, получавших химиотерапию, были почти одинаковыми. 16 Таблица 2 Исследования по аллогенной ТГСК по сравнению с химиотерапией у детей, с впервые установленным ОМЛ Исследования, количество Группы пациентов для Безрецидивная Общая выживаемость, пациентов проведения ТГСК в первую выживаемость, % % ремиссию AIEOP LAM87-92 Все пациенты Нет данных Общее число – 388 Алло-ТГСК – 78 64±6 Ауто-ТГСК – 110 55±5 химиотерапия – 89 [26, 109] 28±5 AML BFM87 и -93 Пациенты группы высокого Нет данных Общее число – 356 риска Алло- ТГСК – 39 62±3 Ауто- ТГСК +химиотерапии – 317 64±8 [58] LAME 89/91 Все пациенты Общее число – 244 Алло ТГСК – 74 57±7 71±7 химиотерапия – 170 [110] 52±4 55±4 CCG 2891 Все пациенты Общее число – 537 Алло-ТГСК – 181 55±9 60±9 Ауто- ТГСК – 177 42±8 48±8 химиотерапия – 179 [60] 47±8 53±8 17 Продолжение таблицы 2 CCG-251, -213, -2861, -2891, -2941 Общее число – 1464 Алло- ТГСК – 373 химиотерапия – 1091 [78] CCG 2961 Общее число – 633 Алло- ТГСК – 170 химиотерапия – 463 [73] UK-MRC AML 10 Общее число – 315 Алло- ТГСК – 85 химиотерапия – 230 [26,64, 114, ] Все пациенты 47±6 37±4 54±6 45±4 60±8 50±5 67±8 62±5 Все пациенты Все пациенты 68 59 18 В последние годы стала активно изучаться биология опухолевой клетки, поэтому третьим и достаточно перспективным путем лечения детей, больных ОМЛ, является направление, состоящее из изучения и применения новых препаратов, влияющих на развитие опухоли (Таблица 3). Исследования в области молекулярной генетики сыграли важную роль в расшифровке молекулярного патогенеза ОМЛ. Генные мутации, дисрегуляция экспрессии генов или набора генов позволяют выделить огромное разнообразие генетических подмножеств острых лейкозов. Результаты этих исследований могут оказаться важными для прогноза, а, следовательно, для определения тактики лечения и применения препаратов целенаправленного действия (Таблица 4) [33]. Таблица 3 Молекулярные таргетные препараты, использующиеся при лечении ОМЛ [71] Класс Мишень Препараты Эпигенетические Гистон деацетилазы Вальпроевая кислота, препараты вариностат ДНК-метилтрансферазы Азацитидин, децитабин Ингибиторы киназ FLT3 Мидостаурин, леустауртиниб, сорафениб, АС220 Гемтузумаб озагомицин, CLS123 Моноклональный антитела CD33 Протеосомальные ингибиторы Ингибиторы mTOR NF-κB-рецептор Бортезомиб Белок mTOR Ингибиторы фарнесилтрансферазы Фарнесилтрансфераза Сиролимус, темсиролимус Типифарниб 19 Таблица 4 Прогностическое значение и ответ на терапию отдельных молекулярных маркеров при ОМЛ Биомаркер Прогностическое значение Ответ на лечение CBF-ОМЛ: t(8;21(q22;q22); RUNX1RUNX1T1 и inv(16)(p13.1q22) или t(16;16)(p13,1q22); CBFB-MYH11 Благоприятный прогноз ОМЛ с inv(16) при наличии трисомии 22 выше риск рецидива Вторичные мутации KIT и FLT3 связаны с плохим ответом на лечение в некоторых исследованиях Высокая вероятность рецидива у пациентов с персистенцией молекулярных маркеров [29, 45, 75] FLT3-ITD Неблагоприятный прогноз Очень плохой прогноз при ОМЛ с высоким числом диких мутантных аллелей FLT3-ITD ОМЛ с FLT3-ITD расположенным вне юкстамембранного домена (около 30% от всех данных случаев), по всей видимости, связано с наихудшим прогнозом [52, 63,104] Генотип «мутация NPM1 без FLT3-ITD» связан с благоприятным прогнозом Сочетание с другими мутациями, такими как IDH1, IDH2, DNMT3A, TET2 генов, в настоящее время NPM1 Индукция с высокими дозами цитарабина считается стандартом терапии для пациентов с CBF-ОМЛ В целом, пациенты не являются кандидатами для аллогенной ТГСК, хотя ТГСК может быть рассмотрена для пациентов с плохим ответом на индукционную терапию, ТГСК связана с низкой трансплантационной смертностью Добавление анти-CD33 к терапии улучшило общую выживаемость KIT-ингибиторы (дазатиниб) в сочетании с химиотерапией проходят 2 фазу клинических исследований [29, 47] Аллогенная ТГСК, вероятно, улучшает прогноз Пациенты должны включаться в клинические исследования с применением FLT3 тирозинкиназ ингибиторов [41, 76] При наличии нормальной цитогенетики хороший ответ при стандартной химиотерапии с применением высоких доз цитарабина 20 Продолжение таблицы 4 изучается NPM1 мутации связаны с хорошим ответом на лечение даже у пациентов старше 70 лет [29, 42,44] CEBPA IDH1, IDH2 WT1 Не рекомендовано проведение алогенной ТГСК в первую ремиссию, вопрос о проведении ТГСК, должен быть рассмотрен индивидуально Наличие других мутаций (IDH1, IDH2, DNMT3A) не должно влиять на выбор терапии [68, 69] Благоприятный прогноз Хороший ответ при стандартной Выделяют две подгуппы: одиночная и двойные химиотерапии с применением высоких доз мутации CEBPA [34, 79] цитарабина, не рекомендовано проведение аллогенной ТГСК в первую ремиссию Прогностическое значение варьирует в зависимости от Ответ на лечение не известен типа мутации IDH IDH ингибиторы в стадии доклинических IDH1 мутации – более высокий риск рецидива, низкая исследований общая выживаемость, однако в различных [72] молекулярных субтипах эффект так же различается IDH2 мутации редко встречаются в сочетании с другими (NPM1, CEBPA, FLT3-ITD), связаны с низким достижением ремиссии Вопрос о прогностическом значение IDH2 остается спорным [9, 48, 49] Прогностическое значение остается спорным, хотя Ответ на лечение не известен большинство исследований сообщают о плохом прогнозе Дополнительные исследования, преимущественно в большой группе пациентов, необходимы для изучения прогноза при различной постремиссионной терапии Продолжение таблицы 4 RUNX1 TET2 DNMT3A 21 WT1 SNP мутация в экзоне 7 в одном исследовании показан хороший прогноз при ОМЛ нормальной цитогенетикой [63,74, 97] Прогностическое значение не ясно В части исследований показана связь RUNX1 мутации с низкой вероятностью достижения ремиссии и плохим прогнозом [14, 89] Прогностическое значение не известно В исследовании CALGB обнаружено неблагоприятное влияние на прогноз при наличии благоприятного типа ОМЛ с мутацией NPM1 без FLT3-ITD; в исследовании AMLSG таких данных не получено [101, 102] Прогностическое значение не известно DNMT3A мутации относятся к группе промежуточного риска по цитогенетическим аномалиям ( как группа с нормальным кариотипом) [32] Ответ на лечение не известен В исследовании AMLSG предположено, что проведение аллогенной ТГСК может улучшить результат [90] Ответ на лечение не известен Ответ на лечение не известен 22 Поиск новой терапии для излечения и улучшения качества жизни детей, больных ОМЛ, главная задача в настоящее время. Учитывая, то что у большинства детей с ОМЛ выявляется нормальный кариотип опухолевых клеток, необходим поиск общих механизмов, контролирующих их жизнеспособность. Именно эпигенетические сигналы накладываются на генетику клеток и по-своему решают реализацию генетической информации [1]. Эпигенетические изменения играют важнейшую роль в патогенезе многих видов злокачественных опухолей, включая лейкоз, влияние на эти механизмы является новым направлением в лечении. Эпигенетика имеет несколько значений. C. Waddington изучал эпигенез, как «генотип во времени своего развития, приводящий к формированию фенотипа» [119]. В отличие от него, A. Riggs et al. определяли эпигенетику, как «изучение митотически и/или мейотически наследуемых изменений функции генов, которые не могут быть объяснены изменениями последовательности ДНК» [17]. Эти определения существенно различаются, хотя часто путаются, но относятся к одному явлению. Термин C. Waddington охватывает всю онтогенетику, которая изучает, изменения активности генов в процессе развития фенотипа, но он устарел. Определение, выдвинутое A. Riggs et al., указывает, что эпигенетика не является наследуемым мутационным изменением [91]. Нынешнее определение эпигенетики – это наследственное изменение экспрессии генов, которое происходит независимо от нарушений в первичной последовательности ДНК. Большинство из этих наследственных изменений определяются при дифференцировке клеток и стабильно поддерживаются через несколько циклов деления клеток, что позволяет клеткам иметь отличительные черты и содержать одинаковую генетическую информацию. Молекулярными основами эпигенетики являются: метилирование ДНК, посттрансляционные модификации белков гистонов, а также ремоделирование нуклеосом [1, 95]. Имея нормальный генотип, нарушения эпигенетических механизмов могут сделать человека склонным к развитию опухоли или другого хронического заболевания. Так, например однояйцевые близнецы, которые наследуют 23 одинаковый набор генов от родителей, могут, в конечном итоге, различаться физическими характеристиками, поведением, и предрасположенностью к болезням. Возможно, эти различия возникают из-за эпигенетических факторов, влияющих на активацию определенных генов, но когда и где эти гены становятся активными неизвестно. Метилирование ДНК и модификация гистонов два основных примера процессов, в результате которых возможны эпигенетические модификации генома. Изменения, связанные с обоими этими процессами накапливаются на протяжении всей жизни человека и могут передаваться от одного поколения к другому [43]. Ученые университета John Hopkins, установили, что профиль эпигенетических маркеров изменяется по мере старения человека, причем степень изменений схожа среди членов одной семьи, что объясняет возрастающую с возрастом предрасположенность к развитию различных заболеваний [24]. В этом исследовании изучалась степень метилирования гистона. Уровень метилирования значительно варьирует между разными людьми. Было обнаружено, что примерно у 30% участников исследования в течение 11 лет степень метилирования значительно изменилась: у одной части наблюдаемых общий уровень метилирования повысился, в то время как у другой – снизился. Таким образом, было получено доказательство того, что эпигенетика человека действительно изменяется с возрастом [12, 31, 55]. Исследования однояйцевых близнецов показали, что в детстве близнецы имеют аналогичную степень метилирования ДНК, в то время как в более старшем возрасте появляются существенные различия. Возможно, что эти негенетические возрастные особенности приводят к различной предрасположенности к заболеваниям [36]. Исследование степени изменения уровня метилирования ДНК в течение 16 лет у 126 индивидуумов, являющихся представителями 2-3 поколений одних и тех же семей показало, что для членов одной семьи изменения метилирования обычно имеют похожий характер. Weaver I. C. показал, что при отсутствии естественного вскармливания у людей в дальнейшей жизни наблюдается снижение количества рецепторов метилирования [37,120, 121]. 24 Эпигенетические модификации, которые изменяют структуру хроматина можно разделить на четыре основные категории: 1) метилирование ДНК, 2) ковалентная модификация гистонов, 3) нековалентные механизмы, 4) некодирующие РНК, влияющие на экспрессию генов. Взаимодействие этих изменений создает "эпигенетический пейзаж", который регулирует способность генома проявляться в различных типах клеток их стадий развития и различных заболеваний, в том числе опухолевых [56, 57]. Метилирование ДНК – эпигенетический механизм, используемый для долгосрочного глушения экспрессии генов. Механизм метилирования создается во время развития плода и, как правило, поддерживается в течение всей жизни человека. Однако в ДНК людей старшего возраста часто можно увидеть отклонения от предполагаемых образцов метилирования, что может привести к менее строгому контролю экспрессии генов и увеличению нестабильности генома. Такие эпигенетические механизмы могут играть важную роль в инициировании и поддержании злокачественных клеток опухолей [5, 33]. Метилирование ДНК имеет важнейшую роль в контроле деятельности генов и строении ядра клетки. Метилирование ДНК заключается в присоединении метильной группы к цитозину, в составе CpG-динуклеотида в позиции С5 цитозинового кольца. CpG- участки не являются случайно распределенными в геноме, неметилированные CpG-динуклеотиды сгруппированы в так называемые «CpG-островки», которые охватывают 5' концы регуляторной области многих генов. Метилирование CpG - островков приводит к деактивации гена [16, 40, 43]. Метилирование ДНК в клетке контролирует все генетические процессы, в том числе такие как: транскрипция, репликация, рекомбинация, репарация, транспозиция генов. Было установлено, что система метилирования в злокачественных клетках значительно отличается по сравнению с нормальными клетками, причем тотальное деметилирование генома сопровождается увеличением активности метилтрансферазы и локальным гиперметилированим CpG – островков. Так как метилирование в нормальных клетках контролирует все генетические процессы, 25 то эти нарушения ведут к изменению структуры хроматина и функции ДНК, внося тем самым вклад в создание генетической и фенотипической изменчивости опухолевой клетки [17]. Тотальное гипометилирование генома было обнаружено, как одно из первичных нарушений метилирования ДНК в злокачественных клетках. Уменьшение количества метильных групп является одним из ранних событий в клеточной трансформации, зачастую еще до появления сформированной опухоли. Роль гипометилирования ДНК была доказана на основании данных о том, что содержание грызунов на безметиониновой диете, ведущей к дефициту метильных групп, вызывает гипометилирование ДНК и образование опухолей печени. Но конкретные механизмы канцерогенного эффекта до сих пор неясны [16, 123]. Наряду со снижением общего уровня метилирования ДНК, в опухолях обычно наблюдаются участки ДНК с ненормально повышенным уровнем метилирования. располагается предполагается, Так внутри что как большая часть CpG-островков именно эти в неметилированных регуляторных цитозины цитозинов участках подвергаются генов, аберрантному метилированию в опухолевых клетках [17]. Это предположение подтверждается тем, что неметилированный тканях CpG-островок в промоторе гена кальцитонина, расположенного на коротком плече хромосомы 11 (11р), метилирован с высокой плотностью при солидных опухолях, лейкозах и клетках, трансформированных различными вирусами [18, 35, 43]. А гены супрессоры опухолей инактиврованы аберрантным метилированием CpG-островков промоторной области. Была выдвинута гипотеза, что данный участок хромосомы 11p является горячей точкой метилирования CpG-островков при злокачественных опухолях, а аберрантное метилирование является важным механизмом выключения генов-супрессоров. Конкретные гены могут быть промоутерами гиперметилирования определенного вида опухоли, в настоящее время разрабатываются профили метилирования для различных видов опухолей [35]. ДНК метилтрансферазы способны приводить к превращению цитозина в урацил в случае недостатка S-аденозилметионина. Участие ДНК метилтрансфераз 26 в мутагенезе подразумевает наличие повышенной экспрессии фермента, пониженной концентрации соответствующей системы S-аденозилметионина репарации. Было и снижения показано, что активности активность метилтрансфераз повышена в опухолевых клеточных линиях в 4-3000 раз по сравнению с нормальными тканями, однако наличие биохимических условий необходимых для превращения цитозина в тимин или урацил пока неясно [17, 18, 91]. Метилирование ДНК – является основой геномного импринтинга. Геномный импринтинг является формой неменделевского эпигенетического наследования, которое характеризуется дифференциальной экспрессией гена в зависимости от его родительского происхождения – матери или отца. Последующие генетические модификации могут привести к тому, что изменения в экспрессии генов будут стабильно передаваться в процессе развития клеточных поколений [16, 30]. В последние годы интенсивно исследуется эффект геномного импринтинга в связи с различной патологией у человека. Примеров заболеваний, в основе которых лежит расстройство функции импринтированных участков генома, довольно много, поэтому можно говорить об особом классе заболеваний человека — «болезнях импринтинга». Наиболее убедительные данные получены при синдроме Прадера-Вилли, Беквита-Видемана. Кластер генов, локализованный в коротком плече 11 хромосомы, участвует в развитии синдрома БеквитаВидемана, ассоциирующегося с эмбриональными опухолями, такими как опухоль Вильмса. Опухоль этого типа развивается у детей в раннем возрасте из метанефрических бластных клеток. Было обнаружено, что в 70% случаев в опухолях Вильмса имеется аберрантное деметилирование материнского аллея и биаллельная экспрессия гена инсулиноподобного фактора роста IGF2, который при сверхэкспрессии проявляет свойства онкогена. Биаллельная экспрессия IGF2 часто наблюдается в фенотипически нормальных тканях окружающих опухоль, то есть является ранним событием при возникновении опухоли Вильмса [38, 43]. Нарушение системы метилирования проявляется на ранних стадиях злокачественной трансформации клеток. С медицинской точки зрения это 27 открывает возможности для ранней диагностики и лечения заболевания. Тем более, что в отличие от мутаций, которые принципиально необратимы, модификации ДНК, хотя и весьма стабильны, но принципиально обратимы [3, 33, 35, 38, 77]. Метилирование ДНК модификацией белков модификации гистонов происходит гистонов. Наиболее являются параллельно с химической распространенными метилирование, видами ацетилирование, фосфорилирование, убиквитинирование (соединение С-конца убиквитина с боковыми остатками лизина в субстрате) и сумоилирование (ковалентное связывание эпсилон-аминогруппы полипептида, с белком лизина, SUMO, расположенной представляющим из на себя С-конце компонент убиквитиновой системы) [16]. Гистоны (ядерные белки, необходимые для сборки и упаковки нитей ДНК в хромосомы) формируют октамерные структуры, вокруг которых закручивается нить ДНК, образуя, таким образом, нуклеосомы. Существует пять различных типов гистонов: H1/H5, H2A, H2B, H3, H4. Гистоны не просто участвуют в ДНКупаковке белков, но входят в молекулярные структуры, которые участвуют в регуляции экспрессии генов. Модификация гистонов происходит в различных белках. Аминокислотные посттрансляционным остатки модификациям: гистонов могут ацетилированию, подвергаться фосфорилированию, метилированию. Модификации аминокислотных остатков гистоновых белков происходят, в основном, в N-терминальных участках, которые расположены за пределами компактного клеточных сигналов. В октамера, и зависимости подвергаются от типа и действию места различных модификаций аминокислотных остатков, каждая нуклеосома имеет свой «гистоновый код», регулирующий активность транскрипции. Ацетилирование и метилирование гистонов оказывают прямое воздействие на различные ядерные процессы, включая транскрипцию генов, репарацию ДНК, репликацию ДНК, и формирование хромосом. Функциональные последствия метилирования гистонов зависят от типа аминокислоты – лизин (возможно моно-, ди-, триметилирование) 28 или аргинин (возможно моно- и диметилирование) и сайта модификации, где происходит метилирование (Таблица 5), в результате происходит репрессия или активация транскрипции [46]. Метилирование гистона Н3 в K4 тесно связано с транскрипционной активацией, в то время как метилирование гистона H3 в K9 или K27, гистона H4 в K20 связано с транскрипционной репрессий. Таблица 5 Роль основных модификаций в регуляции транскрипции Модификация Роль в транскрипции Сайты модифицирования Ацетилирование Активация Н3 (К9, К14, К18, К56) Н4 (К5, К8, К12, К16) Н2А (?) Н2В (К6, К7, К16, К17) Фосфорилирование Активация Н3 (S10) Метилирование Активация Н3 (К4, К36, К79) Репрессия Н3 (К9, К27) Н4 (К20) Метилирование ДНК и модификация гистонов имеют важное значение для физиологической деятельности клеток и тканей [39]. Усиление или потеря ацетилирования и метилирования лизина гистонов наблюдаются в опухолевых клетках, при этом общая картина в геноме нарушается [16, 18]. В опухолевых клетках были обнаружены потери ацетилирования гистона H4 (K16) и триметилирования гистона Н4 (K20) и, как следствие, нарушение основной структуры гетерохроматина [123]. Конечно, модели модификации гистонов не идентичны в различных видах злокачественных опухолей и даже на различных этапах их развития. Классификация типов опухолей в различных эпигенетических моделях могла бы помочь дифференцировать их. Диагностика опухолей на основе анализа модели модификации гистона возможна в будущем [46, 61]. 29 Утрата ацетилирования лизина, а не увеличение метилирования гистонов, было определено в качестве первого шага в глушении генов, а воздействие на активность гистондеацетилазы (ГДАЦ), которая отвечает за удаление ацетильной групп гистонов, стало основой таргетной эпигенетической терапии [123]. Потеря ацетилирования не только приводит к глушению генов, но также может привести к снижению уровня восстановления ДНК, ускоряя молекулярные события, приводящие к развитию опухоли [61, 65]. Ингибиторы ГДАЦ имеют специфический противоопухолевый эффект. Так как механизм их действий остается неясным, возможно, что эти препараты действуют на неопознанные цели, а не только на ацетилирование гистонов. Поиск новых гистон деметилаз является областью интенсивного исследования, так как, вполне вероятно, что каждый фермент связывается с конкретным остатком лизина или аргинина в различной степени метилирования и, даже может быть, возможно, что эти деметилазы взаимодействуют с определенными факторами транскрипции [1, 35, 61]. Представляется, что прекращение ацетилирования гистонов является основным событием, ведущим к глушению генов, тогда как усиление метилирования Н3(K9) играет второстепенную роль. На данный момент не определена вся группа ферментов гистондеметилаз и значение роли каждого фермента в составе всей эпигенетической модели полностью не понята, а целенаправленное действие каждого фермента устанавливается [1, 18, 19, 99]. Помимо хорошо изученных эпигенетических процессов (метилирование ДНК, модификация гистонов) в настоящее время так же изучается роль в туморогенезе и других эпигенетических модификаций таких как нековалентные механизмы и микро-РНК. Нековалентные механизмы, такие как ремоделирование нуклеосом и замена классических белков гистонов со специализированными гистоновыми вариантами, играют важную роль в структуре хроматина в качестве регулировки активности генов. Нуклеосомы так же регулируют экспрессию генов путем изменения доступности регуляторных последовательностей ДНК транскрипционных факторов. Полногеномный анализ нуклеосом позволил 30 выявить общие организационные системы с точным позиционированием нуклеосом вокруг гена промоторов. Взаимодействие нуклеосомного аппарата ремоделирования с метилированием ДНК и модификацией гистонов играет ключевую роль в создании глобальной генной экспрессии и архитектуры хроматина [1, 18, 56, 57]. Микро-РНК – это некодирующие РНК, которые регулируют экспрессию гена через посттранскрипционное глушение генов-мишеней. Микро-РНК экспрессируются в ткани определенным образом и контролируют широкий спектр биологических процессов, включая пролиферацию клеток, апоптоз и дифференцировку. Список микро-РНК определенных в геноме человека и их потенциальных генов-мишеней быстро растет, демонстрируя свою обширную роль в поддержании глобальной экспрессии генов, они могут также влиять на другие эпигенетические механизмы внутри клетки путем направленного влияния на ферменты, ответственные за метилирование ДНК и модификацию гистонов. Такое взаимодействие между различными компонентами эпигенетического аппарата вновь подчеркивает комплексный характер эпигенетических механизмов, участвующих в поддержании глобальной экспрессии генов [16, 18, 95]. Эпигенетическое глушение различных генов может привести к прямой генетической нестабильности, оно было признано в качестве третьего пути удовлетворяющего гипотезе, выдвинутой A. Knudson, которая предполагает необходимость двух ударов для развития опухоли (первый – наследуемая мутация, то есть мутация в клетках зародышевой линии, и второй удар – соматическая мутация) [1]. Таким образом, рождение стволовой лейкозной клетки происходит в результате образования химерного или мутантного гена и в результате эпигенетической активации генов, отвечающих за пролиферацию опухоли, происходит накопление клеток-предшественников и злокачественных клеток, которые не подвергаются апоптозу [38]. При ОМЛ характерным является высокий уровень экспрессии ДНКметилтрансфераз, что сопровождается гиперметилированием промотеров генов - 31 опухолевых супрессоров и ингибиторов циклинзависимой киназы (CDKN2A и CDKN2B), гена эстрогенового рецептора I (ESR1), и гена ретинобластомы (RB1) [2, 66]. В последние годы появились данные целого ряда исследований, доказывающих, что ДНК-метилтрансферазы участвуют в образовании белковых комплексов, подавляющих транскрипцию, которая возникает при участии химерных протеинов, таких, как PML-RARα и AML/ETO [2, 11, 28, 35, 54]. Участие гистондеацетилаз в лейкемогенезе хорошо изучено на классических примерах острого промиелоцитарного лейкоза (ОПЛ) и ОМЛ с транслокацией t(8;21) или сопровождающихся аномалиями 16 хромосомы. При ОПЛ в 95% случаев образуется транслокация t(15;17), ведущая к появлению химерного белка PML-RARα, способствующего синтезу патологического гена Pml-RARα, который блокирует клеточную дифференцировку путем ингибирования рецептора к ретиноидам. Лейкозные клетки нормально дифференцируются до промиелоцитов, после чего происходит полная остановка дифференцировки. Этот блок может быть преодолен назначением ингибитора гистондеаценилазы [2, 4]. Осуществляемое ГДАЦ деацетилирование гистонов служит также сигналом для активации других ферментов: гистоновых метилтрансфераз и ДНК- метилтрансфераз, также включающихся в репрессирующий комплекс. Таким образом, осуществляется эпигенетической взаимодействие регуляции: между модификацией двумя структуры механизмами гистонов и метилированием ДНК. Фармакологические концентрации ретиноевой кислоты, в тысячи раз превышающие физиологические, способны вызвать диссоциацию репрессирующего комплекса и запустить механизм дифференцировки [11]. Одновременно ретиноевая кислота приводит к активации «лиганда смерти» TRAIL, являющегося ключевым компонентом механизма апоптоза [108]. Тот факт, что эпигенетические аномалии, в отличие от генетических мутаций, потенциально обратимы и могут быть восстановлены, делает изучение данного направления терапии достаточно перспективным [95]. Полностью трансретиноевая кислота (ATRA), как «дифференцирующий агент» в терапии ОПЛ представляет собой типичный пример препарата с 32 эпигенетическим действием: не изменяя структуру ДНК, она приводит к разрушению репрессирующего комплекса, деацетилирующего гистоны регуляторных элементов генов, ответственных за дифференцировку клеток. В незначительном числе случаев ОПЛ, с транслокацией t(11;17) образуются химерный гена PLZF/RARA или STAT5b/RAR. Химерные протеины при которых образуют более прочную связь с репрессирующим комплексом за счет дополнительных сайтов в структуре второго участника химерного белка продукта генов PLZF или STAT5. Эта связь не разрушается в присутствии высоких концентраций ретиноевой кислоты именно за счет дополнительных связей репрессирующего комплекса [2, 5]. Следует отметить, что в эксперименте in vitro комбинация полностью трансретиноевой кислоты и ингибиторов гистондеацетилаз, разрушающих дополнительные связи химерных протеинов, позволяет добиться дифференцировки лейкемических клеток [88]. При ОМЛ, сопровождающихся t(8;21) или аномалиями 16 хромосомы с вовлечением генов ЕТО(MTG8), AML1(RUNX1) и CBFβ, происходит нарушение образования нормального транскрипционного комплекса CBF. Этот комплекс состоит из продуктов генов CBFα, AML1(RUNX1) и CBFβ и обладает гистонацетилазной активностью, потенцируя экспрессию большой группы генов, отвечающих за нормальную дифференцировку гемопоэтических клеток. При возникновении различных генетических аномалий, изменяющих нормальную структуру любого из белков комплекса CBF, нарушается его связь с факторомгистонацетилазой , а также с коактиватором и ядерным рецептором TIF2 [2, 106]. В последнее время исследуется множество химических соединений, обладающих функцией ингибирования гистондеацетилаз, однако, большинство из них пока проходят первую или вторую фазу клинических испытаний [65]. Весьма сложными нарушениями сопровождаются повреждения нормального функционирования гена MLL, кодирующего протеин, который обладает гистонметилтрансферазной активностью. Основной функцией этого белка является участие в огромном транскрипционном комплексе, в состав которого входят, по меньшей мере, 29 других белков, вовлеченных как в ацетилирование и 33 деацетилирование гистонов, так и в метилирование гистонов и ДНК [13]. Этот комплекс осуществляет регуляцию транскрипции генов гомеобокса (генов, вовлеченные в регуляцию развития организма), участвующих в эмбриональном развитии и экспрессирующихся в ранних предшественниках гемопоэза. В различных транслокациях с участием MLL (которых известно около 60 для всех видов лейкозов) образуются химерные белки, потерявшие метилтрансферазную активность за счет отсутствия соответствующего домена в составе химерных генов, кодирующих их [28]. Как уже отмечалось, в отличие от генетических аномалий, которые являются необратимыми, эпигенетические изменения могут быть изменены фармакологически. Следовательно, возможно эпигенетические препараты, имеют сильный терапевтический потенциал в лечении ОМЛ [95].Так как они действуют только на делящиеся клетки, можно утверждать, что лечение этими препаратами должно быть в основном нацелено на быстро делящиеся опухолевые клетки и иметь минимальное воздействие на медленное деление нормальных клеток. Этот аргумент подтвержден исследованиями, демонстрирующими минимальные побочные эффекты препаратов с эпигенетическим действием [23]. Один из препаратов, ингибирующий деацетилазы, является вальпроевая кислота (ВК) – антиконвульсант. это короткоцепочная Показано, что она жирная индуцирует кислота, пероральный дифференцировку и трансформацию гемопоэтических клеток-предшественников и лейкозных бластов в костном мозге Предположительно, и периферической ВК оказывает крови больных противоопухолевое ОМЛ [94, действие 116]. путем протеосомальной деградации гистондеацетилазы (ГДАЦ), в частности, ГДАЦ 2. Кроме того, при ОМЛ с t(8;21), ВК может влиять на химерный ген AML1/ETO. ВК стимулирует глобальную диссоциацию AML1/ETO- ГДАЦ комплекса из промотора генов AML1/ETO, и вызывает значительное торможение деятельности гистона H3 и гиперацетелирование гистона H4, РНК-полимеразы II, что приводит к транскрипционной реактивация генов-мишеней, т.е. подавление гибридного 34 белка AML1/ETO. Эти фармакологические эффекты приводят к подавлению лейкемической активности. Другим препаратом, влияющим на ацетилирование и метилирование гистонов является полностью транс-ретиноевая кислота (ATRA), эффективность которой подтверждена при лечении ОПЛ. Полностью транс-ретиноевая кислота (ATRA) – природный метаболит ретинола, принадлежащий к классу ретиноидов, который включает природные и синтетические аналоги. ATRA может индуцировать дифференцировку и апоптоз опухолевых клеток при разных вариантах ОМЛ. Ryningen A. et al. исследовали биологических эффектов ATRA в монотерапии у 22 пациентов с ОМЛ (исключены больные ОПЛ), которые в течение двух дней получали монотерапию ATRA. Проводился анализ образцов крови до и после двух дней приема ATRA у 12 пациентов, которые имели в периферической крови циркулирующие лейкозные клетки. Так было отмечено, что при приеме ATRA увеличился процент циркулирующих бластных клеток в фазе G0/G1 для 9 из 12 пациентов. Циркулирующие опухолевые клетки, полученные во время терапии, имели увеличенный внутриклеточный уровень P21, и снижение уровня траскрипционных факторов. Таким образом, in vivo лечение ATRA при ОМЛ влияет на морфологию лейкозных клеток, регуляцию клеточного цикла и апоптоз, и, возможно, также микрососудистые эндотелиальные функции клеток [50]. Tonnelly et al. показали, что ВК индуцирует сильное торможение роста клеток на клеточных линиях острого миелобластного лейкоза. Исследования in vitro показали, что ВК может активировать p53-независимые G1 клеточный цикл и апоптоз в MLLmut ОМЛ. Помимо ГДАЦ ингибирования, ВК также синергично с полностью транс-ретиноевой кислотой (ATRA) индуцирует дифференцировку бластов при ОМЛ in vitro [102, 105]. Несколько клинических испытаний проводимых с назначением ГДАЦ у пациентов с миелодиспластическим синдромом (МДС) и/или ОМЛ показали низкий уровень ответа, как правило, в диапазоне 10-20% [54, 62, 71, 111]. 35 Наиболее распространенные виды токсичности это усталость, тошнота, рвота и диарея, которые и являлись наиболее общими причинами, ограничивающим дозу токсичности [111, 115, 117]. Увеличение ацетилирования гистонов было обнаружено большинством исследований, но никакой корреляции с клиническим ответом не было установлено [51, 115]. Существует обоснование использования ГДАЦ с другими агентами, такими как гипометилирующие вещества и обычные химиотерапевтические препараты. Исследования in vitro и in vivo показали, что ВК более эффективна в сочетании с различными препаратами. Siitonen T. et al. исследовали воздействие ВК в качестве монотерапии с цитарабином (Ara-C) и этопозидом (VP-16) на линии острого миелобластного лейкоза. Они отметили, резкое увеличение цитотоксичности при сочетании ВК и Ara-C [96]. SanchezGonzalez B. et al. изучали клеточные и молекулярные изменения при использовании сочетания антрациклина идарубицина с ВК клеточных линиях ОМЛ. В этом исследовании было показано, что сочетание с антрациклинами дает синергический эффект in vitro, что должно приводить к значительной клинической активности у пациентов с лейкозом [15, 96]. Так же исследования in vitro показали, что препарат, ингибирующий ГДАЦ в сочетании с ДНК гипометилирующим веществом действуют синергически, вызывая ретроэкспрессию и замалчивание генов, отвечающих за пролиферацию в опухолевых клетках [82, 99]. Kuendgen A. et al. исследовали эффективность ВК в монотерапии и в сочетании с ATRA. У 75 больных с МДС (n=43) и вторичным ОМЛ (n=32), у девяти пациентов с начала лечения была включена ATRA. Ответ оценивался согласно критериям Международной Рабочей Группы по МДС и был достигнут у 35% пациентов. В группе больных ОМЛ у 16% пациентов [85, 111]. Soriano A. et al. проводили изучение сочетания децитабина с ВК и ATRA у пациентов с ОМЛ и с высоким риском миелодиспластического синдрома. В исследование было включено 53 пациента, средний возраст составил 69 лет, из них 49 (92%) больных ОМЛ и 4 (8%) – МДС. При количестве бластов в костном 36 мозге менее 5%, восстановлении в периферической крови нейтрофилов (>1×10 9/л) и тромбоцитов (>100×109/л) фиксировалась ремиссия; при количестве бластов в костном мозге менее 5%, восстановлении в периферической крови нейтрофилов (>1×109/л), сохраняющейся тромбоцитопении (˂100×109/л) – частичная ремиссия; при сохраняющейся панцитопении, но количестве бластов в костном мозге менее 5% – костномозговой ответ. Продолжительность ремиссии, которая рассчитывалась от момента установления ремиссии до диагностики рецидива, составила в среднем 26 недель. В это исследование было включено три пациента в возрасте 5, 15 и 18 лет с рецидивом/рефрактерным течением ОМЛ. У больного 15 лет был зафиксирован по данным пунктата костного мозга ответ на лечение. Не было значимой токсичности у данной группы пациентов [92]. Garcia-Manero G. et al. проводили исследование сочетания децитабина и ВК у 54 пациентов с ОМЛ, средний возраст которых составил 60 лет. Ответ наблюдался у 22% пациентов, в том числе у 19% полная ремиссия, общая выживаемость составила 15,3 месяцев. В исследование было включено семь детей в возрасте от 4 до 21 лет с рецидивами / рефракторным течением ОМЛ. Хотя ни полная, ни частичная ремиссия не была достигнута, у 3 больных был ответ по костному мозгу, у одного больного отмечалось снижение бластных клеток в костном мозге до 6%. Не было описано значительной токсичности у детей, связанных с терапией [105]. Следовательно, комбинация ВК с децитабином и ATRA требует дальнейшего изучения в педиатрической практике [92]. Nervi C. et al. исследовали сочетание ВК и ATRA. Они наблюдали только один случай гематологического улучшения. Однако, у всех пациентов (n=10) были, морфологические, цитохимические и иммунофенотипические изменения дифференцировки, что было подтверждено путем демонстрации цитогенетических нарушений в созревании клеток [117]. Мы предполагая, что блокирование генов, способствующих пролиферации бластов позволит увеличить дифференцировку «созревание» опухолевых клеток, путем добавления к химиотерапии препаратов с эпигенетическим действием. 37 Таким образом, использование ВК и ATRA в сочетании с химиотерапией, возможно, позволит увеличить частоту полных ремиссий по сравнению только с химиотерапией и увеличить выживаемость больных. 38 ГЛАВА 2. ХАРАКТЕРИСТИКА ПАЦИЕНТОВ, МЕТОДОВ ИССЛЕДОВАНИЯ И ЛЕЧЕНИЯ В исследование был включен 121 ребенок, с подтвержденным диагнозом ОМЛ. Из них 51(42,1%) больной получил лечение по протоколу ОМЛ НИИ ДО 2002, 70 (57,9%) – по протоколу ОМЛ НИИ ДОГ 2007. Пациенты лечились в 8 онкологических стационарах: 1) отделении химиотерапии гемобластозов НИИ ДОГ ФГБУ РОНЦ РАМН им. Н.Н. Блохина – 66 (54,0%) больных; 2) отделении онкологии онкодиспансера г. Балашиха – 29 (24,0%); 3) онкологическом отделении ГБУЗ ДККБ г. Краснодара – 8 (6,6%); 4) в детском онкогематологическом центре ГУЗ ОДБ г. Ростов-на-Дону – 7(5,8%); 5) отделении детской онкологии и гематологии ГУЗ областной детской клинической больницы г. Екатеринбурга – 1 (0,8%); 6) отделении онкогематологии ДГКБ №1 г. Санкт-Петербурга – 6 (5,0%); 7) отделении химиотерапии ДОКБ им. П.Г. Выжлецова г. Архангельска – 2 (1,7%); 8) онкогематологическом отделении БУЗ Воронежской областной детской клинической больницы № 1 – 2 (1,7%) в период с февраля 2006 по февраль 2012гг. Результаты оценивались на 31 мая 2013г. Наблюдаемые дети были в возрасте от 2,5мес. до 17 лет. Средний возраст больных составил 7,8±0,5лет. Из них мальчиков было 68 (52,2%), девочек 53 (43,8%). Распределение больных в зависимости от пола и возраста в различных протоколах представлены в таблицах 6 и 7. Средний возраст детей, больных ОМЛ, лечившихся по протоколу ОМЛ НИИ ДО 2002, был 8,9±0,8 лет, из них мальчиков – 26 (51,0%), девочек – 25 (49,0%). Мальчиков было несколько больше, но они преобладали только в возрастных группах от 1 года до 3х лет и от 7 лет до 14 (р=0,19). 39 Таблица 6 Распределение больных, лечившихся по протоколу ОМЛ НИИ ДО 2002, в зависимости от пола и возраста Возраст Пол Общее количество (годы) Мальчики больных Девочки N % N % N % ˂1 1 25,0 3 75,0 4 7,8 1,1 – 3 5 71,4 2 28,6 7 13,7 3,1- 7 4 36,4 7 63,6 11 21,6 7,1-14 14 63,6 8 36,4 22 43,1 >14 2 28,6 5 71,4 7 13,7 Всего 26 51,0 25 49,0 51 100 Таблица 7 Распределение больных, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от пола и возраста Возраст Пол Общее (годы) Мальчики количество Девочки больных N % N % N % ˂1 7 50,0 7 50,0 14 20,0 1,1 – 3 7 53,8 6 46,2 13 18,6 3,1- 7 9 81,8 2 18,2 11 15,7 7,1-14 14 63,6 8 36,4 22 31,4 >14 5 50,0 5 50,0 10 14,3 Всего 42 60,0 28 40,0 70 100 Средний возраст детей, больных ОМЛ, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, был 7,1±0,7 лет, из них мальчиков – 42 (60,0%), девочек – 28 (40,0%). В большинстве групп преобладали мальчики, только в возрасте младше 1 40 года и старше 14 лет – количество больных мальчиков и девочек было одинаково (р=0,48). Сравнивая распределение пациентов, получавших лечение по двум протоколам, не было выявлено достоверной разницы в распределении больных по полу и возрастным группам (p=0,32 и p=0,23 соответственно). Диагноз устанавливался на основании морфологических, цитохимических, иммунологических и цитогенетических параметров бластных клеток, обнаруживаемых в периферической крови и костном мозге. Морфологическая идентификация бластных клеток проводилась по мазкам пунктатов костного мозга и периферической крови, окрашенных по методу Романовского-Гимза. Типирование бластных клеток осуществлялось по критериям международной классификации FAB. Иммунофенотипирование бластных клеток костного мозга проводилось методом непрямой моноклональных иммунофлюоресценции антител. с Оценку результатов использованием проводили комплекса на проточном цитометре Fascan (Becton, Dickinson, США) в гейте бластных клеток, идентифицируемых на основании светорассеяния. Антигенположительным считался случай с экспрессией маркера на более, чем 20% бластных клеток. Исследование иммунофенотипической характеристики бластных клеток костного мозга проводилось вед.н.сотр, профессором Н.Н.Тупицыным и ст.н.сотр, к.м.н. Л.Ю. Гривцовой в лаборатории клинической радиоиммунологии (зав. лабораторией д.м.н., профессор З.Г.Кадагидзе). Распределение больных по вариантам ОМЛ представлены в таблице 8. Чаще всего у больных был диагностирован FAB-М2 вариант – 32 (26,4%) ребенка, FABМ4 – 29 (24,0%) и FAB-М1 – 25 (20,7%) детей. Среди, получивших лечение по протоколу ОМЛ НИИ ДО 2002, большую часть составили больные с FAB-М1 – 19 (37,3%) и FAB-М2 – 13 (25,5%) вариантами. В группе, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, с FAB-М2 – 19 (27,1%) и FAB-М4 – 20 (28,6%). 41 Таблица 8 Распределение больных, в зависимости от FAB морфологического варианта и протокола лечения (р=0,002) Вариант НИИ ДО ОМЛ НИИ ДОГ ОМЛ Общее ОМЛ 2002 2007 количество больных N % N % N % М0 3 5,9 2 2,9 5 4,1 М1 19 37,3 6 8,6 25 20,7 М2 13 25,5 19 27,1 32 26,4 М4 9 17,6 20 28,6 29 24,0 М5 4 7,8 17 24,3 21 17,4 М6 2 3,9 2 2,9 4 3,3 М7 1 2,0 4 5,7 5 4,1 Всего 51 100 70 100 121 100 У 84 больных был проведен хромосомный анализ с использованием общепринятого метода краткосрочного (24 и 48 ч) культивирования клеток костного мозга и/или периферической крови. Распределение пациентов, у которых удалось провести цитогенетическое исследование, представлено в таблице 9. Таблица 9 Распределение детей с ОМЛ в зависимости от кариотипа и протокола лечения Кариотип ОМЛ НИИ ДО ОМЛ НИИ Всего 2002 ДОГ 2007 N % N % N % 46XX/46XY 8 20,5 8 17,8 16 19,0 t(8;21) 8 20,5 3 6,7 11 13,1 t(8;21) + 3 7,7 4 8,9 7 8,3 другие аномалии inv16/t(16;16) 4 10,3 7 15,6 11 13,1 t(6;9) 1 2,2 1 1,2 t(9;11) 2 5,1 2 4,4 4 4,8 Более 3 3 7,7 3 6,7 6 7,1 аномалий t(6;11) 1 2,6 1 1,2 42 Продолжение таблицы 9 Другие аномалии 47XX/47XY (+21) del 11(q23) Всего 7 17,9 3 7,7 39 16 35,6 1 47 100 2,2 100 23 27,4 3 3,6 1 84 1,2 100 Данные цитогенетического исследования опухолевых клеток подтвердили высокую гетерогенность ОМЛ у детей. ОМЛ с повторяющимися генетическими аномалиями был диагностирован у 39 пациентов из 84 (46,5%). Не повторяющиеся генетические аномалии были обнаружены у 23 (27,4%) больных, а нормальный кариотип у 16 (19,0%) (р=0,2). Все дети с ОМЛ, лечившиеся по двум протоколам, были стратифицированы на группы риска: 1. Стандартный риск. В группу стандартного риска включены дети больные ОМЛ с t(8;21), inv(16) или t(16;16). Морфологические варианты не учитывались. 2. Средний риск. Больные с морфологическими FAB-М1, М2 или М4 вариантами, с нормальным кариотипом или утратой половой хромосомы, с вовлечением 11q23, исключая t(10;11), +8, аномалией длинного плеча хромосомы 3 или эритроидных маркеров. 3. Высокий риск. Согласно FAB классификации М0, М5, М6, М7, ОМЛ с мультилинейной дисплазией без предшествующего миелодиспластического синдрома и с предшествующим миелодиспластическим синдромом. Морфологические варианты FAB-М1, FAB-М2, FAB-М4 в сочетании с t(6;9), t(10;11), t(9;22), del(7q-), del(5q-), -7, -5, t(3;5); t(3;3), более 3 хромосомными аномалиями исключая стандартные. Больные с более чем 25% бластов в миелограмме на 15 день от начала индукции ремиссии стандартного и среднего риска. Распределение по группам риска представлено в таблице 10. 43 Таблица 10 Распределение больных ОМЛ по группам риска и протоколу лечения Группа риска ОМЛ НИИ ДО ОМЛ НИИ Всего ДОГ 2007 2002 N % N % N % Стандартный 13 25,5 13 18,6 26 21,5 Средний 25 49,0 22 31,4 47 38,8 Высокий 13 25,5 35 50,0 48 39,7 Всего 51 100 70 100 121 100 Наибольшее число больных вошло в среднюю и высокую группы риска: 47 (38,8%) и 48 (39,7%) соответственно. В протоколе ОМЛ НИИ ДОГ 2007 большая часть больных были отнесены к группе высокого риска, а в протоколе ОМЛ НИИ ДО 2002 к группе среднего риска (р=0,02). После объединения пациентов средней и высокой групп риска в неблагоприятную (Таблица 11), достоверной разницы между больными в двух протоколах не было получено (р=0,36). Таблица 11 Распределение больных по группам риска и протоколу лечения Группа риска ОМЛ НИИ ДО 2002 ОМЛ НИИ ДОГ 2007 N % N % Стандартная 13 25,5 13 18,6 Неблагоприятная 38 74,5 57 81,4 (средний риск и высокий) Всего 51 100 70 100 Лечение Всем больным проводилось лечение, состоящее из индукции ремиссии, консолидации, интенсификации, поддерживающей терапии. 44 Протокол ОМЛ НИИ ДО 2002 Согласно протоколу ОМЛ НИИ ДО 2002 больным со стандартным риском была проведена индукция ремиссии длительностью 8 дней, состоявшая из цитозинарабинозида (Ara-C) 100мг/м2/сут внутривенно (в/в) капельно 24 часа 1 – 2 дни, 100мг/м2 в/в капельно в течение 1 часа каждые 12 часов 3 – 8 дни, идарубицина (IDA) 12мг/м2/сут в/в капельно 1 час 3 – 5 дни, и вепезида (VP-16) 150мг/м2/сут в/в капельно 2 часа 6 – 8 дни. При количестве бластных клеток в миелограмме более 25%, на 15 день от начала лечения продолжалась терапия согласно высокой группе риска. При количестве бластных клеток до 15% на 15 день от начала лечения, терапия консолидации ремиссии откладывалась до восстановления абсолютного содержания гранулоцитов (АСГ) более 0,5×109/л и тромбоцитов более 50×109/л по данным анализа периферической крови (АПК), но не позднее 21-го дня от окончания индуктивного лечения. С целью профилактического поражения ЦНС вводился Ara-C интратекально в дозах в зависимости от возраста в 1-й день химиотерапии (Таблица 12). Таблица 12 Дозы цитозинарабинозида в зависимости от возраста Возраст Ara-C <1года 20мг 1-2 года 26мг 2-3 года 34мг >3 лет 40мг При лейкемическом поражении ЦНС, которое подтверждалось клинически и/или морфологически, также с помощью компьютерной томографии вводились интратекально 3 препарата, дозы препаратов в таблице 13. 45 Таблица 13 Дозы химиопрепаратов в зависимости от возраста. Возраст Ara-C Мtx <1года 1-2 года 2-3 года >3 лет 20мг 26мг 34мг 40мг 5 8 10 12 Преднизолон 4 6 8 10 С 21-го дня от окончания индукции ремиссии начиналась терапия консолидации ремиссии. При количестве бластов в миелограмме на 15 день от начала химиотерапии от 15,1% до 25%, терапия консолидации ремиссии начиналась на 16 день при условии отсутствия инфекционного процесса и возможности адекватного обеспечения больного препаратами крови. При количестве бластов более 25% – лечение согласно протоколу высокого риска. Терапия консолидации ремиссии состояла из: Ara-C 75мг/м2/сут в/в струйно 3 – 6, 10 – 13, 17 – 20 и 24 – 27 дни, рубомицина (даунорубицина) (RUB) 30мг/м2/сут в/в капельно в течение 2 часов 1, 8, 15 и 22 дни, 6-меркаптопурин (6-МП) 60мг/м2/сут внутрь ежедневно 1 – 27 дни. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились в первый и 15 дни консолидации ремиссии в ранее указанных дозировках. По окончании курса консолидации и восстановлении АСГ более 0,5×109/л и тромбоцитов более 100×109/л проводилось 2 курса интенсификации с интервалом в 2 недели, состоящие из высоких доз Ara-C 1000мг/м2 в/в капельно каждые 12 часов 1 – 3 дни, и VP-16 125мг/м2/сут в/в капельно 2 – 5 дни. В дальнейшем больным проводилось облучение ЦНС в суммарно очаговой дозе (СОД) 12Гр при отсутствии поражения ЦНС и 18 Гр при инициальном поражении ЦНС, и поддерживающая терапия. Поддерживающая терапия, состояла из Ara-C 40мг/м2/сут подкожно один раз в день 4-дневным курсом каждые 28 дней, и 6-МП 40мг/м2/сут внутрь ежедневно в течение 78 недель (Рисунок 1). 46 Рисунок 1. Дизайн терапии больных стандартной группы риска протокол ОМЛ НИИ ДО 2002 Больным средней группы риска, проводился курс индукции, такой же, как и у больных группы стандартного риска. При количестве бластных клеток в миелограмме более 25%, на 15 день от начала лечения продолжалась терапия согласно высокой группе риска. С 21-го дня от окончания индукции ремиссии начиналась терапия консолидации ремиссии (аналогичная группе стандартного риска). Затем больные получали один курс интенсификации, после которого проводилась мобилизация и сбор ГСК. После второго курса интенсификации проводилась высокодозная химиотерапия с ауто-трансплантации ГСК (аутоТГСК). При наличии полностью совместимого донора костного мозга планировалась аллогенная (алло) ТГСК. Все больные, кроме тех, кому была проведена ТГСК, после интенсификации II получали поддерживающую терапию (Рисунок 2). Рисунок 2. Дизайн терапии больных средней группы риска протокол ОМЛ НИИ ДО 2002 47 Пациенты с высоким риском ОМЛ или со стандартным и средним риском и уровнем бластов более 25% на 15 день от начала индукции ремиссии получали альтернирующие курсы химиотерапии. Индукция включала два курса химиотерапии, консолидация состояла из аналогичного лечения. Первый и третий курсы – HAM, состоящие из ARA-C 1000мг/м2 каждые 12 часов в/в капельно в течение 3 часов 1-3 дни и митоксантрона 10мг/м2 2-4 дни в/в инфузия в течение 30 мин. Второй и четвертый: флюдарабин 30мг/м2 в/в капельно в течение 2 часов 1-5 дни, VP-16 100мг/м2 в/в капельно через 1 час после окончания инфузии. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились на 6 день курса в ранее указанных дозировках (Рисунок 3). Всем больным, у которых была достигнута ремиссия планировалось проведение консолидации ремиссии с последующей алло-ТГСК (при наличии полностью совместимого донора костного мозга) или ауто- ТГСК при отсутствии полностью совместимого донора костного мозга. Все больные, кроме тех, кому была проведена ТГСК, получали облучение ЦНС (СОД=12 Гр) и поддерживающую терапию, состоявшую из Ara-C 40мг/м2/сут подкожно один раз в день 4-дневным курсом каждые 28 дней, и 6-МП 40мг/м2/сут внутрь ежедневно в течение 78 недель. Рисунок 3. Дизайн терапии больных высокой группы риска протокол ОМЛ НИИ ДО 2002 48 Протокол ОМЛ НИИ ДОГ 2007 Больные, получавшие лечение по протоколу ОМЛ НИИ ДОГ 2007, были стратифицированы по группам риска, так же как и в протоколе ОМЛ НИИ ДОГ 2002. Пациентам стандартной группы риска в протоколе ОМЛ НИИ ДОГ 2007 эпигенетические препараты были включены только в поддерживающую терапию. Индукция ремиссии состояла из Ara-C 100мг/м2/сут в/в капельно 24 часа 1 – 2 дни и 100мг/м2 в/в капельно каждые 12 часов 3 – 8 дни, VP-16 150мг/м2/сут в/в капельно 2 часа 6 – 8 дни, идарубицина (IDA) 12мг/м2/сут в/в капельно 1 час 3 – 5 дни. С целью профилактического поражения ЦНС вводился Ara-C интратекально в дозах в зависимости от возраста в 1-й день химиотерапии (Таблица12). При лейкемическом поражении ЦНС, которое подтверждалось клинически и/или морфологически, также с помощью компьютерной томографии вводились интратекально 3 препарата, дозы препаратов в таблице 13. При количестве бластных клеток в миелограмме более 25% на 15 день от начала лечения начиналась терапия согласно высокой группе риска. При количестве бластных клеток менее 25% в костном мозге на 15 день от начала лечения терапия консолидации ремиссии откладывалась до восстановления абсолютного содержания гранулоцитов (АСГ) более 0,5×109/л и тромбоцитов более 50×109/л по данным анализа периферической крови (АПК), но не позднее 21-го дня от окончания индуктивного лечения. Терапия консолидации ремиссии состояла из: Ara-C 75мг/м2/сут в/в струйно 3 – 6, 10 – 13, 17 – 20 и 24 – 27 дни, RUB 30 мг/м2/сут в/в капельно в течение 2 часов 1, 8, 15 и 22 дни, 6-МП 60 мг/м2/сут внутрь ежедневно 1 – 27 дни. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились в первый и 15 дни консолидации ремиссии в ранее указанных дозировках. По окончанию курса консолидации и восстановлении АСГ более 0,5×109/л и тромбоцитов более 100×109/л проводились 2 курса ранней интенсификации (hAE) с интервалом в 2 недели: Ara-C 1000 мг/м2 в/в капельно каждые 12 часов 1 – 3 дни 49 и VP-16 125 мг/м2/сут в/в капельно в течение 2 часов 2 – 5 дни. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились на 5 день курса в ранее указанных дозировках. После окончания интенсификации II при инициальном лейкозном поражении ЦНС проводилось облучение головного мозга в суммарной очаговой дозе (СОД) 18 Гр. Облучение ЦНС проводилось больным с t(16;16) или inv(16) без инициального поражения ЦНС СОД=12 Гр, РОД=2Гр. Далее больные получали поддерживающую терапию, включавшая 6-меркаптопурин 40 мг/м2/сут ежедневно до 78 недели, Ara-C 40 мг/м2/сут один раз в день 4-дневным курсом каждые 28 дней, депакин 25 мг/кг ежедневно до 78 недели, весаноид (ATRA) 25 мг/м2 ежедневно сразу после окончания интенсификации II в течение 43 дней, 14дневным курсом (Рисунок 4). Рисунок 4. Дизайн терапии больных стандартной группы риска протокол ОМЛ НИИ ДОГ 2007 Пациенты со средним и высоким риском ОМЛ с первого дня получали эпигенетическое лечение (депакин 25 мг/кг/сут внутрь ежедневно в течение всего курса терапии и весаноид 25 мг/ м2/ день внутрь 1–45 дни) вместе с химиотерапией (Рисунок 5,6). Индукция ремиссии включала 2 курса химиотерапии AIE и НАМ. Крус AIE (Ara-C 100 мг/м2/сут в/в капельно 24 часа 1 – 2 дни и 100 мг/м2 в/в капельно каждые 12 часов 3 – 8 дни, идарубицин 12 50 мг/м2/сут в/в капельно 3 – 5 дни, VP-16 150 мг/м2/сут в/в капельно 6 – 8 дни). С целью профилактического поражения ЦНС вводился Ara-C интратекально в дозах в зависимости от возраста в 1-й день химиотерапии (Таблица12). При лейкемическом поражении ЦНС, которое подтверждалось клинически и/или морфологически, также с помощью компьютерной или магнитно-резонансной томографии вводились интратекально 3 препарата, дозы препаратов в таблице 13. При количестве бластных клеток менее 15% на 15 день от начала лечения, второй курс индукции НАМ (Ara-C 3000 мг/м2 в/в капельно каждые 12 часов 1–3 дни, митоксантрон 12 мг/м2/день в/в капельно 2–4 дни) начинался при восстановлении АСГ более 0,5×109/л и тромбоцитов более 100×109/л. Курс НАМ проводился в «timing» режиме (на 16 день от начала лечения) при количестве бластных клеток более 15% на 15 день от начала индукции. Режим «timing» не соблюдался только при констатации тяжелой инфекции. Консолидация ремиссии проводилась больным, у которых по окончанию индукции ремиссии в костном мозге было менее 15% бластных клеток. Она включала введение гранулоцитарный колониестимулирующий фактор (Г-КСФ) 5 мкг/кг в день подкожно 1–7 дни, Ara-C 2000 мг/м2/сут в/в капельно 2–6 дни, флударабин 30 мг/м2 в/в капельно 2–6 дни, депакин 25 мг/кг внутрь ежедневно, весаноид 25 мг/м2 ежедневно 1–14 дни. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились на 6 день курса в ранее указанных дозировках. Курс ранней интенсификации проводился больным достигшим ремиссию к 21 дню от окончания блока индукции, и состоял из Ara-C 3000 мг/м2 в/в капельно каждые 12 часов 1 – 3 дни, VP-16 125 мг/м2/сут в/в капельно, депакина 25 мг/кг ежедневно без перерывов внутрь, весаноида 25 мг/м2/день внутрь ежедневно 1 – 14 дни. С профилактической и лечебной целью эндолюмбально химиопрепараты вводились на 5 день курса в ранее указанных дозировках. Пациентам высокой группы риска с полным или частичным ответом на химиотерапию была продолжена консолидация ремиссии и планировалась аллоТГСК (при наличии полностью совместимого донора костного мозга). 51 После окончания основного лечения проводилась поддерживающая терапия, включавшая прием 6-МП 40 мг/м2/сут ежедневно до 78 недели от начала терапии индукции ремиссии, Ara-C 40 мг/м2/сут один раз в день подкожно 4дневным курсом каждые 28 дней, депакина 25 мг/кг ежедневно внутрь до 78 недель от начала терапии индукции ремиссии, весаноида 25 мг/м 2 ежедневно внутрь 14-дневными курсами в течение 43 дней. Поддерживающую терапию не получали больные, которым была проведена ТГСК. Рисунок 5. Дизайн терапии больных средней группы риска протокол ОМЛ НИИ ДОГ 2007 Рисунок 6. Дизайн терапии больных высокой группы риска протокол ОМЛ НИИ ДОГ 2007 52 На 15 день от начала лечения оценивался ответ на терапию соответственно следующим критериям: 1. Полный ответ (М-1) – менее 5% бластов в костном мозге, при отсутствии бластов в анализе периферической крови. 2. Частичный ответ (М-2) – по данным костного мозга количество бластов 6 - 25%, полное отсутствие бластов в анализе периферической крови. 3. Отсутствие ответа (М-3) – количество бластов в костном мозге более 25% или развитие экстрамедуллярного очага болезни. Критерии ремиссии были следующими: количество бластов в костном мозге менее 5%; восстановлении абсолютного числа нейтрофилов более 1,0×10 9/л, тромбоцитов более 100×109/л, независимости от переливаний эритроцитарной взвеси и отсутствие внекостномозговых опухолевых очагов. 53 Статистическая обработка данных Оценка параметрических данных проводилась посредством сравнения средних величин с использованием коэффициента Стьюдента. Разница считалась значимой при p˂0,05. Непараметрические данные сравнивались при помощи построения таблиц сопряженности с использованием критерия χ2 Пирсона. Разница между группами считалась достоверной при p˂0,05. Оценка возможной выживаемости проводилась при помощи построения кривых по методу Kaplan-Meier. В данном исследовании проводилась оценка безрецидивной выживаемости (БРВ), бессобытийной выживаемости (БСВ) и общей выживаемости (ОВ). Сравнение кривых выживаемости проводилось по методу long-rank. Разница между кривыми считалась достоверной при p˂0,05. БРВ – от момента начала лечения до момента возникновения рецидива или отсутствие ответа на терапию. БСВ – от начала лечения до момента прекращения ремиссии не зависимо от причины. ОВ – от начала лечения до смерти больного. 54 ГЛАВА 3. РЕЗУЛЬТАТЫ ЛЕЧЕНИЯ ДЕТЕЙ, ПОЛУЧАВШИХ ТЕРАПИЮ СОГЛАСНО ПРОТОКОЛАМ ОМЛ НИИ ДО 2002 И ОМЛ НИИ ДОГ 2007 Частота достижений ремиссий На 15 день от начала лечения оценивался ответ на индукционную химиотерапию по данным пунктатов костного мозга. Два пациента из 121 погибли до 15 дня из-за тяжелого сепсиса: один получал лечение по протоколу ОМЛ НИИ ДО 2002, один – по протоколу ОМЛ НИИ ДОГ 2007. Полного ответа к 15 дню лечения удалось достичь у 94 (77,7%) больных из 121. При анализе ответа на терапию в зависимости от протокола лечения, в протоколе ОМЛ НИИ ДО 2002 из 51 ребенка, включенных в исследование, у 39 (76,5%) детей – зафиксирован ответ М-1, у 6 (11,8%) – М-2 и у 5 (9,8%) – М-3. В протоколе ОМЛ НИИ ДОГ 2007 из 70 пациентов у 55 (78,6%) к 15 дню был достигнут полный ответ, у 7 (10,0%) – частичный и у 7 (10,0%) – не было ответа(p=0,95) (Рисунок 7). Рисунок 7. Ответ на 15 день от начала индукции в двух протоколах. 55 В группе стандартного риска у 12 (92,3%) больных, получивших лечение по протоколу ОМЛ НИИ ДО 2002, на 15 день от начала лечения диагностирован М-1 ответ, у одного ребенка (7,7%) – М-2. У всех 13 (100%) детей, вошедших в группу стандартного риска, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, удалось достичь полного ответа к 15 дню от начала лечения (p=0,3). Среди пациентов группы среднего риска с ответом М-1 к 15 дню от начала индукционной терапии в протоколе ОМЛ НИИ ДО 2002 было 16 (64,0%) больных, М-2 – 5 (20,0%) и М-3 – 4 (16,0%). В протоколе ОМЛ НИИ ДОГ 2007 у 18 (81,8%) зафиксирован полный ответ (М-1), у 2 (9,1%) – частичный ответ (М-2) и у одного (4,5%) пациента не было ответа(p=0,24). В группе среднего риска, благодаря добавлению к химиотерапии препаратов с эпигенетическим действием, достигнуто больше полных ответов на 15 день лечения. В группе высокого риска количество детей с ответом М-1, получивших лечение по протоколу ОМЛ НИИ ДО 2002, составило 11 (84,6%) больных и у одного (7,7%) – ответа не было. При лечении по протоколу ОМЛ НИИ ДОГ 2007 у 24 (68,6%) пациентов зафиксирован полный ответ, у 5 (14,3%) – частичный и у 6 (17,1%) – ответа не было (p=0,1). Вероятнее всего большее количество полных ответов к 15 дню лечения в протоколе ОМЛ НИИ ДО 2002 связано с более интенсивной индукционной химиотерапией. Объединив группы среднего и высокого риска в одну – неблагоприятная группа риска, дополнительно проведено сравнение ответов на лечение к 15 дню в зависимости от протокола. Среди детей, получивших лечение по протоколу ОМЛ НИИ ДО 2002, из неблагоприятной группы, полный ответ на 15 день терапии был достигнут у 27 (71,1%), частичный – у 5 (13,2%), не было ответа – у 5 (13,2%). При лечении по протоколу ОМЛ НИИ ДОГ 2007, в неблагоприятной группе риска, ответ M-1 зафиксирован у 42 (73,7%) больных, ответ M-2 – у 7 (12,3%), ответ M-3 – у 7 (12,3%) пациентов (p=0,98). Таким образом, при добавлении к терапии препаратов с эпигенетическим действием отмечена тенденция к увеличению числа полных ответов к 15 дню лечения у пациентов 56 неблагоприятной группы риска. Распределение больных в зависимости от ответа на 15 день терапии представлено в таблице 14. Таблица 14 Оценка ответа на 15 день лечения детей с ОМЛ в зависимости от группы риска, получавших лечение по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Группа риска Ответ на лечение Ответ на лечение Ответ на лечение Р М-1 М-2 м-3 ОМЛ ОМЛ ОМЛ ОМЛ ОМЛ ОМЛ НИИ НИИ НИИ НИИ НИИ НИИ ДО ДОГ ДО ДОГ ДО ДОГ 2002 2007 2002 2007 2002 2007 N N N % N % N % N % 1 0 0 0 0 0 4 % % Стандартная 12 92,3 13 100 Средняя 16 64,0 18 81,8 5 20,0 2 9,1 Высокая 11 84,6 24 68,6 0 0 5 Неблагоприятная 27 71,1 42 73,7 5 13,2 7 7,7 0 0,3 16,0 1 4,5 0,2 14,3 1 7,7 17,1 0,1 12,3 5 13,2 7 6 12,3 0,98 По окончанию курса индукционной химиотерапии у 107 (88,4%) больных удалось достичь ремиссии, у 9 (7,4%) – не была достигнута ремиссия и 5 (4,1%) – погибло от инфекционных осложнений. Анализ данных пунктатов костного мозга по окончании индукционной химиотерапии у пациентов, получавших лечение согласно протоколу ОМЛ НИИ ДО 2002, показал, что у 43 (84,3%) достигнута ремиссия, у 6 (11,8%) – ремиссии не было и 2 (3,9%) ребенка умерли от сепсиса. В протоколе ОМЛ НИИ ДОГ 2007 у 64 (91,4%) пациентов зафиксирована ремиссия, у 3 (4,3%) – нет и 3 (4,3%) ребенка умерло от инфекционных осложнений (р=0,3). 57 Рисунок 8. Частота достижения ремиссий к окончанию индукционной химиотерапии, протокол ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007. В группе стандартного риска в обоих протоколах у всех больных после курса индукции зафиксирована ремиссия. В группе среднего риска в протоколе ОМЛ НИИ ДО 2002 диагностирована ремиссия у 21 (84,0%) пациента, а у 4 (16,0%) ремиссию достичь не удалось. В протоколе ОМЛ НИИ ДОГ 2007 в группе среднего риска у 19 (86,4%) больных – зафиксирована ремиссия, у одного (4,5%) – ремиссия не была достигнута (р=0,48). Среди пациентов группы высокого риска в протоколе ОМЛ НИИ ДО 2002 по окончании индукционной химиотерапии у 9 (69,2%) установлена ремиссия, у двух (15,4%) ремиссии не было. При лечении по протоколу НИИ ДОГ 2007 у 32 (91,4%) больных группы высокого риска достигнута ремиссия и только у двух (5,7%) ее не было (р=0,05). В группе неблагоприятного риска так же удалось увеличить частоту ремиссий с 78,9% (30 больных) в протоколе ОМЛ НИИ ДО 2002 до 89,5% (51 пациент) протокол ОМЛ НИИ ДОГ 2007, р=0,23 (Таблица15). 58 Таблица 15 Оценка ответа по окончании индукционной химиотерапии в зависимости от группы риска протокол ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Группа риска Ремиссия Нет ремиссии Индукционная смертность ОМЛ ОМЛ ОМЛ ОМЛ ОМЛ ОМЛ НИИ ДО НИИ НИИ ДО НИИ НИИ НИИ 2002 ДОГ 2002 ДОГ ДО ДОГ 2007 2007 2002 2007 N % N % N % N % N % N % Р Стандартная 13 100 Средняя 21 84,0 19 86,4 4 16,0 1 4,5 - - 2 9,1 0,48 Высокая 9 69,2 32 91,4 2 15,4 2 5,7 2 15,4 1 4,3 0,05 78,9 51 89,5 6 15,8 3 5,3 2 5,3 5,3 0,23 Неблагоприятная 30 13 100 3 Следовательно, благодаря сочетанию химиотерапии с эпигенетической терапией в группе среднего и неблагоприятного риска отмечена тенденция к увеличению частоты ремиссий, а в группе высокого риска нам достоверно удалось увеличить количество ремиссий к 21 дню от окончания индукционной терапии. Результаты выживаемости У пациентов, получавших лечение по протоколу ОМЛ НИИ ДО 2002, пятилетняя БРВ составила 43,6±7,2%, при средней продолжительности наблюдения 56,3±7,9 мес; БСВ – 39,2±7,0% при средней продолжительности наблюдения 51,1±7,6 мес; ОВ – 47,4±7,0% при средней продолжительности наблюдения 62,6±7,9 мес. При терапии по протоколу ОМЛ НИИ ДОГ 2007 показатели выживаемости оказались выше, так БРВ составила 56,1±7,0% (p=0,09), при средней продолжительности наблюдения 45,1±3,8 мес; БСВ – 51,7±7,0% (p=0,75), при средней продолжительности наблюдения 39,6±3,9 мес; ОВ – 59 54,7±6,0% (p=0,39), при средней продолжительности наблюдения 43,2±3,9 мес (Рисунок 9-11). Рисунок 9. БРВ детей, больных ОМЛ, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 10. БСВ детей, больных ОМЛ, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 60 Рисунок 11. ОВ детей, больных ОМЛ, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 В соответствии с принятыми в педиатрии возрастными периодами, больные ОМЛ, включенные в наше исследование были разделены на 5 возрастных групп. достоверной разницы среди по клинико-морфологическим данным у детей до 14 лет в двух протоколах не было (р≥0,4) , в то время как группы больных старше 14 лет достоверно различались (р≤0,02). В протоколе ОМЛ НИИ ДОГ 2007 большая часть пациентов старше 14 лет (9 из 10) были отнесены к неблагоприятной прогностической группе. У пациентов младше одного года, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007, достоверно удалось увеличить пятилетнюю БРВ до 91,7±8,1%; БСВ до 81,8±11,2% и ОВ до 83,9±10,4%; по сравнению с протоколом ОМЛ НИИ ДО 2002, где пятилетняя БРВ составила – 25,0±21,3% (p=0,002); БСВ – 25,0±22,4%(p=0,015) и ОВ – 25,0±22,0% (p=0,01). Именно у детей младше одного года сочетание эпигенетической и химиотерапии позволило достоверно увеличить выживаемость. Вероятнее всего, препараты с эпигенетическим 61 действием у маленьких детей способствуют дифференцировке бластных клеток и делают их более чувствительными к химиопрепаратам. Так же удалось повысить пятилетнюю выживаемости (БРВ, БСВ, ОВ) в группах пациентов от 1 года до 14 лет при лечении по протоколу ОМЛ НИИ ДОГ 2007. Результаты показателей пятилетней выживаемости детей старше 14 лет, получавших лечение по протоколу ОМЛ НИИ ДО 2002, были выше: БРВ составила 85,7±13,1%; БСВ – 85,7±13,1%; ОВ – 87,7±13,0%; а у больных, лечившихся по протоколу ОМЛ НИИ ДОГ 2007: 25,0±15,0% (p=0,02); 25,0±15,0% (p=0,03) и 25,6±15,3% (p=0,007) соответственно (Таблица 16-18). Что объясняется тем, что в протоколе ОМЛ НИИ ДОГ 2007 пациенты старше 14 лет были отнесены к неблагоприятной прогностической группе. Таблица 16 Пятилетняя БРВ у детей, больных ОМЛ, лечения Возраст БРВ по протоколу ОМЛ НИИ ДО 2002 (%) ˂ 1 года 25,0±21,3 (n=4) 1,1 – 3 года 21,4±18,1 (n=7) 3,1 – 7 лет 36,4±15,4 (n=11) 7,1 – 14 лет 41,5±11,0(n=22) Старше 14 лет 85,7±13,1 (n=7) в зависимости от возраста и протокола БРВ по протоколу ОМЛ НИИ ДОГ 2007 (%) 91,7±8,1 (n=14) 49,4±15,4 (n=13) 45,0±21,5 (n=11) 61,1±11,3 (n=22) 25,0±15,0 (n=10) Р 0,002 0,29 0,45 0,16 0,02 Таблица 17 Пятилетняя БСВ у детей, больных ОМЛ, в зависимости от возраста и протокола лечения Возраст БСВ по БСВ по Р протоколу ОМЛ протоколу ОМЛ НИИ ДО 2002 НИИ ДОГ 2007 (%) (%) ˂ 1 года 25,0±22,4 (n=4) 81,8±11,2 (n=14) 0,015 1,1 – 3 года 14,3±13,5 (n=7) 45,5±15,6 (n=13) 0,23 3,1 – 7 лет 36,4±15,7 (n=11) 37,5±19,3 (n=11) 0,67 7,1 – 14 лет 36,4±10,0(n=22) 51,3±11,7 (n=22) 0,26 Старше 14 лет 85,7±13,1 (n=7) 25,0±15,0 (n=10) 0,03 62 Таблица 18 Пятилетняя ОВ у детей, больных ОМЛ, в зависимости от возраста и протокола лечения Возраст ОВ по протоколу ОВ по протоколу Р ОМЛ НИИ ДО ОМЛ НИИ ДОГ 2002 (%) 2007 (%) ˂ 1 года 25,0±22,0 (n=4) 83,9±10,4 (n=14) 0,01 1,1 – 3 года 28,6±17,0 (n=7) 49,9±15,2 (n=13) 0,23 3,1 – 7 лет 43,6±16,3 (n=11) 53,3±17,4 (n=11) 0,55 7,1 – 14 лет 47,7±11,7(n=22) 55,8±11,8 (n=22) 0,37 Старше 14 лет 87,7±13,0 (n=7) 25,6±15,3 (n=10) 0,007 В исследовании была проанализирована пятилетняя выживаемость (БРВ, БСВ, ОВ) у детей с ОМЛ, в зависимости от FAB морфологического варианта и протокола лечения (Таблица 19). Учитывая то, что группы больных при разделении на FAB-варианты оказались немногочисленными и достоверно различимыми, нами проанализирована выживаемость больных с различными FAB-вариантами в каждом протоколе отдельно. При анализе пятилетней БРВ больных, получавших лечение по протоколу ОМЛ НИИ ДО 2002, в зависимости от FAB морфологического варианта, лучшая получена у детей с FAB-M2 (75,5±12,3%) и FAB-M1 вариантами (39,0±11,0%); хуже с FAB-M4 (33,3±16,7%) и FAB-M5 вариантами (33,3±27,4%); пациенты с FAB-M0 (n=3), FAB-M6 (n=2) и FAB-M7 вариантами (n=1) умерли от прогрессирования/рецидива основного заболевания в течение 12 месяцев (Рисунок 12). Анализ достоверности различий попарно между морфологическими вариантами представлен в таблице 20. Достоверная разница была выявлена между группами больных с FAB-M0 и FAB-M1, FAB-M2, FAB-M4, FAB-M5 вариантами ОМЛ; между FAB-M1 и FAB-M2, FAB-M6 вариантами; FAB-M2 и FAB-M4, FABM6 вариантами; FAB-M4 и FAB-M6 вариантами; FAB-M5 и FAB-M6 вариантом ОМЛ. 63 Таблица 19 Пятилетняя БРВ и ОВ детей с ОМЛ, в зависимости от FAB морфологического варианта Вариант ОМЛ M1 Протокол ОМЛ НИИ ДО 2002 Количество БРВ БСВ ОВ больных (n) (p=0,22) (p=0,09) (p=0,34) 3 Все дети умерли от сверхраннего рецидива ОМЛ в течение 12 месяцев 19 39,0±11,0% 36,8±11,9% 43,1±12,2% M2 13 75,5±12,3% M4 9 M5 M0 Протокол ОМЛ НИИ ДОГ 2007 Количество БРВ БСВ ОВ больных (n) (p=0,22) (p=0,09) (p=0,34) 2 Жив 1 ребенок в течение 12 месяцев 6 50,0±20,3% 50,0±20,3% 50,0±20,3% 69,2±13,9% 84,6±10,0% 19 53,0±14,8% 52,8±14,1% 71,8±10,5% 33,3±16,7% 33,3±16,0% 44,4±17,7% 20 64,3±12,3% 52,5±12,9% 49,0±12,5% 4 33,3±27,4% 25,0±22,3% 33,3±27,4% 17 53,0±14,1% 53,0±14,1% 53,0±14,1% M6 2 2 M7 1 Все дети умерли от прогрессирования/ рецидива ОМЛ в течение 12 месяцев Ребенок умер от прогрессирования основного заболевания в течение 12 месяцев Один ребенок жив, без признаков заболевания в течение 12 месяцев 66,7±27,4% 50,0±25,0% 66,7±27,4% 4 64 Достоверно выявлена лучшая БРВ больных, лечившихся по протоколу ОМЛ НИИ ДО 2002, с FAB-M2 вариантом, чем с FAB-M0, FAB-M1, FAB-M4, FAB-M6; у детей с ОМЛ FAB-M4 вариантом, чем с FAB-M0 и FAB-M6; у больных с ОМЛ FAB-M1, чем с FAB-M0 и FAB-M6 вариантами; у пациентов с FAB-M5 вариантом лучше, чем FAB-M0 и FAB-M6 вариантами. Рисунок 12. БРВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДО 2002, в зависимости от FAB-варианта ОМЛ (p˂0,0001) Таблица 20 Результаты значимой разницы БРВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДО 2002. p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 0,000 0,001 0,021 0,01 M1 0,000 0,052 0,63 0,88 M2 0,001 0,052 0,04 0,22 M4 0,021 0,63 0,037 0,69 M5 0,01 0,88 0,22 0,69 M6 0,41 0,000 0,003 0,05 0,025 M7 0,16 0,82 0,78 0,67 65 Такая же тенденция оказалась и при исследовании БСВ. Так пятилетняя БСВ у больных с FAB-M2 вариантом была 69,2±13,9%, FAB-M1 – 36,8±11,9%; FAB-M4 – 33,3±16,0%; FAB-M5 – 25,0±22,3%; а у пациентов FAB-M0, FAB-M6 и FAB-M7 0% (Рисунок 13). Достоверность разницы БСВ при попарном сравнении между детьми с различными морфологическими вариантами, получивших лечение по протокол у ОМЛ НИИ ДО 2002 представлена в таблице 21. Выявлена достоверная разница БСВ у детей с FAB-M0 и FAB-M1, FAB-M2, FAB-M4, FABM5 вариантами; между больными с FAB-M1 и FAB-M6, FAB-M7 морфологическими вариантами; FAB-M4 и FAB-M6 вариантами ОМЛ, а также между пациентами с FAB-M5 и FAB-M6, FAB-M7 морфологическими вариантами. Достоверно выявлена лучшая БСВ больных, лечившихся по протоколу ОМЛ НИИ ДО 2002, с FAB-M2 вариантом, чем с FAB-M0, FAB-M6, FAB-M7; у детей с ОМЛ FAB-M4 вариантом, чем с FAB-M0 и FAB-M6; у больных с ОМЛ FAB-M1, чем с FAB-M0, FAB-M6, FAB-M7-вариантами; у пациентов с FAB-M5 вариантом лучше, чем FAB-M0, FAB-M6 и FAB-M7 вариантами. Рисунок 13. БСВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДО 2002, в зависимости от FAB-варианта ОМЛ (p˂0,0001) 66 Таблица 21 Результаты значимой разницы БСВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДО 2002 p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 M1 M2 M4 M5 M6 M7 0,000 0,000 0,001 0,02 0,01 0,41 0,16 0,12 0,59 0,45 0,000 0,004 0,001 0,12 0,08 0,11 0,003 0,02 0,021 0,59 0,08 0,95 0,05 0,19 0,01 0,45 0,11 0,95 0,03 0,05 Анализ пятилетней ОВ детей, лечившихся по протоколу ОМЛ НИИ ДО 2002, в зависимости от FAB морфологического варианта, так же показал лучшую выживаемость больных с FAB-M2 вариантом (84,6±10,0%) ОМЛ, хуже с FAB-M4 (44,4±17,7%), FAB-M1 (43,1±12,2%) и FAB-M5 (33,3±27,4%), все больные FABM0, FAB-M6, FAB-M7 вариантами ОМЛ умерли от прогрессирования/рецидива основного заболевания в течение года (Рисунок 14). Достоверность разницы ОВ при попарном сравнении между детьми с различными морфологическими вариантами, получившими лечение по протоколу ОМЛ НИИ ДО 2002, представлена в таблице 22. Достоверная разница ОВ была выявлена между больными с FAB-M2 вариантом и всеми другими морфологическими вариантами. Так же для FAB-M0 варианта достоверная разница была установлена с FAB-M1 и FAB-M2; для FAB-M1 с FAB-M2, FAB-M6, FAB-M7 вариантами ОМЛ; для FABM4 с FAB-M6, FAB-M7 вариантами; и между FAB-M5 и FAB-M7 вариантом. Достоверно выявлена лучшая ОВ больных, лечившихся по протоколу ОМЛ НИИ ДО 2002, с FAB-M2 вариантом, чем с другими морфологическими вариантами; у детей с ОМЛ FAB-M4 вариантом, чем с FAB-M6 и FAB-M7; у больных с ОМЛ FAB-M1, чем с FAB-M6 и FAB-M7 вариантами; у пациентов с FAB-M5 вариантом лучше, чем FAB-M7 вариантом. 67 Рисунок 14. ОВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДО 2002, в зависимости от FAB-варианта ОМЛ (p˂0,0001) Таблица 22 Результаты значимой разницы ОВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДО 2002 p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 M1 M2 M4 M5 M6 M7 0,000 0,000 0,000 0,09 0,15 0,79 0,08 0,052 0,78 0,26 0,000 0,000 0,000 0,05 0,05 0,02 0,001 0,000 0,09 0,78 0,05 0,55 0,02 0,07 0,15 0,26 0,02 0,55 0,11 0,05 При анализе пятилетней БРВ больных, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от FAB морфологического варианта, лучшая БРВ отмечалась у больных с FAB-M7 (66,7±27,4%) и FAB-M4 вариантами (64,3±12,3%); хуже с FAB-M2 (53,0±14,8%), FAB-M5 (53,0±14,1%) и FAB-M1 (50,0±20,3%); с FAB-M0 и FAB-M7 морфологическими вариантами было по двое детей, из них живы по одному ребенку (длительность наблюдения 12 месяцев), но 68 данные не достоверны p=0,83 (Рисунок 15). Достоверной разницы БРВ попарно между морфологическими вариантами в протоколе ОМЛ НИИ ДОГ 2007 не было (Таблица 23). Рисунок 15. БРВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от FAB-варианта ОМЛ (p=0,83) Таблица 23 Результаты значимой разницы БРВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007 p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 M1 M2 M4 M5 M6 M7 0,51 0,51 0,19 0,38 0,12 0,32 0,16 0,72 0,62 0,73 0,47 0,48 0,19 0,72 0,98 0,79 0,58 0,61 0,38 0,62 0,98 0,97 0,47 0,67 0,12 0,73 0,79 0,97 0,49 0,53 Лучшая пятилетней БСВ у детей, получивших лечение по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от FAB морфологического варианта, отмечалась у 69 больных с FAB-M5 (53,0±14,1%) ОМЛ, хуже с FAB-M2 (52,8±14,1%), FAB-M4 (52,5±12,9%), FAB-M1 (50,0±20,3%), FAB-M7 (50,0±25,0%) (Рисунок 16). Как было указано выше с FAB-M0 и FAB-M6 вариантами ОМЛ лейкоза живы по одному ребенку в течение 1 года (p=0,88). Достоверной разницы БСВ попарно между морфологическими вариантами в протоколе ОМЛ НИИ ДОГ 2007 не было (Таблица 24). Рисунок 16. БСВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от FAB-варианта ОМЛ (p=0,88) Таблица 24 Результаты значимой разницы БСВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007 p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 0,51 0,2 0,49 0,12 M1 0,51 0,73 0,93 0,73 M2 0,2 0,73 0,54 0,79 M4 0,48 0,93 0,54 0,62 M5 0,12 0,73 0,79 0,62 M6 0,81 0,68 0,31 0,73 0,5 M7 0,45 0,89 0,85 0,94 0,94 70 Пятилетняя ОВ у детей, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от морфологического варианта, составила для FAB-M2 – 71,8±10,5%; FAB-M7 – 66,7±27,4%; FAB-M5 – 53,0±14,1%; FAB-M1 – 50,0±20,3%; FAB-M4 – 49,0±12,5%; FAB-M0 и FAB-M6 – 0% (в настоящее время живы по 1 ребенку в течение 12 месяцев) (Рисунок 17). Анализ сравнения ОВ попарно между морфологическими вариантами представлен в таблице 25. Достоверная разница ОВ была выявлено только между группами больных с FAB-M0 вариантом и FABM2, FAB-M5. Таким образом, у детей с FAB-M2 морфологическим вариантом, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, достоверно лучше ОВ, чем у больных с FAB-M0 вариантом, получавших лечение по данному протоколу (p=0,02). И у пациентов с FAB-M5 вариантом ОМЛ общая выживаемость достоверно выше, чем у больных с FAB-M0 вариантом (p=0,05). Рисунок 17. ОВ больных ОМЛ, получивших лечение по протоколу ОМЛ НИИ ДОГ 2007, в зависимости от FAB-варианта ОМЛ (p=0,5) 71 Таблица 25 Результаты значимой разницы ОВ для детей с различными FAB-вариантами ОМЛ, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007 p Морфологи P ческий Морфологический вариант вариант M0 M1 M2 M4 M5 M0 0,68 0,02 0,54 0,05 M1 0,68 0,34 0,89 0,88 M2 0,02 0,34 0,12 0,18 M4 0,54 0,89 0,12 0,75 M5 0,05 0,88 0,18 0,75 M6 0,81 0,68 0,07 0,58 0,23 M7 0,45 0,99 0,46 0,85 0,99 Следовательно, при лечении по протоколу ОМЛ НИИ ДО 2002 важное прогностическое значение имел FAB морфологический вариант, подобных данных при анализе выживаемости детей, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, не получено. Так же в протоколе ОМЛ НИИ ДО 2002 достоверно хуже была выживаемость больных с FAB-М5, FAB-М6, FAB-М7 вариантами, а в протоколе ОМЛ НИИ ДОГ 2007 неблагоприятное значение данных вариантов ОМЛ утратило свое влияние. Среди больных стандартной группы риска, лечившихся по протоколу ОМЛ НИИ ДО 2002 (n=13), 5-летняя БРВ составила 69,2±12,7%, при средней продолжительности наблюдения 82,8±12,6 мес; БСВ – 69,2±12,7%, при средней продолжительности наблюдения 82,8±12,6 мес; ОВ составила 75,5±12,3%, при средней продолжительности наблюдения 91,0±11,0 мес. У детей, лечившихся по протоколу ОМЛ НИИ ДОГ 2007 (n=13) БРВ составила 71,3±14,1% (p=0,68), при средней продолжительности наблюдения и 58,3±5,1 мес; БСВ – 71,3±14,1% (p=0,68) при средней продолжительности наблюдения 39,6±3,9 мес; ОВ – 90,0±9,8% (p=0,29) при средней продолжительности наблюдения 65,4±3,0 мес (Рисунок 18-20). 72 Рисунок 18. БРВ детей, больных ОМЛ, стандартной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 19. БСВ детей, больных ОМЛ, стандартной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 73 Рисунок 20. ОВ детей, больных ОМЛ, стандартной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Пятилетняя безрецидивная выживаемость, больных, включенных в среднюю группу риска, лечившихся по протоколу ОМЛ НИИ ДО 2002 (n=25), составила 35,9±10,8%, при средней продолжительности наблюдения 47,0±10,7 мес; БСВ – 32,0±7,3%, при средней продолжительности наблюдения 51,1±7,6 мес; ОВ – 40,5±10,6%, при средней продолжительности наблюдения 56,0±11,2 мес. Применяя протокол ОМЛ НИИ ДОГ 2007 (n=22), удалось повысить: БРВ – 52,9±11,3% (p=0,3), средняя продолжительность наблюдения 40,7±7,0 мес; БСВ – 47,8±12,8% (p=0,26), средняя продолжительность наблюдения 39,6±3,9 мес и ОВ составила 47,6±11,5% (p=0,69), средняя продолжительность наблюдения 38,4±6,5 мес (Рисунок 21-23). 74 Рисунок 21. БРВ детей, больных ОМЛ, средней группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 22. БСВ детей, больных ОМЛ, средней группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 75 Рисунок 23. ОВ детей, больных ОМЛ, средней группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Среди больных высокой группы риска, лечившихся по протоколу ОМЛ НИИ ДО 2002(n=13), 5-летняя БРВ составила 30,8±10,9%, средняя продолжительность наблюдения 37,0±14,9 мес; БСВ – 23,1±12,7%, средняя продолжительность наблюдения 51,1±7,6 мес; ОВ – 30,8±13,9%, средняя продолжительность наблюдения 36,9±13,5 мес. Протокол ОМЛ НИИ ДОГ 2007 (n=35), позволил увеличить БРВ, БСВ и ОВ у пациентов высокой группы риска: БРВ составила 53,4±10% (p=0,04), средняя продолжительность наблюдения 38,9±1,0 мес; БСВ – 45,0±9,8% (p=0,2), средняя продолжительность наблюдения 39,6±3,9 мес; ОВ – 45,1±10,7% (p=0,16), средняя продолжительность наблюдения 35,2±4,8 мес (Рисунок 24-26). 76 Рисунок 24. БРВ детей, больных ОМЛ, высокой группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 25. БСВ детей, больных ОМЛ, высокой группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 77 Рисунок 26. ОВ детей, больных ОМЛ, высокой группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 При сравнении показателей выживаемости больных группы неблагоприятного риска (средняя и высокая), удалось достоверно улучшить БРВ и БСВ, при лечении по протоколу ОМЛ НИИ ДОГ 2007, n=57 (Рисунок 27-29). Пятилетняя выживаемость в протоколе ОМЛ НИИ ДО 2002 (n=38) составила: БРВ – 34,0±8,1%, средняя продолжительность наблюдения 44,6±8,9 мес; БСВ – 28,9±7,9%, средняя продолжительность наблюдения 39,9±8,4 мес; ОВ – 37,7±8,0%, средняя продолжительность наблюдения 50,9±9,0 мес. При анализе показателей выживаемости в протоколе ОМЛ НИИ ДОГ 2007, БРВ составила 53,0±8,5%, средняя продолжительность наблюдения 41,9±4,5 мес (p=0,04); БСВ – 47,6±8,9%, средняя продолжительность наблюдения 38,2±4,4 мес (p=0,03); ОВ – 45,9±8,2%, средняя продолжительность наблюдения 37,7±4,1 мес (p=0,28). 78 Рисунок 27. БРВ детей, больных ОМЛ, неблагоприятной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 28. БСВ детей, больных ОМЛ, неблагоприятной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 79 Рисунок 29. ОВ детей, больных ОМЛ, неблагоприятной группы риска, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Следовательно, применяя химиотерапию в сочетании с препаратами, влияющими на состояние активности генов, удалось повысить БРВ, БСВ, ОВ детей, включенных в группу высокого риска по сравнению с пациентами, получавшими только химиотерапию. Не было получено достоверного увеличения показателей БРВ, БСВ и ОВ у больных, включенных в группу среднего и стандартного риска, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007 по сравнению с пациентами, получавшими лечение по протоколу ОМЛ НИИ ДО 2002. Достоверно удалось повысить БРВ и БСВ у детей из неблагоприятной группы риска (средняя и высокая). Нами проведен анализ выживаемости в зависимости от ответа на индукционную терапию. У 39 (76,5%) пациентов, получавших терапию по протоколу НИИ ДО ОМЛ 2002, с ответом M-1 на 15 день терапии, БРВ составила 80 52,1±8,9%; БСВ – 52,1±8,9%; ОВ – 55,1±8,7%. В протоколе НИИ ДОГ ОМЛ 2007 у 55 (78,6%) детей был полный ответ на 15 день от начала терапии, в этой группе БРВ составила 65,9±8,5% (p=0,2); БСВ – 65,9±8,5% (p=0,13); ОВ – 66,0±7,7% (p=0,42) (Рисунок 30, 31). Рисунок 30. Выживаемость (БРВ, БСВ) больных с ответом М-1 на 15 день от начала курс индукции, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 31. ОВ больных с ответом М-1 по костному мозгу на 15 день от начала курса индукции, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 81 Результаты выживаемости у детей с частичным ответом (M-2) на 15 день терапии были значительно хуже в обоих протоколах (Рисунок 32). У пациентов, получавших лечение по протоколу ОМЛ НИИ ДО 2002, с ответом M2 (n=6), выживаемость (БРВ, БСВ, ОВ) составила 16,7±15,9%; а в протоколе ОМЛ НИИ ДОГ (n=7) – 21,4±18,7% (p>0,6). Следовательно, у больные с частичным ответом на 15 день от начала индукции должны быть переведены в высокую группу риска и терапия индукции должна проводиться в режиме «timing». Рисунок 32. Общая выживаемость больных с ответом М-2 по костному мозгу на 15 день от начала курса индукции, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 На 15 день от начала курса индукции у 12 детей не было ответа терапию, у 5 (9,8%) пациентов, получивших лечение по протоколу ОМЛ НИИ ДО 2002, и у 7 (10,0%), по протоколу ОМЛ НИИ ДОГ 2007. Пятилетняя выживаемость у больных c M-3 ответом на 15 день от начала курса индукции (Рисунок 33, 34), по протоколу ОМЛ НИИ ДО 2002, составила: БРВ – 20,0±18,9%; БСВ – 20,0±18,9%; ОВ – 40,0±22,3%. У детей, лечивших по протоколу ОМЛ НИИ ДОГ 2007, несколько выше: БРВ – 34,3±19,7% (p=0,2); БСВ – 34,3±19,7% (p=0,33); ОВ – 82 35,0±17,2% (p=0,7). Следовательно, благодаря интенсификации индукционной химиотерапии при отсутствии ответа на 15 день в протоколе ОМЛ НИИ ДОГ 2007 удалось повысить выживаемость данной группы больных. Рисунок 33. Безрецидивная и бессобытийная выживаемость больных с ответом М-3 по костному мозгу на 15 день от начала курса индукции, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 Рисунок 34. Общая выживаемость больных с ответом М-3 по костному мозгу на 15 день от начала курс индукции, лечившихся по протоколам ОМЛ НИИ ДО 2002 и ОМЛ НИИ ДОГ 2007 83 По окончании индукции: у 43 (84,3%) больных, лечившихся по протоколу НИИ ДО ОМЛ 2002 и у 64 (91,4%) – по протоколу НИИ ДОГ ОМЛ 2007. Пятилетняя безрецидивная выживаемость данной группы пациентов в протоколе НИИ ДО ОМЛ 2002 составила 51,7±8,0%; БСВ – 43,8±7,6% и ОВ – 54,5±7,8%. В протоколе НИИ ДОГ ОМЛ 2007 пятилетняя БРВ – 60,7±7,3% (р=0,2); БСВ – 53,8±7,3 (р=0,3); ОВ – 60,2±6,9% (р=0,4) (Рисунок 35-37). Рисунок 35. БРВ больных, достигших ремиссии к окончанию индукции в протоколе НИИ ДО ОМЛ 2002 и НИИ ДОГ ОМЛ 2007 Рисунок 36. БСВ больных, достигших ремиссии к окончанию индукции в протоколе НИИ ДО ОМЛ 2002 и НИИ ДОГ ОМЛ 2007 84 Рисунок 37. ОВ больных, достигших ремиссии к окончанию индукции в протоколе НИИ ДО ОМЛ 2002 и НИИ ДОГ ОМЛ 2007 После курса индукционной химиотерапии не было ответа на лечение у 9 (7,4%) больных, из них 6 (11,8%) детей получили лечение по протоколу ОМЛ НИИ ДО 2002, в настоящее время один ребенок жив без признаков основного заболевания, максимальный срок наблюдения 73 месяца. У 3(4,3%) пациентов, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, не удалось достигнуть ремиссии после индукционной химиотерапии, из них жив один ребенок в течение 23 месяцев. Таким образом, благодаря использованию протокола ОМЛ НИИ ДОГ 2007 удалось значительно увеличить выживаемость пациентов до одного года, а так же у больных достигших ремиссию. Существенно увеличилась выживаемость детей, включенных в среднюю и высокую группы риска, по сравнению с протоколом НИИ ДО ОМЛ 2002. 85 Переносимость терапии Токсичность эпигенетической терапии оценивалась по шкале NCI CTC Version 2.0 от апреля 1999. Добавление к терапии препаратов с эпигенетическим действием к химиотерапии не ухудшило переносимость и не увеличило токсичность лечения. Основным побочным эффектом приема ATRA была головная боль, требовавшая назначение анальгетиков у 7 (13,7%) детей с ОМЛ. «Синдром ATRA» наблюдался в индукции ремиссии только у одного пациента, клинически он проявлялся лихорадкой, инфильтрацией легких, задержкой жидкости с развитием отеков, был купирован отменой ATRA и назначением дексаметазона. Тяжелых токсических проявлений от приема ВК нами не было выявлено. Из возможных тяжелых проявлений нежелательных эффектов от добавления к терапии ВК была миелосупрессия и удлинение длительности нейтропении. При анализе длительности нейтропении, при лечении по протоколу ОМЛ НИИ ДО 2002, в среднем она составила 21,3±3,7 дня; при лечении по протоколу ОМЛ НИИ ДОГ 2007, в среднем 22,1±4,2 дня. Инфекционные осложнения У всех больных, получавших лечение по двум протоколам, как следствие химиотерапии, развивалась нейтропения, тромбоцитопения, анемия. С заместительной целью все дети получали трансфузии тромбоконцентрата, эритроцитарной взвеси. Следствием длительной нейтропении является развитие инфекционных осложнений. В протоколе ОМЛ НИИ ДО 2002 индукционная смертность составила 3,9%; смертность в ремиссии – 13,7%. В протоколе ОМЛ НИИ ДОГ 2007 смерть в индукции составила 4,3%; а в ремиссии – 7,1% (Рисунок 38). 86 14 12 10 8 ОМЛ НИИ ДО 2002 6 ОМЛ НИИ ДОГ 2007 4 2 0 Индукционная смертность Смерть в ремиссии Рисунок 38. Смертность на протоколах Наиболее тяжелые инфекционные осложнения развиваются после курса индукционной химиотерапии, так как пациенты поступают в стационар уже с тяжелой инфекцией, развившейся из-за депрессии нормального кроветворения и бластной метаплазии костного мозга. Нами проанализированы осложнения инфекционного характера у 82 (67,8%) больных после индукционной химиотерапии при лечении по двум протоколом, данные достоверны р=0,02. При лечении по протоколу ОМЛ НИИ ДО 2002, наиболее частым проявлением инфекции были фебрильная нейтропения – у 30 (62,5%) пациентов и сепсис – у 6 (12,5%), так же у 5 (10,4%) была диагностирована пневмония, у 4 (8,3%) нейтропенический этероколит, и одного ребенка (2,1%) инфекция мягких тканей, у двоих детей (4,2%) не было инфекционных осложнений. После индукционной химиотерапии у больных, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, были диагностированы следующие инфекционные осложнения: пневмония – у 12 (35,3%) пациентов, фебрильная нейтропения – у 10 (29,4%), нейтропенический энтероколит – у 5 (14,7%), сепсис – у 3 (8,8%) и инфекция мягких тканей у 3 (8,8%), у одного ребенка (2,9%) инфекционных осложнений не было. Двенадцати (17,1%) пациентам в протоколе НИИ ДОГ ОМЛ 2007 в связи с тяжелым инфекционным 87 процессом была проведена трансфузия гранулоцитов, благодаря чему удалось купировать признаки инфекции у всех больных. У пациентов, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007, чаще был диагностирован локализованный очаг инфекции, в отличие от структуры инфекционных осложнений у больных, получавших терапию по протоколу ОМЛ НИИ ДО 2002, где чаще диагностировалась фебрильная нейтропения и сепсис (p=0,02). Данные различия связаны с оптимизацией противомикробной терапии и улучшению лабораторно-инструментальных методов диагностики. Для определения влияния на частоту и структуру инфекционных осложнений при добавлении препаратов с эпигенетическим действием к интенсивной химиотерапии нами была проанализирована структура инфекционных осложнений в группах среднего и высокого риска (Таблица 26), так как именно у этих пациентов ATRA и ВК назначались с первого курса химиотерапии. Таблица 26 Структура инфекционных осложнений у больных ОМЛ средней и высокой групп риска после курса индукционной химиотерапии Инфекционные Группа среднего риска Группа высокого риска осложнения ОМЛ НИИ ОМЛ НИИ ОМЛ НИИ ОМЛ НИИ ДОГ ДО 2002, ДОГ 2007, ДО 2002, 2007, N (%) N (%) N (%) N (%) Фебрильная нейтропения Пневмония Нейтропеничес кий энтероколит Инфекция мягких тканей Сепсис Отсутствие признаков инфекции Всего больных P 14 (58,3%) 2 (18,2%) 7 (63,6%) 5 (27,8%) 2 (8,3%) 2 (8,3%) 4 (36,4%) 3 (27,3%) 2 (18,2%) 1(9,1%) 7 (38,9%) 1 (5,6%) 0 0 0 3(16,7%) 4 (16,7%) 2 (8,3%) 1 (9,1%) 1 (9,1%) 1 (9,1%) 0 2 (11,1%) 0 24 0,81 11 11 0,28 18 88 Структура инфекционных осложнений в группе среднего риска различна, так при лечении по протоколу ОМЛ НИИ ДО 2002, наибольшее число составило больных с фебрильной нейтропенией 14 (58,3%) и сепсисом – 4 (16,7%), а при лечении по протоколу ОМЛ НИИ ДОГ 2007, у пациентов чаще диагностировалась пневмония – 4 (36,4%) и нейтропенический энтероколит 3 (27,3%). Процент детей с ОМЛ, перенесших индукционную химиотерапию без инфекционных осложнений был практически одинаков (8,3% по протоколу ОМЛ НИИ ДО 2002 и 9,1% по протоколу ОМЛ НИИ ДОГ 2007). В группе детей высокого риска структура инфекционных осложнения, при лечении по протоколу ОМЛ НИИ ДО 2002, чаще диагностировалась фебрильная нейтропения – у 7 (63,6%) и пневмония – у 2 (18,2%) больных; при лечении по протоколу ОМЛ НИИ ДОГ 2007: пневмония – у 7 (38,9%) и фебрильная нейтропения – 5 (27,8%) детей. Таким образом, структура инфекционных осложнений не зависит от добавления к лечению препаратов с эпигенетическим действием (ATRA и ВК), а более частое обнаружение локализованных инфекционных очагов, при лечении по протоколу ОМЛ НИИ ДОГ 2007, связано, вероятно, с более совершенными методами инструментальной диагностики. 89 ГЛАВА 4. РОЛЬ ВЫСОКОДОЗНОЙ ХИМИОТЕРАПИИ И ТРАНСПЛАНТАЦИИ ГЕМОПОЭТИЧЕСКИХ СТВОЛОВЫХ КЛЕТОК В ЛЕЧЕНИИ ДЕТЕЙ С ОМЛ Согласно протоколу НИИ ДО ОМЛ 2002 в группе больных среднего риска планировалось проведение ВДХТ с ауто- ТГСК, а в группе высокого риска ВДХТ с алло- ТГСК или при отсутствии совместимого донора ауто- ТГСК, при отказе родителей от ВДХТ была продолжена поддерживающая терапия. Таким образом, в протоколе НИИ ДО ОМЛ 2002 тринадцати (25,5%) пациентам была проведена ВДХТ с ауто- ТГСК и одному (1,9%) алло-ТГСК. В протоколе НИИ ДОГ ОМЛ 2007 в группе высокого риска планировалось проведение ВДХТ с алло-ТГСК при наличии полностью совместимого родственного донора, но так как доноров не было, всем пациентам была продолжена химиотерапия и поддерживающая терапия. У трех (5,7%) пациентов в протоколе НИИ ДОГ ОМЛ 2007 не была достигнута ремиссия к окончании индукционной химиотерапии, им был проведен курс FLAG, а затем ВДХТ и частично совместимая родственная ТГСК. Характеристика пациентов представлена в таблице 27. 90 Таблица 27 Характеристика пациентов, которым проведена ВДХТ с ТГСК Возраст (годы) Пол Группа риска FABвариант лейкоза Ответ на 15 день индукции Ответ на индукцию Цитогенетика Исход НИИ ДО ОМЛ 2002 1 14,0 Муж Средняя М2 М-1 Ремиссия 46xy умер 2 14,0 Муж Средняя М1 М-1 Ремиссия 47xxy жив 3 9,0 Муж Средняя М1 М-2 47xy (+21) 4 4,0 Муж Стандартная М1 М-2 5 9,0 Муж Средняя М2 М-1 Нет ремиссии Нет ремиссии Ремиссия умер (рецидив) умер (рецидив) жив 6 2,0 Жен Высокая М4 М-1 Ремиссия 7 4,0 Жен Средняя М1 М-1 Ремиссия 46x(-x), -2, -11, +t(x;2) (q22;q21), +t(2;11), +m Нет данных 8 7,0 Жен Средняя М1 М-1 Ремиссия 46xx 9 13,0 10 6,0 Муж Жен Средняя Стандартная М1 М2 М-1 М-1 t(8;21) t(8;21)(q22;q22) 11 14,0 Жен Средняя М1 М-3 Ремиссия Нет ремиссии Ремиссия умер (рецидив) умер (рецидив) умер (рецидив) жив жив Нет данных жив 47xy (+21) t(8;21) 91 Продолжение таблицы 27 12 2,0 Муж Средняя М1 М-1 Ремиссия 13 2 мес. Жен Высокая М5 М-1 Ремиссия 14 2,0 Муж Средняя М1 М-1 Ремиссия НИИ ДОГ ОМЛ 2007 1 4,0 Муж Высокая М2 М-1 Нет ремиссии Нет ремиссии Нет ремиссии 2 11,0 Муж Средняя М2 М-3 3 15,0 Муж Высокая М4 М-2 48xy, +8, +19, del(7)(q34), add(12)(p12) Comp, 50xx, +8, -10, -11, +16, +t(10;11;12) (p12;q13;q23;q15), +t(10;11) (p12;q13), +del(12)(q15) Нет данных умер (рецидив) жив 46xy, add(11)(q23) жив 47ху (+8) жив 46xy умер (рецидив) жив (рецидив) 92 При анализе показателей выживаемости у пациентов, лечившихся по протоколу ОМЛ НИИ ДО 2002, в группе детей, которым было рекомендовано проведение ТГСК, но не была сделана, пятилетняя БРВ и БСВ составили 46,2±14,7%, средняя продолжительность наблюдения 58,8±13,1 мес, а ОВ – 57,7±15,7%, средняя продолжительность наблюдения 72,2±13,1 мес. У тех больных, которым была проведена ТГСК, согласно протоколу ОМЛ НИИ ДО 2002, БРВ составила 46,4±14,9%, средняя продолжительность наблюдения 69,9±8,6 мес (p=0,21); БСВ – 42,9±13,5%, средняя продолжительность наблюдения 69,9±8,6 мес (p=0,22); ОВ – 55,0±14,2%, средняя продолжительность наблюдения 71,3±14,9 мес (p=0,11). Результаты представлены на рисунках 39, 40, 41. Рисунок 39. БРВ больных ОМЛ, в зависимости от типа терапии, согласно протоколу ОМЛ НИИ ДО 2002 93 Рисунок 40. БСВ больных ОМЛ, в зависимости от типа терапии, согласно протоколу ОМЛ НИИ ДО 2002 Рисунок 41. Рисунок. ОВ больных ОМЛ, в зависимости от типа терапии, согласно протоколу ОМЛ НИИ ДО 2002 94 Следовательно, не получено разницы в БРВ, БСВ и ОВ среди групп больных, которым была проведена ВДХТ с ауто-ТГСК и тех, кто получал только химиотерапию, хотя было показано проведение ВДХТ с ауто-ТГСК. Проведение ВДХТ с алло-ТГСК в протоколе ОМЛ НИИ ДОГ 2007 было показано 32 (45,7%) пациентам, из них 28, в связи с отсутствием донора была продолжена ХТ, пятилетняя БРВ в этой группе составила 58,0±11,1%; БСВ – 53,3±10,7% и ОВ – 51,1±10,9%. Таким образом, благодаря сочетанию химиотерапии с эпигенетическим лечение нам удалось без проведения аллогенной ТГСК достичь высоких показателей выживаемости у детей с ОМЛ. Высокодозная химиотерапия с дальнейшей родственной частично совместимой ТГСК была проведена 3 (4,3%) пациентам, не достигшим ремиссию после индукционной ХТ, 2-летняя выживаемость (БРВ, БСВ, ОВ) составила 33,3±27,8%. Таким образом, алло-ТГСК является абсолютно необходимым компонентом терапии у пациентов, которые не достигли ремиссию к окончанию индукционной химиотерапии. 95 ЗАКЛЮЧЕНИЕ Острый лейкоз представляет собой наиболее распространенный вид опухоли в детском и подростковом возрасте, на которые приходится 26% всех опухолей диагностируемых у детей [4]. Применяя современную химиотерапию, у 90% детей с ОМЛ удается достичь ремиссию [109], но общая выживаемость остается низкой [112]. Современные исследования дают аналогичные оценки бессобытийной выживаемости (41% - 56%) наиболее частой причиной прекращения лечения является ранний рецидив, что требует разработки новых эффективных методов терапии [6, 59, 83]. Лечение ОМЛ состоит из индукции с использованием интенсивной химиотерапии, периода консолидации, с использованием трансплантации гемопоэтических стволовых клеток или химиотерапии. Доказано отсутствие улучшения выживаемости больных при увеличении числа курсов химиотерапии, увеличение токсичности как острой, так и поздней при увеличении доз химиопрепаратов [21, 27]. Вопрос о проведении ТГСК у детей, больных ОМЛ в первую ремиссию остается достаточно спорным. Частота рецидивов после аллогеной ТГСК значительно ниже, чем после химиотерапии, но при этом токсичность при ТГСК значительно выше [7, 22, 73]. Кроме того, вероятность достичь второй ремиссии после аллогеной ТГСК зачастую ниже, чем после химиотерапии. Поэтому достаточно перспективный путь лечения детей, больных ОМЛ, является применение новых препаратов, влияющих на развитие опухоли. Исследования в области молекулярной генетики сыграли важную роль в расшифровке молекулярного патогенеза ОМЛ. Генные мутации, дисрегуляция экспрессии генов или набора генов позволяют выделить огромное разнообразие генетических подмножеств острых лейкозов. Поиск новой терапии для излечения и улучшения качества жизни детей больных ОМЛ – главная задача в настоящее время. Учитывая, что у большинства детей с ОМЛ выявляется нормальный кариотип опухолевых клеток, необходим поиск общих элементов, контролирующих их работу. Именно эпигенетические 96 сигналы накладываются на генетику и по-своему решают реализацию генетической информации [1]. В отличие от генетических аномалий, которые являются необратимыми, эпигенетические изменения могут быть изменены фармакологически. Следовательно, возможно эпигенетические препараты, имеют сильный терапевтический потенциал в лечении ОМЛ [95]. Сочетание вальпроевой кислоты и полностью транс-ретиноевой кислоты (ATRA), как дифференцирующих агентов, представляют собой препараты с эпигенетическим противоопухолевое действием. действие ВК предположительно, оказывает путем протеосомальной деградации гистондеацетилазы. ATRA может индуцировать дифференцировку и апоптоз опухолевых клеток при разных вариантах ОМЛ. Лечение ATRA при ОМЛ влияет на морфологию лейкозных клеток, регуляцию клеточного цикла и апоптоз, и, возможно, также микрососудистые эндотелиальные функции клеток [50]. Исследования in vitro и in vivo у пациентов с рецидивами/рефрактерными видами ОМЛ показали, что ВК более эффективна в сочетании с различными препаратами [92, 105, 117]. Для того чтобы увеличить число ремиссий и выживаемость детей, больных острым миелоидным лейкозом, нами к химиотерапии были добавлены препараты с эпигенетическим действием. Проводился сравнительный анализ результатов лечения пациентов с ОМЛ, получавших химиотерапевтическое лечение согласно протоколу ОМЛ НИИ ДО 2002 с результатами, полученными при включении в протокол (ОМЛ НИИ ДОГ 2007) эпигенетической терапии (вальпроевая и полностью транс-ретиноевая кислоты). В нашем исследовании мы оценивали частоту ремиссий в результате терапии индукции у детей с ОМЛ, было проведено сравнение выживаемости больных с ОМЛ, получавших лечение по двум протоколам. Была проанализирована эффективность поддерживающей терапии с эпигенетическим лечением и без него у больных со стандартным риском ОМЛ, а так же токсичность вальпроевой кислоты и полностью транс-ретиноевой кислот в протоколе ОМЛ НИИ ДОГ 2007. В исследование был включен 121 ребенок, с подтвержденным диагнозом 97 ОМЛ. Из них 51(42,1%) больной получил лечение по протоколу ОМЛ НИИ ДО 2002, 70 (57,9%) – по протоколу ОМЛ НИИ ДОГ 2007. Пациенты лечились в 8 онкологических стационарах. Наблюдаемые дети были в возрасте от 2,5мес. до 17 лет. Средний возраст больных составил 7,8±0,5 лет. Мальчиков было немного больше 68 (52,2%), чем девочек 53 (43,8%). Не было выявлено достоверной разницы в распределении больных по полу и возрастным группам (p=0,32 и p=0,23 соответственно). Диагноз устанавливался на основании морфологических, цитохимических, иммунологических и цитогенетических параметров бластных клеток, обнаруживаемых в периферической крови и костном мозге. У 84 пациентов был проведен хромосомный анализ, который подтвердил высокую гетерогенность данного заболевания, так у 27,4% пациентов были диагностированы различные не повторяющиеся генетические аномалии, а у 19% больных нормальный кариотип опухолевых клеток. Все пациенты в двух протоколах были стратифицированы на группы риска (стандартная, средняя, высокая). В протоколе ОМЛ НИИ ДОГ 2007 половина детей вошли в группу высокого риска, а в протоколе ОМЛ НИИ ДО 2002 в группу среднего риска (р=0,02). Учитывая наличие достоверной разницы при данной стратификации пациентов, больные среднего и высокого риска были объединены в неблагоприятная группу риска. В протоколе ОМЛ НИИ ДО 2002 в нее вошло 38 (74,5%) детей, а в протоколе ОМЛ НИИ ДОГ 2007 – 57 (81,4%) больных (р=0,36). Препараты с эпигенетическим действием в протоколе ОМЛ НИИ ДОГ 2007 были включены в поддерживающее лечение у больных стандартной группы риска, у пациентов средней и высокой групп с начала курса индукции. К 15 дню от начала индукции число полных ответов в обоих протоколах было одинаково: у 55 (78,6%) пациентов в протоколе ОМЛ НИИ ДОГ 2007, и у 39 (76,5%) в протоколе ОМЛ НИИ ДО 2002. Лечение больных группы среднего риска до 15 дня отличалось только включением эпигенетических препаратов в протоколе ОМЛ НИИ ДОГ 2007, благодаря чему количество полных ответов 98 увеличилось до 81,4% (18 пациентов) с 64,0% (16 больных) в сравнении только с химиотерапевтическим лечением в протоколе ОМЛ НИИ ДО 2002 (р=0,24). У пациентов, получавших лечение согласно протоколу ОМЛ НИИ ДО 2002, группы высокого риска число полных ответов к 15 дню было больше – 84,6% (11 больных), чем у, лечившихся по протоколу ОМЛ НИИ ДОГ 2007 – 68,6% (24 ребенка), р=0,1. Это связано с более интенсивным первым курсом индукционной химиотерапии в протоколе ОМЛ НИИ ДО 2002, где суммарная доза ARA-C составила 3000 мг/м2 и была больше, чем в протоколе ОМЛ НИИ ДОГ 2007 (суммарная доза ARA-C=1400 мг/м2). Частота полных ответов у больных неблагоприятного риска в двух протоколах была одинаковым – в 73,7% (42 пациента) протокол ОМЛ НИИ ДОГ 2007 и в 71,1% (27 больных) – протокол ОМЛ НИИ ДО 2002), р=0,98. Сочетание эпигенетического лечения с химиотерапией позволило в протоколе ОМЛ НИИ ДОГ 2007 по окончании индукционной химиотерапии у 64 (91,4%) пациентов достичь ремиссию, что значительно выше, чем при лечение согласно протоколу ОМЛ НИИ ДО 2002 – 43 (84,3%) ребенка (р=0,3). Включение эпигенетического лечения у больных высокой группы риска позволило достоверно увеличить число ремиссий до 91,4% (32 пациента) в протоколе ОМЛ НИИ ДОГ 2007 с 69,2% (19 детей) – протокол ОМЛ НИИ ДО 2002 (р=0,05). В протоколе ОМЛ НИИ ДОГ 2007 удалось улучшить показатели пятилетней выживаемости (БРВ, БСВ, ОВ) детей до 56,1±7,0%; 51,7±7,0%; 54,7±6,1% против 43,6±7,2% (р=0,09); 39,2±7,0% (р=0,75); 47,4±7,0% (р=0,39) соответственно при лечении по протоколу ОМЛ НИИ ДО 2002. Благодаря добавлению эпигенетического лечения в протоколе ОМЛ НИИ ДОГ 2007 удалось значительно и достоверно повысить показатели выживаемости детей младше года: пятилетнюю БРВ до 91,7±8,1%; БСВ до 81,8±11,2% и ОВ до 83,9±10,4%; по сравнению с протоколом ОМЛ НИИ ДО 2002, где пятилетняя БРВ составила – 25,0±21,3% (p=0,002); БСВ – 25,0±22,4% (p=0,015) и ОВ – 25,0±22,0% (p=0,01). Больные до года в обоих протоколах были из высокой группы риска. Вероятнее всего препараты с эпигенетическим действием именно у маленьких 99 детей способствуют дифференцировке бластных клеток и делают их более чувствительными к химиопрепаратам. Показатели выживаемости больных старше 14 лет оказались хуже при лечении по протоколу ОМЛ НИИ ДОГ 2007: БРВ – 25,0±15,0%; БСВ – 25,0±15,0%; ОВ – 25,6±15,3%, чем в протоколе ОМЛ НИИ ДО 2002: 85,7±13,1(p=0,02); 85,7±13,1%(p=0,03), 87,7±13,0%(p=0,007) соответственно. Что объясняется тем, что в протоколе ОМЛ НИИ ДОГ 2007 все пациенты старше 14 лет были плохой прогностической группы. При лечении по протоколу ОМЛ НИИ ДО 2002 важное прогностическое значение имел FAB морфологический вариант, подобных данных при анализе выживаемости детей, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, не получено. В протоколе ОМЛ НИИ ДОГ 2007 утратило свое неблагоприятное значении FAB-М5, FAB-М6, FAB-М7 вариантов ОМЛ. Безрецидивная и бессобытийная пятилетняя выживаемость детей, включенных в стандартную группу риска, в протоколе ОМЛ НИИ ДОГ 2007 составила 71,3±14,1% против 69,2±12,7% в протоколе ОМЛ НИИ ДО 2002 (p=0,68) и ОВ – 90,0±9,8% против 75,5±12,3% (p=0,29) соответственно. Разница в показателях выживаемости получена незначительная, но, все таки, можно говорить о наличии тенденции к её увеличения у больных, получавших эпигенетические препараты в поддерживающей терапии (протокол НИИ ДОГ ОМЛ 2007). Пятилетняя безрецидивная выживаемость, больных средней группы риска, лечившихся по протоколу ОМЛ НИИ ДОГ 2007, составила 52,9±11,3%; БСВ – 47,8±12,8% и ОВ – 47,6±11,5% против 35,9±10,8% (p=0,3); 32,0±7,3% (p=0,26) и 40,5±10,6% (p=0,69) соответственно в протоколе ОМЛ НИИ ДО 2002. В группе высокого риска благодаря сочетанию химиотерапии и эпигенетической терапии в протоколе ОМЛ НИИ ДОГ 2007, БРВ составила 53,4±10,1%; БСВ – 45,0±9,8%; ОВ – 45,1±10,7%; что значительно выше показателей, полученных при лечении протоколу ОМЛ НИИ ДО 2002, где БРВ была 30,8±10,9% (p=0,04), БСВ – 23,1±12,7% (p=0,2) и ОВ – 30,8±13,9% (p=0,16). Нашим исследованием подтверждена важность оценки ответа на 15 день от начала курс индукционной химиотерапии. При полном ответе к 15 дню индукции 100 в протоколе ОМЛ НИИ ДОГ 2007, БРВ больных составила 65,9±8,5%; БСВ – 65,9±8,5% и ОВ – 66,0±7,7% против 52,1±8,9% (p=0,2); 52,1±8,9% (p=0,13); 55,1±8,7% (p=0,42) в протоколе ОМЛ НИИ ДО 2002. Показатели выживаемости у больных с М-2 и М-3 ответом оказались значительно хуже. Сочетание эпигенетического лечения с химиотерапией позволило у пациентов с частичным ответом увеличить пятилетнюю БРВ, БСВ и ОВ до 21,4±18,7% в протоколе ОМЛ НИИ ДОГ 2007 с 16,7±15,9% (протокол ОМЛ НИИ ДО 2002), p>0,6. А у больных с М-3 ответом была достигнута пятилетняя БРВ – 34,3±19,7%; БСВ – 34,3±19,7% и ОВ – 35,0±17,2% в протоколе ОМЛ НИИ ДОГ 2007 против 20,0±18,9% (p=0,2); 20,0±18,9% (p=0,33); 40,0±22,3% (p=0,7) соответственно в протоколе ОМЛ НИИ ДО 2002. Таким образом, пациентам с частичным или отсутствием ответа на 15 день индукциии необходимо решение вопроса о проведение ВДХТ с последующей аллогеной ТГСК, в том числе родственной частично совместимой ТГСК. Благодаря включению эпигенетических препаратов не только в индукционную химиотерапию, а так же в курсы консолидации, интенсификации и поддерживающее лечении, у больных достигших ремиссии к окончанию индукции в протоколе ОМЛ НИИ ДОГ 2007 удалось увеличить пятилетнюю БРВ до 60,7±7,3%; БСВ – 53,8±7,3; ОВ – 60,2±6,9% в сравнении с протоколом НИИ ДО ОМЛ 2002, где БРВ составила 51,7±8,0% (р=0,2); БСВ – 43,8±7,6% (р=0,3) и ОВ – 54,5±7,8% (р=0,4). Не выявлено достоверной разницы в выживаемости детей, которым была проведена ВДХТ с ауто-ТГСК и теми, кто получал химиотерапевтическое лечение согласно протоколу ОМЛ НИИ ДО 2002. Пятилетняя БРВ и БСВ пациентов, получивших ХТ, составили 46,2±14,7% и ОВ – 57,7±15,7%; а больных, которым была проведена ВДХТ с ауто-ТГСК БРВ – 46,4±14,9% (p=0,21); БСВ – 42,9±13,5% (p=0,22); ОВ – 55,0±14,2% (p=0,11). Возможно, проведение ВДХТ с ауто-ТГСК у пациентов с отсутствием минимальной остаточной болезнью позволит сократить длительность лечения, а, следовательно, токсичность и стоимость всего лечения. Благодаря проведению родственной частично совместимой ТГСК, в группе детей 101 с крайне неблагоприятным прогнозом (рефрактерными формами лейкоза), получавших терапию по протоколу ОМЛ НИИ ДОГ 2007, достигнута 2-летняя выживаемости 33,3±27,8%, что дает возможность рассматривать проведение аллогенной родственной частично совместимой ТГСК, как опцию при лечении данной группы больных. Добавление к терапии препаратов с эпигенетическим действием к химиотерапии не повлияло на ухудшение переносимости или усиление токсичности лечения. Таким образом, благодаря применению препаратов, воздействующих на активность генов, посредством снижения уровня гиперметелирования ДНК удалось увеличить количество ремиссий в результате индукции ремиссии, также БСВ, БРВ и ОВ. Не было отмечено увеличения длительности аплазии костного мозга и увеличения какой-либо токсичности при лечении химиопрепаратами в сочетании с вальпроевой и ретиноевой кислотами. Кроме того, доказана необходимость проведения алло-ТГСК больным ОМЛ, у которых отмечался медленный ответ на индуктивное лечение. 102 ВЫВОДЫ 1. Количество ремиссий было больше у пациентов, получавших лечение по протоколу ОМЛ НИИ ДОГ 2007 – 91,4%, чем у детей, лечившихся по протоколу ОМЛ НИИ ДО 2002 – 84,3% (р=0,3). Увеличена частота ремиссий в результате терапии индукции у детей с высоким риском ОМЛ, лечившихся по протоколу ОМЛ НИИ ДОГ 2007 – 91,4%, чем у детей, получавших лечение без эпигенетической терапии по протоколу ОМЛ НИИ ДО 2002 – 69,2% (р=0,05). 2. Благодаря включению препаратов с эпигенетическим действием удалось улучшить безрецидивную, бессобытийную и общую выживаемость детей с ОМЛ: 56,1±7,0%; 51,7±7,0%; 54,7±6,1%, по сравнению с пациентами, получавшими только химиотерапию 43,6±7,2% (р=0,09); 39,2±7,0% (р=0,75)%; 47,4±7,0% (р=0,39) соответственно. 3. Достоверно увеличена безрецидивная выживаемость при сочетании эпигенетической и химиотерапии у больных ОМЛ высокой группы риска – 53,4±10,1%; чем по протоколу ОМЛ НИИ ДО 2002 – 30,8±10,9% (p=0,04), а так же безрецидивная и бессобытийная у больных из неблагоприятной группы риска 53,0±8,5% и 47,6±8,9%, по сравнению с протоколом ОМЛ НИИ ДО 2002 – 34,0±8,1% (p=0,04) и 28,9±7,9% (p=0,03) соответственно. 4. Включение эпигенетических препаратов в поддерживающую терапию у больных ОМЛ стандартной группы риска выявило тенденцию к улучшению безрецидивной, бессобытийной 71,3±14,1% и общей выживаемости 90,0±9,8%, по сравнению только с химиотерапией 69,2±12,7% (p=0,68) и 75,5±12,3% (p=0,29) соответственно в протоколе ОМЛ НИИ ДО 2002. 5. Доказана высокая эффективность эпигенетической терапии у детей до одного года в протоколе ОМЛ НИИ ДОГ 2007: безрецидивная выживаемость составила 91,7±8,1%; бессобытийная 81,8±11,2% и общая 83,9±10,4%; по 103 сравнению с протоколом ОМЛ НИИ ДО 2002 – 25,0±21,3% (p=0,002); 25,0±22,4% (p=0,015) и 25,0±22,0% (p=0,01) соответственно. 6. Препараты с эпигенетическим действием не ухудшают переносимость и не увеличивают токсичность при добавлении их к химиотерапии. 104 СПИСОК ЛИТЕРАТУРЫ 1. Ванюшин Б.Ф. Материализация эпигенетики, или небольшие изменения с большими последствиями // Химия и жизнь. 2004. № 2. С.32-37. 2. Демидова И. А. Эпигенетические нарушения при острых лейкозах // Клиническая онкогематология. 2008. Т.1(1). С. 16-20. 3. Ковалев Р.А., Штамм Т.А. Эпигенетическая терапия рака с применением ингибитора деацетилаз. Роль онкосупрессорного белка р53 // Молекулярная биология. 2008. С.23. 4. Лейкозы у детей / под ред.: Г.Л. Менткевич, С.А. Маякова. М: Практическая медицина, 2009. 346 с. 5. Структурные особенности метилирования генома человека при лейкозах / Е.Н. Вшивцева [и др.] // Молекулярная биология. 2008. С.9-10. 6. Эпигенетическая терапия индукции ремиссии у детей, больных острым миелоидным лейкозом / А. В. Попа [и др.] // Клиническая онкогематология. 2008. Т.1(1). С. 34-38. 7. A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission / W.G. Woods [et al.] // Blood. 2001. vol. 97(1). PP.56-62. 8. A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia / D. Niewerth [et al.] // Blood. 2010. vol. 116(13). PP. 2205-2213. 9. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value / S. Abbas [et al.] // Blood. 2010. vol. 116(12). PP. 2122-2126. 10. Acute myeloid leukemia with myelodysplasia-related changes in WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. 4th edition / D.A. Arber [et al.] // International Agency for Research on Cancer (IARC), Lyon, France. 2008b. PP. 124-126. 11. Acute myeloid leukemia: Therapeutic impact of epigenetic drugs / L. Altucci [et al.] // Int Journ of Biochem and Cell Biol. 2005. vol. 37. PP. 1752-1762. 105 12.Age, gene/environment susceptibility-Reykjavik Study: multidisciplinary applied phenomics / T.B. Harris [et al.] // Am J Epidemiol. 2007. vol. 165(9). PP. 1076-87. 13. ALL-1 s a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulations / T. Nakamura [et al.] // Mol Cell. vol. 10. PP. 1119-1128. 14. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations / J.L. Tang [et al.] // Blood. 2009. vol. 114(26). PP. 5352-5361. 15. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor / B. Sanchez-Gonzalez [et al.] //Blood. 2006. vol. 108(4). PP. 1174–1182. 16. Bernstein B.E., Meissner A., Lander E. S. The mammalian epigenome // Cell. 2007. vol. 128. PP. 669–681. 17. Bird A. Perceptions of epigenetics // Nature. 2007. vol. 447. PP. 396-398. 18. Bonetta L. Epigenomics: Detailed analysis // Nature. 2008. vol. 454(7205). PP.795798. 19. Bonetta L. Getting up close and personal with your genome // Cell. 2008. vol. 133(5). PP. 753-756. 20. Bonnet D., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell // Nat Med. 1997. vol. 3(7). PP. 730–737. 21.Can anthracycline therapy for pediatric malignancies be less cardiotoxic? / J.M. Fulbright [et al.] // Curr Oncol Rep. 2010. vol. 12(6). PP. 411-419. 22. Causes of death–other than progressive leukemia–in childhood acute lymphoblastic (ALL) and myeloid leukemia (AML): the Dutch Childhood Oncology Group experience / A.M. Slats [et al.] // Leukemia. 2005. vol. 19(4). PP. 537-544. 23. Comment on ‘‘Chromosomal instability and tumors promoted by DNA hypomethylation’’ and ‘‘Induction of tumors in nice by genomic hypomethylation’’ / A.S. Yang [et al.] // Science. 2003. vol. 14. PP.489-492. 106 24. Comprehensive methylome map of lineage commitment from hematopoietic progenitors / H. Ji [et al.] // Nature. 2010. vol. 467(7313). PP.338-42. 25. Constantinides P.G., Jones P.A., Gevers W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment // Nature. 1997. vol. 267. PP. 364–366. 26. Creutzig U., Reinhardt D. Current controversies: which patients with acute myeloid leukemia should receive a bone marrow transplantation? – European review // Br J Hematol. 2002. vol. 118(2). PP.365- 377. 27.Daunorubicin versus mitoxantrone versus idarubicin as induction and consolidation chemotherapy for adults with acute myeloid leukemia: The EORTC and GIMEMA Groups Study AML-10 / F. Mandelli [et al.] // JCO. 2009. vol. 27(32). PP. 53975403. 28. Di Croce L. Chromatin modifying activity of leukemia associated fusion proteins // Human Mol Genet. 2005. vol. 14. PP. 77-84. 29. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet / H. Do¨hner [et al.] //Blood. 2010. vol. 115(3). PP. 453-474. 30. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome / M. Weber [et al.] // Nat Genet. 2007. vol. 39. PP.457-466. 31. DNA methyltransferase 1 and 3B activate BAG-1 expression via recruitment of CTCFL/BORIS and modulation of promoter histone methylation / L. Sun [et al.] // Cancer Res. 2008. vol. 68(8). PP.2726-35. 32. DNMT3A mutations in acute myeloid leukemia / T.J. Ley [et al.] // N Engl J Med. 2010. vol. 363(25). PP. 2424-2433. 33. Döhner Н., Gaidzik V. I. Impact of genetic features on treatment decisions in AML // Hematology. American Society of Hematology Education Program Book. 2011. PP.36-41. 107 34. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome / B.J. Wouters [et al.] // Blood. 2009. vol. 113(13). PP.3088-3091. 35. Ducasse M., Brown M. Epigenetic aberrations and cancer // Molecular Cancer. 2006. vol. 5 PP. 60-70. 36. Epigenetic differences arise during the lifetime of monozygotic twins / M.F. Fraga [et al.] // Proc. Natl Acad. Sci. USA. 2005. vol. 102(30). PP. 10604–10609. 37. Epigenetic programming by maternal behavior / I. C. Weaver [et al.] // Nat Neurosci. 2004 vol. 7(8). PP.847-854. 38. Epigenetics in human disease and prospects for epigenetic therapy / G. Egger [et al.] // Nature. 2004. vol. 429(6990). PP. 457-63. 39. Esteller M. Epigenetics in Cancer // N Engl J Med. 2008. vol. 358. PP. 1148-59. 40. Esteller M. Epigenetics in changes cancer // Biol Rep. 2011. PP. 1-6. 41. Fathi H., Levis M. FLT3 inhibitors: a story of the old and the new // Curr Opin Hematol. 2011. vol. 18(2). PP.71-76. 42. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study / H. Becker [et al.] // J Clin Oncol. 2010. vol. 28(4). PP. 596-604. 43. Feinberg A. P. Epigenomics reveals a functional genome anatomy and a new approach to common disease // Nat Biotechnol. 2010. vol. 28(10). PP. 1049–1052. 44. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from AMLSG trial AML HD98B / R.F. Schlenk [et al.] // Haematologica. 2009. vol. 94(1). PP. 54-60. 45. Grimwade D., Vyas P., Freeman S. Assessment of minimal residual disease in acute myeloid leukemia // Curr Opin Oncol. 2010. vol. 22(6). PP. 656-663. 108 46. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2´-deoxycytidine / C.T. Nguyen [et al.] // Cancer Res. 2002. vol. 62. PP. 6456–6461. 47. Identification of patients with acute myeloid leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial / Burnett A.K. [et al.] // J Clin Oncol. 2011. vol. 29(4). PP. 369-377. 48. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B Study / G. Marcucci [et al.] // J Clin Oncol. 2010. vol. 28(14). PP. 2348-2355. 49. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia (AML) and confer adverse prognosis in cytogenetically normal AML with NPM1 mutation without FLT3-ITD / P. Paschka [et al.] // J Clin Oncol. 2010. vol. 28(22). PP. 3636-3643. 50. In vivo biological effects of ATRA in the treatment of AML / A. Ryningen [et al.] // Expert Opin Investing Drugs. 2008. vol. 17(11). PP. 1623-33. 51. Increase in platelet count in older, poor-risk patients with acute myeloid leukemia or myelodysplastic syndrome treated with valproic acid and all-trans retinoic acid / C. Pilatrino [et al.] // Cancer. 2005. vol. 104. PP. 101–109. 52. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome / S. Kayser [et al.] // Blood. 2009. vol. 114(12). PP. 2386-2392. 53.Interleukin-2 for the treatment of advanced acute myelogenous leukemia patients with limited disease: updated experience with 20 cases / G. Meloni [et al.] // Leuk Lymphoma. 1996. vol. 21(5-6). PP.429-35. при поддержке на рецидивах рефрактерных 54. Interplay of RUNX1/MTG8 and DNA methyltransferase 1in acute myeloid leukemia / S. Liu [et al.] // Cancer Res. 2005. vol. 65. PP.1277-1284. 55. Intra-individual change over time in DNA methylation with familial clustering / H.T. Bjornsson [et al.] // JAMA. 2008. vol. 299(24). PP. 2877-2883. 109 56. Jones P.A., Baylin S.B. The epigenomics of cancer // Cell. 2007. vol. 128(4). PP. 683-92. 57. Jones P.A., Baylin S.B. The fundamental role of epigenetic events in cancer // Nat Rev Genet. 2002. vol. 3(6). PP. 415-28. 58. Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98 / U. Creutzig [et al.] // J Clin Oncol. 2006. Vol. 24(27). PP. 4499-4506. 59. Long-term results in children with AML: NOPHO-AML Study Group – Report of three consecutive trials / S.O. Lie [et al.] // Leukemia. 2005. vol. 19(12). 2090-2100. 60. Long-term results of children with acute myeloid leukemia: a report of three consecutive Phase III trials by the Children’s Cancer Group: CCG 251, CCG 213 and CCG 2891 / F.O. Smith [et al.] // Leukemia. 2005. vol. 19(12). PP. 2054-2062. 61. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer / M.F. Fraga [et al.] // Nat. Genet. 2005. vol. 37.PP. 391– 400. 62. Malfoy B. The revival of DNA methylation // J Cell Sci. 2000. vol. 113. PP. 38873888. 63. Marcucci G., Haferlach T., Do¨hner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications // J Clin Oncol. 2011. vol. 29(5). PP.475-486. 64. Marked improvements in outcome with chemotherapy alone in pediatric acute myeloid leukemia: results of the United Kingdom Medical Research Council’s 10th AML trial. MRC Childhood Leukaemia Working Party / R.F. Stevens [et al.] // Br J Haematol. 1998. vol. 101(1). PP. 130-140. 65. Marks P.A, Miller T., Richon V.M. Histone deacetylases // Curr Opin Pharmacol. 2003. vol. 3. PP. 344-351 66. Melki J.R., Vincent P.C., Clark S.J. Concurent DNA hypermethylation of multiple genes in acute myeloid leukemia // Cancer Res. 1999. vol. 59. PP. 3730-3740. 110 67. Meshinchi S., Arceci R.J. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia // Oncologist. 2007. vol. 12(3). PP. 341–355. 68. Monitoring of minimal residual disease in NPM1 mutated acute myeloid leukemia: A study of the German-Austrian AML Study Group (AMLSG) / J. Kro¨nke [et al.] // J Clin Oncol. 2011. vol. 29(19). PP. 2709-2716. 69.Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia / R.F. Schlenk [et al.] // N Engl J Med. 2008. vol. 358(18). PP. 1909-1918. 70. New Classification of Acute Myeloid Leukemia and Precursor-related Neoplasms: Changes and Unsolved Issues / B.M. Falini [et al.] // Discov Med. 2010. vol. 10(53). PP. 281-92. 71. Novel targeted drug therapies for the treatment of childhood acute leukemia / Brown P. [et al.] // Expert Rev Hematol. 2009. vol. 1; 2(9). PP.145-146. 72. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutaratedependent dioxygenases / W. Xu [et al.] // Cancer Cell. 2011. vol. 19(1). PP.17-30. 73. Outcomes in CCG-2961, a children’s oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children’s oncology group / B.J. Lange [et al.] // Blood. 2008. vol. 111(3). PP. 1044-1053. 74. Owen C., Fitzgibbon J., Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia // Hematol Oncol. 2010. vol. 28(1). PP.13-19. 75. Paschka P. Core-binding factor acute myeloid leukemia // Semin Oncol. 2008. vol. 35(4). PP. 410-417. 76. Phase I AML study of AC220, a potent and selective second generation FLT3 receptor tyrosine kinase inhibitor / J.E. Cortes [et al.] // Blood. 2008. vol. 112(11). PP.767. 77. Plimack E.R., Kantarjian H.M., Issa J.P. Decitabine and its role in the treatment of hematopoietic malignancies // Leuk Lymphoma. 2007. vol. 48(8). PP.1472-81. 111 78. Postremission therapy for children with acute myeloid leukemia: the children’s cancer group experience in the transplant era / T.A. Alonzo [et al.] // Leukemia. 2005. vol. 19(6). PP. 965- 970. 79. Prognostic impact, concurrent genetic mutations and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML: further evidence for CEBPA double mutant AML as a distinctive disease entity / E. Taskesen [et al.] // Blood. 2011. vol. 117(8). PP. 2469-2475. 80. Pui C.H., Evans W.E. Treatment of acute lymphoblastic leukemia // N Engl J Med. 2006. vol. 354(2). PP.166–178. 81. Pui C.H., Relling M.V. Downing J.R. Acute lymphoblastic leukemia // N Engl J Med. 2004. vol. 350(15). PP.1535–1548. 82. Quintas-Cardama A., Santos F.P., Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia // Leukemia. 2010. vol. 25(2). PP. 226-235. 83. Response-Guided Induction Therapy in Pediatric Acute Myeloid Leukemia With Excellent Remission Rate / J. Abrahamsson [et al.] // J Clin Oncol. 2011. vol. 29(3). PP. 310-315. 84. Results of 58872 and 58921 trials in acute myeloblastic leukemia and relative value of chemotherapy vs allogeneic bone marrow transplantation in first complete remission: the EORTC Children Leukemia Group report / N. Entz-Werle [et al.] // Leukemia. 2005. vol. 19(12). PP.2072-2081. 85. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia / A. Kuendgen [et al.] // Ann Hematol. 2005. vol. 84. PP. 61-66. 86. Results of a randomized trial in children with Acute Myeloid Leukemia: Medical Research Council AML12 trial / Brenda E. S. [et al.] // British Journal of Hematology. 2011. vol. 115. PP. 366–376. 112 87. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95 / A. Moricke [et al.] // Blood. 2008. vol. 111(9). PP. 4477–4489. 88. Role of histone deacetylase complex in acute promyelocytic leukemia / R.G. Lin [et al.] // Nature. 1991. vol. 391. PP. 811-814. 89.RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis / S. Schnittger [et al.] // Blood. 2011. vol. 117(8). PP. 2348- 2357. 90. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML Study Group / V.I. Gaidzik [et al.] // J Clin Oncol. 2011. vol. 29(10). PP.1364-1372. 91. Russo. V. E., Martienssen. R. A., Riggs. A. D. Epigenetic Mechanisms of Gene Regulation // Genes & Development. 1996. vol. 11 (11). PP. 1357-1492. 92. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome / A.O. Soriano [et al.] // Blood. 2007. vol. 110. PP. 2302–2308. 93.Second induction with high-dose cytarabine and mitoxantrone: different impact on pediatric AML patients with t (8;21) and with inv (16) / U. Creutzig [et al.] // Blood. 2011. vol. 118(20). PP. 5409-5415. 94. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia / Cimino G. [et al.] // Cancer Res. 2006. vol. 66. PP. 8903–8911. 95. Sharma S., Kelly T.K., Jones P.A. Epigenetics in cancer // Carcinogenesis. 2010. vol. 31(1). PP. 27-36. 96. Siitonen T., Koistinen P., Savolainen E.R. Increase in Ara-C cytotoxicity in the presence of valproate, a histone deacetylase inhibitor, is associated with the concurrent expression of cyclinD1 and p27 in acutemyeloblastic leukemia cells // Leukemia Research. 2005. vol. 29(11). PP.1335–1342. 113 97. Single nucleotide polymorphism in the mutational hotspot of WT1 predicts a favorable outcome in patients with cytogenetically normal acute myeloid leukemia / F. Damm [et al.] // J Clin Oncol. 2010. vol. 28(4). PP.578-585. 98. Successive clinical trials for childhood acute myeloid leukemia at St Jude Children’s Research Hospital, 1980 through 2000 / R.C. Ribeiro [et al.] // Leukemia. 2005. vol. 19(12). 2125-2129. 99. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer / Cameron E.E. [et al.] // Nat Genet. 1999. vol. 21. PP. 103–107. 100. Tefferi A., Thiele J.,Vardiman J.W. The 2008 World Health Organization Classification System for Myeloproliferative Neoplasms // Cancer. 2009. PP. 3842– 3847. 101. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: A Cancer and Leukemia Group B Study / K.H. Metzeler [et al.] // J Clin Oncol. 2011. vol. 29(10). PP. 1373-1381. 102. TET2 mutations in acute myeloid leukemia (AML): Results on 783 patients treated within the AML HD98A Study of the AML Study Group (AMLSG) / V.I. Gaidzik [et al.] // Blood. 2010. vol. 116(21). PP. 97. 103. The histone deacetylase inhibitor valproic acid alters sensitivity towards all trans retinoic acid in acute myeloblastic leukemia cells / M.R. Trus [et al.] // Leukemia. 2005. vol. 19. PP. 1161–1168. 104. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status / C.L. Green [et al.] // Blood. 2010. vol. 116(15). PP. 2779-2782. 105. The role of HDACs inhibitors in childhood and adolescence acute leukemias / R. Masetti [et al.] // J Biomed Biotechnol. 2011. vol. 11. PP. 9. 106. The t(8;21) fusion protein contacts co-repressors and histone deacetylases to repress the transcription of the p14ARF tumor repressor / S.W. Hiebert [et al.] // Blood Cells Mol Dis. 2003. vol. 30. PP. 177-183. 114 107. Tomizawa D., Tabuchi K., Kinoshita A. Repetitive cycles of high-dose cytarabine are effective for childhood acute myeloid leukemia: long-term outcome of the children with AML treated on two consecutive trials of Tokyo Children’s Cancer Study Group // Pediatric Blood Cancer. 2007. vol. 49(2). PP. 127-132. 108. TRAIL: At the center of drug able anti-tumor pathways / N. Clarke [et al.] //Cell Cycle. 2005. vol. 4(7). PP. 914-918. 109. Treatment and long-term results in children with acute myeloid leukaemia treated according to the AIEOP AML protocols / A. Pession [et al.] // Leukemia. 2005. vol. 19(12). PP.2043-2053. 110. Treatment of childhood acute myeloblastic leukemia. Dose intensification improves outcome and maintenance therapy is of no benefit. Multicenter studies of the French LAME (Leuce´mie Aigue¨ Mye´loblastique Enfant) Cooperative Group / Y. Perel [et al.] // Leukemia. 2005. vol. 19(12). 2082-2089. 111. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid / A. Kuendgen [et al.] // Blood. 2004. vol. 104. PP. 1266–1269. 112. Treatment strategies and long-term results in pediatric patients treated in four consecutive AML-BFM trials / U. Creutzig [et al.] // Leukemia. 2005. vol. 19(12). 2030-2042. 113. Treatment strategy and and long-term results in pediatric patients treated in two consecutive AML-GATLA-trials / H. Armendariz [et al.] // Leukemia 2005. vol. 19(12). PP. 2063-2071. 114. Treatment strategy and long-term results in pediatric patients treated in consecutive UKAML trials / B.E. Gibson [et al.] // Leukemia. 2005. vol. 19(12). PP. 2130-2138. 115. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia / E. Raffoux [et al.] // Haematologica. 2005. vol. 90. PP. 986–988. 115 116. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells / M. Gottlicher [et al.] // Embo J. 2001. vol. 20. PP. 6969–6978. 117. Valproic acid plus retinoic acid induce differentiation in chemotherapy resistant acute myeloid leucemia patients / С.Nervi [et al.] // Blood. 2004. vol. 10 (2) PP. 104. 118. Vardiman J.W., Harris N.L., Brunning R.D. The World Health Organization (WHO) classification of the myeloid neoplasms // Blood. 2002. vol. 100(7). PP.292302. 119. Waddington C.H. Preliminary notes on the development of the wings in normal and mutant strains of drosophila. USA: Proc Natl Acad Sci. 1939. 307 p. 120. Weaver I.C. Epigenetic effects of glucocorticoids // Semin Fetal Neonatal Med. 2009. vol. 14(3). PP.143-150. 121. Weaver I.C. Shaping adult phenotypes through early life environments // Birth Defects Res C Embryo Today. 2009. vol. 87(4). PP.314-326. 122. Yin C.C., Medeiros L.J., Bueso-Ramos C.E. Recent advances in the diagnosis and classification of myeloid neoplasms – comments on the 2008 WHO classification // Int. Jnl. Lab. Hem. 2010. vol. 32. PP. 461–476. 123. Yoo C. B., Jones P. A. Epigenetic therapy of cancer: past, present and future // Nat Rev Drug Discov. 2006. vol.5(1). PP. 37-50.