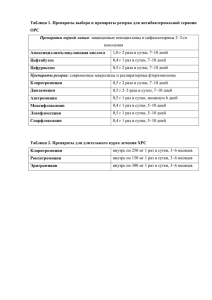
ОГЛАВЛЕНИЕ Авторский коллектив . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Список сокращений . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Введение . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Предмет, задачи и методология фармакологии . . . . . . . . . . . . . . . . Источники получения лекарственных веществ . . . . . . . . . . . . . . . . Этапы создания новых лекарственных средств . . . . . . . . . . . . . . . . Доклинические испытания . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Клинические исследования . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Исследования биоэквивалентности лекарственных препаратов . . . Регистрация лекарственного препарата . . . . . . . . . . . . . . . . . . . . . . Пострегистрационные испытания . . . . . . . . . . . . . . . . . . . . . . . . . . . История отечественной фармакологии . . . . . . . . . . . . . . . . . . . . . . . Названия лекарственных средств . . . . . . . . . . . . . . . . . . . . . . . . . . . . Классификации лекарственных средств . . . . . . . . . . . . . . . . . . . . . . 12 13 15 15 16 16 19 19 20 21 21 21 33 35 ЧАСТЬ I. ОБЩАЯ ФАРМАКОЛОГИЯ . . . . . . . . . . . . . . . . . . . . . . . . . 37 Глава 1. Фармакокинетика . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 1.1. Трансцеллюлярный транспорт лекарственных веществ . . . . . 38 1.2. Парацеллюлярный транспорт лекарственных веществ . . . . . . 47 1.3. Всасывание лекарственных веществ . . . . . . . . . . . . . . . . . . . . . . 49 1.4. Распределение лекарственных веществ в организме . . . . . . . . 65 1.5. Депонирование лекарственных веществ в организме . . . . . . . 72 1.6. Биотрансформация лекарственных веществ . . . . . . . . . . . . . . . 74 1.7. Выведение лекарственных веществ из организма . . . . . . . . . . 86 1.8. Математическое моделирование фармакокинетических процессов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92 1.9. Оптимизация дозирования лекарственных веществ . . . . . . . . 99 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 103 Глава 2. Фармакодинамика . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104 2.1. Фармакологические эффекты, локализация и механизмы действия лекарственных веществ . . . . . . . . . . . . . . . . . . . . . . . 104 2.2. Виды действия лекарственных веществ . . . . . . . . . . . . . . . . . . 122 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 124 Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику лекарственных веществ . . . . . . . . . . . 126 3.1. Свойства лекарственных веществ. Лекарственные формы . . 126 3.2. Свойства организма . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129 3.3. Режим назначения лекарственных веществ . . . . . . . . . . . . . . 132 3.4. Хронофармакология . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 144 4 Оглавление Глава 4. Понятие о фармакопрофилактике и фармакотерапии. Виды лекарственной терапии . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 5. Побочные и токсические действия лекарственных веществ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.1. Побочные действия лекарственных веществ . . . . . . . . . . . . . 5.2. Токсические действия лекарственных веществ . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . ЧАСТЬ II. ЧАСТНАЯ ФАРМАКОЛОГИЯ . . . . . . . . . . . . . . . . . . . . . Нейротропные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Средства, влияющие на периферическую нервную систему . . . . . . . Средства, действующие на афферентную иннервацию . . . . . . . . . . . Глава 6. Средства, угнетающие афферентную иннервацию . . . . . . 6.1. Местноанестезирующие средства (местные анестетики) . . . 6.2. Вяжущие, обволакивающие и адсорбирующие средства . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 7. Средства, стимулирующие окончания афферентных нервов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, действующие на эфферентную иннервацию . . . . . . . . . . . . Глава 8. Средства, действующие на холинергические синапсы . . . 8.1. Средства, стимулирующие холинергические синапсы . . . . . 8.2. Средства, блокирующие холинергические синапсы . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 9. Средства, действующие на адренергические синапсы . . . 9.1. Средства, стимулирующие адренергические синапсы . . . . . 9.2. Средства, блокирующие адренергические синапсы . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, влияющие на центральную нервную систему . . . . . . . . . . . Глава 10. Средства для наркоза (общие анестетики) . . . . . . . . . . . . 10.1. Средства для ингаляционного наркоза . . . . . . . . . . . . . . . . . 10.2. Средства для неингаляционного наркоза . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 11. Снотворные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.1. Снотворные средства с ненаркотическим типом действия . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.2. Снотворные средства с наркотическим типом действия . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 12. Противоэпилептические средства . . . . . . . . . . . . . . . . . . . 12.1. Блокаторы потенциалозависимых натриевых каналов . . . . 145 146 147 147 148 151 153 153 153 154 155 155 165 168 170 172 174 179 181 203 224 227 230 250 275 277 283 284 289 294 295 297 302 306 307 310 Оглавление 12.2. Средства, повышающие эффективность ГАМК-ергической системы . . . . . . . . . . . . . . . . . . . . . . . . . . . 12.3. Средства, угнетающие действие возбуждающих аминокислот . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12.4. Блокаторы кальциевых каналов Т-типа . . . . . . . . . . . . . . . . . 12.5. Средства, обладающие комбинированным действием . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 13. Противопаркинсонические средства . . . . . . . . . . . . . . . . . 13.1. Средства, стимулирующие дофаминергическую передачу . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13.2. Средства, угнетающие холинергическую передачу . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 14. Анальгезирующие средства (анальгетики) . . . . . . . . . . . . 14.1. Средства преимущественно центрального действия . . . . . . 14.2. Анальгезирующие средства преимущественно периферического действия (нестероидные противовоспалительные средства) . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 15. Психотропные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15.1. Антипсихотические средства . . . . . . . . . . . . . . . . . . . . . . . . . . 15.2. Антидепрессанты . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15.3. Нормотимические средства (соли лития) . . . . . . . . . . . . . . . 15.4. Анксиолитические средства (транквилизаторы) . . . . . . . . . 15.5. Седативные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15.6. Психостимуляторы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15.7. Ноотропные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 16. Аналептики . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, влияющие на функции исполнительных органов и систем . . . Глава 17. Средства, влияющие на функции органов дыхания . . . . 17.1. Стимуляторы дыхания . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17.2. Противокашлевые средства . . . . . . . . . . . . . . . . . . . . . . . . . . . 17.3. Отхаркивающие средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17.4. Средства, применяемые при бронхиальной астме . . . . . . . . 17.5. Средства, применяемые при хронических обструктивных болезнях легких . . . . . . . . . . . . . . . . . . . . . . . 17.6. Препараты сурфактантов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, влияющие на сердечно-сосудистую систему . . . . . . . . . . . 5 313 316 316 317 318 320 322 326 327 329 332 345 346 348 348 361 371 373 377 378 382 384 388 391 392 392 393 394 396 401 416 418 419 422 6 Оглавление Глава 18. Антиаритмические средства . . . . . . . . . . . . . . . . . . . . . . . . 18.1. Класс I — блокаторы натриевых каналов . . . . . . . . . . . . . . . . 18.2. Класс II — β-адреноблокаторы . . . . . . . . . . . . . . . . . . . . . . . . 18.3. Класс III — блокаторы калиевых каналов . . . . . . . . . . . . . . . 18.4. Класс IV — блокаторы кальциевых каналов . . . . . . . . . . . . . 18.5. Другие средства, применяемые при тахиаритмиях и экстрасистолии . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 19. Средства, применяемые при недостаточности коронарного кровообращения . . . . . . . . . . . . . . . . . . . . . . 19.1. Средства, применяемые при стенокардии (антиангинальные средства) . . . . . . . . . . . . . . . . . . . . . . . . . . 19.2. Средства, применяемые при остром коронарном синдроме . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 20. Средства, применяемые при артериальной гипертензии (антигипертензивные средства) . . . . . . . . . . 20.1. Антигипертензивные средства нейротропного действия . . . 20.2. Средства, снижающие активность ренинангиотензиновой системы . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20.3. Антигипертензивные средства миотропного действия . . . . 20.4. Мочегонные средства (диуретики) . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 21. Средства, повышающие артериальное давление (гипертензивные средства) . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые при сердечной недостаточности . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22.1. Кардиотонические средства . . . . . . . . . . . . . . . . . . . . . . . . . . . 22.2. Средства, применяемые при сердечной недостаточности . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 23. Средства, применяемые при нарушении мозгового кровообращения . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23.1. Блокаторы кальциевых каналов . . . . . . . . . . . . . . . . . . . . . . . 23.2. Производные алкалоидов барвинка . . . . . . . . . . . . . . . . . . . . 23.3. Производные алкалоидов спорыньи . . . . . . . . . . . . . . . . . . . 23.4. Производные никотиновой кислоты . . . . . . . . . . . . . . . . . . . 23.5. Производные ксантина . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23.6. Средства, применяемые при мигрени . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 423 433 440 441 444 446 447 449 451 467 468 469 470 480 484 490 492 494 495 496 496 509 519 521 522 524 524 525 525 527 528 Оглавление Глава 24. Средства, применяемые при атеросклерозе . . . . . . . . . . . 24.1. Гиполипидемические средства (антигиперлипопротеинемические средства) . . . . . . . . . . . . 24.2. Антиоксиданты . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 25. Ангиопротекторы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, влияющие на систему крови . . . . . . . . . . . . . . . . . . . . . . . . . Глава 26. Средства, регулирующие кроветворение . . . . . . . . . . . . . . 26.1. Средства, влияющие на эритропоэз . . . . . . . . . . . . . . . . . . . . 26.2. Средства, влияющие на лейкопоэз . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 27. Средства, влияющие на гемостаз и тромбообразование . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27.1. Средства, снижающие агрегацию тромбоцитов (антиагреганты) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27.2. Средства, влияющие на свертывание крови . . . . . . . . . . . . . 27.3. Средства, влияющие на фибринолиз . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 28. Мочегонные средства (диуретики) . . . . . . . . . . . . . . . . . . . 28.1. Средства, влияющие на функцию эпителия почечных канальцев . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28.2. Осмотические диуретики . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28.3. Другие диуретики . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 29. Средства, влияющие на тонус и сократительную активность миометрия . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29.1. Средства, повышающие тонус и сократительную активность миометрия . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29.2. Средства, снижающие тонус и сократительную активность миометрия . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29.3. Средства, понижающие тонус шейки матки . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 30. Средства, влияющие на функции органов пищеварения . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30.1. Средства, влияющие на аппетит . . . . . . . . . . . . . . . . . . . . . . . 30.2. Рвотные и противорвотные средства . . . . . . . . . . . . . . . . . . . 30.3. Антацидные средства и средства, понижающие секрецию пищеварительных желез (антисекреторные средства) . . . . . 30.4. Гастроцитопротекторы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 529 535 543 545 547 551 553 553 553 559 560 562 566 584 604 609 611 614 622 623 625 626 627 631 633 633 635 635 637 639 645 8 Оглавление 30.5. Средства, используемые при нарушении экскреторной функции желудка, печени и поджелудочной железы . . . . . . 30.6. Ингибиторы протеолиза . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30.7. Желчегонные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30.8. Гепатопротекторные средства . . . . . . . . . . . . . . . . . . . . . . . . . 30.9. Холелитолитические средства . . . . . . . . . . . . . . . . . . . . . . . . . 30.10. Стимуляторы моторики желудочно-кишечного тракта и прокинетические средства . . . . . . . . . . . . . . . . . . . . . . . . . 30.11. Слабительные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30.12. Антидиарейные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30.13. Средства, восстанавливающие нормальную микрофлору кишечника . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, регулирующие процессы обмена веществ . . . . . . . . . . . . . . Глава 31. Препараты гормонов, их синтетических заменителей и антагонистов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31.1. Гормональные препараты пептидной структуры . . . . . . . . . 31.2. Гормональные средства стероидной структуры . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 32. Витамины . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32.1. Препараты жирорастворимых витаминов . . . . . . . . . . . . . . . 32.2. Препараты водорастворимых витаминов . . . . . . . . . . . . . . . . 32.3. Витаминоподобные вещества . . . . . . . . . . . . . . . . . . . . . . . . . 32.4. Растительные витаминные препараты . . . . . . . . . . . . . . . . . . 32.5. Витаминные препараты животного происхождения . . . . . . 32.6. Поливитаминные препараты . . . . . . . . . . . . . . . . . . . . . . . . . . 32.7. Цитамины . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Средства, угнетающие воспаление и регулирующие иммунные процессы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Глава 33. Противовоспалительные средства . . . . . . . . . . . . . . . . . . . 33.1. Стероидные противовоспалительные средства . . . . . . . . . . 33.2. Нестероидные противовоспалительные средства . . . . . . . . 33.3. Медленнодействующие противоревматоидные средства . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 34. Средства, применяемые при подагре (противоподагрические средства) . . . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . Глава 35. Средства, регулирующие иммунные процессы (иммунотропные средства) . . . . . . . . . . . . . . . . . . . . . . . . . 646 649 650 654 654 655 656 659 660 661 663 663 665 702 737 741 745 749 754 754 754 755 755 756 757 757 760 760 765 768 769 772 773 Оглавление 9 35.1. Иммуносупрессивные средства . . . . . . . . . . . . . . . . . . . . . . . . 773 35.2. Иммуностимулирующие средства (иммуностимуляторы) . . . 780 35.3. Противоаллергические средства . . . . . . . . . . . . . . . . . . . . . . . 789 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 798 Противомикробные, противовирусные и противопаразитарные средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 801 Глава 36. Антисептические и дезинфицирующие средства . . . . . . . 802 36.1. Галогены и галогенсодержащие соединения . . . . . . . . . . . . . 804 36.2. Окислители . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 807 36.3. Кислоты и щелочи . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 808 36.4. Соли тяжелых металлов . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 809 36.5. Альдегиды и спирты . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 811 36.6. Соединения ароматического ряда . . . . . . . . . . . . . . . . . . . . . . 813 36.7. Красители . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 813 36.8. Детергенты . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 815 36.9. Производные нитрофурана . . . . . . . . . . . . . . . . . . . . . . . . . . . 816 36.10. Антисептики из других групп . . . . . . . . . . . . . . . . . . . . . . . . . 817 36.11. Антисептики растительного происхождения . . . . . . . . . . . 818 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 819 Химиотерапевтические средства, применяемые при инфекционных заболеваниях . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 821 Глава 37. Антибактериальные химиотерапевтические средства . . . 823 37.1. Антибиотики, нарушающие синтез клеточной стенки . . . . 831 37.2. Антибиотики, нарушающие синтез белков . . . . . . . . . . . . . . 857 37.3. Антибиотики, нарушающие проницаемость цитоплазматической мембраны . . . . . . . . . . . . . . . . . . . . . . . 875 37.4. Антибиотики, нарушающие синтез РНК . . . . . . . . . . . . . . . . 876 37.5. Побочные эффекты антибиотиков . . . . . . . . . . . . . . . . . . . . . 879 37.6. Синтетические антибактериальные средства . . . . . . . . . . . . 880 37.7. Противосифилитические средства . . . . . . . . . . . . . . . . . . . . . 905 37.8. Противотуберкулезные средства . . . . . . . . . . . . . . . . . . . . . . . 906 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 912 Глава 38. Противогрибковые средства . . . . . . . . . . . . . . . . . . . . . . . . 914 38.1. Противогрибковые антибиотики . . . . . . . . . . . . . . . . . . . . . . 915 38.2. Синтетические противогрибковые средства . . . . . . . . . . . . . 919 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 931 Глава 39. Противовирусные средства . . . . . . . . . . . . . . . . . . . . . . . . . 933 39.1. Средства, подавляющие репродукцию вирусов герпеса (противогерпетические средства) . . . . . . . . . . . . . . . . . . . . . . 938 10 Оглавление 39.2. Средства, подавляющие репродукцию вирусов иммунодефицита человека (средства против ВИЧ-инфекции) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 946 39.3. Средства, подавляющие репродукцию вирусов гриппа . . . 954 39.4. Другие противовирусные средства . . . . . . . . . . . . . . . . . . . . . 959 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 966 Глава 40. Средства для лечения протозойных инфекций . . . . . . . . 968 40.1. Противомалярийные средства . . . . . . . . . . . . . . . . . . . . . . . . . 968 40.2. Препараты для лечения амебиаза, лейшманиоза, трихомониаза и других протозойных инфекций . . . . . . . . . 974 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 979 Глава 41. Противоглистные (антигельминтные) средства . . . . . . . 981 41.1. Противонематодозные препараты . . . . . . . . . . . . . . . . . . . . . 982 41.2. Противоцестодозные препараты . . . . . . . . . . . . . . . . . . . . . . . 983 41.3. Препараты, применяемые при внекишечных гельминтозах . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 984 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . . 985 Средства, применяемые при злокачественных новообразованиях . . . 987 Глава 42. Противоопухолевые средства . . . . . . . . . . . . . . . . . . . . . . . 987 42.1. Общие принципы противоопухолевой химиотерапии . . . . 987 42.2. Цитотоксические средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . 992 42.3. Алкилирующие средства . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 997 42.4. Антибиотики . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 999 42.5. Гормоны и их антагонисты . . . . . . . . . . . . . . . . . . . . . . . . . . . 1003 42.6. Препараты моноклональных антител . . . . . . . . . . . . . . . . . . 1006 42.7. Другие препараты . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1009 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . 1010 Глава 43. Общие принципы лечения отравлений. Плазмозамещающие и дезинтоксикационные растворы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1013 43.1. Удаление токсических веществ и задержка их всасывания в кровь . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1013 43.2. Уменьшение концентрации всосавшегося яда в крови и удаление его из организма . . . . . . . . . . . . . . . . . . . . . . . . . . 1014 43.3. Устранение действия токсических веществ, попавших в кровь . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1015 43.4. Восстановление жизненно важных функций . . . . . . . . . . . 1016 43.5. Плазмозамещающие и дезинтоксикационные растворы . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1016 Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . 1017 Оглавление Глава 44. Системы доставки лекарственных средств . . . . . . . . . . . 44.1. Модификация существующих путей доставки . . . . . . . . . . 44.2. Новые системы доставки (полимерные системы) . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . Глава 45. Различные средства аптечного ассортимента . . . . . . . . . 45.1. Гомеопатические средства . . . . . . . . . . . . . . . . . . . . . . . . . . . 45.2. Биологически активные добавки к пище . . . . . . . . . . . . . . . 45.3. Корректоры метаболизма костной и хрящевой ткани . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . Глава 46. Основные лекарственные формы . . . . . . . . . . . . . . . . . . . 46.1. Твердые лекарственные формы . . . . . . . . . . . . . . . . . . . . . . . 46.2. Мягкие лекарственные формы . . . . . . . . . . . . . . . . . . . . . . . 46.3. Жидкие лекарственные формы . . . . . . . . . . . . . . . . . . . . . . . 46.4. Лекарственные формы для инъекций . . . . . . . . . . . . . . . . . . 46.5. Лекарственные формы для ингаляций . . . . . . . . . . . . . . . . . Вопросы и задания для самоконтроля . . . . . . . . . . . . . . . . . . . . . . 11 1019 1019 1025 1033 1034 1034 1043 1047 1053 1054 1054 1056 1057 1060 1061 1061 Ответы к вопросам и заданиям для самоконтроля . . . . . . . . . . . . . 1063 Предметный указатель . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1066 Алфавитный указатель препаратов . . . . . . . . . . . . . . . . . . . . . . . . . . 1076 АВТОРСКИЙ КОЛЛЕКТИВ Аляутдин Ренад Николаевич — профессор, заведующий кафедрой фармакологии Технологического университета MARA (Малайзия), заместитель директора Центра экспертизы безопасности лекарственных средств (ЦЭБЛС) ФГБУ «Научный центр экспертизы средств медицинского применения» Минздрава России. Бондарчук Наталия Геннадьевна — ассистент кафедры фармакологии лечебного факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Давыдова Ольга Николаевна — доцент кафедры фармакологии фармацевтического факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Еникеева Дилара Ахметовна — доцент кафедры фармакологии лечебного факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Зацепилова Тамара Анатольевна — доцент кафедры фармакологии фармацевтического факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Королева Лидия Робертовна — доцент кафедры фармакологии фармацевтического факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Крендаль Феликс Петрович — доцент кафедры фармакологии фармацевтического факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Петров Валерий Евгеньевич — доцент кафедры фармакологии лечебного факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Романов Борис Константинович — д-р мед. наук, директор Центра экспертизы безопасности лекарственных средств (ЦЭБЛС) ФГБУ «Научный центр экспертизы средств медицинского применения» Минздрава России. Халин Игорь Владимирович — доцент кафедры фармакологии медицинского факультета Малазийского национального университета обороны (Малайзия). Чубаров Владимир Николаевич — доцент кафедры фармакологии фармацевтического факультета ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России. Якушева Елена Николаевна — профессор, зав. кафедрой фармакологии Рязанского медицинского университета им. И.П. Павлова. СПИСОК СОКРАЩЕНИЙ ♠ — торговые наименования лекарственных средств — лекарственные средства, не зарегистрированные в РФ АД — артериальное давление АДФ — аденозиндифосфат АПК — антигенпрезентирующие клетки Апо — аполипопротеины АПФ — ангиотензин-превращающий фермент АТФ — аденозинтрифосфат БАВ — биологически активные вещества БАД — биологически активные добавки ВИЧ — вирус иммунодефицита человека ВМедА — Военно-медицинская академия ГАМК — гамма-аминомасляная кислота ГДФ — гуанозиндифосфат ГИП — инсулинотропный полипептид ГК — глюкокортикоиды ГПП — глюкагоноподобный пептид ГТФ — гуанозинтрифосфат ГЭБ — гематоэнцефалический барьер ДГФР — дигидрофолатредуктаза ДОФА — диоксифенилаланин ЕД — единица действия ЖКТ — желудочно-кишечный тракт ИЛ — интерлейкины ИФ — интерфероны ИФН — интерферон КОМТ — катехол-орто-метилтрансфераза ЛВ — лекарственное вещество ЛГ — лютеинизирующий гормон ЛПВП — липопротеины высокой плотности ЛПНП — липопротеины низкой плотности ЛПОНП — липопротеины очень низкой плотности ЛППП — липопротеины промежуточной плотности ЛС — лекарственное средство МАО — моноаминоксидаза МКАТ — моноклональные антитела МНН — международное непатентованное название МПКТ — минеральная плотность костной ткани 14 НМГ НПР НПВС ОКС ОПСС ПАБК ПФП СТГ Пг ТГ ТТГ ФОС ХОБЛ ХСН цАМФ цГМФ ФЛ фМет ФНО ФСГ ХС ЦНС ЦОГ ЧСС ЭКГ ЭРП ЭЭГ AMPA Список сокращений — низкомолекулярные гепарины — нежелательная побочная реакция — нестероидные противовоспалительные средства — острый коронарный синдром — общее периферическое сопротивление сосудов — пара-аминобензойная кислота — продукты функционального питания — соматотропный гормон — простагландины — триглицериды — тиреотропный гормон — фосфорорганические средства — хроническая обструктивная болезнь легких — хроническая сердечная недостаточность — циклический аденозинмонофосфат — циклический гуанозинмонофосфат — фосфолипиды — формилированный метионин — фактор некроза опухоли — фолликулостимулирующий гормон — холестерин — центральная нервная система — циклооксигеназа — частота сердечных сокращений — электрокардиограмма — эффективный рефрактерный период — электроэнцефалограмма — альфа-амино-3-гидрокси-5-метил-4-изоксазолпропионовая кислота EGFR (аббр. от англ. — epidermal growth factor receptor) — рецепторы к эпидермальному фактору роста Ig — иммуноглобулин МНС (аббр. от англ. — Major Histocompatibiliy Complex) — главный комплекс гистосовместимости MRSA — метициллин-резистентный Staphylococcus aureus NMDA — n-метил-d-аспартат — период полуэлиминации лекарственного вещества t½ VEGF (аббр. от англ. — vascular endothelial growth factor) — фактор роста эндотелия сосудов ВВЕДЕНИЕ Предмет, задачи и методология фармакологии Фармакология (от греч. pharmacon — лекарство, яд и logos — учение) — наука о взаимодействии лекарственных веществ (ЛВ) и организма. Основные задачи фармакологии — создание лекарственных средств (ЛС) и обоснование рационального их применения. Фармакология, с одной стороны, самостоятельная наука, а с другой — неотъемлемая часть современной терапии, объединяющая теоретические знания и практическую медицину. Фармакология — среда активного информационного обмена между естественнонаучной основой медицины (биологией, химией, физиологией и морфологией), клиническими дисциплинами и фармацией. Значение фармакологии как связующего звена научных и клинических дисциплин огромно. Исследование механизмов действия ЛВ помогает расширить представления о химической сущности процессов, происходящих в живых клетках, а также о механизмах функционирования всех систем человеческого организма. В этом случае ЛВ выступают в роли фармакологических «зондов», помогающих оценить наличие, направленность и выраженность ответных реакций со стороны клеток, тканей, органов и систем. Выделяют теоретическую, экспериментальную и клиническую фармакологию. Теоретическая и экспериментальная фармакология составляют фундаментальный раздел науки. Экспериментальная фармакология — связующее звено между теоретической и клинической фармакологией. Основные задачи экспериментальной фармакологии: ● моделирование механизмов взаимодействия ЛС и биологических систем (организм человека или экспериментальная модель) на различных уровнях (субклеточный, тканевой, органный или системный); ● изучение эффектов взаимодействия организма и вещества. Существуют 3 основных методических подхода экспериментальной фармакологии (как основы для решения задач фармакологической науки): биохимический, физиологический и морфологический. Используя биохимический подход, фармакологи изучают природу реакций взаимодействия между ЛВ и биологическими молекулами. Физиологический и морфологический подходы применяют для анализа вызыва- 16 Введение емых фармакологическим воздействием изменений функционирования и строения органов и систем. Источники получения лекарственных веществ ● Минеральные соединения (магния сульфат; натрия сульфат). ● Ткани и органы животных (инсулин; препараты гормонов щитовидной железы; ферментные препараты; вещества, регулирующие пищеварение). ● Растения (сердечные гликозиды; морфин; резерпин). ● Микроорганизмы (антибиотики: пенициллины, цефалоспорины, макролиды и др.). В 40-х годах ХХ в. была разработана технология получения антибиотиков из почвенных грибов, а в 80-х годах — метод генной инженерии (человеческие инсулины). ● Химический синтез♠ (сульфаниламиды; парацетамол; кислота вальпроевая; новокаин , кислота ацетилсалициловая). С середины ХIХ в. ЛВ активно получают химическим путем. Большинство современных ЛС — продукты химического синтеза. ● Получение специфических белков (моноклональные антитела: мышиные, химерные, хуманизированные, человеческие). Этапы создания новых лекарственных средств Разработку новых ЛС осуществляют, используя многие отрасли науки, при этом основная роль принадлежит специалистам в области химии, фармакологии и фармации. Создание нового ЛС представляет ряд последовательных этапов. Каждый из этапов должен отвечать определенным положениям и стандартам, утвержденным такими государственными учреждениями, как Фармакопейный комитет, Фармакологический комитет, Управление МЗ РФ по внедрению новых ЛС. Процесс создания новых ЛС регулируется международными стандартами. ● GLP (GoodLaboratory Practice — «Качественная лабораторная практика»). ● GMP (Good Manufacturing Practice — «Качественная производственная практика»). ● GCP (Good Clinical Practice — «Качественная клиническая практика»). Существует три основных направления получения новой активной субстанции (действующего вещества или комплекса веществ). Этапы создания новых лекарственных средств 17 Химический синтез лекарственных веществ ● Эмпирический путь: скрининг, случайные находки. ● Направленный синтез: воспроизведение структуры эндогенных веществ, химическая модификация известных молекул. ● Целенаправленный синтез (рациональный дизайн химического соединения): основан на понимании зависимости «химическая структура — фармакологическое действие». Основу эмпирического пути (от греч. empeiria — опыт) создания ЛВ составляет метод «проб и ошибок». Для этого фармакологи с помощью набора биологических тестов (на молекулярном, клеточном, органном уровнях) обнаруживают наличие или отсутствие определенной фармакологической активности у заранее выбранных химических соединений. Так, наличие противомикробной активности определяют у микроорганизмов; спазмолитической активности — на изолированных гладкомышечных органах (ex vivo); гипогликемическое действие — по способности снижать уровень сахара в крови испытуемых животных (in vivo). Затем из всех исследуемых химических соединений выбирают наиболее активные и сравнивают степень их фармакологического действия и токсичность с существующими ЛС (их используют в качестве стандарта). Такой путь отбора активных веществ получил название «лекарственного скрининга» (от англ. screen — просеивать, сортировать). Внедрение в медицинскую практику некоторых препаратов произошло в результате случайных находок. Так обнаружили противомикробную активность азокрасителя с сульфаниламидной боковой цепью (красный стрептоцид), в результате была создана целая группа химиотерапевтических средств — сульфаниламидов. Другой путь создания ЛВ состоит в получении соединений с определенным видом фармакологической активности. Это направленный синтез ЛВ. Первый этап синтеза заключается в воспроизведении веществ, образующихся в живых организмах: адреналин♠, норадреналин♠, некоторые гормоны, простагландины, витамины. Химическая модификация известных молекул позволяет создать ЛВ, обладающие более выраженным фармакологическим эффектом и минимальным побочным действием. Так, изменив химическую структуру ингибиторов карбоангидразы, создали тиазидные диуретики, обладающие более сильным мочегонным действием. После введения дополнительных радикалов и фтора в молекулу налидиксовой кислоты получили новую группу противомикробных средств с расширенным спектром противомикробного действия — фторхинолоны. 18 Введение Целенаправленный синтез ЛС подразумевает создание веществ с заранее заданными фармакологическими свойствами. Синтез новых структур с предполагаемой активностью чаще всего проводят в том классе химических соединений, где уже найдены вещества, обладающие определенной направленностью действия. Примером может служить создание блокаторов Н2-гистаминовых рецепторов. Исследователи знали, что гистамин — мощный стимулятор секреции хлористоводородной кислоты в желудке, а противогистаминные средства (применяемые при аллергических реакциях) не устраняют этот эффект. Затем был сделан вывод о существовании подтипов гистаминовых рецепторов, выполняющих различные функции. Следовательно, должны существовать вещества, различные по химической структуре, блокирующие подтипы гистаминовых рецепторов. Фармакологи выдвинули гипотезу: модификация молекулы гистамина позволит создать селективные антагонисты гистаминовых рецепторов желудка. Результат рационального дизайна молекулы гистамина (в середине 70-х годов ХХ в.) — противоязвенное средство циметидин — первый блокатор Н2-рецепторов. Выделение лекарственных веществ из тканей и органов животных, растений и минералов Таким путем получены гормоны, галеновые и новогаленовые средства, органопрепараты и минеральные вещества. Выделение лекарственных веществ — продуктов жизнедеятельности грибов и микроорганизмов методами клеточной и генной инженерии Биотехнология — отрасль человеческих знаний, изучающая, в частности, выделение ЛВ — продуктов жизнедеятельности грибов и микроорганизмов. Биотехнология использует в промышленном масштабе биологические системы и биологические процессы. Обычно применяют микроорганизмы, культуры клеток, культуры тканей растений и животных. С помощью биотехнологических методов создают полусинтетические антибиотики. Большой интерес представляет получение в промышленном масштабе инсулина человека методом генной инженерии. Разработаны биотехнологические методы синтеза соматостатина, фолликулостимулирующего гормона, тироксина, стероидных гормонов. После создания новой активной субстанции и определения ее основных фармакологических свойств следует этап доклинических исследований. Клинические исследования 19 Доклинические испытания Помимо изучения специфической активности, во время доклинических исследований (в опытах на животных) полученное вещество тестируют на токсичность; определяют его эмбриотоксичность, тератогенность, канцерогенность, мутагенность. Эти исследования (на животных) проводят в соответствии со стандартами GLP. Далее определяют среднюю эффективную (ЕД50 — доза, оказывающая эффект на 50% животных) и среднюю летальную дозы (ЛД50 — доза, вызывающая гибель 50% животных). Клинические исследования Планирование и клинические исследования ЛС проводятся на здоровых добровольцах или на пациентах на основе международного стандарта GCP. В Российской Федерации на основе правил GCP разработан стандарт «Правила проведения качественных клинических испытаний». Правила GCP — свод положений, основа для планирования и выполнения клинических исследований, анализа и обобщения полученных результатов. В соответствии с этими правилами получают достоверные результаты, позволяющие избавить пациентов от необоснованного риска, соблюсти их права и конфиденциальность. Другими словами, GCP объясняет способы получения правильных научных данных, заботясь при этом о благополучии участников медицинских исследований. Фазы клинических исследований I фазу клинических исследований проводят на небольшом числе здоровых добровольцев (4–24 человека) для установления безопасности лекарственных препаратов. В ходе I фазы получают предварительные данные о безопасности препарата, создают первое описание его фармакокинетики и фармакодинамики у человека. Параметры, изучаемые в ходе I фазы: ● фармакодинамика и фармакокинетика одной дозы и нескольких доз препарата при разных путях введения; ● биодоступность; ● метаболизм активной субстанции; ● влияние возраста, пола, пищи, функций печени и почек на фармакокинетику и фармакодинамику активной субстанции; ● взаимодействие активного вещества с другими ЛС. 20 Введение II фаза клинических исследований проводится на 100–200 испытуемых для подбора оптимальных дозировок лекарственного препарата и курса лечения пациентов с профильным заболеванием, оптимальных доз и схем вакцинации иммунобиологическими ЛС здоровых добровольцев. III фаза клинических исследований проводится для установления безопасности ЛС и его эффективности для пациентов с определенным заболеванием, профилактической эффективности иммунобиологических лекарственных препаратов для здоровых добровольцев. Как правило, проводят сравнение эффективности и безопасности разрабатываемого препарата с другими препаратами этой группы. В таких исследованиях обычно принимают участие от нескольких сотен до нескольких тысяч человек (в среднем 1000–3000). При проведении III фазы определяют оптимальные схемы введения препаратов, изучают наиболее частые нежелательные реакции, клинически значимые лекарственные взаимодействия, влияние возраста, сопутствующих состояний и др. Условия исследований максимально приближают к реальным условиям использования ЛС. Вначале проводят открытые (open) исследования. При этом исследователи информируют врача и больного о свойствах препарата (новый, контрольный или плацебо). Дальнейшие исследования выполняют одинарным слепым (single-blind) методом: пациент не знает, какой препарат принимает (новый, контрольный или плацебо), двойным слепым (double-blind) методом: используемый препарат не известен ни врачу, ни больному. Данные, полученные в клинических испытаниях III фазы, — основа для создания инструкции по применению препарата. По результатам исследований в официальных инстанциях принимают решение о регистрации данного ЛС. Исследования биоэквивалентности лекарственных препаратов Оценка биологической эквивалентности — основной вид клинических исследований для воспроизведенных препаратов (дженериков) — препаратов, содержащих то же ЛВ в той же дозе и лекарственной форме, что и оригинальный препарат, и пpедназначенные для внесосудистого (пpием внутpь, накожная аппликация, ректальное введение и дp.), но не паpентеpального введения, пpи условии, что их действие опосpедовано появлением ЛВ в системном кpовотоке. Препараты, имеющие одинаковую лекарственную форму, считают биоэквивалентными, если они обеспечивают одинаковую биодоступность ЛВ при введении здоровым добровольцам (за исключением токсичных История отечественной фармакологии 21 препаратов), и при этом их концентрации в крови при одном и том же пути введения в одно и то же время различаются не более чем на 5%. Регистрация лекарственного препарата Полученные в ходе исследований данные документируют и направляют в регуляторные органы, регистрирующие данный препарат и дающие разрешение на его медицинское применение. В Российской Федерации лекарственные препараты регистрирует Министерство здравоохранения. Пострегистрационные испытания После регистрации препарата исследования его фармакологических свойств продолжаются. Существует IV фаза клинических испытаний — пострегистрационные исследования. Таким образом, IV фазу клинических исследований проводят (после начала продажи препарата) для выявления ранее неизвестных побочных действий зарегистрированных лекарственных препаратов. Это позволяет разработать более полную стратегию использования препарата и определить отдаленные результаты лечения. В исследованиях принимают участие большое количество пациентов (для обнаружения ранее неизвестных и редко встречающихся побочных действий). Исследования IV фазы также помогают оценить сравнительную эффективность и безопасность препаратов. Полученные данные оформляют (отчет) и направляют в организацию, разрешившую выпуск и применение препарата. Если после регистрации препарата проводят дополнительные клинические испытания для изучения новых, незарегистрированных свойств, показаний, методов применения или комбинаций ЛВ, то такие клинические исследования рассматривают как исследование нового лекарственного препарата. Исследования зарегистрированных препаратов, применяемых по зарегистрированным показаниям («наблюдательные исследования»), не требуют получения разрешения Министерства здравоохранения России. История отечественной фармакологии История фармакологии тесно связана с историей человечества. В окружающей природе первобытные люди интуитивно искали вещества, облегчающие страдания при заболеваниях и травмах. Такая терапия, ос- 22 Введение нованная на простых наблюдениях и личном опыте, получила название эмпирической. На Руси готовили лекарства и лечили болезни знахари, а позже монахи. Они использовали минералы, настои и отвары растений. Со временем в монастырях начали собирать и систематизировать сведения о лечебных травах, создавать рукописные труды по лекарствоведению, например травник «Изборник Святослава» (1073). В конце XV в. возникло феодальное Московское государство, объединившее разрозненные русские княжества. Во многих городах открыли учреждения аптечного типа — зелейные лавки. Владельцы этих лавок, зелейники, готовили и продавали порошки, мази, настойки и другие лекарства. Нередко это были высокообразованные люди, прекрасно знавшие свойства и действия ЛС, в основном растений. Их знания систематизированы в рукописных книгах-травниках, или зелейниках, а также в вертоградах (вертоград — цветник, сад). Наибольшую известность получил «Благопрохладный вертоград» (1534). В 1581 г. по указу царя Ивана IV в Москве была открыта первая аптека, которая обслуживала только царя и его придворных. Для руководства медицинским делом в начале XVII в. был создан Аптекарский приказ. В функции приказа входили заготовка лекарственных растений; обучение лекарей и специалистов по приготовлению лекарств; обеспечение армии медицинской и лекарственной помощью; проверка медицинских знаний у докторов и аптекарей, приезжавших в Россию. Во многих городах появились аптекарские огороды, где выращивали лекарственные растения. В эпоху Петра I в фармакологии и аптечном деле произошли существенные изменения: повсеместное открытие аптек, зарождение фармацевтической промышленности, базирующейся на аптекарских огородах в Петербурге на Аптекарском острове, в Лубнах, около Полтавы. В 1701 г. Петр I издал указ о закрытии зелейных лавок и открытии в Москве 8 частных аптек. Одновременно государство ввело монополию на аптеки, устраняя конкуренцию. В XVIII в. наряду с вольными, частными аптеками продолжали существовать и расширяться государственные. Продажа ЛС была разрешена только в аптеках. В 1707 г. Аптекарский приказ преобразовали сначала в Аптекарскую канцелярию, а затем — в Медицинскую коллегию и Медицинскую канцелярию. При Аптекарском приказе обучали ремеслу и практическим навыкам лекарей, костоправов, аптекарей. При госпиталях были организованы медицинские школы, в которых изучали медицинские предметы и аптекарское дело. История отечественной фармакологии 23 В 1719 г. в Санкт-Петербурге открыт Аптекарский сад, где выращивали лекарственные растения. На государственном уровне стали осуществлять мероприятия по изысканию новых отечественных ЛС. В 1720–1721 гг. в Санкт-Петербурге создан первый в России завод «казенных врачебных заготовлений» — первое государственное производство по изготовлению ЛС из отечественного сырья. Первым русским профессором-фармакологом можно считать К.И. Щепина (1728–1770), преподавателя московской госпитальной школы, защитившего диссертацию о лечебных свойствах хлебного кваса. Иоганн Христиан Керштенс, доктор медицины и философии, профессор химии и минералогии, — первый профессор медицинского факультета Московского университета. 13 августа 1758 г. он официально открыл медицинский факультет. Керштенс читал курс о врачебных веществах, так называемое врачебное веществословие и гигиену, а в следующем учебном году — курс химии и натуральную историю простых аптекарских лекарств, освещая происхождение и химические свойства ЛВ, относящиеся главным образом к неорганическим соединениям. В 1778 г. в России была издана первая государственная Фармакопея на латинском языке, а в 1866 г. — на русском языке. Профессор Н.М. Максимович-Амбодик (1744–1812) написал первый учебник по врачебному лекарствоведению (веществословию) «Врачебное веществословие или описание целительных растений», изданный в 1783– 1788 гг. в Санкт-Петербурге. В 1835 г. заведующий кафедрой врачебного веществословия Московского университета А.А. Иовский (1796–1884) издал учебники «Начертание общей фармакологии» (1835) и «Начертания фармации» (1838). В учебнике «Начертание общей фармакологии» он отмечает, что предмет фармакологии включает все, что охватывает врачебное веществословие: фармакогнозию, фармацевтическую химию, рецептуру во всем ее объеме, фармакодинамику и фармакотерапию. По его мнению, краеугольный камень фармакологии — изучение болезни и организма человека для успешного оказания помощи ЛС при заболевании. Зарубежные ученые Мажанди (Mageandie), Орфилла Бернард (Orfilla Bernard), исследуя действия ЛВ, положили начало научной фармакологии. В середине XIX в. благодаря внедрению экспериментального метода фармакология стала самостоятельной наукой. Экспериментальная фармакология впервые возникла в Юрьевском университете в 1847 г.: Р. Бухгейм (1820–1879) создал первую в мире лабораторию экспериментальной фармакологии. 24 Введение В дальнейшее развитие науки внесли значительный вклад русские ученые: А.П. Нелюбин, И.А. Двигубский, Е.В. Пеликан, А.А. Соколовский, И.В. Забелин, В.И. Дыбковский, И.М. Сеченов, И.П. Павлов, Н.П. Кравков. Профессор фармации Императорской медико-хирургической академии Александр Петрович Нелюбин (1785–1858) был сторонником экспериментального изучения действия лекарств и ядов на животных. Он определял фармакологию как систему точных знаний, основанную на изучении химических и физических свойств ЛВ, методов синтеза, приготовления лекарственных форм и их действия в зависимости от состояния организма. А.П. Нелюбин предложил раздельное преподавание фармакологии и фармации, а в 1827 г. написал руководство по лекарствоведению «Фармакография, или химико-врачебное предписание приготовления и употребления новейших лекарств», выдержавшее 5 изданий. Профессор Московского университета Иван Алексеевич Двигубский (1772–1839) способствовал формированию русской ботанической терминологии, развитию отечественного лекарствоведения, внедрению в лечебную практику отечественных лекарственных растений. Главная его работа — «Изображение растений преимущественно российских, упот ребляемых в лекарствах, и таких, которые наружным видом с ними сходны и часто за них принимаются, но лекарственных сил не имеют» Иван Алексеевич Двигубский (ч. I, II, 1823, 1829). Профессор Евгений Венцеславович Пеликан (1824–1884) на кафедре судебной химии и токсикологии Санкт-Петербургской медико-хирургической академии изучал действие кураре и строфанта. Ему принадлежат такие труды, как «Опыт приложения современных физико-химических исследований к учению о ядах», «Исследование о спорынье» (1865), «Руководство к токсикологии» (1879). Развитию фармакологии помогли труды одного из основоположников отечественной экспериментальной физиологии Алексея Матвеевича Филомафитского (1807–1849). Он первым ввел лекционные демонстрации опытов на животных, утверждая экспериментальный метод в изучении физиологических и патологических процессов. В это время медицин- История отечественной фармакологии 25 ский факультет Московского университета исследовал физиологические действия наркотических средств для выработки показаний, противопоказаний и техники применения. А.М. Филомафитский изучал действие эфира и хлороформа. Анализу механизма действия наркотических веществ посвящена его важнейшая для фармакологов и клиницистов работа «Физиологический взгляд на употребление эфиров, хлороформа и бензина как притупляющих нервную деятельность», в которой рассмотрена Алексей Матвеевич последовательность выключения функ- Филомафитский ций различных отделов нервной системы при постепенном усилении наркоза вплоть до наступления смерти. В марте 1847 г. под председательством А.М. Филомафитского создан Наркозный комитет, состоявший из двух комиссий: экспериментальной и клинической. В его деятельности участвовали: фармакологи Т.П. Анке и Т.Е. Лясковский, хирурги В.А. Басов, Ф.И. Иноземцев, А.И. Поля, терапевты И.В. Варвинский, А.И. Овера, акушер М.В. Рихтер, химик Г.А. Гивартовский и др. Труды комитета повлияли на развитие общей анестезии в нашей стране и послужили основой для большой исследовательской работы, проведенной медицинским факультетом во второй половине XIX в. В 1814 г. видный терапевт Устин Евдокимович Дядьковский (1784– 1841) начал преподавать курс ботаники и фармакологии при Московской медико-хирургической академии, а двумя годами позже защитил докторскую диссертацию «Рассуждение об образе действия лекарств на человеческое тело». Одновременно он читал лекции по общей патологии, терапии, фармакологии и рецептуре. Ученый утверждал: при определенных внешних влияниях и реакциях организма и яд может оказаться лекарством, и лекарство — ядом. Талантливый ученик У.Е. Дядьковского адъюнкт К.В. Лебедев также читал курс фармакологии и рецептуры на медицинском факультете. Именно он считал, что лечить надо не болезни, а больного; свои взгляды на медицину он изложил в книге «Практическая фармакология» (1842– 1848). Хирург Н.И. Пирогов (1810–1881) в эксперименте изучал наркотическое действие эфира, а затем ввел эфирный наркоз в хирургическую практику. В.И. Дыбковский (1830–1870) в Киевском университете изучал кардиотропные вещества и положил начало экспериментальной фарма- 26 Введение кологии. Профессор кафедры фармакологии Казанского университета И.М. Догель (1830–1916) исследовал влияние ЛВ на сердечно-сосудистую систему. Согласно «Общему уставу императорских Российских университетов» 1863 г., кафедра врачебного веществословия медицинского факультета Московского университета была разделена на две самостоятельные кафедры: кафедру фармакологии и кафедру фармакогнозии и фармации. С этого времени лекарствоведение на II курсе преподавали на кафедре фармакогнозии и фармации, а на III курсе — на кафедре теоретической и экспериментальной фармакологии с рецептурой и учением о минеральных водах. Первым заведующим кафедрой фармакологии Московского университета был назначен Алексей Андреевич Соколовский (1822–1891). Он сопровождал чтение лекций по фармакологии экспериментами на животных, организовав для этого специальный фармакологический кабинет, превращенный им же впоследствии в экспериментальную лабораторию, а после его смерти — в фармакологический институт. А.А. Соколовский — автор учебников «Курс органической фармакодинамики, основанной на химико-физиологических началах» (1869), «Неорганическая фармакология, основанная на химико-физиологических началах» (1871), «Руководство общей фармакологии и рецептуры» (1873), «Руководство частной фармакологии» (1875), «Основы общей и частной фармакологии» (1878). Иван Михайлович Сеченов (1829–1905) — основоположник русской физиологии и научной психологии, выпускник медицинского факультета Московского университета, в 1860 г. защитил докторскую диссертацию «Материалы для будущей физиологии алкогольного опьянения». Позже он изучал действие нейротропных веществ на мышечную систему. В Медико-хирургической академии он заведовал кафедрой, где создал физиологическую лабораторию. И.М. Сеченов написал учебник «Физиология нервной системы», перевел учебник Германа «Основы физиологии». В 1876 г. ученый был приглашен на кафедру физиологии Петербургского университета, одновременно читал лекции на Бестужевских женских курсах. В 1889 г. стал приват-доцентом Московского университета. Основные его труды: Иван Михайлович Сеченов История отечественной фармакологии 27 «Рефлексы головного мозга» (1866, переиздание 1961), «Психологические этюды: сборник статей» (1873), «Автобиографические записки» (1907), «Физиологические очерки» (1923), «Избранные труды» (1935). Иван Петрович Павлов (1849–1936) научную деятельность начал в клинике С.П. Боткина, где в течение 11 лет руководил экспериментальной лабораторией. Он изучал действие сердечных гликозидов и жаропонижающих средств. С 1890 по 1895 г. И.П. Павлов возглавлял кафедру фармакологии в Военно-медицинской академии в Санкт-Петербурге. Его научная деятельность охватывает три направления исследований: в области кровообращения, пищеварения и физиологии высшей нервной деятельности. Под руководством ученого впервые было Иван Петрович Павлов исследовано влияние бромидов и кофеина на высшую нервную деятельность, а также влияние горечей, кислот, щелочей, спирта этилового на систему пищеварения. И.П. Павлов разработал метод «изолированного сердца», имеющий огромное значение для экспериментальной медицины. Именно он положил начало психофармакологии, а его труды изданы в шести томах «Полного собрания сочинений» (1951–1952). Начало XX в. отмечено новыми успехами в развитии фармакологии: русскими учеными внедрен метод исследования ЛС на изолированных органах. В 1904 г. профессор физиологии Томского университета А.А. Кулябко (1866–1930) в работе «Фармакологические и токсикологические исследования на вырезанном сердце» сообщил об оживлении изолированных сердец животных и человека и об их длительной работе в искусственных условиях вне организма. Основателем отечественной фармакологии принято считать Н.П. Кравкова (1865–1924). В 1884 г., завершив обучение в гимназии, он стал студентом естественного отделения физико-математического факультета Санкт-Петербургского университета, а в 1888 г. поступил на II курс Военно-медицинской академии (ВМедА). Руководство ВМедА оставило молодого выпускника для научной деятельности. Первоначально Н.П. Кравков работал в лабораториях известных ученых — И.М. Сеченова и В.В. Пашутина. В 1894 г., успешно защитив докторскую диссертацию, он был направлен руководством ВМедА в загранкомандировку, 28 Введение где за два года познакомился с работой многих лабораторий знаменитых европейских ученых. В 1898 г. Н.П. Кравков получил в ВМедА звание приват-доцента (внештатного преподавателя) по общей и экспериментальной патологии, а в 1899 г. его назначили экстраординарным профессором кафедры фармакологии ВМедА. И только в 1904 г. он получил звание ординарного (штатного) профессора и одновременно стал заведующим кафедрой фармакологии ВМедА, котоНиколай Павлович Кравков рую возглавлял в течение 25 лет. Своими фундаментальными открытиями в области фармакологии он обогатил русскую и мировую науку, внес большой вклад в развитие биологии, физиологии и патологии. Н.П. Кравков — основатель ряда разделов фармакологии, в частности сравнительной эволюционной фармакологии патологических процессов, основоположник отечественной промышленной и военной токсикологии. Центральные проблемы его научных поисков — изучение механизма физиологического действия ЛВ и соотношения такого действия с этиологией и симптоматикой патологических состояний. В 1904 г. Н.П. Кравков приступил к изучению действия фармакологических веществ на изолированных органах. Будучи глубоко убежденным, что фармаколог-экспериментатор должен знать этиологию, патогенез, течение и исход болезней не только человека, но и животных, он говорил: «Обладая этими знаниями, исследователь в экспериментах может нарушать в нужном направлении нормальную жизнь животного и впоследствии ее восстанавливать». Метод изолированных органов лег в основу теории фазового действия ЛВ. Н.П. Кравков связал воедино все стадии взаимодействия химических веществ и тканей органов, изучил и объяснил последовательность стадий. В лаборатории кафедры фармакологии ВМедА им впервые показана зависимость характера эффекта от различного фазового действия ядов, введено понятие стадий вхождения и выхода яда из организма. Его научные работы затрагивают проблемы общей фармакологии: выяснение зависимости биологического эффекта от концентрации ЛВ, влияние температурных факторов на действие веществ. Большое внимание уделено зависимости фармакологического действия веществ от их химического строения. В лаборатории Н.П. Кравкова изучали действие наркоза История отечественной фармакологии 29 и снотворных средств различных химических групп. Под руководством ученого изучены комбинированное действие ЛВ и возможность изменения чувствительности к ним, позже положенные в основу метода комбинированного наркоза. Разработанные Н.П. Кравковым методики внутривенного и базисного наркоза нашли широкое применение в медицинской практике во всех странах мира. Именно в трудах этого выдающегося ученого положено начало современным методам базис-наркоза, парентерального и внутривенного наркоза. Во всем мире внутривенный гедоналовый наркоз известен как «русский метод» обезболивания. В последние годы своей жизни Н.П. Кравков изучал некоторые проблемы эндокринологии. Важно отметить, что первая в России диссертация о функциях щитовидной железы защищена в 1910 г. именно в его лаборатории. Практический результат экспериментов Н.П. Кравкова на эндокринных железах — выделение из перфузата поджелудочной железы гормона, названного им панкреотоксином. По фармакологическим свойствам выделенный гормон подобен инсулину. Панкреотоксин был получен в сухом виде и успешно применен для лечения сахарного диабета. Следует подчеркнуть, что панкреотоксин стали использовать для практических целей еще тогда, когда не было известно о попытке получения зарубежными учеными инсулина из поджелудочной железы. Это дает основание утверждать, что Н.П. Кравков и канадские ученые Бантинг и Бест открыли инсулин независимо друг от друга. К сожалению, из-за смерти ученого многие работы в области эндокринологии не были закончены. Педагогическая деятельность Н.П. Кравкова занимает одну из славных страниц в истории отечественной фармакологии. Его перу принадлежит двухтомное руководство «Основы фармакологии», выдержавшее 14 переизданий. Этот учебник был востребован во всех медицинских вузах страны. Н.П. Кравков основал отечественную школу фармакологов, его учениками стали: С.В. Аничков, В.В. Закусов, М.П. Николаев Титульный лист руководи др. Ведущие направления научной школы ства Н.П. Кравкова «ОсноН.П. Кравкова — изучение и поиск новых вы фармакологии» 30 Введение ЛВ, средств симптоматической и патогенетической терапии. В 1926 г. Н.П. Кравкову посмертно присуждена премия им. В.И. Ленина. С.В. Аничков (1892–1981) — ученик Н.А. Кравкова. Посещая лекции И.П. Павлова в ВМедА, он заинтересовался экспериментальной работой и фармакологией, позже заведовал кафедрами фармакологии в ВМедА им. С.М. Кирова и Ленинградского санитарно-гигиенического института. Под его руководством развернулись систематические исследования по фармакологии вегетативной нервной системы. Он возглавлял отдел фармакологии в институте экспериментальной медицины АМН СССР (1948–1981). Основное направление работ его научной школы — создание и изучение новых фармакологических препаратов на основе их структурного сходства с природными соединениями. Василий Васильевич Закусов (1903–1986) работал на кафедре фармакологии ВМедА им. С.М. Кирова. В 1936 г. защитил докторскую диссертацию «Рефлексы на дыхание при действии ядов на сосуды различных сосудистых областей», заведовал кафедрами фармакологии I и III Ленинградских медицинских институтов, Куйбышевской военно-медицинской академии, I ММИ им. И.М. Сеченова. На протяжении 25 лет был директором основанного им Института фармакологии АМН СССР, в 1948 г. избран членом-корреспондентом, а в 1952 г. — действительным членом АМН СССР. В.В. Закусов создал синаптическую теорию действия фармакологических веществ, успешно развиваемую в работах его многочисленных учеников и сотрудников, — фундамент современных предВасилий Васильевич Закусов ставлений этой области науки. За цикл исследований по фармакологии синаптической передачи В.В. Закусов (совместно с С.В. Аничковым) удостоен в 1976 г. Ленинской премии. Большинство работ В.В. Закусова разрабатывает два направления: фармакологию нервной и сердечно-сосудистой системы. Ближайший ученик и последователь профессора В.В. Закусова — академик РАМН Дмитрий Александрович Харкевич, автор многочисленных трудов в области фармакологии нейротропных средств. С 1964 по 1998 г. Д.А. Харкевич возглавлял кафедру фармакологии лечебного История отечественной фармакологии 31 и медико-профилактического факультетов I ММИ, затем ММА им. И.М. Сеченова. В.В. Николаев (1871–1950) посвятил фармакологии 55 лет жизни. В 1895 г. он с отличием окончил медицинский факультет Казанского университета и стал работать лаборантом на кафедре фармакологии. Склонность к научной работе он обнаружил еще в студенческие годы. Начинал он в 1892 г. в физиологической лаборатории, а спустя год в фармакологической лаборатории провел свою первую экспериментальную работу «К вопросу об иннервации сердца лягушки», подтвердившую гипотезу профессора Юрьевского университета Ф.Г. Биддера о том, что парасимпатический нерв сердца имеет двухнейронное строение и в толще миокарда есть особые структуры, названные впоследствии парасимпатическими ганглиями. В 1921 г. Владимир Васильевич Николаев В.В. Николаев был избран профессором на кафедре фармакологии в Московском государственном университете, в 1922 г. он стал заведующим кафедрой фармакологии лечебного факультета. Расширению сферы научных интересов Владимира Васильевича Николаева способствовали его зарубежные научные командировки. В начале 1930-х годов он работал в лабораториях фармакологии профессоров Шмидеберга в Страсбурге и Томса в Берлине, изучал методы преподавания фармакологии у известных немецких профессоров Штрауба и Тренделенбурга, Гойбнера, Фюнера, Борнштейна и др. В 1922 г. возглавил только что организованное Московское научное общество фармацевтов. При его участии вышли VI, VII и VIII издания Государственной Фармакопеи, в которой ему принадлежат «Таблицы противоядий и пособий при отравлениях», «Список ядовитых и сильнодействующих веществ». В.В. Николаев был ответственным редактором раздела «Фармакология» 1-го издания БМЭ и главным редактором журнала «Фармация и фармакология». Он организовал кафедру фармакологии Смоленского медицинского института (1923–1928), III Московского медицинского института (1934–1939), Московского стоматологического института (1937–1939). В открытом в 1936 г. Московском фармацевтическом институте (в настоящее время фармацевтический факультет ММА 32 Введение им. И.М. Сеченова) В.В. Николаев создал кафедру фармакологии и стал первым ее заведующим (1936–1937). В.В. Николаев изучал седативные свойства синюхи, кардиотоническое действие желтушника, фармакологические эффекты хлороформа, атропина, мускарина, никотина, бромидов. С целью дальнейшего развития отечественной фармакологической школы и значительного увеличения объема педагогической работы в 1936 г. В.В. Николаев пригласил работать на кафедру М.П. Николаева (1893–1949). Он был переведен на кафедру фармакологии I Московского медицинского института из Ленинградской ВМедА им. С.М. Кирова в качестве заведующего кафедрой фармакологии санитарно-гигиенического факультета, где работал профессором до конца своих дней. Одновременно он получил назначение на заведывание отделом фармакологии и токсикологии ВИЭМ им. А.М. Горького. Михаил Петрович Николаев — ученик и последователь основоположника отечественной фармакологии Н.П. Кравкова. В 1910 г., после окончания с золотой медалью гимназии, он поступил в ВМедА в Петербурге. Будучи студентом, М.П. Николаев принимал активное участие в работе руководимого Н.П. Кравковым «Кружка теоретической медицины» и в 1912–1913 гг. выполнил там свою первую экспериментальную работу «Влияние питуитрина на рост саркомы у мышей». М.П. Николаевым было написано более 140 работ, посвященных фармакологии сердечно-сосудистых и эндокринных Михаил Петрович Николаев препаратов, а также патологической фармакологии. М.П. Николаев — автор ряда ценных научных работ и двух руководств: «Экспериментальные основы фармакологии и токсикологии» (1941) и «Учебник фармакологии» (1948) для студентов фармацевтических факультетов. Он был также редактором журнала «Фармакология и токсикология». По инициативе М.П. Николаева в 1937 г. под его председательством была организована первая Всесоюзная конференция фармакологов и токсикологов. С 1940 по 1946 г. М.П. Николаев возглавлял кафедру фармакологии фармацевтического факультета. Большой вклад в дело подготовки провизоров внесла профессор М.М. Николаева. Прекрасный педагог и организатор учебно-методиче- Названия лекарственных средств 33 ской работы, она много сил и энергии отдала совершенствованию учебного процесса в фармацевтическом институте. М.М. Николаева окончила Высшие женские курсы в Петербурге, а в 1925 г. — I Ленинградский медицинский институт. После окончания института она в течение 12 лет работала под руководством видного фармаколога В.В. Савича в отделе фармакологии Института экспериментальной медицины, занимая должности препаратора, врача-лаборанта, младшего и старшего научного сотрудника. С 1943 г. М.М. Николаева преподавала на кафедре I ММИ им. И.М. Сеченова, а с 1950 г. руководила кафедрой фармакологии фармацевтического факультета, внесла значительный вклад в совершенствование высшего фармацевтического образования в нашей стране. Профессор М.М. Николаева — автор 45 научных работ. Она провела оригинальное научное исследование о локализации действия снотворных и наркотических веществ в центральной нервной системе (ЦНС). Большой вклад в развитие фармакологии и фармацевтического образования внес талантливый ученик В.В. Николаева и М.П. Николаевой академик РАЕН Александр Николаевич Кудрин. Он заведовал кафедрой фармакологии фармацевтического факультета I ММИ им. И.М. Сеченова с 1961 по 1998 г. А.Н. Кудрин разработал химико-фармацевтическое направление в фармакологии, включающее изыскание новых ЛС и теорию их целенаправленного создания; отбор наиболее активных соединений и первоначальное изучение характера и механизма их действия; биологический контроль качества и безопасности применения ЛС. В настоящее время основы клеточной и молекулярной фармакологии разрабатывают не только фармакологи, но и представители самых различных наук и специальностей: физиологи, биологи, математики, генетики. Названия лекарственных средств Номенклатура ЛС включает 3 основных названия: ● химическое название; ● международное непатентованное наименование; ● коммерческое (торговое) наименование. Химическое название Химическое название отражает состав и структуру ЛВ. Химические названия редко употребляют в практическом здравоохранении. Как правило, эти названия приводят в аннотациях к препаратам. Существуют 34 Введение специальные справочные издания, содержащие химические названия всех ЛС. Примеры: 1,3-диметил-ксантин, 5-этил-5-фенилбарбитуровая кислота и т.д. Международное непатентованное наименование Международное непатентованное наименование (МНН) — наименование ЛВ, рекомендованное Всемирной организацией здравоохранения. МНН используют в учебной и научной литературе для удобства идентификации препарата по принадлежности к определенной фармакологической группе и во избежание ошибок. Иногда МНН отражает химическое строение ЛВ. Примеры: ацетилсалициловая кислота, ацетаминофен. В последнее время в связи развитием так называемой «биологической» фармакологии проведена унификация препаратов на основе моноклональных антител. Так, окончание –mab указывает на то, что это моноклональное антитело. На источник получения указывает слог перед окончанием: -u- (человеческое), -mo- (мышиное), -zu- (гуманизированное) или -xi- (химерное). Мишень (орган или процесс) характеризует следующий от конца слог: -lim- (иммуннитет), -kin- (цитокины), -vir- (вирусы), -cir- (сердечно-сосудистая система), -tu (m) - (опухоль), -col- (толстый кишечник). Например, abciximab (ab-ci-xi-mab) — это моноклональное антитело, химерное, мишень — сердечно-сосудистая система; trastuzumab (trast-tu-zu-mab) — моноклональное антитело, гуманизированное, мишень — опухоль. Коммерческое (торговое) наименование (brand name) Торговое название — коммерческая собственность (охраняемая патентом торговая марка) фармацевтической фирмы, производящей данный конкретный оригинальный лекарственный препарат. Примеры: аспирин♠ (ацетилсалициловая кислота), лазикс♠ (фуросемид), вольтарен♠ (диклофенак). Фирмы-производители используют торговые названия для маркетинговых целей, для продвижения лекарственных препаратов на рынке и повышения их конкурентоспособности. Если у фирмы-разработчика закончился срок действия патента, то другие компании могут производить данное ЛС и продавать его под международным названием. Такие препараты называют воспроизведенными, или дженерическими препаратами. Стоимость дженериков, как правило, ниже стоимости оригинальных препаратов, так как затраты на разработку и клинические испытания ЛС в цену не включают. Препараты, содержащие одно и то же ЛВ в одинаковых дозах и в одной лекарственной форме, разные производители иногда выпускают под раз- Классификации лекарственных средств 35 ными торговыми названиями (препараты-синонимы). Поэтому при отсутствии в аптеке ЛС провизор может предложить пациенту заменить один препарат другим (препаратом-синонимом). Классификации лекарственных средств Классификация по алфавиту В основу этой классификации положен принцип размещения наименований ЛС в алфавитном порядке (на русском и латинском языках). Химическая классификация Основа данной классификации — химическая структура ЛВ. ● Производные имидазола: —бендазол; —клотримазол; —метронидазол. ● Производные фенотиазина: —хлорпромазин; —этапиразин. ● Производные метилксантина: —кофеин; —теофиллин; —теобромин. ЛВ, близкие по химической структуре, могут оказывать на организм разные действия. Фармакологическая классификация Фармакологическая классификация — комбинированная. Согласно этой классификации, ЛС делят на разряды — большие блоки, соответствующие системе организма, подвергающейся действию ЛС. Например, ЛВ, оказывающие эффекты на сердечно-сосудистую, пищеварительную системы, ЦНС и т.д. Разряды подразделяют на классы. Класс определяет характер фармакологического действия ЛС. Например, в разряд «ЛС, действующие на сердечно-сосудистую систему» входят классы: «Антиаритмические средства», «Кардиотонические средства», «Антигипертензивные (гипотензивные) средства» и др. Классы делят на группы. Например, в классе «Антиаритмические средства» выделяют четыре группы: блокаторы натриевых каналов; препараты, замедляющие реполяризацию; β-адреноблокаторы; блокаторы кальциевых каналов. Группы подразделяют на подгруппы. Например, 36 Введение группу β-адреноблокаторов составляют неселективные и селективные препараты. Таким образом, фармакологическая классификация имеет многоступенчатый характер. Фармакотерапевтическая классификация Основу фармакотерапевтической классификации составляют заболевания (для их лечения применяют конкретные препараты). Например, «Средства для лечения язвенной болезни желудка и двенадцатиперстной кишки», «Средства для лечения бронхиальной астмы». В фармакотерапевтические группы ЛС входят препараты, относящиеся к разным разрядам, классам и группам. Данную классификацию используют в основном врачи. Классификация Chemical Abstracts Service Классификация Chemical Abstracts Service — однозначный идентификатор химических субстанций. Каждой химической структуре в данной классификации присвоен регистрационный номер. Например, номер CAS азитромицина — 83905-01-5. Фармацевтические и медицинские справочники всего мира содержат регистрационные номера ЛВ. Часть I ОБЩАЯ ФАРМАКОЛОГИЯ В этом разделе приведены сведения об общих закономерностях фармакокинетики и фармакодинамики ЛВ. Фармакокинетика — это всасывание, распределение в организме, депонирование, биотрансформация (метаболизм) и выведение ЛВ. Фармакодинамика включает такие понятия, как фармакологические эффекты, механизмы действия, локализация действия и виды действия ЛВ. Отдельно рассматриваются факторы, влияющие на фармакокинетику и фармакодинамику ЛВ, а также общие закономерности побочного и токсического действий ЛВ. Кроме того, обсуждаются основные виды лекарственной терапии. Глава 1 ФАРМАКОКИНЕТИКА Фармакокинетические процессы: всасывание, распределение, депонирование, биотрансформация и выведение — связаны с проникновением ЛВ через биологические мембраны, в основном через цитоплазматические мембраны клеток — трансцеллюлярный транспорт и через межклеточные промежутки — парацеллюлярный транспорт. 1.1. ТРАНСЦЕЛЛЮЛЯРНЫЙ ТРАНСПОРТ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Существуют следующие способы проникновения веществ через биологические мембраны: ● пассивная диффузия; ● перенос веществ через мембраны с помощью транспортных систем: —активный транспорт; —облегченная диффузия; ● пиноцитоз; ● рецептор-опосредованный эндоцитоз. 1.1.1. Пассивная диффузия Путем пассивной диффузии вещества проникают через мембраны по градиенту концентрации (если концентрация вещества с одной стороны мембраны выше, чем с другой, вещество перемещается через мембрану от большей концентрации к меньшей). Этот процесс не требует затраты энергии. Поскольку биологические мембраны в основном состоят из липидов, через них, как правило, легко проникают липофильные неполярные вещества (рис. 1.1). Такие вещества, покидая водную среду (например, экстрацеллюлярную жидкость), проникают в мембрану, растворяются в ее липидной фазе и, диффундируя через два слоя липидов, высвобождаются в водную среду (цитоплазму) с другой стороны мем- Пассивная диффузия через водные поры АТФ Pi АДФ Рис. 1.1. Основные способы проникновения веществ через биологические мембраны Pi Б Активный транспорт Белокпереносчик Внутриклеточное пространство Биологическая мембрана Внеклеточное пространство Пассивная диффузия через липиды Глава 1. Фармакокинетика 39 40 Часть I. Общая фармакология браны. При этом скорость пассивной диффузии веществ через липиды зависит от их относительной липофильности, т.е. от коэффициента распределения веществ между органическим растворителем (октанол) и водой. В определенных пределах вещества с высоким коэффициентом распределения проникают через липиды мембран лучше веществ с низкими значениями этого коэффициента. Несмотря на то, что липофильность — определяющее свойство для проникновения веществ через липидные слои мембраны, необходимо, чтобы липофильное вещество также обладало некоторой способностью растворяться в воде, что позволяет ему, покидая мембрану, переходить в водную среду (экстрацеллюлярную жидкость, цитозоль и др.). Такую способность органическим соединениям придает наличие в их структуре электроотрицательных атомов кислорода, азота и серы, которые могут взаимодействовать с водой с образованием водородных связей. Очень высокая липофильность и отсутствие растворимости в воде могут удерживать соединение в липидной фазе мембраны. Скорость пассивной диффузии веществ через мембраны зависит также от молекулярной массы (обратно пропорциональна квадратному корню молекулярной массы), формы молекулы вещества, температуры среды, толщины и площади мембраны, через которую проникает вещество. Все эти факторы учитываются законом Фика, согласно которому: dQ / dt = (С1–С2) Площадь мембраны Коэфф. диффузии / / Толщина мембраны, где dQ/dt — скорость диффузии, (С1–С2) — градиент концентрации. Коэффициент диффузии в основном определяется степенью липофильности вещества, т.е. его коэффициентом распределения между октанолом и водой (другие факторы имеют меньшее значение). Пассивная диффузия соединений, имеющих заряд, осложняется влиянием на этот процесс потенциала мембраны. Кроме того, заряженные частицы окружаются молекулами воды, и такая водная оболочка препятствует их проникновению через липиды. Поэтому заряженные соединения, хорошо растворимые в водной среде и малорастворимые в липидах, т.е. гидрофильные полярные вещества, плохо проникают через липидные слои мембраны. Пассивная диффузия гидрофильных полярных веществ возможна через водные поры (аквапорины), гликопротеины клеточных мембран, проницаемые для воды и растворенных в ней веществ (см. рис. 1.1). Однако такая пассивная диффузия (пассивная диффузия в водной среде) не имеет существенного значения для проникновения ЛВ через мембраны. Это Глава 1. Фармакокинетика 41 объясняется тем, что диаметр водных пор невелик (приблизительно 0,3– 0,4 нм), и через них проникают только вода и небольшие гидрофильные молекулы (например, мочевина или глицерин). Диаметр молекул большинства гидрофильных ЛВ превышает 1 нм, поэтому они не проходят через водные поры в мембране и, следовательно, не проникают в клетки путем пассивной диффузии. Многие ЛВ являются слабыми кислотами или слабыми основаниями, т.е. слабыми электролитами. В водной среде такие вещества частично ионизированы. Поскольку путем пассивной диффузии через липиды мембран легко проходят только неионизированные молекулы (как правило, растворимые в липидах), проникновение слабых кислот и слабых оснований зависит от степени их ионизации. Степень ионизации слабых кислот и слабых оснований определяется значениями рН среды и константой ионизации (Ка) веществ. Слабые кислоты в большей степени ионизированы в щелочной среде, а слабые основания — в кислой. Ионизация слабых кислот: HA H + + A– щелочная среда Ионизация слабых оснований: BH + B + H + кислая среда Константа ионизации (Ка) характеризует способность вещества к ионизации при определенном значении рН среды (численно равна концентрации водородных ионов в среде, при которой ионизирована половина молекул данного вещества). На практике для характеристики способности веществ к ионизации используют показатель рКа, который является отрицательным логарифмом Ка (–lgКa). Показатель pКа численно равен значению рН среды, при котором ионизирована половина молекул данного вещества. Значения рКа слабых кислот и слабых оснований варьируют в широких пределах (табл. 1.1). Чем меньше рКа слабой кислоты, тем легче она ионизируется даже при относительно низких значениях рН среды. Так, ацетилсалициловая кислота (рКа=3,5) при рН=4,5 ионизирована более чем на 90%, а степень ионизации аскорбиновой кислоты (рКа=11,5) при том же значении рН составляет доли процента (рис. 1.2). Для слабых оснований существует обратная зависимость. Чем выше рКа слабого основания, 42 Часть I. Общая фармакология тем в большей степени оно ионизировано даже при относительно высоких значениях рН среды. Таблица 1.1. Константы ионизации некоторых лекарственных веществ Слабые кислоты Ампициллин Аспирин Аскорбиновая кислота Варфарин Леводопа Парацетамол Теофиллин Фенобарбитал Фуросемид 100 Слабые основания Адреналин Атропин Амфетамин Диазепам Кодеин Морфин Лидокаин Хлордиазепоксид Тербуталин А 90 Степень ионизации, % рКа 2,5 3,5 11,5 5,0 2,3 9,5 8,8 7,4 3,9 рКа 8,7 9,7 9,8 3,0 8,2 7,9 7,9 4,6 10,1 Б 80 70 60 50 40 30 20 10 0 0 1 2 3 3,5 4 5 6 7 8 9 10 11,5 12 13 pH Рис. 1.2. Зависимость степени ионизации слабых кислот от рН среды и рКа соединений: А — ацетилсалициловая кислота (рКа=3,5); Б — аскорбиновая кислота (рКа=11,5) Степень ионизации слабой кислоты или слабого основания можно рассчитать по формуле Гендерсона–Гассельбальха: Глава 1. Фармакокинетика 43 ● для слабых кислот: lg [A – ] = pH – pKa, [HA] ● для слабых оснований: lg [B] = pH – pKa. [BH +] Эта формула позволяет определить степень проникновения ЛВ (слабых кислот или слабых оснований) через мембраны, разделяющие среды организма с различными значениями рН, например при всасывании ЛВ из желудка (рН=1,0–2,0) в плазму крови (рН=7,4) или при реабсорбции ЛВ из почечных канальцев (рН=5,0–8,0). Изменяя рН среды, можно изменить (увеличить и уменьшить) степень проникновения слабых кислот и слабых оснований через мембраны. Это может быть использовано в определенных клинических ситуациях, например для ускорения выведения некоторых ЛВ почками. Для того чтобы ускорить выведение слабых кислот, следует повысить рН почечного фильтрата, так как щелочная среда, способствуя ионизации слабых кислот, препятствует их реабсорбции в почечных канальцах. С этой целью обычно вводят гидрокарбонат натрия. Для ускорения выведения слабых оснований уменьшают рН почечного фильтрата, создавая кислую среду с помощью хлорида аммония. 1.1.2. Перенос веществ через мембраны с помощью специальных транспортных систем Специальные транспортные системы — это белковые молекулы (белки-переносчики, или транспортеры), которые пронизывают клеточную мембрану и имеют специфические места связывания для определенных веществ, что обеспечивает их избирательный транспорт через мембраны. В основном белки-переносчики транспортируют вещества, которые не проникают через мембраны клеток путем пассивной диффузии вследствие гидрофильности и больших размеров молекул. Белки-переносчики облегчают проникновение в клетки необходимых для их жизнедеятельности веществ (нутриентов). Другая функция белков-переносчиков — удаление из клеток чужеродных соединений и токсических веществ эндогенного происхождения. 44 Часть I. Общая фармакология Активный транспорт Процесс переноса веществ через мембраны может происходить против градиента концентрации. Такой процесс требует затраты энергии и обозначается как активный транспорт. В зависимости от природы источника энергии различают: ● первичный активный транспорт, использующий энергию, выделяемую при гидролизе АТФ; ● вторичный активный транспорт, при котором перенос вещества через мембрану против градиента концентрации часто связан с движением ионов Na+ по градиенту концентрации, создаваемому Na+, K+-ATФазой. При этом вещество может перемещаться: —в одном направлении с ионами Na+ — котранспорт, или симпорт; —в противоположном направлении — антипорт. Активный транспорт веществ осуществляется следующим образом. Вещество связывается с белком-переносчиком с одной стороны мембраны. При участии энергии происходят изменение конформации белковой молекулы и перенос вещества через мембрану. Затем уменьшение силы связывания между переносчиком и транспортируемым веществом приводит к его высвобождению (см. рис. 1.1) в окружающую среду (цитоплазму, интерстициальную жидкость и др.). Активный транспорт веществ через мембраны характеризуется: ● специфичностью (с белками-переносчиками связываются лишь вещества, имеющие определенную структуру); ● насыщаемостью (при достижении определенной концентрации транспортируемого вещества все места связывания на белках-переносчиках оказываются занятыми этим веществом, и скорость его транспорта достигает предельной величины); ● потреблением энергии, так как происходит против градиента концентрации. Облегченная диффузия Перенос веществ через мембраны по градиенту концентрации (от большей концентрации к меньшей) называется облегченной диффузией. При этом изменение конформации белка-переносчика и, следовательно, перенос и высвобождение вещества (например, глюкозы) с другой стороны мембраны происходят при связывании вещества с переносчиком без потребления энергии. Подобно активному транспорту, облегченная диффузия — специфичный по отношению к определенным веществам и насыщаемый процесс. Глава 1. Фармакокинетика 45 Активный транспорт и облегченная диффузия обеспечивают транспорт через клеточные мембраны таких необходимых для жизнедеятельности клеток веществ, как аминокислоты, сахара, пиримидиновые и пуриновые основания, железо, витамины. И только немногие ЛВ, близкие к ним по химической структуре, способны проникать через клеточные мембраны с помощью тех же (специфичных) транспортных систем. Например, транспорт леводопы (диоксифенилаланина) через гематоэнцефалический барьер (ГЭБ) происходит при участии транспортного белка, переносящего через мембраны ароматические аминокислоты (см. главу «Противопаркинсонические средства»). Белки-переносчики, участвующие в транспорте ЛВ через мембраны, в основном относятся к двум суперсемействам: АВС (АТФ-связывающие кассетные переносчики; транспортеры, использующие энергию АТФ) и SLC (переносчики растворенных веществ; транспортеры, вовлеченные в процессы облегченной диффузии и вторичного активного транспорта). Эти транспортеры находятся в эпителии кишечника и почечных канальцев, гепатоцитах, эндотелии капилляров мозга, где они осуществляют обмен веществ между клетками и окружающей средой. При этом SLCтранспортеры могут переносить вещества в двух направлениях (в клетку и из клетки), а АВС-транспортеры избирательно участвуют только в однонаправленном транспорте веществ через мембраны (или в клетку, или из клетки). К транспортерам, удаляющим чужеродные соединения (в том числе ЛВ) из клеток, относят АТФ-зависимые транспортные белки, такие как P-гликопротеин, известный также как белок множественной лекарственной резистентности 1 (МDRP1), MDRP2, белки резистентности рака молочной железы. Р-гликопротеин — наиболее распространенный вид таких белков, обнаружен в мембранах энтероцитов, гепатоцитов, эндотелиальных клеток, эпителиальных клеток почечных канальцев. При его участии происходят выкачивание липофильных веществ из энтероцитов в просвет кишечника, секреция их в желчь и в просвет почечных канальцев, а также удаление из эндотелиальных клеток в просвет сосуда, что препятствует всасыванию, проникновению через гистогематические барьеры и ускоряет выведение этих ЛВ из организма. Этот «выкачивающий» механизм призван защищать клетки организма от чужеродных липофильных соединений. Впервые Р-гликопротеин и другие белки лекарственной резистентности были обнаружены в мембранах клеток злокачественных опухолей. При участии этих белков противоопухолевые вещества удаляются из этих 46 Часть I. Общая фармакология клеток, что нередко является причиной неэффективности противоопухолевой терапии. Некоторые ЛВ снижают (хинидин, лидокаин, верапамил) или повышают (морфин , дексаметазон , препараты зверобоя) активность Р-гликопротеина. Так, хинидин, ингибируя Р-гликопротеин, переносящий дигоксин из энтероцитов в просвет кишечника, повышает его концентрацию в крови, что увеличивает риск интоксикации этим препаратом. Ингибиторы Р-гликопротеина повышают способность ряда веществ (иммунодепрессанта циклоспорина, противовирусного средства саквинавира) проникать через плаценту, что увеличивает их токсическое воздействие на плод. Напротив, индукторы Р-гликопротеина могут снизить эффективность ЛВ, которые являются его субстратами, например препараты зверобоя (обладающие легким антидепрессивным эффектом) могут уменьшить эффективность антидепрессанта амитриптилина. Синтез Р-гликопротеина и других белков-переносчиков контролируется генами, экспрессия которых варьирует в зависимости от пола (экспрессия гена Р-гликопротеина более чем в 2 раза выше у мужчин, чем у женщин) и подвержена полиморфизму. Это может быть причиной половых и индивидуальных различий в фармакокинетике ЛВ, являющихся субстратами этого транспортера. 1.1.3. Пиноцитоз При пиноцитозе (от греч. pino — пью) крупные молекулы вещества соприкасаются с наружной поверхностью мембраны и окружаются ею с образованием пузырька (вакуоли или везикулы), который отделяется от мембраны и погружается внутрь клетки. При этом клеткой захватывается («выпивается») небольшое количество экстрацеллюлярной жидкости. Процесс захвата вещества клеткой называется эндоцитозом. После захвата вещество в составе вакуоли может быть доставлено в лизосомы, где метаболизируется под действием ферментов (рис. 1.3А) или транспортируется через цитоплазму и высвобождается наружу путем экзоцитоза (при слиянии везикулярной и цитоплазматической мембран). Такой чрезклеточный транспорт вещества получил название трансцитоз (рис. 1.3Б). 1.1.4. Рецептор-опосредованный эндоцитоз При рецептор-опосредованном эндоцитозе вещество связывается и образует комплексы с мембранными рецепторами, внутриклеточный участок которых контактирует с белком адаптином. Несколько таких ком- Глава 1. Фармакокинетика 47 Внутриклеточное пространство мембрана 2 мембрана Б 1 3 А Рис. 1.3. Пиноцитоз (везикулярный транспорт): 1 — молекулы вещества; 2 — везикула; 3 — лизосома (пояснения в тексте) плексов объединяются в агрегаты и захватываются клеткой с образованием везикулы, которая снаружи окружается белком адаптином и оболочкой из специального цитоплазматического белка клатрина. Везикула диффундирует внутрь клетки и после потери клатриновой оболочки и адаптина сливается с эндосомой. Вещество после его диссоциации из комплекса с рецептором может доставляться эндосомой к мишеням внутри клетки (лизосомы, ядро, аппарат Гольджи) и/или высвобождаться наружу путем экзоцитоза (рис. 1.4). С помощью рецептор-опосредованного эндоцитоза некоторые высокомолекулярные вещества (инсулин, липопротеины низкой плотности) могут проникать внутрь клеток. Рецептор-опосредованный процесс обладает специфичностью и переносит вещества через мембрану независимо от градиента концентрации. 1.2. ПАРАЦЕЛЛЮЛЯРНЫЙ ТРАНСПОРТ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Большинство гидрофильных ЛВ всасывается, распределяется по органам и тканям и выводится из организма, не проникая через мембраны клеток. Гидрофильные вещества, растворяясь в интерстициальной жидкости, способны проникать в кровь, а из крови — в интерстициальную жидкость через промежутки между эндотелиальными клетками. Такой способ проникновения зависит от величины межклеточных промежутков и обозначается как парацеллюлярный транспорт. 48 Часть I. Общая фармакология 1 2 9 3 8 pH 5 11 7 4 6 10 5 Внеклеточное пространство лизосома Внутриклеточное пространство Внеклеточное пространство Рис. 1.4. Рецептор-опосредованный эндоцитоз (из Lullmann H. et al. Color atlas of pharmacology. — Stuttgart, NY, 2005, c изм.): 1 — молекулы вещества; 2 — рецептор; 3 — белок адаптин; 4 — клатриновая оболочка; 5 — везикула, окруженная адаптином и клатриновой оболочкой; 6 — потеря клатриновой оболочки; 7 — отделение адаптина; 8 — слияние везикулы с «ранней» эндосомой; 9 — диссоциация рецепторов из комплекса с веществом и возвращение рецепторов в мембрану; 10 — доставка вещества эндосомой в клеточные органеллы; 11 — высвобождение вещества из клетки путем экзоцитоза Вещества могут перемещаться через межклеточные промежутки в водной среде по градиенту концентрации путем пассивной диффузии в водной фазе. Если перемещение гидрофильных веществ происходит под давлением (гидростатическим или осмотическим), используют термин «фильтрация». При этом исключается проникновение веществ, диаметр молекул которых превышает размер межклеточных промежутков. Межклеточные промежутки в разных тканях не одинаковы по величине, поэтому гидрофильные ЛВ при различных путях введения всасываются в неодинаковой степени и неравномерно распределяются в организме. Промежутки между эпителиальными клетками слизистой оболочки кишечника невелики (между клетками имеются так называемые плотные контакты), что затрудняет всасывание гидрофильных ЛВ из кишечника в кровь. Эпителий других отделов пищеварительного тракта также препятствует всасыванию гидрофильных соединений. Аналогичными свойствами обладает цилиарный эпителий дыхательных путей и эпителий почечных канальцев, поэтому гидрофильные соеди- Глава 1. Фармакокинетика 49 нения плохо всасываются с поверхности легких и не реабсорбируются в почечных канальцах. Таким образом, эпителиальный барьер создает определенную преграду для проникновения многих гидрофильных соединений. Хотя эпителий не является преградой для проникновения липофильных соединений, которые легко проходят через мембраны клеток путем пассивной диффузии, существует механизм, ограничивающий этот процесс. После проникновения через мембрану липофильное вещество может быть удалено из клетки (например, из эпителиальных клеток в просвет кишечника) при участии Р-гликопротеина и некоторых других мембранных транспортеров. Промежутки между эндотелиальными клетками сосудов периферических тканей (скелетных мышц, подкожной клетчатки, внутренних органов) имеют достаточно большие размеры (приблизительно 2 нм и более) и пропускают большинство гидрофильных ЛВ, обеспечивая достаточно быстрое их проникновение из тканей в кровь и обратно (рис. 1.5Б). Эндотелиальные клетки сосудов головного мозга, наоборот, тесно прилегают друг к другу, образуя так называемые плотные контакты, препятствующие проникновению в ЦНС гидрофильных полярных веществ (см. рис. 1.5А). Этот слой клеток составляет основу гематоэнцефалического барьера (ГЭБ), который защищает ткани мозга от чужеродных соединений и поддерживает в ЦНС постоянный состав внутренней среды. Проникновение гидрофильных полярных веществ ограничено и через другие гистогематические барьеры, такие как гематоофтальмический, гематотестикулярный, плацентарный. 1.3. ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Всасывание (абсорбция, от лат. absorbeo — всасываю) — процесс, в результате которого ЛВ поступает с места введения в кровеносную и/ или лимфатическую систему. Всасывание ЛВ начинается сразу после его введения. От пути введения ЛВ зависят скорость и степень его всасывания, и в конечном итоге — скорость развития фармакологического эффекта, его величина и продолжительность. 1.3.1. Пути введения лекарственных средств Различают энтеральные (через пищеварительный тракт) и парентеральные (минуя пищеварительный тракт) пути введения ЛС. 50 Часть I. Общая фармакология Б А Рис. 1.5. Проникновение веществ через стенки капилляров мозга (А) и капилляров скелетных мышц (Б) Энтеральные пути введения К энтеральным (от греч. еnto — внутри и enteron — кишка) путям введения относят: ● сублингвальный (под язык); ● трансбуккальный (за щеку); ● пероральный (внутрь, per os); ● ректальный (через прямую кишку, per rectum). Глава 1. Фармакокинетика 51 Сублингвальное и трансбуккальное введение При сублингвальном и трансбуккальном путях введения через слизистую оболочку ротовой полости хорошо всасываются (путем пассивной диффузии) липофильные неполярные и незначительно — гидрофильные полярные вещества. Сублингвальный и трансбуккальный пути введения ЛВ имеют следующие преимущества перед другими путями введения: ● простота и удобство для больного; ● отсутствие воздействия на ЛВ хлористоводородной кислоты; ● поступление ЛВ в общий кровоток, минуя печень, предотвращает их преждевременное разрушение и выделение с желчью (отсутствие эффекта первого прохождения через печень); ● быстрота всасывания ЛВ вследствие хорошего кровоснабжения слизистой оболочки полости рта, а следовательно, быстрота развития терапевтического эффекта (возможность применения при неотложных состояниях). Однако из-за небольшой всасывающей поверхности слизистой оболочки полости рта эти пути введения приемлемы лишь для высокоактивных веществ, применяемых в малых дозах, таких как нитроглицерин, некоторых стероидных гормонов. Так, для устранения приступа стенокардии сублингвально применяют таблетки, содержащие 0,5 мг нитроглицерина, — эффект развивается через 1–2 мин. Сублингвально принимают клонидин и каптоприл при гипертензивном кризе, лоперамид — при острой диарее. Во всех этих случаях рассчитывают на быстрое всасывание ЛВ в общий кровоток и быстрое развитие эффекта. Пероральное введение При введении внутрь ЛВ всасываются преимущественно из желудка и/или тонкой кишки. При этом пути введения всасывание из других отделов желудочно-кишечного тракта (ЖКТ) ограничено в связи с непродолжительным контактом ЛВ с эпителием этих областей (ротовая полость, пищевод) или незначительной концентрацией ЛВ в месте всасывания (прямая кишка). Вещества в основном всасываются путем пассивной диффузии. Таким путем наиболее легко всасываются липофильные неполярные вещества. Всасывание гидрофильных полярных веществ ограничено небольшой величиной межклеточных промежутков в эпителии ЖКТ. Некоторые гидрофильные ЛВ (например, леводопа) всасываются при участии транспортных белков. Несмотря на то, что липофильность является определяющим свойством, необходимым для проникновения веществ через мембраны эпителия, способность растворяться в воде также 52 Часть I. Общая фармакология оказывает влияние на всасывание ЛВ. Для достижения места всасывания вещество должно раствориться в содержимом кишечника (в водной среде). Поэтому из ЖКТ лучше всасываются вещества, обладающие не только липофильными (что облегчает их проникновение через мембраны), но и, в некоторой степени, гидрофильными свойствами. Так, прием внутрь слабых кислот и оснований в виде солей улучшает их всасывание, так как соли лучше растворяются в воде. Влияние рН среды. Всасывание слабокислых соединений (ацетилсалициловой кислоты, барбитуратов и др.) начинается в желудке, в кислой среде которого бульшая часть слабых кислот не ионизирована. Однако основным местом всасывания всех ЛВ, включая слабые кислоты, является кишечник. Этому способствуют большая всасывающая поверхность слизистой оболочки (200 м2), меньшая толщина стенки кишечника и ее интенсивное кровоснабжение. Из кишечника лучше всасываются слабые кислоты со значениями рКа больше 3,0. Так, например, ацетилсалициловая кислота (рКа=3,5) в таблетированной лекарственной форме на 90% всасывается в тонкой кишке. Слабые основания хорошо всасываются в кишечнике, так как в щелочной среде кишечника (в двенадцатиперстной кишке рН=5–6, постепенно увеличиваясь до 9–11 в подвздошной кишке) слабые основания в основном находятся в неионизированной форме, что облегчает их проникновение через мембраны эпителиальных клеток. Более полному всасыванию слабых оснований в кишечнике способствуют значения рКа меньше 8,0. Слабые основания практически не всасываются в кислой среде желудка (за исключением соединений с очень низкими значениями рКа). Влияние лекарственной формы. В большинстве случаев наилучшие условия для более полного и быстрого всасывания ЛВ создаются при их введении в виде жидких лекарственных форм (водных растворов, суспензий, микстур). Некоторые малорастворимые в воде вещества назначают в виде водно-спиртовых растворов. Всасывание ЛВ при их назначении в форме суспензий зависит от размера частиц вещества. Чем выше степень диспергирования вещества (и, следовательно, меньше размер частиц), тем выше скорость его всасывания. Степень диспергирования вещества повышается при добавлении к суспензии поверхностно-активных веществ. Большинство ЛС для приема внутрь применяют в виде твердых лекарственных форм (таблеток, капсул). В этом случае процесс всасывания ЛВ во многом зависит от скорости, с которой твердые лекарственные формы распадаются в кишечнике. Быстрая распадаемость (дезинтеграция) таблеток или капсул способствует более полному и быстрому всасыванию ЛВ. Для ускорения распадаемости таблеток в их состав включают специальные Глава 1. Фармакокинетика 53 дезинтегрирующие вещества, способствующие разрушению таблеток. После дезинтеграции таблеток (или капсул) процесс всасывания ЛВ зависит от скорости его растворения в содержимом кишечника и поступления к месту всасывания. Лимитирующим фактором является скорость диффузии ЛВ в водной среде содержимого кишечника. Увеличение количества и уменьшение размеров микрочастиц способствуют более быстрой диффузии и растворению ЛВ в окружающей среде, ускоряя его всасывание. Этому же способствует использование водорастворимых солей ЛВ. Поскольку таблетки в ЖКТ распадаются достаточно медленно, различие в скорости и степени всасывания одного и того же вещества при введении его в виде таблеток и растворов может быть достаточно существенным. Так, через 30 мин после приема ацетилсалициловой кислоты в виде таблеток ее концентрация в плазме крови оказывается в 2 раза ниже, чем при применении этого вещества в той же дозе в виде раствора. Для замедления всасывания и создания более постоянной концентрации ЛВ в крови используют лекарственные формы с замедленным (контролируемым) высвобождением ЛВ. Таким образом можно получить препараты пролонгированного действия, действующие гораздо дольше (блокатор кальциевых каналов нифедипин в обычных лекарственных формах назначают 3 раза в сутки, а его пролонгированные формы — 1–2 раза в сутки). В ЖКТ ЛВ подвергаются воздействию хлористоводородной кислоты и пищеварительных ферментов. Например, бензилпенициллин разрушается хлористоводородной кислотой желудочного сока, а инсулин и другие вещества полипептидной структуры — протеолитическими ферментами. Чтобы избежать разрушения некоторых веществ в кислой среде желудка (или предотвратить воздействие раздражающих веществ на его стенку), их назначают в специальных лекарственных формах (таблетках или капсулах с кислотоустойчивым покрытием). Такие лекарственные формы без изменения проходят через желудок и распадаются лишь в тонком кишечнике (так называемые кишечнорастворимые лекарственные формы). Влияние содержимого желудочно-кишечного тракта. Количество и качественный состав содержимого ЖКТ также влияют на всасывание ЛВ. Пища механически препятствует контакту ЛВ со всасывающей поверхностью желудка и кишечника и, как правило, замедляет всасывание ЛВ. Кроме того, некоторые компоненты пищи могут ускорять перистальтику кишечника, и это отрицательно сказывается на степени всасывания некоторых ЛВ. В то же время, стимулируя кровоток в стенке кишечника, пища может ускорить всасывание ЛВ. Влияние пищи на всасывание ЛВ во многом зависит от характера веществ, входящих в ее состав. Жирная пища, задерживая опорожнение же- 54 Часть I. Общая фармакология лудка и, следовательно, поступление ЛВ в кишечник, замедляет и уменьшает всасывание большинства ЛВ. Исключение составляют липофильные плохо растворимые в воде вещества (например, жирорастворимые витамины, гризеофульвин, спиронолактон), лучше всасывающиеся в присутствии желчи, секреция которой повышается под воздействием жиров. Некоторые компоненты пищи способны нарушать всасывание ЛВ. Так, например, ионы кальция, содержащиеся в молочных продуктах, образуют с антибиотиками тетрациклинового ряда плохо всасывающиеся комплексы, а танин чая таким же образом нарушает всасывание кодеина, папаверина и других алкалоидов. Сок грейпфрута содержит фуранокумарины (дигидроксибергамоттин и др.), которые ингибируют фермент (СYP3A4) энтероцитов, метаболизирующий многие ЛВ. В результате при употреблении этого сока снижается метаболизм ряда ЛВ (нифедипина, циклоспорина, некоторых статинов и других субстратов СYP3A4) в стенке кишечника и повышается их всасывание в системный кровоток. Это приводит к чрезмерному увеличению концентрации этих ЛВ в крови, создавая опасность развития токсических эффектов. Поскольку фуранокумарины грейпфрута ингибируют фермент необратимо, их действие достаточно продолжительно. По этой причине не рекомендуется сочетать прием таких препаратов с соком грейпфрута, а также с соком и плодами лайма и плодами помело, которые также содержат вещества, ингибирующие СYP3A4. В соответствии с общей рекомендацией, во избежание возможного взаимодействия ЛВ с компонентами пищи многие ЛВ следует принимать за 40 мин –1 ч до или через 1,5–2 ч после еды. Если необходимо быстро создать высокую концентрацию ЛВ в крови (при условии, что оно не оказывает раздражающего действия, и отсутствии противопоказаний), рекомендуют принимать препарат натощак, запивая стаканом воды. Это обеспечивает быстрый контакт вещества со слизистой оболочкой ЖКТ. Однако в связи с тем, что влияние пищи на всасывание ЛВ неоднозначно, вопрос о времени приема зависит от свойств ЛВ и характера пищи. Всасывание липофильных ЛВ, таких как пропранолол, спиронолактон, гризеофульвин, нитрофурантоин, повышается в присутствии пищи, поэтому их принимают во время еды. Пищеварительные ферменты и витамины также лучше принимать во время еды. Многие препараты можно принимать независимо от приема пищи. Некоторые ЛВ существенно влияют на всасывание других ЛВ, принимаемых одновременно. Активированный уголь и другие энтеросорбенты подавляют всасывание практически всех ЛВ. Антациды препятствуют всасыванию противомикробных средств группы фторхинолонов, так как магний, кальций и алюминий, входящие в состав антацидов, обра- Глава 1. Фармакокинетика 55 зуют с фторхинолонами невсасывающиеся хелатные комплексы. Кроме того, антациды и антисекреторные средства (снижающие секрецию соляной кислоты), повышая рН содержимого желудка, нарушают всасывание слабых кислот, но улучшают всасывание слабых оснований. ЛВ, ингибирующие Р-гликопротеин и/или ферменты стенки кишечника (МАО, СYP3A4), могут увеличить всасывание других ЛВ, которые являются их субстратами. Влияния одного ЛВ на всасывание другого можно избежать, если между их приемами сделать перерыв (2 ч и более). Изменение нормальной микрофлоры ЖКТ может влиять на степень всасывания ЛВ, которые метаболизируются под действием некоторых микроорганизмов, присутствующих в кишечнике в норме. Известно, что около 40% принятого внутрь дигоксина метаболизируется Eubacterium Lentum, представителем нормальной микрофлоры кишечника; одновременный прием антибиотиков и дигоксина может привести к подавлению этого эффекта и увеличению всасывания дигоксина. Повышение концентрации дигоксина в крови может вызвать развитие интоксикации, так как этот препарат обладает небольшой широтой терапевтического действия. Влияние моторики ЖКТ. Поскольку кишечник является основным местом всасывания веществ, большинство ЛВ, особенно слабые основания (пропранолол, кодеин), всасываются более интенсивно при ускорении опорожнения желудка (например, при применении прокинетика метоклопрамида или макролидов). Наоборот, снижение скорости эвакуации содержимого желудка (чему способствуют прием М-холиноблокаторов, высокая кислотность желудочного химуса, температура пищи выше или ниже физиологической) замедляет всасывание таких ЛВ и задерживает начало их действия. На время эвакуации содержимого желудка влияет также характер пищи (жидкости эвакуируются быстрее, чем твердая пища, углеводы — быстрее, чем белки, а белки — быстрее, чем жиры), калорийность (высокая калорийность снижает скорость эвакуации), количество пищи и размер ее частиц (крупные частицы пищи стимулируют опорожнение желудка). Усиление моторики кишечника может нарушить всасывание медленно всасывающихся ЛВ, например дигоксина, поэтому его всасывание резко уменьшается при одновременном приеме с прокинетиком метоклопрамидом. При приеме ЛВ, угнетающих перистальтику кишечника, таких как М-холиноблокаторы (атропин), всасывание медленно всасывающихся веществ (дигоксина, препаратов железа) может резко увеличиться, повышая риск возникновения их токсических эффектов. Пища тоже оказывает влияние на перистальтическую активность кишечника. Скорость продвижения содержимого по кишечнику снижается при употреблении большого количества пищи, в особенности пищи, богатой жирами. 56 Часть I. Общая фармакология Влияние эффекта первого прохождения через печень. Из тонкого кишечника вещества всасываются в воротную вену, с током крови поступают сначала в печень и лишь затем в системный кровоток (рис. 1.6). В печени многие ЛВ частично метаболизируются (большинство веществ при этом инактивируется) и/или выделяются с желчью в просвет кишечника, поэтому в системный кровоток поступает лишь часть всосавшегося вещества. Этот процесс называется эффектом первого прохождения через печень, или элиминацией при первом прохождении через печень (термин «элиминация» включает биотрансформацию и выведение ЛВ). Печень Пепсин Желчный пузырь HСl Лекарственный препарат Поджелудочная железа Портальная вена Лимфатический проток Всасывание Рис. 1.6. Всасывание лекарственных веществ при введении внутрь Глава 1. Фармакокинетика 57 Большинство ЛВ оказывает действие только после достижения системного кровотока и последующего распределения по органам и тканям. О количестве ЛВ, которое достигло места своего действия, судят по общему количеству неизмененного вещества, поступившего в системный кровоток (считается, что существует прямая корреляция между количеством вещества в системном кровотоке и в органах и тканях, где оно оказывает свое действие). Для этого используют термин «биодоступность». Биодоступность определяют как часть введенной дозы ЛВ, которая в неизмененном виде достигла системного кровотока, и обычно выражают в процентах. Биодоступность вещества при внутривенном введении принимают равной 100%. Биодоступность ЛВ при приеме внутрь, как правило, меньше 100%. В справочной литературе обычно приводят значения биодоступности ЛВ при введении внутрь. Уменьшение биодоступности ЛВ при приеме внутрь может быть связано с различными причинами. Одни ЛВ частично разрушаются под влиянием хлористоводородной кислоты и/или пищеварительных ферментов ЖКТ, другие неполностью всасываются в кишечнике (например, гидрофильные полярные соединения) или подвергаются метаболизму в стенке кишечника (например, леводопа, которая под действием диоксифенилаланина (ДОФА)-декарбоксилазы превращается в дофамин), либо, являясь субстратами Р-гликопротеина, удаляются из энтероцитов. Все перечисленные выше факторы, уменьшающие всасывание ЛВ в портальный кровоток, снижают его биодоступность. Кроме того, многие вещества подвергаются интенсивной элиминации при первом прохождении через печень (метаболизируются ферментами печени и выводятся с желчью в просвет кишечника), в связи с чем имеют низкую биодоступность. Соответственно, дозы таких ЛВ при введении внутрь обычно превышают дозы, необходимые для достижения того же эффекта при парентеральном или сублингвальном введениях. Так, например, нитроглицерин (практически полностью всасывающийся из кишечника, при первом прохождении через печень элиминирует более чем на 90%) назначают сублингвально в дозе 0,5 мг, а внутрь в дозе 6,4 мг. Причиной низкой биодоступности препарата может быть неполное высвобождение действующего вещества из таблетированных лекарственных форм (или капсул). Поэтому даже препараты, содержащие одно и то же вещество в одинаковой дозе и в одной лекарственной форме, при одном и том же пути введения (т.е. препараты, обладающие так называемой фармацевтической эквивалентностью) могут иметь разную биодоступность. Фармацевтически эквивалентные препараты, производимые в различных условиях, могут различаться не только по биодоступности (степени 58 Часть I. Общая фармакология всасывания) ЛВ, но и по скорости всасывания (характеризуется константой скорости абсорбции). Некоторые препараты могут иметь одинаковую биодоступность, но разную константу скорости абсорбции. Время достижения максимальной концентрации вещества в крови при введении таких препаратов может существенно различаться. Это, как правило, связано с различиями физических свойств субстанции (размеров микрочастиц, степени гидратации и др.) и/или технологии производства препарата (в том числе способов таблетирования, характера и количества вспомогательных веществ и т.д.). В связи с этим для характеристики идентичности фармацевтически эквивалентных препаратов из различных партий и, в особенности, препаратов, производимых различными фармацевтическими предприятиями, введено понятие биоэквивалентность. Два препарата считают биоэквивалентными, если они обеспечивают одинаковую биодоступность ЛВ и одинаковую скорость достижения его максимальной концентрации в крови (т.е. одинаковую скорость поступления ЛВ в системный кровоток из места введения). Сравнение по биоэквивалентности имеет большое значение при получении воспроизведенных лекарственных препаратов, или «дженериков», при этом воспроизведенный препарат должен быть эквивалентным оригинальному, выпускаемому фирмой-разработчиком. Пероральный путь введения, так же как сублингвальный, имеет ряд преимуществ перед парентеральными путями введения: он наиболее прост и удобен для больного, не требует стерильности препаратов и специально обученного персонала. Однако внутрь можно назначать вещества, не разрушающиеся в ЖКТ в значительной степени. На степень всасывания оказывает также влияние относительная липофильность ЛВ. К недостаткам этого пути введения можно также отнести зависимость всасывания ЛВ от состояния слизистой оболочки и моторики кишечника, рН среды и состава содержимого кишечника (возможность взаимодействия с компонентами пищи и другими ЛВ), а также частичную инактивацию многих ЛВ при первом прохождении через печень. Кроме того, сами ЛВ могут оказывать влияние на процесс пищеварения и всасывания компонентов пищи, в том числе на усвоение витаминов. Например, осмотические слабительные средства затрудняют всасывание питательных веществ из кишечника, а антацидные средства, нейтрализуя хлористоводородную кислоту желудочного сока, нарушают процесс переваривания белков. У некоторых больных назначение ЛВ внутрь невозможно (например, при отказе больного от лечения, нарушении акта глотания, отсутствии сознания, у детей раннего возраста). В этих случаях ЛВ можно вводить Глава 1. Фармакокинетика 59 с помощью тонкого желудочного зонда через носовые ходы или через рот в желудок и/или двенадцатиперстную кишку. Ректальное введение Введение ЛВ в прямую кишку (ректально) используют при невозможности перорального приема (например, при рвоте), при неприятном вкусе и запахе ЛВ или его разрушении в желудке и верхних отделах кишечника. Этот путь введения часто используется в педиатрической практике. Ректально ЛВ назначают в форме суппозиториев или в лекарственных клизмах (средний объем 50 мл). Если ЛВ обладает местнораздражающим действием на слизистую оболочку прямой кишки, его предварительно смешивают со слизями. Из прямой кишки ЛВ быстро поступает в системный кровоток, при этом около 50% введенной дозы всасывается в среднюю и нижнюю ректальные вены и через нижнюю полую вену попадает в системный кровоток, минуя печень. Остальная часть из верхнего отдела прямой кишки через верхнюю ректальную вену попадает в систему воротной вены и подвергается пресистемной элиминации в печени. Ректальный путь не используют для введения высокомолекулярных ЛВ (белков, жиров, полисахаридов). Некоторые вещества вводят ректально для местного воздействия на слизистую оболочку прямой кишки (например, ректальные суппозитории с бензокаином). Ректальный путь введения имеет определенные недостатки, связанные с небольшой всасывающей поверхностью, кратковременностью контакта вещества со слизистой оболочкой, возможным неполным всасыванием ЛВ и непостоянными значениями биодоступности, а также высокой чувствительностью слизистой оболочки к раздражающему действию веществ. Парентеральные пути введения К парентеральным путям введения относят: ● внутривенный; ● внутриартериальный; ● интрастернальный (в грудину); ● внутримышечный; ● подкожный; ● внутрибрюшинный; ● под оболочки мозга и др. Внутривенное введение При этом пути введения ЛВ сразу попадают в системный кровоток, что объясняет короткий латентный период их действия. 60 Часть I. Общая фармакология В вену вводят только водные растворы ЛВ. Во избежание резкого повышения концентрации в артериях, сердце и легких большинство ЛВ вводят медленно (в течение 1 мин), часто после предварительного разведения, например, раствором натрия хлорида. Большие объемы растворов вводят капельным (инфузионным) способом. В этих случаях используют специальные системы с капельницами, позволяющие регулировать скорость введения (обычно составляет 20–60 капель в минуту, что соответствует приблизительно 1–3 мл раствора в минуту). Таким способом вводят ЛВ с коротким периодом полуэлиминации, например лидокаин. Внутривенно можно вводить гипертонические растворы. Из-за риска закупорки сосудов (эмболии) недопустимо внутривенное введение масляных растворов, суспензий, водных растворов с пузырьками газа. Возможно введение жировых ультраэмульсий заводского производства. Основной недостаток внутривенного введения — действие вещества, попавшего в системный кровоток, не может быть быстро прекращено в случае необходимости (например, при передозировке или непереносимости препарата). К недостаткам относят также опасность ифицирования тканей и тромбоз вен в месте инъекции, болевые ощущения. Неудобство этого метода —необходимость использования стерильных лекарственных форм и наличие подготовленного персонала. Внутривенный путь введения обычно используют при оказании неотложной медицинской помощи, но можно применять также планово и для курсового лечения в условиях стационара и амбулаторно. Внутриартериальное введение Введение ЛВ в артерию, кровоснабжающую определенный орган, дает возможность создать в нем высокую концентрацию действующего вещества. Внутриартериально вводят рентгеноконтрастные и противоопухолевые препараты. В некоторых случаях внутриартериально вводят антибиотики. Интрастернальное введение Интрастернальное введение (введение в грудину) применяют при невозможности внутривенного введения, например, у детей, пациентов старческого возраста. Внутримышечное введение ЛВ обычно вводят в ягодичную мышцу (верхний наружный квадрант ягодицы). Внутримышечно вводят как липофильные, так и гидрофильные соединения. Всасывание гидрофильных ЛВ происходит в основном Глава 1. Фармакокинетика 61 путем фильтрации через межклеточные промежутки в эндотелии сосудов скелетных мышц. Липофильные ЛВ всасываются в кровь путем пассивной диффузии через мембраны эндотелиальных клеток. Скорость всасывания зависит от интенсивности кровотока в месте введения. Мышечная ткань имеет хорошее кровоснабжение, поэтому всасывание ЛВ происходит довольно быстро, позволяя в большинстве случаев создать достаточно высокую концентрацию ЛВ в крови через 10–30 мин. Вещества, связывающиеся с белками мышечной ткани (диазепам, фенитоин), всасываются медленнее. Внутримышечно вводят водные растворы (до 10 мл), а для обеспечения длительного (пролонгированного) эффекта — масляные растворы и суспензии, создающие в месте введения депо препарата, из которого медленно высвобождается ЛВ (рис. 1.7). Внутримышечно нельзя вводить гипертонические растворы и раздражающие вещества. Несмотря на использование стерильных препаратов, при внутримышечном введении существует опасность развития инфекции тканей в месте введения и возникновения абсцессов. Рис. 1.7. Парентеральные пути введения лекарственных веществ 62 Часть I. Общая фармакология Подкожное введение При введении под кожу липофильные и гидрофильные вещества всасываются такими же способами, что и при внутримышечном введении (т.е. путем пассивной диффузии и фильтрации), однако в связи с менее интенсивным кровоснабжением из подкожной клетчатки (см. рис. 1.7) вещества всасываются медленнее, чем из мышечной ткани. Для ускорения всасывания ЛВ используют согревающие компрессы и местный массаж, стимулирующие кровоток в месте введения. Для ускорения всасывания можно одновременно с ЛВ вводить гиалуронидазу — фермент, разрушающий мукополисахариды соединительной ткани (при этом увеличивается площадь всасывания ЛВ). При подкожном введении веществ, всасывание которых является нежелательным (например, местных анестетиков), одновременно вводят сосудосуживающие вещества (адреналин♠), уменьшающие кровоток в месте введения. Подкожно вводят водные растворы (до 2 мл) и с осторожностью масляные растворы и суспензии, обеспечивающие более медленное всасывание ЛВ в кровь. В подкожную клетчатку можно имплантировать силиконовые контейнеры, таблетированные стерильные лекарственные формы. Благодаря медленному высвобождению веществ из этих лекарственных форм достигается достаточно постоянная концентрация ЛВ в крови в течение недель и даже месяцев. Таким способом вводят некоторые контрацептивы, препараты тестостерона. Подкожно нельзя вводить вещества с раздражающим действием и гипертонические растворы. Подкожное введение раздражающих (кальция хлорид) или сосудосуживающих веществ (норадреналин♠) может вызвать некроз подкожной жировой клетчатки. Внутрибрюшинное введение Вещества вводят в полость брюшины между ее париетальным и висцеральным листками. Этот путь используют, например, для введения антибиотиков во время хирургических вмешательств на органах брюшной полости. Введение под оболочки мозга ЛВ можно вводить субарахноидально или эпидурально. Так при инфекционных поражениях тканей и оболочек мозга вводят антибиотики, плохо проникающие через ГЭБ. Субарахноидальное введение местных анестетиков используют для спинномозговой анестезии. Внутривенно, внутриартериально, интрастернально, внутримышечно, подкожно и под оболочки мозга вводят только стерильные лекарствен- Глава 1. Фармакокинетика 63 ные формы; введение осуществляет квалифицированный медицинский персонал. Ингаляционное введение Ингаляционно (от лат. inhalare — вдыхать) вводят газообразные вещества, пары легко испаряющихся жидкостей, аэрозоли (воздушные взвеси мелкодисперсных частиц жидких или твердых веществ диаметром обычно от 1 до 10 мкм). Всасывание ЛВ в кровь с большой поверхности легких происходит очень быстро, при этом лучше всасываются липофильные неполярные соединения. Этот способ используют при введении средств для ингаляционного наркоза (газообразных веществ и легко испаряющихся жидкостей). Ингаляционное введение в виде аэрозолей используют в основном для местного воздействия на слизистую оболочку и гладкие мышцы дыхательных путей, при этом мелкие частицы (менее 2 мкм) достигают альвеол, а более крупные (6 мкм и более) задерживаются эпителием терминальных бронхиол и верхних дыхательных путей. Ингаляционное введение — один из самых распространенных способов введения бронхорасширяющих средств и препаратов глюкокортикоидов при лечении бронхиальной астмы (в данном случае всасывание веществ в кровь нежелательно, так как приводит к появлению системных побочных эффектов). Из дыхательных путей частицы веществ удаляются с помощью мукоцилиарного транспорта, при этом значительное количество вещества достигает ротовой полости, проглатывается и может всасываться из кишечника. Поэтому для предупреждения резорбтивного действия веществ при ингаляционном введении в виде аэрозолей используют плохо всасывающиеся гидрофильные соединения (например, ипратропия бромид) или вещества, подвергающиеся интенсивной пресистемной элиминации, такие как сальбутамол или глюкокортикоиды беклометазон, будесонид и др. Интраназальное введение ЛВ вводят в полость носа в виде капель или специальных интраназальных спреев. Всасывание происходит со слизистой оболочки полости носа путем пассивной диффузии. В эпителии носовой полости есть ферменты, метаболизирующие некоторые ЛВ (изоформы цитохрома Р-450 — CYP1A1, CYP4В1, CYP2B1), что может уменьшить степень интраназального всасывания. На интраназальное всасывание могут влиять кровоснабжение слизистой оболочки, вязкость слизи, рН, влажность, температура и другие факторы. 64 Часть I. Общая фармакология Интраназально вводят препараты некоторых пептидных гормонов, назначаемых в малых дозах. Например, десмопрессин, аналог антидиуретического гормона задней доли гипофиза, применяют интраназально при несахарном диабете в дозе 10–20 мкг. Трансдермальное введение Некоторые липофильные ЛВ в виде дозированных мазей или пластырей (трансдермальные терапевтические системы) наносят на кожу. Они всасываются с ее поверхности, попадают в системный кровоток, минуя печень, и оказывают резорбтивное действие. Таким путем вводят нитроглицерин для предупреждения приступов стенокардии, скополамин при морской и воздушной болезнях, ривастигмин при болезни Альцгеймера, фентанил для снятия боли, никотин для отвыкания от курения. С помощью трансдермальных лекарственных форм можно длительно поддерживать постоянную терапевтическую концентрацию ЛВ в крови и таким образом обеспечить продолжительный лечебный эффект. Так, пластыри, содержащие нитроглицерин, оказывают антиангинальное действие (лечебный эффект при стенокардии) в течение 12 ч. Чрескожное всасывание ЛВ представляет собой более сложный процесс, чем проникновение веществ через слой эпителиальных клеток или эндотелий сосудов. Кожа состоит из нескольких слоев. Эпидермис, верхний роговой слой которого состоит из кератина, препятствует проникновению гидрофильных ЛВ. Дерма, представленная соединительной и железистой тканями с пронизывающей их капиллярной сетью, в достаточной степени проницаема как для липофильных, так и для гидрофильных веществ, которые, проникая в капилляры подкожной области, попадают в системный кровоток. Кроме того, гидрофильные полярные вещества и ионизированные молекулы могут проникать в кровоток через придатки кожи — потовые и сальные железы, волосяные фолликулы, площадь которых невелика (составляет около 1% от общей площади поверхности тела). Поэтому такой путь всасывания гидрофильных ЛВ не имеет существенного значения. На чрескожное всасывание веществ оказывают влияние многие факторы, в том числе состояние кожи (нарушение целостности верхнего рогового слоя может повысить всасывание ЛВ, включая гидрофильные вещества), место и площадь контакта вещества с кожей, частота применения препарата, факторы окружающей среды (температура, влажность). На всасывание ЛВ может влиять присутствие в коже ферментов (изоферментов CYP1A1, CYP2A1, CYP1B1, UDP-глюкуронозилтрансферазы), метаболизирующих некоторые ЛВ. Глава 1. Фармакокинетика 65 Всасывание ЛВ, в том числе гидрофильных веществ с поверхности кожи, значительно повышается под действием спирта и других органических растворителей, сурфактантов (натрия лаурилсульфат), диметилсульфоксида (димексида♠), применяемого иногда вместе с мазями и кремами, содержащими противовоспалительные средства. Возможно введение ионизированных ЛВ с помощью ионофореза (ионофоретическое введение). Всасывание таких веществ после нанесения их на кожу или слизистые оболочки происходит под воздействием слабого электрического поля. Ионофоретический способ введения нередко применяют в стоматологии. Кроме того, ЛВ наносят на кожу или слизистые оболочки для получения местного действия в составе специальных лекарственных форм для наружного применения (мазей, кремов, растворов для наружного применения и т.д.). Всасывание ЛВ в кровь в таких случаях нежелательно. Другие способы введения ЛВ можно вводить также в полость плевры (противотуберкулезные средства), в полость суставной сумки (гидрокортизон при ревматоидном артрите), в тело и в просвет органа (введение окситоцина в шейку и тело матки для остановки послеродовых кровотечений, внутривлагалищное введение препаратов прогестерона с целью контрацепции). Способ введения ЛВ во многом определяет степень и скорость их всасывания. Выбор пути введения зависит от многих факторов, в том числе от свойства ЛВ, таких как полярность, растворимость в воде или липидах. Знания об особенностях всасывания гидрофильных и липофильных соединений при различных путях введения имеют большое значение для правильного выбора лекарственной формы и пути введения ЛВ в каждом конкретном случае. 1.4. РАСПРЕДЕЛЕНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ В ОРГАНИЗМЕ После поступления в системный кровоток ЛВ распределяются в различные органы и ткани. Скорость и характер распределения веществ зависит от: ● кровоснабжения органов и тканей; ● способности ЛВ растворяться в воде или липидах (т.е. относительной гидрофильности/липофильности веществ). Интенсивность кровоснабжения органов и тканей имеет основное значение для распределения ЛВ в организме. Кровоток во многом опре- 66 Часть I. Общая фармакология деляет скорость доставки ЛВ к органам и тканям. Имеют значение также путь и место введения препарата. ЛВ распределяются быстрее в хорошо кровоснабжаемые органы, такие как сердце, легкие, печень, почки, головной мозг, и достаточно медленно — в ткани с относительно плохим кровоснабжением (подкожную клетчатку, жировую и костную ткани). Из капилляров органов и тканей ЛВ проникают в интерстициальную жидкость. Гидрофильные вещества, растворенные в водной среде, перемещаются через промежутки между эндотелиальными клетками под гидростатическим давлением. Липофильные вещества проникают через мембраны эндотелиальных клеток путем пассивной диффузии по градиенту концентрации. Проникновению веществ способствуют низкая скорость кровотока и большая суммарная площадь эндотелия капиллярной сети. Гидрофильные полярные вещества распределяются в организме неравномерно. Большинство гидрофильных ЛВ не проникают внутрь клеток и распределяются в плазме крови и интерстициальной жидкости. Относительно равномерно в организме распределяются липофильные неполярные вещества. Путем пассивной диффузии они проникают через мембраны клеток и распределяются как во внеклеточной, так и во внутриклеточной жидкостях организма. Распределение слабых кислот и слабых оснований между внеклеточной (экстрацеллюлярной) и внутриклеточной жидкостями организма зависит от их рКа и рН среды. рН внутриклеточной жидкости равен приблизительно 7,0 и незначительно отличается от рН внеклеточной жидкости (7,4), поэтому проникновение слабых электролитов через мембраны клеток, а следовательно, и характер их распределения в значительной степени определяются значениями рКа. Слабые кислоты со значениями рКа менее 8,0 остаются во внеклеточной жидкости, а слабые основания со значениями рКа более 6,0 могут проникать через мембраны и накапливаться внутри клеток. Подобное распределение характерно для местного анестетика лидокаина, который, являясь слабым основанием (рКа=7,8), в неионизированной форме проникает через клеточную мембрану внутрь аксона и, частично ионизируясь в более кислой внутриклеточной среде, задерживается в этой среде и оказывает свое действие (см. раздел «Местноанестезирующие средства»). 1.4.1. Проникновение лекарственных веществ через гистогематические барьеры Гистогематические барьеры —разделительные барьеры между кровью и тканями и органами, защита которых от воздействия чужеродных Глава 1. Фармакокинетика 67 агентов необходима для их нормального функционирования. Основной элемент структуры гистогематического барьера — эндотелий сосудов, состоящий из непрерывного слоя эндотелиальных клеток и обладающий избирательной проницаемостью для различных веществ. Гистогематические барьеры, с одной стороны, обеспечивают и регулируют доставку в соответствующие органы и ткани веществ, необходимых для их жизнедеятельности, а с другой стороны, защищают эти органы от токсического воздействия эндогенных (продуктов метаболизма) и экзогенных веществ. Таким образом, гистогематические барьеры независимо от места их расположения осуществляют транспортную и защитную функции. Гистогематические барьеры имеют определенные различия в своей структуре в зависимости от морфологических и физиологических особенностей органа и ткани, который они окружают. Гематоэнцефалический барьер Барьер между кровью и тканями мозга, так называемый гематоэнцефалический барьер, имеет важнейшее значение для функционирования ЦНС. В эндотелии капилляров головного мозга межклеточные промежутки отсутствуют, а между стенками клеток находятся специфические белки, образующие плотные контакты, которые исключают парацеллюлярный транспорт гидрофильных веществ. Такой непрерывный слой эндотелиальных клеток вместе с базальной мембраной, перицитами и астроцитами образует гематоэнцефалический барьер, препятствующий распределению гидрофильных полярных веществ в ткани мозга (см. рис. 1.5). Некоторые гидрофильные ЛВ проникают через ГЭБ с помощью транспортных белков (леводопа) или рецептор-зависимого эндоцитоза (инсулин). В капиллярах мозга пиноцитоз не выражен. Липофильные вещества проникают через мембраны эндотелиоцитов мозга путем пассивной диффузии, но многие из них, являясь субстратами АТФ-зависимого транспортного белка Р-гликопротеина, выкачиваются из клеток в просвет сосуда. Р-гликопротеин ограничивает поступление в ткани мозга дигоксина, циклоспорина, домперидона, лоперамида и многих других ЛВ. Ограничение поступления в мозг лоперамида (агониста опиоидных рецепторов) позволяет использовать его периферические эффекты (снижение перистальтики кишечника) для лечения диареи, при этом отсутствуют побочные эффекты, связанные со стимуляцией опиоидных рецепторов в ЦНС (угнетение дыхания, эйфория). Сочетание приема лоперамида с приемом других ЛВ, ингибиторов Р-гликопротеина (например, верапамила), может привести к возникновению нежелатель- 68 Часть I. Общая фармакология ных центральных эффектов этого препарата вследствие повышения его концентрации в тканях мозга. Домперидон (гастрокинетическое и противорвотное средство) выгодно отличается от метоклопрамида — препарата с таким же механизмом действия — отсутствием центральных побочных эффектов (связанных с блокадой D2-рецепторов в ЦНС), так как он удаляется из головного мозга Р-гликопротеином. Ограничивать поступление ЛВ в ЦНС могут и другие АТФ-зависимые транспортные белки, которые, как и Р-гликопротеин, относятся к так называемым выкачивающим транспортерам, а также некоторые SLC-транспортеры органических анионов. Антагонист Н1-рецепторов гистамина 2-го поколения фексофенадин (противоаллергическое средство) не оказывает центрального седативного действия (нежелательный эффект), так как является субстратом транспортных белков, удаляющих его из мозга. Таким образом, ткани мозга защищены от воздействия чужеродных соединений, включая ЛВ, не только непрерывным слоем эндотелиальных клеток и других структурных элементов, непроницаемых для большинства гидрофильных веществ, но и системой транспортеров, не пропускающих в ЦНС различные, в основном липофильные, вещества. С другой стороны, ограниченный доступ в ткани мозга ЛВ, терапевтическое применение которых основано на их центральном действии, может привести к снижению концентрации этих ЛВ ниже терапевтического уровня и отсутствию необходимого эффекта. На проницаемость ГЭБ влияют различные факторы. Проницаемость ГЭБ может повышаться: ● при увеличении осмотического давления в сосудах мозга и повышении АД; ● при воздействии радиации и микроволн, медиаторов воспаления — гистамина и брадикинина; ● при различных патологических состояниях (черепно-мозговой травме, ишемии мозга, рассеянном склерозе, болезни Альцгеймера, энцефалите и менингите); ● при воспалении мозговых оболочек (ГЭБ становится более проницаемым для гидрофильных ЛВ, что позволяет вводить внутривенно бензилпенициллин для лечения бактериального менингита). Для улучшения проникновения в ЦНС некоторых ЛВ с целью их воздействия на мозг в разное время использовались различные методы, в том числе повышение осмотического давления под действием маннитола (осмотическое «открывание» ГЭБ), в результате чего происходят нарушение структуры плотных межклеточных контактов эндотелия капилляров мозга и увеличение парацеллюлярного транспорта. При злокачественных опухолях мозга этот метод позволяет доставлять противоопухолевые пре- Глава 1. Фармакокинетика 69 параты (метотрексат, прокарбазин) непосредственно в пораженные области мозга. Более щадящим способом увеличения доставки ЛВ в мозг является использование пролекарств, способствующих транспорту в ЦНС гидрофильных соединений. Пролекарство (неактивное соединение) специфически связывается с мембранным транспортером, переносящим его в мозг (леводопа), или в результате изменения структуры активного ЛВ (присоединения определенных группировок) приобретает способность растворяться в липидах и проникать через мембрану (габапентин). И в том, и другом случаях пролекарство после доставки в мозг превращается в активное вещество. Для увеличения проникновения в ЦНС липофильных соединений, являющихся субстратами Р-гликопротеина и других транспортеров, можно использовать их ингибиторы. Так, ингибитор Р-гликопротеина верапамил повышает доставку в мозг противогрибкового препарата итраконазола и ингибиторов протеазы ВИЧ. Последним словом в решении проблемы направленного транспорта ЛВ в мозг явилось использование наночастиц (размером от 10 до 1000 нм), приготовленных из биодеградируемых полимерных материалов, мало растворимых в воде и совместимых с тканями человека. Транспортируемые вещества могут быть включены внутрь наночастиц, связаны с ними ковалентно или сорбированы на их поверхности. В экспериментах наночастицы оказались способны переносить через ГЭБ соединения пептидной структуры (даларгин♠), вещества, удаляемые из ЦНС Р-гликопротеином (лоперамид). На стадии клинических испытаний находится нанотехнологический противоопухолевый препарат доксорубицина. Некоторые участки головного мозга не защищены ГЭБ. Пусковая зона рвотного центра доступна для действия веществ, не проникающих через ГЭБ, таких как антагонист дофаминовых D2-рецепторов домперидон, что позволяет применять этот препарат в качестве противорвотного и гастрокинетического средства, не оказывающего влияния на другие отделы головного мозга. Гематоплацентарный барьер Гематоплацентарный барьер представляет собой сложную транспортную систему, разделяющую кровообращение матери и плода. Он состоит из монослоя клеток (синцитиотрофобластов), апикальная мембрана которых контактирует с материнской кровью, а базальная — с кровью плода. Проникновение веществ через этот барьер происходит так же как через все биологические мембраны. Липофильные вещества могут преодолевать этот барьер путем пассивной диффузии, а некоторые гидрофильные вещества — с помощью транспортных белков или (редко) пиноцитоза. 70 Часть I. Общая фармакология Во время беременности плацента регулирует обмен веществ (эндогенных и экзогенных, в том числе ЛВ) между материнским организмом и плодом. Обеспечивая доставку в плодные ткани питательных веществ (глюкозы, аминокислот, витаминов и др.), плацента также осуществляет удаление продуктов метаболизма из системы кровообращения плода в систему кровообращения материнского организма. Проникновение в плод чужеродных соединений, включая ЛВ, регулируется плацентой избирательно. Скорость пассивной диффузии через гематоплацентарный барьер липофильных веществ зависит от степени их липофильности и размера молекул. Легко диффундируют через мембрану трофобластов вещества с молекулярной массой ≤500 Д (дальтон), а вещества с большей молекулярной массой (но ≤1 000 Д) проникают не полностью и медленнее. Вещества с еще большой молекулярной массой практически не проходят через плацентарную мембрану. Проникновение слабых кислот и слабых оснований зависит от степени их ионизации и, следовательно, от рН крови матери и плода. В обычных условиях рН плазмы крови плода несколько ниже (7,0–7,2), чем рН крови матери (7,4), но даже такое небольшое различие способствует удерживанию в тканях плода слабоосновных соединений. Гидрофильные вещества могут проникать через плаценту с помощью специфических транспортных белков. Облегченная диффузия, существующая для переноса через мембрану трофобластов эндогенных субстратов, мало участвует в транспорте ЛВ. Активный транспорт в ткани плода ЛВ, имеющих структурное сходство с эндогенными веществами (аминокислотами и др.), имеет большее значение. При этом может быть задействован как первичный (АТФ-зависимый), так и вторичный (зависимый от трансмембранного градиента Na+ и др. катионов) транспорт веществ. Некоторые транспортеры (относящиеся к SLC-семейству) участвуют не только в переносе веществ из материнской крови в плаценту, но и обратно. Многие липофильные соединения удаляются из плаценты в кровообращение матери при участии так называемых выкачивающих транспортеров, работающих только в одном направлении. Это в первую очередь АТФ-зависимый АВС-транспортер Р-гликопротеин, локализованный в апикальной мембране (с материнской стороны) трофобласта. При его участии происходит удаление из плаценты таких потенциально токсичных для плода ЛВ, как рифампицин, ингибиторы протеазы ВИЧ, верапамил, циклоспорин, и некоторых других липофильных веществ. Ингибиторы Р-гликопротеина повышают способность ряда веществ (иммунодепрессанта циклоспорина, противовирусного средства саквинавира) проникать через плаценту, увеличивая их токсическое воздействие на плод. Глава 1. Фармакокинетика 71 В апикальной мембране присутствуют и другие АВС-транспортеры, в том числе белок резистентности рака груди, препятствующий проникновению в плод противоопухолевых препаратов (например, доксорубицина). Таким образом, гематоплацентарный барьер обладает несколькими механизмами, препятствующими токсическому воздействию на плод чужеродных соединений: гидрофильные вещества поступают через плаценту избирательно, а липофильные соединения, способные преодолеть барьер, удаляются из плаценты с помощью специальных транспортеров. Следует добавить, что проницаемость плацентарного барьера изменяется в течение беременности. Истончение плаценты и повышение ее проницаемости наблюдается с 32 по 35 неделю беременности. Наиболее опасным временем для приема ЛВ считается период органогенеза, когда по возможности следует отказаться от приема всех ЛС. Некоторые ЛВ оказывают повреждающее действие именно в этот период (например, прием солей лития в период формирования сердечной трубки). Потенциальная опасность токсического воздействия некоторых ЛВ на плод существует даже при их приеме в период, предшествующий зачатию. Это касается ЛВ, способных к кумуляции (например, ретиноидов), которые могут присутствовать в организме беременной в период органогенеза. Но и в другие периоды беременности ЛВ могут оказывать нежелательное действие на плод, поэтому прием препаратов беременными должен находиться под строгим врачебным контролем. Другие гистогематические барьеры Другие гистогематические барьеры также осуществляют защитную функцию, в том числе в отношении гидрофильных соединений. К ним относят: ● гематоофтальмический барьер, не пропускающий гидрофильные полярные ЛВ в среды глаза; ● гематотестикулярный барьер (между кровью и содержимым семенных канальцев); ● гематофолликулярный (гематоовариальный) барьер. Гематотестикулярный барьер, через который липофильные вещества могут проникать путем пассивной диффузии, имеет защитный механизм в виде АТФ-зависимых транспортных белков (Р-гликопротеина и некоторых других), ограничивающих проникновение некоторых липофильных ЛВ, в том числе оказывающих отрицательное воздействие на сперматогенез (циклоспорин). Показано также значение Р-гликопротеина для функции овариального барьера, защищающего яичники от воздействия липофильных ЛВ. 72 Часть I. Общая фармакология Таким образом, особенности распределения липофильных и гидрофильных ЛВ в органах и тканях определяются их различной способностью проникать через клеточные мембраны, в том числе через гистогематические барьеры, а также зависят от структурных и биохимических характеристик этих барьеров. 1.5. ДЕПОНИРОВАНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ В ОРГАНИЗМЕ При распределении в организме некоторые ЛВ могут задерживаться и накапливаться в различных тканях. Происходит это вследствие обратимого связывания ЛВ с белками, фосфолипидами и нуклеопротеинами клеток. Этот процесс носит название депонирование. Вещества могут депонироваться в различных тканях, что отчасти зависит от физико-химических свойств ЛВ. В соединительной ткани могут накапливаться полярные соединения, жировая ткань — основное место депонирования липофильных веществ. Концентрация вещества в месте его депонирования может быть очень высокой. Так, концентрации противомалярийного средства хлорохина в печени, где он избирательно накапливается, в 1 000 раз превышают его концентрации в плазме крови. Некоторые вещества, избирательно накапливаясь в определенных органах и тканях, оказывают там специфическое действие. Например, йод, необходимый для синтеза тиреоидных гормонов, концентрируется в щитовидной железе, а фтор, принимающий участие в формировании костной ткани, накапливается в костях и зубах. Депонирование некоторых ЛВ может привести к развитию побочных эффектов. Тетрациклины, связываясь с кальцием, накапливаются в костной ткани, в том числе в зубах, что может привести к нарушению формирования скелета при внутриутробном развитии плода и повреждению зубов у маленьких детей. Поэтому назначение тетрациклинов противопоказано беременным и детям до 8 лет. Однако действие большинства ЛВ развивается не в местах их депонирования. Из депо вещества постепенно высвобождаются в кровь и распределяются в другие органы и ткани, достигая места своего действия. При этом депонирование может привести к удлинению (пролонгированию) действия препарата или к возникновению эффекта последействия. Эффект последействия возникает, например, при введении средства для внутривенного наркоза тиопентала натрия, высоколипофильного соединения, в большом количестве накапливающегося в жировой ткани. Сразу после Глава 1. Фармакокинетика 73 введения тиопентал распределяется в головной мозг и вызывает непродолжительный наркоз (около 15 мин), после которого развивается посленаркозный сон (в течение 2–3 ч), связанный с медленным высвобождением препарата из жирового депо в ЦНС, где он оказывает угнетающее действие. Самый распространенный вид депонирования ЛВ — связывание с белками плазмы крови. Слабокислые соединения (нестероидные противовоспалительные средства, сульфаниламиды) связываются с альбуминами (основной белок плазмы крови), а слабые основания — с α1-кислым гликопротеином и другими белками плазмы крови. Некоторые вещества (глюкокортикоиды, препараты железа) избирательно связываются с определенными плазменными белками (транскортином, трансферрином). Связывание ЛВ с белками плазмы крови — обратимый процесс (происходит при участии водородных и Ван-дер-Ваальсовых связей), может быть представлен следующим образом: ЛВ + белок комплекс ЛВ–белок. Комплексы ЛВ–белок не проникают через мембраны клеток и межклеточные промежутки в эндотелии сосудов и служат своеобразным резервуаром (депо) данного вещества в крови (рис. 1.8А). Связанные с белками ЛВ не достигают места своего действия и не проявляют фармакологической активности. Однако в связи с тем, что это связывание обратимо, часть ЛВ постепенно, по мере снижения концентрации свободного вещества в плазме крови, высвобождается из комплекса с белком и поступает к месту своего действия (рис. 1.8Б). Комплексы ЛВ с плазменными белками не проникают в клетки печени и не фильтруются в почечных клубочках, что приводит к снижению скорости биотрансформации и выведения ЛВ и, следовательно, к пролонгированию их действия. Для большинства ЛВ связывание с белками плазмы крови неспецифично. Разные ЛВ могут связываться с одними и теми же белками с достаточно высоким аффинитетом, при этом они конкурируют за места связывания на белковых молекулах и могут вытеснять друг друга. В таких случаях большое значение имеет степень связывания веществ с белками при их терапевтических концентрациях в крови. Например, толбутамид (гипогликемическое средство, применяемое при сахарном диабете) приблизительно на 96% связывается с белками плазмы крови, т.е. в свободном (активном) состоянии в крови находится только около 5% вещества. При одновременном назначении сульфаниламидов, также интенсивно связывающихся с белками плазмы крови, происходит быстрое вытеснение толбутамида из мест связывания, что приводит к значительному повышению концентрации свободного вещества в крови. В результате толбутамид 74 Часть I. Общая фармакология Плазма крови Ткани ЭК 1 3 2 А 1 3 2 Б Рис. 1.8. Связывание лекарственных веществ с белками плазмы крови: А — молекулы лекарственного вещества (1) связаны с белками плазмы крови (2) и поэтому практически не проникают в ткани к месту своего действия (3 — клетки тканей); ЭК — эндотелиальные клетки; Б — молекулы лекарственного вещества (1) после высвобождения из комплекса с белками плазмы крови (2) проникают через эндотелий сосудов к месту своего действия (3 — клетки тканей) вызывает чрезмерно выраженную, но менее продолжительную гипогликемию, так как одновременно ускоряются его биотрансформация и выведение из организма. Особую опасность представляет одновременное назначение сульфаниламидов и антикоагулянта варфарина, связывающегося с белками плазмы крови на 99%. Быстрое повышение концентрации свободного варфарина (препарата с малой широтой терапевтического действия) может привести к резкому снижению свертываемости крови и кровотечениям. Вытеснение из связи с белками не приводит к клинически значимому изменению концентрации свободного вещества в крови, если ЛВ связывается с белками менее чем на 90%. Значение имеют также и другие факторы, такие как медленное вытеснение вещества, депонирование вещества в тканях, что уменьшает концентрацию свободного ЛВ в крови и, следовательно, устраняет причину его токсического действия. По этим причинам вытеснение лишь немногих ЛВ из связи с белками плазмы крови приводит к клинически значимым последствиям. 1.6. БИОТРАНСФОРМАЦИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Биотрансформация (метаболизм) — изменение химической структуры и физико-химических свойств ЛВ под действием ферментов организма. Основная направленность этого процесса — удаление чужеродных со- Глава 1. Фармакокинетика 75 единений, в том числе ЛВ, из организма путем превращения неполярных липофильных веществ в полярные гидрофильные соединения. В связи с тем, что полярные гидрофильные вещества в отличие от липофильных не реабсорбируются в почечных канальцах, они быстро выводятся почками, а некоторые из них выводятся с желчью в просвет кишечника. Биотрансформация липофильных ЛВ происходит под действием ферментов печени, локализованных в мембране эндоплазматического ретикулума гепатоцитов. Эти ферменты называют микросомальными, так как они оказываются связанными с мелкими субклеточными фрагментами гладкого эндоплазматического ретикулума (микросомами), которые образуются при гомогенизации печеночной ткани или тканей других органов и могут быть выделены центрифугированием (осаждаются в так называемой микросомальной фракции). Основное место локализации микросомальных ферментов — гепатоциты, но они обнаружены также и в других органах (кишечнике, почках, легких, головном мозге). В плазме крови, в печени, стенке кишечника, почках, легких, коже, слизистых оболочках и других тканях имеются немикросомальные ферменты, локализованные в цитозоле или митохондриях. Различают два основных вида метаболизма ЛВ: ● несинтетические реакции (метаболическая трансформация); ● биосинтетические реакции (биосинтетическая трансформация, или конъюгация). Большинство ЛВ сначала метаболизируется при участии реакций метаболической трансформации с образованием реакционно-способных метаболитов, вступающих затем в реакции конъюгации. При конъюгации к ЛВ или их метаболитам присоединяются остатки эндогенных соединений (глюкуроновой кислоты и др.) или химические группировки (ацетильные, метильные), поэтому реакции конъюгации обозначают термином «биосинтетическая трансформация». 1.6.1. Метаболическая трансформация Реакции метаболической трансформации включают: ● окисление; ● восстановление; ● гидролиз. Окисление Многие липофильные соединения подвергаются окислению в печени под действием микросомальной системы ферментов, известных как окси- 76 Часть I. Общая фармакология дазы смешанных функций (или монооксигеназы), основной компонент которых — цитохром Р-450 (гемопротеин, связывающий ЛВ и кислород в своем активном центре). Реакция протекает при участии цитохром-Р450-редуктазы и никотинамидадениндинуклеотидфосфата — НAДФН, донора электронов. В результате после восстановления молекулярного кислорода происходят присоединение одного атома кислорода к субстрату (ЛВ) с образованием окисленного метаболита и включение другого атома кислорода в молекулу воды: RH + O2 + НAДФН + H+ ROH + H2O + НAДФ+, где RH — лекарственное вещество, ROH — метаболит. Кислород может быть включен в молекулу субстрата в составе гидроксильной группы (реакция гидроксилирования), эпоксидной группы (реакция эпоксидации), может замещать аминогруппу (реакция дезаминирования) или атом серы. В реакциях дезалкилирования метаболиты образуются при включении кислорода в алкильную группу, отделяющуюся от молекулы субстрата. Примеры реакций микросомального окисления приведены в табл. 1.2. Оксидазы смешанных функций в целом обладают низкой субстратной специфичностью и могут метаболизировать многие химические соединения. В то же время отдельные изоформы (изоферменты) цитохрома Р-450 (Cytochrome P-450, CYP) отличаются определенной специфичностью (каждая из них участвует в метаболизме относительно небольшого количества веществ). В настоящее время известно более тысячи изоферментов цитохрома Р-450, подразделяемых на семейства и подсемейства. Изоформы, имеющие более 40% общего аминокислотного состава, объединены в семейства и обозначаются арабскими цифрами (CYP1, CYP2, CYP3 и т.д.). Подсемейства, обозначаемые латинскими буквами, объединяют изоформы с идентичностью аминокислотного состава более 55% (CYP2D, CYP3A и т.д.). Отдельные изоферменты обозначают арабскими цифрами, следующими за латинскими буквами (CYP1A2, CYP2D6, CYP3A4). ЛВ могут быть субстратами двух и более изоферментов, при этом различные изоферменты способны метаболизировать одно вещество в различных участках его молекулы. В табл. 1.3 приведены основные изоферменты цитохрома Р-450 печени человека, принимающие участие в метаболизме ЛВ, и примеры ЛВ, являющихся субстратами этих изоферментов. Наибольшее количество ЛВ метаболизируется в печени при участии CYP3A4. Микросомальное окисление ЛВ под действием изоферментов цитохрома Р-450 происходит и в других органах: кишечнике, легких, почках. Глава 1. Фармакокинетика 77 По своей метаболической активности эти органы уступают печени, хотя вносят определенный вклад в биотрансформацию некоторых ЛВ. Среди микросомальных ферментов стенки кишечника наиболее велико содержание CYP3A4 (70% от содержания всех изоферментов цитохрома Р-450). Этот изофермент наряду с Р-гликопротеином участвует в пресистемной элиминации многих ЛВ, снижая их биодоступность. Таблица 1.2. Основные реакции метаболизма (биотрансформации) лекарственных веществ Реакции биотрансформации Лекарственные вещества Микросомальное окисление Ароматическое гидроксилирование Фенобарбитал, фенитоин, пропранолол, варфарин Алифатическое гидроксилирование Толбутамид, ибупрофен, дигитоксин, барбитураты N-окисление Морфин, хинидин, парацетамол S-окисление Хлорпромазин, циметидин Дезаминирование Диазепам, амфетамин, эфедрин Дезалкилирование Морфин, кодеин, кофеин, теофиллин Немикросомальное окисление Окислительное дезаминирование Норэпинефрин, серотонин, эпинефрин Ароматическое гидроксилирование Аллопуринол Декарбоксилирование Леводопа Восстановление Нитрогруппы Хлорамфеникол, нитразепам Карбонильной группы Налоксон Дегалогенирование Галотан Гидролиз Сложных эфиров Прокаин, ацетилсалициловая кислота, эналаприл, суксаметония бромид Амидов Прокаинамид, индометацин Биосинтетические реакции Конъюгация с остатком глюкуроновой Парацетамол, хлорамфеникол, кислоты (образование эфиров, диазепам, морфин, дигоксин тиоэфиров или амидов глюкуроновой кислоты) Конъюгация с остатком серной кислоты Парацетамол, стероиды (образование сульфатов) Конъюгация с глицином Салициловая кислота Конъюгация с глутатионом Этакриновая кислота, доксорубицин Ацетилирование Сульфаниламиды, изониазид Метилирование Катехоламины, каптоприл 78 Часть I. Общая фармакология Таблица 1.3. Основные изоферменты цитохрома Р-450, участвующие в метаболизме лекарственных веществ, их индукторы и ингибиторы Субстраты Индукторы CYP1A2 Изоферменты Кофеин, теофиллин, парацетамол, варфарин, тамоксифен, кломипрамин Фенобарбитал, омепразол, рифампицин, вещества, содержащиеся в сигаретном дыме и жареной пище (бензопирены, метилхолантрены), броколли, брюссельская капуста Ципрофлоксацин, циметидин, кларитромицин, эритромицин CYP2C9 Ибупрофен, фенитоин, толбутамид, варфарин Рифампицин, фенобарбитал Диклофенак, сульфаниламиды, циметидин CYP2C19 Диазепам, напроксен, пропранолол, омепразол, клопидогрел Рифампицин, фенобарбитал Омепразол, флуоксетин CYP2D6 Кодеин, клозапин, Не известны омепразол, метопролол, тимолол, галоперидол, трициклические антидепрессанты Амиодарон, галоперидол, флуоксетин, хинидин, циметидин CYP2E1 Этанол, парацетаЭтанол (хроничемол, галотан, энфлу- ский прием), изоран ниазид Дисульфирам, ритонавир CYP3A4/3A5 Амиодарон, варфарин, верапамил, диазепам, дилтиазем, кетоконазол, кортикостероиды, кокаин, ловастатин, лидокаин, лозартан, макролиды, мидазолам, нифедипин, прогестерон, ритонавир, спиронолактон, сульфаметоксазол, тестостерон, циклоспорин, хинидин, этинилэстрадиол Кетоконазол, метронидазол, омепразол, циметидин, хинидин, ципрофлоксацин, эритромицин, кларитромицин, хинидин, фуранокумарины сока грейпфрута Барбитураты, рифампицин, фенитоин, карбамазепин, глюкокортикоиды, фенилбутазон, макролиды, трава зверобоя (гиперфорин) Ингибиторы Глава 1. Фармакокинетика 79 Окисление некоторых ЛВ происходит также при участии немикросомальных ферментов, локализованных в цитозоле, митохондриях, лизосомах и цитоплазматических мембранах клеток. Для этих ферментов характерна субстратная специфичность. Так, моноаминоксидаза типа А (МАО-А) осуществляет окислительное дезаминирование катехоламинов (норадреналина, адреналина, серотонина и др.), под действием алкогольдегидрогеназы этанол окисляется до ацетальдегида, под действием ксантиноксидазы происходит гидроксилирование пуриновых соединений (аллопуринола, теофиллина). Восстановление Восстановление ЛВ заключается в присоединении к его молекуле атома водорода или удалении атома кислорода. Эти реакции могут протекать при участии микросомальных (восстановление хлорамфеникола) и немикросомальных (восстановление хлоралгидрата) ферментов. Некоторые ЛВ (например, месалазин) восстанавливаются в кишечнике под действием редуктаз, продуцируемых кишечными бактериями. Гидролиз Гидролиз большинства ЛВ осуществляют немикросомальные ферменты (эстеразы, амидазы, фосфатазы) в плазме крови и тканях (в основном в печени). Вследствие присоединения воды происходит разрыв эфирных, амидных и фосфатных связей в молекулах ЛВ. Гидролизу подвергаются сложные эфиры (суксаметоний, прокаин, бензокаин, ацетилсалициловая кислота) и амиды (прокаинамид, индометацин). Некоторые ЛВ гидролизуются под действием микросомальных ферментов, например амидаз (местные анестетики из группы амидов). Микросомальный фермент эпоксидгидролаза гидролизует высокореактивные метаболиты, образующиеся при микросомальном окислении некоторых ЛВ (например, карбамазепина) с образованием неактивных соединений. В процессе метаболической биотрансформации обычно происходит снижение активности и токсичности исходных веществ. Однако метаболиты, образуемые в результате несинтетических реакций, могут обладать такой же, а иногда и более высокой активностью, чем исходные соединения. Образование активных метаболитов обеспечивает длительное действие некоторых ЛВ (например, диазепама). Активные метаболиты образуются при биотрансформации верапамила, имипрамина, кодеина, хлорохина, лидокаина и некоторых других ЛВ. Примером ЛВ, неактивных в исходном состоянии и активируемых в процессе метаболизма, являются предшественники лекарств — про- 80 Часть I. Общая фармакология лекарства. Пролекарства часто используют с целью улучшения фармакокинетических свойств ЛВ, обычно с целью повышения всасывания и, следовательно, биодоступности активного вещества. Например, антигипертензивные средства из группы ингибиторов ангиотензин-превращающего фермента, содержащие карбоксильную группу (эналаприл и др.), гидролизуются в организме с образованием активных соединений. При приеме внутрь всасывание эналаприла составляет 60%, в то время как его активный метаболит эналаприлат всасывается всего лишь на 10%. Присоединение остатка аминокислоты валина к ацикловиру (препарат валацикловир) повышает его биодоступность более чем на 30%, так как валацикловир является субстратом транспотера олигопептидов и всасывается путем активного транспорта, превращаясь в активное соединение в печени. С помощью пролекарств могут решаться проблемы с доставкой ЛВ к месту его действия. Так, предшественник дофамина леводопа в отличие от дофамина проникает в ЦНС, где под влиянием ДОФА-декарбоксилазы превращается в активное соединение дофамин. В некоторых случаях в процессе метаболической трансформации образуются токсичные соединения. Примером является образование промежуточного токсичного метаболита (N-ацетил-пара-бензохинонимина) при микросомальном окислении анальгетика парацетамола под действием изофермента CYP2Е1 в печени и в меньшей степени в почках. Инактивация этого метаболита происходит в результате его связывания с глутатионом, однако при истощении запасов глутатиона, причиной которого может быть повышенное образование метаболита (вследствие передозировки препарата или индукции CYP2Е1 при хроническом приеме алкоголя), он оказывает токсическое действие на печень и почки. 1.6.2. Биосинтетическая трансформация В процессе биосинтетических реакций к функциональным группировкам (аминогруппам, гидроксильным, карбоксильным группам) молекул ЛВ или их метаболитов присоединяются остатки эндогенных соединений (глюкуроновой или серной кислот, глутатиона, глицина и др.) или высокополярные химические группы (ацетильные, метильные). Эти реакции протекают при участии ферментов (в основном трансфераз) печени, а также ферментов других тканей (стенки кишечника, легких, почек). Данные ферменты содержатся в эндоплазматическом ретикулуме гепатоцитов (микросомальные ферменты) или в цитозольной фракции. Глава 1. Фармакокинетика 81 Наиболее общей реакцией является конъюгация с глюкуроновой кислотой. Присоединение остатков глюкуроновой кислоты (образование глюкуронидов) происходит при участии микросомального фермента уридиндифосфат-глюкуронилтрансферазы, обладающего низкой субстратной специфичностью, вследствие чего он метаболизирует многие ЛВ, содержащие разные функциональные группы, а также их метаболиты и некоторые эндогенные вещества (например, билирубин, гормоны щитовидной железы). При этом, как правило, образуются полярные неактивные глюкурониды, быстро выводящиеся из организма. Исключение составляет морфин-6-глюкуронид, обладающий, как и морфин, анальгетической активностью. Кроме того, конъюгация с глюкуроновой кислотой может приводить к активации некоторых канцерогенов. Коньюгаты с глюкуроновой кислотой (глюкурониды) выводятся с желчью в просвет кишечника и подвергаются энтерогепатической циркуляции. Под действием цитозольного фермента сульфотрансферазы в печени и других органах происходит присоединение к субстрату (в основном к фенольным соединениям) остатка серной кислоты (реакция сульфатирования) с образованием сложных эфиров серной кислоты. Кроме того, сульфатированию подвергаются такие эндогенные соединения, как катехоламины, стероидные гормоны, гормоны щитовидной железы. При сульфатировании миноксидила происходит образование активного метаболита миноксидила сульфата. Реакции сульфатирования и образования глюкуронидов протекают также в стенке кишечника и почках, хотя активность метаболизирующих ферментов в этих тканях, как правило, гораздо ниже, чем в печени. Метаболизм в стенке кишечника имеет существенное значение для пресистемной элиминации некоторых ЛВ. Так, например, сульфатирование тербуталина и изопреналина происходит в основном в стенке кишечника, а не в печени. Конъюгация с глутатионом происходит под действием фермента глутатион SH-S-трансферазы, локализованного в цитозоле. В реакцию конъюгации с глутатионом вступают некоторые реакционно-способные вещества (эпоксиды, хиноны), в том числе промежуточный метаболит, образующийся в результате микросомального окисления парацетамола, в результате чего резко снижается его токсичность. В процессе конъюгации образуются высокополярные гидрофильные соединения, быстро выводящиеся почками или с желчью в просвет кишечника. Конъюгаты, за некоторым исключением (миноксидил, морфин), менее активны и, как правило, менее токсичны, чем исходные ЛВ или их метаболиты. 82 Часть I. Общая фармакология 1.6.3. Факторы, влияющие на биотрансформацию лекарственных веществ Активность ферментов, метаболизирующих ЛВ, а следовательно, и скорость их биотрансформации зависят от пола, возраста, состояния организма, одновременного назначения других ЛС, а также от некоторых веществ, содержащихся в продуктах питания. У мужчин активность микросомальных ферментов выше, чем у женщин, так как синтез этих ферментов стимулируется мужскими половыми гормонами. Такие вещества, как этанол, эстрогены, бензодиазепины, салицилаты, метаболизируются быстрее у мужчин, чем у женщин. В эмбриональном периоде большинство ферментов метаболизма ЛВ отсутствует. У новорожденных в первые 2–4 нед жизни активность многих ферментов (в частности, ферментов, участвующих в реакциях конъюгации) снижена и достигает достаточного уровня лишь через 1–6 мес. Поэтому детям в первые недели жизни не рекомендуется назначать такие ЛВ, как хлорамфеникол (вследствие недостаточной активности микросомальных ферментов замедлены процессы конъюгации его токсичного метаболита). В старческом возрасте снижаются активность некоторых микросомальных ферментов, печеночный кровоток и масса печени, вследствие чего уменьшается скорость метаболизма многих ЛВ. Снижение синтеза белков в печени приводит к изменению параметров распределения ЛВ. В связи с этим пациентам старше 60 лет такие ЛВ назначают в меньших дозах. При заболеваниях печени снижается активность микросомальных ферментов и замедляется биотрансформация многих ЛВ, что приводит к усилению и удлинению их действия. Уменьшение скорости кровотока также существенно замедляет метаболизм некоторых ЛВ (морфина, лидокаина), поэтому при сердечной недостаточности обычные дозы этих препаратов могут вызвать токсические эффекты. Нарушения функций щитовидной железы повышают (при гипертиреозе) или снижают (при гипотиреозе) биотрансформацию ЛВ. Сахарный диабет и другие нарушения функции эндокринной системы также влияют на лекарственный метаболизм. Синтез и активность микросомальных ферментов может повышаться под действием различных ЛВ, а также некоторых веществ, содержащихся в продуктах питания, сигаретном дыме, окружающей среде и т.д. Этот процесс называется индукцией ферментов. Индукции могут подвергаться ферменты, участвующие как в несинтетических реакциях, так и в реакциях конъюгации (в основном с глюкуроновой кислотой). Индукторы микросомальных ферментов, как правило, липофильные вещества и могут быть субстратами ферментов, которые они индуцируют. Глава 1. Фармакокинетика 83 При воздействии индукторов микросомальных ферментов повышается скорость биотрансформации ЛВ, метаболизирующихся этими ферментами (см. табл. 1.3), приводя к ослаблению их терапевтического действия. Поскольку CYP3A4 участвует в метаболизме многих ЛВ (более 60% препаратов, применяемых в клинической практике), индукция этого изофермента достаточно часто может иметь нежелательные последствия. Некоторые ЛВ (например, фенобарбитал, рифампицин) являются универсальными индукторами, повышая активность нескольких изоферментов цитохрома Р-450, в том числе CYP3A4, и, как следствие, ослабляют терапевтические эффекты многих ЛВ. Например, эффективность пероральных контрацептивов может снизиться на фоне лечения рифампицином или фенобарбиталом из-за ускорения метаболизма входящих в их состав эстрогенов. Если в процессе биотрансформации ЛВ образуются токсичные метаболиты, индукция метаболизирующих это вещество ферментов приводит к повышению риска его токсического действия. Так, индукция CYP2Е1 при хроническом употреблении алкоголя или приеме противотуберкулезного препарата изониазида (см. табл. 1.3) увеличивает токсичность парацетамола. Токсичные вещества, вызывающие злокачественные новообразования (канцерогены), могут быть образованы в результате метаболизма некоторых веществ, находящихся в пищевых продуктах или окружающей среде, под действием изоферментов CYP1А2 и CYP1А1 (без метаболической активации эти вещества не канцерогенны). Индукция изоферментов противоязвенным препаратом омепразолом повышает опасность образования таких канцерогенов. Как правило, действие индукторов синтеза микросомальных ферментов (в том числе фенобарбитала) развивается медленно (в течение нескольких недель). Рифампицин оказывает более быстрый эффект, существенно повышая активность ферментов, уже через 2–4 дня от начала применения. Препараты некоторых лекарственных растений могут ускорять метаболизм ЛВ. Например, препараты зверобоя, применяемые в качестве легких антидепрессантов, вызывают индукцию изофермента CYP3A4 и поэтому ослабляют или предупреждают действие ЛВ, метаболизирующихся при участии этого изофермента. Полициклические ароматические углеводороды (бензопирены, метилхолантрены), содержащиеся в табачном дыме, некоторые вещества, применяемые в промышленности (например, полихлорированные бифенилы), или побочные продукты химического синтеза (диоксин) вызывают 84 Часть I. Общая фармакология индукцию изофермента CYP1A2. Индукция изофермента CYP2Е1 развивается при хроническом употреблении алкоголя (см. табл. 1.3). В некоторых случаях может увеличиваться скорость метаболизма самого индуктора (аутоиндукция), вследствие чего ослабляются его фармакологические эффекты. Аутоиндукция характерна для барбитуратов (в частности, фенобарбитала) и является причиной развития толерантности при их длительном приеме. Некоторые ЛВ снижают активность микросомальных ферментов, в результате чего повышается концентрация в крови веществ, метаболизируемых этими ферментами. Это может привести к развитию токсических эффектов. Например, циметидин, некоторые макролидные антибиотики, кетоконазол (и другие азолы), ципрофлоксацин, ингибируя изоферменты CYP3A4/3А5, замедляют микросомальное окисление варфарина, что может усилить его антикоагулянтный эффект и спровоцировать кровотечение. Ингибирование этого же изофермента противогрибковыми азолами может усилить проявления нефротоксического действия циклоспорина — иммунодепрессанта, применяемого при трансплантации органов. Токсическое действие теофиллина, метаболизирующегося при участии CYP1A2, резко усиливается при его одновременном назначении с антибактериальным препаратом ципрофлоксацином, ингибирующим этот изофермент. Фуранокумарины, содержащиеся в грейпфрутовом соке, ингибируют изофермент CYP3A4 преимущественно в стенке кишечника, в результате чего в системном кровотоке повышается концентрация многих ЛВ и возрастает риск их токсических эффектов (см. табл. 1.3). Напротив, применение пролекарства одновременно с ингибиторами ферментов, участвующих в образовании его активного метаболита, может привести к уменьшению концентрации активного вещества в крови и снижению терапевтического эффекта этого препарата. Примером такого взаимодействия является снижение эффекта клопидогрела, применяемого для предупреждения тромбозов при его назначении вместе с противоязвенным препаратом омепразолом, ингибитором CYP2C19 (клопидогрел превращается в активный метаболит под действием нескольких изоферментов цитохрома Р-450, в большей степени под действием CYP2C19). В отличие от индукторов метаболизма ингибиторы ферментов действуют быстрее (эффект ингибирования отмечен через 24 ч после приема соответствующего препарата). Следует добавить, что некоторые индукторы и ингибиторы микросомальных ферментов одновременно выполняют такую же функцию в отношении «выкачивающего» транспортера Р-гликопротеина (и некоторых других транспортеров), еще в большей степени изменяя (повышая Глава 1. Фармакокинетика 85 или снижая) эффект ЛВ, являющихся их общими субстратами. Трава зверобоя не только индуцирует изофермент CYP3A4, но и является индуктором Р-гликопротеина, а некоторые компоненты сока грейпфрута ингибируют и этот транспортер и изофермент. При одновременном применении ЛВ с индукторами или ингибиторами транспортеров и/или ферментов метаболизма этих ЛВ необходимо корректировать назначаемые дозы ЛВ. То же необходимо делать и при отмене индукторов или ингибиторов. Изменения активности ферментов метаболизма ЛВ могут определяться генетическими факторами. Такие изменения имеют в своей основе передающиеся из поколения в поколение мутации генов, кодирующих синтез данных ферментов. Данный феномен носит название генетического полиморфизма и имеет следствием значительные межиндивидуальные различия в метаболизме ЛВ. При этом у определенного процента больных, принимающих данное ЛВ, активность метаболизирующих ферментов может быть повышена, в связи с чем процесс биотрансформации ЛВ ускоряется, а его действие снижается. И наоборот, активность ферментов может быть снижена (недостаточность ферментов), вследствие чего биотрансформация ЛВ будет происходить медленнее, а его действие будет усиливаться вплоть до появления токсических эффектов. Межиндивидуальные различия в скорости метаболизма ЛВ позволяют выделить среди популяции группы, различающиеся по активности различных ферментов: ● группа с медленной скоростью метаболизма (медленные метаболизаторы); ● группа с быстрой скоростью метаболизма (быстрые метаболизаторы); ● группа с нормальной скоростью метаболизма. Часто отмечается генетический полиморфизм изоферментов цитохрома Р-450. Известны последствия генетического полиморфизма изофермента CYP2C19, участвующего в метаболической активации антитромботического препарата клопидогрела. При этом может наблюдаться снижение ферментативной активности CYP2C19 вплоть до ее полной потери, что сопровождается снижением терапевтического эффекта препарата, или (реже) — повышение активности фермента и увеличение риска кровотечений. Известны случаи генетического полиморфизма ферментов, не имеющих отношения к системе цитохрома Р-450. Например, при ацетилировании противотуберкулезного препарата изониазида у определенного процента больных в популяции выявляют недостаточность фермента N-ацетилтрансферазы («медленные ацетиляторы»), а у других больных 86 Часть I. Общая фармакология активность этого фермента повышена («быстрые ацетиляторы»). У «медленных ацетиляторов» концентрация изониазида в плазме крови в 4–6 раз выше, чем у «быстрых ацетиляторов», что может стать причиной токсического действия препарата. Примеры влияния генетической недостаточности некоторых ферментов на действие ЛВ приведены в табл. 1.4. Таблица 1.4. Реакции организма на лекарственные вещества при генетической недостаточности некоторых ферментов Фермент Особые реакции Глюкозо-6-фосфатдегидрогеназа эритроцитов Гемолиз эритроцитов вследствие образования хинона. Гемолитическая анемия N-ацетилПовышение трансфераза печени частоты побочных эффектов Псевдохолинэстераза Удлинение плазмы крови расслабляющего действия на скелетные мышцы (6–8 ч вместо 5–7 мин) Лекарственные вещества Хинин, хинидин, сульфаниламиды, хлорамфеникол Распространенность Изониазид, сульфаниламиды, прокаинамид Суксаметоний Европеоиды (до 50% населения) Европеоиды (0,04% населения), эскимосы (1% населения) Тропические и субтропические страны (до 100 млн человек) 1.7. ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ ИЗ ОРГАНИЗМА ЛВ и продукты их биотрансформации выводятся из организма в основном почками (почечная экскреция), а также через ЖКТ (с желчью в просвет кишечника). 1.7.1. Почечная экскреция Выведение ЛВ и их метаболитов почками происходит при участии трех основных процессов: ● клубочковой фильтрации; ● активной секреции в проксимальных канальцах; ● канальцевой реабсорбции. Глава 1. Фармакокинетика 87 Клубочковая фильтрация При фильтрации плазмы крови через фильтрационный барьер почек ЛВ под гидростатическим давлением поступают из капилляров почечных клубочков в просвет капсулы и далее в почечные канальцы. Фильтрационный барьер состоит из слоя эндотелиальных клеток (промежутки между которыми 70–100 нм), базальной мембраны и эпителиальных клеток внутреннего листка капсулы (подоцитов). Базальная мембрана и слой подоцитов препятствуют фильтрации отрицательно заряженных соединений и определяют размер веществ, способных пройти через фильтрационный барьер. По разным данным, они ограничивают фильтрацию веществ, диаметр молекул которых превышает 6 нм, и препятствуют фильтрации молекул более 8 нм в диаметре. Отрицательный заряд и большой размер альбуминов и других белков плазмы крови почти полностью исключают их фильтрацию в почечных клубочках. В связи с этим экскреция веществ, связывающихся с плазменными белками, происходит медленнее, поскольку в связанном состоянии они не проходят через фильтрационный барьер почечных клубочков. Активная канальцевая секреция Путем активной секреции в просвет канальцев выделяются многие вещества, экскретируемые почками. Вещества секретируются в проксимальных канальцах с помощью специальных транспортных систем против градиента концентрации, в результате чего ЛВ удаляется из плазмы крови практически полностью. Существуют отдельные транспортные системы для органических кислот (транспортеры органических анионов) — пенициллинов, салицилатов, сульфаниламидов, тиазидных диуретиков, фуросемида и др., и для органических оснований (транспортеры органических катионов) — морфина, хинина, допамина, серотонина, трициклических антидепрессантов, никотина и др. Транспортеры органических катионов и анионов обеспечивают перенос через мембраны как гидрофильных, так и гидрофобных (липофильных) веществ. Большинство из них относятся к семейству SLC-транспортеров и находятся как на апикальной (обращенной в просвет канальца), так и на базолатеральной мембранах эпителиальных клеток, причем, как правило, через апикальную и базолатеральную мембраны вещества переносят разные транспортеры. Наряду с SLC-транспортерами органических анионов в апикальной мембране функционируют АТФ-зависимые «выкачивающие транспортеры (белки множественной лекарственной резистентности), выводящие из эпителия в просвет канальцев конъюгаты ЛВ с глутатионом и глюкуроновой кислотой. 88 Часть I. Общая фармакология В отличие от клубочковой фильтрации канальцевая секреция обеспечивает эффективное выведение из организма веществ, связанных с белками плазмы крови. Это объясняется тем, что канальцевая секреция ЛВ не сопровождается одновременным выведением воды, поэтому концентрация свободного вещества в плазме крови снижается и происходит диссоциация связанных с белками молекул вещества. Пенициллин, связываемый на 80% с белками плазмы крови, очень медленно фильтруется в клубочках, но практически полностью выводится путем активной секреции в проксимальных канальцах. В процессе секреции как органические кислоты, так и органические основания могут конкурентно вытеснять друг друга из связи с транспортными белками, вследствие чего экскреция вытесняемого вещества снижается, а его концентрация в плазме крови дольше поддерживается на более высоком уровне. Этот вид лекарственного взаимодействия используется для пролонгирования действия пенициллина при введении пробенецида, выводящегося в просвет почечных канальцев тем же транспортным белком, что и пенициллин. Канальцевая реабсорбция Через мембраны почечных канальцев ЛВ реабсорбируются путем пассивной диффузии по градиенту концентрации. Таким путем реабсорбируются липофильные неполярные соединения, так как они легко проникают через мембраны эпителиальных клеток почечных канальцев. Гидрофильные полярные вещества (в том числе ионизированные соединения) в почках практически не реабсорбируются и выводятся из организма. Выведение почками слабых кислот и слабых оснований находится в прямой зависимости от степени их ионизации и, следовательно, от рН почечного фильтрата, который варьирует в пределах от 4,5 до 8,0. Кислая реакция мочи способствует экскреции слабых оснований (алкалоидов никотина, атропина, хинина) и затрудняет выведение слабых кислот (барбитуратов, ацетилсалициловой кислоты). Для ускорения выведения почками слабых оснований следует изменить реакцию мочи в кислую сторону (снизить рН мочи). Обычно в таких случаях вводят аммония хлорид. И, наоборот, с целью увеличения экскреции слабых кислот назначают натрия гидрокарбонат и другие соединения, сдвигающие реакцию мочи в щелочную сторону (повышая рН мочи). Внутривенное введение, в частности, натрия гидрокарбоната используют для ускорения выведения барбитуратов или ацетилсалициловой кислоты в случае их передозировки. Глава 1. Фармакокинетика 89 Реабсорбция некоторых эндогенных веществ (аминокислот, глюкозы, мочевой кислоты) осуществляется путем активного транспорта в дистальных канальцах. 1.7.2. Выведение через желудочно-кишечный тракт Многие ЛВ (дигоксин, тетрациклины, ампициллин, рифампицин, морфин и др.) выделяются с желчью в просвет кишечника в неизмененном виде или в виде метаболитов и конъюгатов. ЛВ проникают в гепатоциты через эндотелий печеночного синуса путем пассивной диффузии (липофильные вущества) или с помощью транспортных белков. ЛВ (полярные и некоторые липофильные вещества), а также эндогенные вещества (лейкотриены, билирубин) захватываются гепатоцитами путем облегченной диффузии или вторичного активного транспорта при участии специальных транспортных систем для органических анионов и катионов, локализованных в синусоидальной (базолатеральной) мембране гепатоцитов. Эти транспортеры могут работать в двух направлениях: не только транспортировать вещества в гепатоциты, но и выводить ЛВ и их метаболиты из гепатоцитов в кровь. Выделение веществ и их метаболитов в желчь происходит с помощью активного транспорта при участии Р-гликопротеина и других АТФзависимых транспортных белков, находящихся в каналикулярной (канальцевой) мембране гепатоцитов (рис. 1.9). Некоторые АТФ-зависимые транспортеры также локализованы в базолатеральной мембране гепатоцитов и опосредуют активный транспорт ЛВ и их метаболитов в кровеносную систему. Таким образом, транспортная система гепатоцитов обеспечивает скоординированный перенос веществ из крови в гепатобилиарную систему и выведение некоторых веществ и их метаболитов в системный кровоток для последующей экскреции почками. Транспорт ЛВ в гепатоциты имеет большое значение для пресистемной элиминации ЛВ, особенно если эти ЛВ интенсивно метаболизируются под действием микросомальных ферментов печени. Захват гепатоцитами некоторых ЛВ, таких как статины (симвастатин, ловастатин, правастатин), при их первом прохождении через печень с помощью транспортера ОАТР1В1 критичен для терапевтического действия этих ЛВ и их системных побочных эффектов (миопатия). В связи с тем, что статины ингибируют синтез холестерина в печени, их терапевтический эффект непосредственно зависит от активности транспортера. Ингибиторы ОАТР1В1 (гемфиброзил, циклоспорин) могут снижать гиполипидемический эффект статинов, препятствуя их проникновению 90 Часть I. Общая фармакология Кровь 1 Транспортеры органических анионов и катионов 4 7 2 гепатоцит 3 6 5 Рис. 1.9. Гепатобилиарный транспорт лекарственных веществ (из Goodman and Gilman’s The pharmacological basis of Therapeutics. — 11th ed., с изм.): 1 — молекулы лекарственных веществ переносятся в гепатоциты транспортерами органических анионов и катионов (SLS-транспортерами); 2 — АТФ-зависимые АВС-транспортеры (Р-гликопротеин и др.) переносят ЛВ и их метаболиты через канальцевую мембрану; 3 — желчный проточек; 4 — АТФ-зависимые транспортеры, «выкачивающие» ЛВ и их метаболиты из гепатоцитов в кровь; 5 — синусоидальная мембрана гепатоцита; 6 — канальцевая мембрана; 7 — лекарственные вещества и их метаболиты в гепатоциты, при этом повышается опасность побочных эффектов этих препаратов в связи с увеличением их концентрации в системном кровотоке. Возможные генетически детерминированные изменения активности этого транспортера (генетический полиморфизм) могут быть ответственны за межиндивидуальные различия в действии статинов. Большинство ЛВ после выделения с желчью в просвет кишечника может неоднократно повторно всасываться в портальный кровоток и вновь выделяться с желчью в просвет кишечника. Этот циклический процесс называется энтерогепатической (кишечно-печеночной) циркуляцией. В конечном итоге часть ЛВ выводится из кишечника. Некоторые ЛВ (морфин, хлорамфеникол, этинилэстрадиол) выделяются с желчью в виде конъюгатов с глюкуроновой кислотой (глюкуронидов), гидролизующихся в кишечнике с образованием активных веществ, которые снова реабсорбируются и поступают в печень. Таким образом, энтерогепатическая циркуляция способствует пролонгированию действия ЛВ. Глава 1. Фармакокинетика 91 Некоторые ЛВ плохо всасываются в ЖКТ и полностью выводятся из организма через кишечник. Их в основном применяют для лечения или профилактики кишечных инфекций и дисбактериоза (например, неомицин, нистатин, ванкомицин). 1.7.3. Другие пути выведения Газообразные и летучие вещества выделяются легкими. Таким путем из организма выводятся средства для ингаляционного наркоза. Некоторая часть этанола также выводится легкими, при этом содержание этанола в выдыхаемом воздухе коррелирует с его концентрацией в плазме крови. Некоторые вещества могут выделяться потовыми, бронхиальными и слюнными железами, железами желудка (хинин, морфин) и кишечника (слабые органические кислоты), слезными железами (рифампицин). При выделении слюнными железами и железами желудка и кишечника ЛВ могут всасываться вновь. Выведение лекарственных веществ слюнными железами ЛВ проникают в ротовую полость из слюнных желез путем пассивной диффузии через мембраны эпителия, а также с потоком жидкости через десневую щель. В связи с тем, что пассивная диффузия является основным механизмом секреции ЛВ, со слюной в основном секретируются липофильные вещества. Известно, что концентрация некоторых ЛВ в слюне коррелирует с их концентрацией в плазме крови, что может быть использовано для фармакокинетических исследований. Выведение слабых кислот и слабых оснований, а следовательно, их концентрация в слюне зависят от рН слюны. Вследствие непостоянства этого показателя данный тест применительно к таким веществам может быть использован ограниченно. Концентрация некоторых ЛВ (парацетамол, лидокаин, соли лития, прокаинамид) в слюне превышает их концентрацию в плазме крови (например, соотношение этих концентраций для прокаинамида составляет 3,5, для парацетамола — 1,4, а для солей лития — 2,85). Выведение лекарственных веществ молочными железами Многие вещества (ацетилсалициловая кислота, барбитураты, антитиреоидные средства, тетрациклины, эритромицин, сульфаниламиды, соли лития, кофеин, этанол, никотин) выделяются молочными железами в период лактации, при этом интенсивность выделения зависит от липофильности веществ и степени их ионизации. Поскольку грудное молоко имеет более кислую реакцию (рН=6,5), чем плазма крови (рН=7,4), кон- 92 Часть I. Общая фармакология центрация слабых оснований в нем достигает более высоких значений (по сравнению с концентрацией слабых кислот), чем в плазме крови. В связи с этим такие вещества, как никотин, морфин, снотворные средства из группы бензодиазепинов и другие слабоосновные соединения могут накапливаться в грудном молоке и во время кормления, выделяясь молочными железами, попасть в организм ребенка. Этанол проникает в грудное молоко и достигает там таких же концентраций, как в плазме крови. Вещества, связывающиеся с жирами, концентрируются в молоке в значительной степени. Концентрация теофиллина в грудном молоке составляет 70%, а хлорамфеникола — 50%. Вещества, интенсивно связывающиеся с белками плазмы крови, секретируются молочными железами медленнее. Однако даже небольшое количество чужеродного вещества в грудном молоке может вызвать у младенца аллергические реакции. Продолжение грудного вскармливания противопоказано при необходимости приема изониазида, хлорамфеникола, солей лития, противоопухолевых и некоторых противовирусных ЛС. Для характеристики совокупности процессов, в результате которых активное ЛВ удаляется из организма, введено понятие элиминация, объединяющее два процесса — биотрансформацию и выведение. Количественно процесс элиминации характеризуется рядом фармакокинетических параметров (см. раздел «Математическое моделирование фармакокинетических процессов»). 1.8. МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ФАРМАКОКИНЕТИЧЕСКИХ ПРОЦЕССОВ Величина и продолжительность фармакологического эффекта во многом зависят от концентрации ЛВ в органах или тканях, где оно оказывает свое действие. Поэтому очень важно поддерживать определенную (терапевтическую) концентрацию ЛВ в месте его действия. Однако в большинстве случаев концентрацию вещества в тканях определить практически невозможно, поэтому при фармакокинетических исследованиях определяют концентрацию ЛВ в плазме крови, которая для большинства веществ коррелирует с их концентрацией в органах-мишенях. В результате всасывания, распределения, депонирования и элиминации (биотрансформации и выведения) ЛВ его концентрация в плазме крови изменяется. Эти изменения могут быть отражены графически. Концентрацию ЛВ измеряют в плазме крови сразу и через определенные промежутки времени после его введения и на основании полученных дан- Глава 1. Фармакокинетика 93 ных строят кривую изменения концентрации ЛВ во времени — фармакокинетическую кривую (рис. 1.10). Для количественной оценки влияния процессов всасывания, распределения, депонирования и элиминации на концентрацию ЛВ в крови используют математические фармакокинетические модели. Различают однокамерные, двухкамерные и многокамерные фармакокинетические модели. В однокамерной модели организм условно представляют в виде камеры, заполненной жидкостью. Вещество может поступать в камеру постепенно, как при приеме внутрь или других внесосудистых путях введения, или мгновенно, как при быстром внутривенном введении (рис. 1.11). После поступления вещества в камеру в количестве D оно, распределяясь мгновенно и равномерно, занимает объем камеры, при этом концентрация вещества в камере обозначается как начальная концентра- Рис. 1.10. Изменение концентрации лекарственного вещества во времени при внутривенном и внесосудистом введениях Рис. 1.11. Однокамерная фармакокинетическая модель: Vd (volume of distribution) — объем распределения вещества в камере; D — количество вещества, введенное в камеру; С0 — начальная концентрация вещества в камере 94 Часть I. Общая фармакология ция — С0. Объем распределения вещества в камере Vd (volume of distribution) равен D/С0. В клинической практике применяют параметр, получивший название «кажущийся объем распределения» (apparent volume of distribution, Vd). Кажущийся объем распределения — гипотетический объем жидкости, в котором ЛВ при его равномерном распределении находится в концентрации, равной его концентрации в плазме крови (Сp). Соответственно, кажущийся объем распределения можно выразить уравнением: Vd = Q / Сp, где Q — количество вещества в организме при его концентрации в плазме крови Сp. Если допустить, что вещество после внутривенного введения в дозе D мгновенно и равномерно распределилось в организме, кажущийся объем распределения можно определить как: Vd = D / С0, где С0 — начальная концентрация вещества в плазме крови. Кажущийся объем распределения позволяет судить о том, в каком соотношении вещество распределяется между жидкостями организма (плазмой крови, интерстициальной, внутриклеточной жидкостями). Так, если величина Vd какого-либо вещества составляет приблизительно 3 л (средний объем плазмы крови), это означает, что данное вещество преимущественно находится в плазме крови. Такой объем распределения характерен для крупномолекулярных соединений, практически не проникающих в клетки крови и через эндотелий сосудов (т.е. веществ, не выходящих за пределы сосудистого русла), например для гепарина, Vd которого приблизительно равен 3,6 л. Если Vd равен 15 л (сумма средних объемов плазмы крови и интерстициальной жидкости), вещество преимущественно содержится в плазме крови и интерстициальной жидкости, т.е. не проникает внутрь клеток. Предположительно — это гидрофильное соединение, не проникащее через клеточные мембраны. К таким ЛВ относятся аминогликозидные антибиотики (гентамицин, тобрамицин). Поэтому эти антибиотики, имея широкий спектр действия, практически не действуют на микроорганизмы, находящиеся внутри клеток, т.е. неэффективны в отношении внутриклеточных инфекций. Неэффективны они также при бактериальном менингите, так как не проникают через ГЭБ. Некоторые ЛВ имеют объем распределения порядка 40–46 л (средний объем всех жидкостей организма). Это означает, что эти ЛВ присутству- Глава 1. Фармакокинетика 95 ,% ют как во внеклеточной, так и во внутриклеточной и трансцеллюлярной жидкостях организма, т.е. проникают через мембраны клеток и гистогематические барьеры. В основном так распределяются в организме липофильные неполярные соединения. Если величина Vd вещества значительно превышает объем жидкостей организма, наиболее вероятно, что это вещество депонировалось в периферических тканях и его концентрация в плазме крови чрезвычайно мала. Большие значения объема распределения характерны для дигоксина (500 л), трициклических антидепрессантов имипрамина и амитриптилина (1600 л), хлорохина (13 000 л). Подобные ЛВ не могут быть эффективно удалены из организма с помощью гемодиализа. Помимо свойств вещества объем распределения зависит от пола, возраста, общего объема жировой ткани и воды в организме. После мгновенного и равномерного распределения в объеме камеры и достижения концентрации С0 вещество удаляется из камеры при участии двух процессов — биотрансформации и экскреции, которые объединяются термином элиминация (см. рис. 1.11). При этом концентрация вещества в камере постепенно снижается с переменной (кинетика 1-го порядка) или постоянной (кинетика нулевого порядка) скоростью. При кинетике 1-го порядка скорость элиминации вещества зависит от его концентрации в крови (чем ниже концентрация вещества, тем меньше скорость элиминации). Постоянной остается часть дозы вещества, которая элиминируется в единицу времени (константа скорости элиминации). В этом случае кривая изменения концентрации вещества во времени имеет экспоненциальный характер (рис. 1.12). Большинство ЛВ элиминируется в соответствии с кинетикой 1-го порядка. Основные параметры, характеризующие процесс элиминации, — конt1/2 станта скорости элиминации (ke1, ke) и период полуэлиминации (t½). Константа скорости элиминации ,t 1-го порядка показывает, какая часть вещества элиминируется из организма в единицу времени (размер- Рис. 1.12. Элиминация вещества, соответствующая кинетике первого ность — мин–1, ч–1). Например, если порядка: k (k ) — константа скороel e kel вещества, введенного внутривен- сти элиминации 1-го порядка; t½ — но в дозе 100 мг, составляет 0,1 ч–1, период полуэлиминации 96 Часть I. Общая фармакология Концентрация ЛВ, % через 1 ч его количество в крови будет составлять 90 мг, через 2 ч — 81 мг и т.д. Элиминация некоторых ЛВ (например, этанола, фенитоина) соответствует кинетике нулевого порядка. Скорость такой элиминации не зависит от концентрации вещества в плазме крови и является постоянной величиной, т.е. в единицу времени элиминируется определенное количество вещества (например, за 1 ч элиминируется 10 г чистого этанола). Это связано с тем, что при терапевтических концентрациях названных веществ в крови происходит насыщение метаболизирующих ферментов или транспортеров, участвующих в их элиминации. Поэтому увеличение концентрации таких веществ в крови не приводит к повышению скорости элиминации. График изменения концентрации вещества во времени (рис. 1.13), характеризующий кинетику нулевого порядка, представляет собой прямую линию, тангенс угла наклона которой к оси абсцисс соответствует константе скорости элиминации нулевого порядка (k0). tg() = –k0 Время, ч Рис. 1.13. Элиминация вещества, соответствующая кинетике нулевого порядка: k0 — константа скорости элиминации 0-го порядка Период полуэлиминации (t½) — время, за которое концентрация вещества в плазме крови снижается на 50% (рис. 1.14). Для большинства ЛВ (элиминация которых подчиняется кинетике 1-го порядка) t½ — величина постоянная в определенных пределах и не зависящая от дозы. Если за один период полуэлиминации из плазмы крови удаляется 50% внутривенно введенного ЛВ, то за 2 периода — 75%, а за 3,3 периода — 90%. Этот параметр используют для подбора интервалов между введениями ЛВ, необходимых для поддержания его постоянной концентрации в крови. Период полуэлиминации связан с константой скорости элиминации следующим соотношением: Глава 1. Фармакокинетика 97 t1/2 = ln 2/ kel = 0,693/ kel. ,% Если сразу после внутривенного введения ЛВ производить измерения его концентрации в плазме крови через короткие интервалы времени, можно получить двухфазный характер изменения концентрации вещества в крови (рис. 1.15). Такой же характер кривой можно получить с помощью двухкамерной фармакокинетической модели (рис. 1.16). В этой модели организм представлен в виде двух сообщающихся между собой камер. Одна из камер этой модели называется центральной (представляет плазму крови и хорошо кровоснабжаемые органы — сердце, печень, почки, легкие), а другая — периферической (представляет плохо кровоснабжаемые ткани — кожу, жировую, мышечную ткани). Вещество вводят в центральную камеру, где оно мгновенно и равномерно распределяется и затем проникает в периферическую камеру. Этот период обозначается как α-фаза, или фаза распределения. Затем вещество перераспределяется из периферической камеры в центральную и удаляется из нее вследствие элиминации. Эта фаза (фаза элиминации) обозначается как β-фаза. α-Фаза характеризуется параметром, который называется периодом полураспределения — t½α а характеристикой β-фазы служит собственно период полуэлиминации, обозначаемый как t½β (см. рис. 1.15). Период полураспределения, как правило, меньше периода полуэлиминации, так как вещество распределяется из центральной камеры в периферическую быстрее, чем элиминируется. Клиренс — фармакокинетический параметр, характеризующий скорость освобождения организма от ЛВ. ,ч Рис. 1.14. Графическое определение периода полуэлиминации: t½ — период полуэлиминации Рис. 1.15. Характер элиминации вещества в двухкамерной модели 98 Часть I. Общая фармакология k2–1 k1–2 Kel Рис. 1.16. Двухкамерная фармакокинетическая модель Поскольку освобождение организма от ЛВ происходит за счет процессов биотрансформации (метаболизма) и экскреции, различают метаболический и экскреторный клиренсы. Метаболический клиренс (Clmet) и экскреторный клиренс (Cexcr) в сумме составляют системный (общий) клиренс (Clt, total clearance): Clmet + Cexcr = Clt. Системный клиренс численно равен объему распределения, освобождаемому от вещества в единицу времени (размерность — объем в единицу времени, например мл/мин, л/ч, иногда с учетом массы тела, например мл/кг/мин): Clt = Vd kel. Значения клиренса прямо пропорциональны скорости элиминации вещества и обратно пропорциональны его концентрации в биологической жидкости (в крови, плазме крови и др.): Clt = Скорость элиминации вещества , C где С — концентрация вещества. В зависимости от путей элиминации ЛВ различают почечный клиренс (Clren), печеночный клиренс (Clhep), а также клиренс, осуществляемый другими органами (легкими, слюнными, потовыми и молочными железами, внепеченочный метаболизм). Наиболее важные составляющие системного клиренса — почечный и печеночный клиренсы. Почечный клиренс численно равен объему плазмы крови, освобождаемому от ЛВ в единицу времени, и зависит от интенсивности процессов Глава 1. Фармакокинетика 99 клубочковой фильтрации, канальцевой секреции и реабсорбции. Почечный клиренс можно определить при постоянной концентрации вещества в плазме крови: Clren = Cu Vu / Cp, где Cu — концентрация вещества в моче, Cp — концентрация вещества в плазме крови, Vu — скорость мочеотделения. Печеночный клиренс зависит от процессов биотрансформации ЛВ и экскреции неизмененного ЛВ с желчью. Значения как почечного, так и печеночного клиренса также находятся в зависимости от объема притекающей крови и скорости кровотока в органе. Поэтому при застойной сердечной недостаточности вследствие снижения печеночного кровотока уменьшается клиренс интенсивно метаболизируемых веществ (например, лидокаина). Значения почечного и печеночного клиренса следует учитывать при назначении ЛВ больным с недостаточностью почек или печени соответственно. 1.9. ОПТИМИЗАЦИЯ ДОЗИРОВАНИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Для достижения оптимального терапевтического эффекта ЛВ необходимо постоянно поддерживать его терапевтическую концентрацию в крови. Постоянно поддерживаемый уровень вещества в плазме крови обозначается как стационарная концентрация (СSS, steady-state concentration). Стационарная концентрация устанавливается при достижении равновесия между процессом поступления вещества в системный кровоток и процессом его элиминации (когда скорость поступления равна скорости элиминации). Наиболее простой способ достижения стационарной концентрации ЛВ — внутривенное капельное введение (рис. 1.17). При внутривенном капельном введении ЛВ величина СSS зависит от скорости введения, которую можно определить по формуле: D/T = Clt Сss, где D — доза, Clt — общий клиренс, T — время введения, СSS — стационарная концентрация. ЛВ необходимо вводить со скоростью, поддерживающей его терапевтическую концентрацию в крови. Существует диапазон терапевтических концентраций (рис. 1.18). Нижняя граница этого диапазона — минимальная эффективная концентрация (СSSmin), ниже которой вещество не оказывает необходимого действия, верхняя граница — максимальная безопасная кон- 100 Часть I. Общая фармакология Рис. 1.17. Достижение стационарной концентрации вещества при внутривенном капельном введении: CSS — стационарная концентрация вещества в крови; Clt — системный клиренс; D/T — скорость введения вещества Концентрация ЛВ в крови Диапазон терапевтических концентраций лекарственного вещества в крови max Css Css min Css Время, ч Рис. 1.18. Диапазон стационарных терапевтических концентраций вещества в крови: СSSmin — минимальная эффективная концентрация; СSSmax — максимальная безопасная концентрация; CSS — средняя терапевтическая концентрация центрация (СSSmax), выше которой находится область токсических концентраций. Обычно поддерживают среднюю концентрацию этого диапазона, т.е. среднюю терапевтическую концентрацию вещества в крови. Значения средних терапевтических концентраций ЛВ приведены в справочной литературе. Время достижения стационарной терапевтической концентрации вещества в крови зависит от его периода полуэлиминации. Через 1 t½ достигается 50%, через 2 t½ — 75% и через 3,3 t½ — 90% от стационарного уровня вещества в крови (как правило, СSS достигается через 4–5 t½). При необходимости получения быстрого терапевтического эффекта, особенно если вещество имеет достаточно большой t½, сначала его вводят большой нагрузочной дозе (для достижения стационарной терапевти- Глава 1. Фармакокинетика 101 ческой концентрации), а затем — инфузионно с определенной скоростью (для поддержания стационарной концентрации). Нагрузочную дозу можно вычислить по формуле: Dн = Vd Css. Однако чаще вещества назначают отдельными дозами через определенные интервалы времени (наиболее часто внутрь). В таких случаях концентрация вещества в крови не остается постоянной, а меняется относительно стационарного уровня, причем эти колебания не должны выходить за пределы диапазона терапевтических концентраций. Поэтому после назначения нагрузочной дозы, обеспечивающей быстрое достижение стационарной терапевтической концентрации, вводят меньшие по величине поддерживающие дозы; при этом допускаются лишь небольшие колебания концентрации вещества в крови относительно его стационарного терапевтического уровня (рис. 1.19). Поддерживающую дозу ЛВ для каждого конкретного больного можно рассчитать по формуле, в которой используются значения клиренса: Dпод = Clt Css T, , где Т — интервал между введениями. При введении ЛВ внутрь учитывают также его биодоступность (часть введенной дозы вещества, которая в неизмененном виде достигла систем- Рис. 1.19. Изменение концентрации лекарственного вещества в плазме крови при многократном введении (через равные промежутки времени вводят поддерживающие дозы вещества): τ — интервал времени между введением поддерживающих доз 102 Часть I. Общая фармакология ного кровотока). В таких случаях поддерживающую дозу необходимо рассчитывать с учетом этого параметра: Dпод = Clt Css T / F = Css Vd Т / 1,44 F t½, где CSS — стационарная концентрация; Т — интервал между введениями, F — биодоступность; 1,44 = 1/ln 2. Биодоступность вещества при введении внутрь зависит от многих факторов и определяется следующим образом. Вещество вводят больному внутривенно и измеряют его концентрации в крови через определенные промежутки времени. На основании полученных данных вычерчивают кривую изменения концентрации вещества во времени при внутривенном введении. Затем тому же больному это вещество вводят внутрь в той же дозе и определяют его концентрации в крови через определенные интервалы времени. По результатам измерения строят кривую изменения концентрации вещества во времени при введении внутрь (рис. 1.20). Затем измеряют площади под кривыми «концентрация–время» (AUC, Area Under the Curve). Биодоступность вещества определяют по формуле: AUCвнутрь 100% , AUCв/в где F — биодоступность (Fraction); AUC — площадь под кривой концентрация–время; в/в — внутривенное введение. ,% F= внутривенное введение в/в Время, ч Рис. 1.20. Определение биодоступности лекарственного вещества при введении внутрь. AUC (Area Under the Curve) — площадь под кривой концентрация–время Глава 1. Фармакокинетика 103 Вопросы и задания для самоконтроля 1. Основной способ всасывания гидрофильных ЛВ при внутримышечном и подкожном введениях: а) пассивная диффузия; б) активный транспорт; в) фильтрация; г) облегченная диффузия; д) пиноцитоз. 2. Что препятствует всасыванию и распределению липофильных ЛВ в организме: а) цитоплазматические мембраны клеток; б) плотные межклеточные контакты; в) АТФ-зависимые транспортеры? 3. ЛВ, связанные с белками плазмы крови: а) не проникают через эндотелий сосудов; б) фильтруются в почечных клубочках; в) быстро выводятся из организма; г) не вызывают фармакологических эффектов. 4. Каковы основные изменения свойств ЛВ под действием микросомальных ферментов печени: а) повышение полярности; б) повышение липофильности; в) повышение гидрофильности; г) снижение полярности? 5. Что повышает почечную экскрецию слабокислых веществ: а) повышение рН почечного фильтрата; б) снижение рН почечного фильтрата; в) введение натрия бикарбоната; г) введение хлорида аммония? Глава 2 ФАРМАКОДИНАМИКА Фармакодинамика включает понятия о фармакологических эффектах, локализации действия и механизмах действия ЛВ (т.е. представления о том, как, где и каким образом ЛВ действуют в организме). К фармакодинамике относится также понятие о видах действия ЛВ. 2.1. ФАРМАКОЛОГИЧЕСКИЕ ЭФФЕКТЫ, ЛОКАЛИЗАЦИЯ И МЕХАНИЗМЫ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Фармакологические эффекты — изменения функции органов и систем организма, вызываемые ЛВ. К фармакологическим эффектам ЛВ относятся, например, повышение частоты сердечных сокращений, снижение артериального давления, повышение порога болевой чувствительности, снижение температуры тела, увеличение продолжительности сна, устранение бреда и галлюцинаций и т.п. Каждое ЛВ, как правило, вызывает ряд определенных, характерных для него фармакологических эффектов. При этом одни фармакологические эффекты ЛВ полезные — благодаря им ЛВ применяют в медицинской практике (основные, или терапевтические, эффекты), а другие не используются и, более того, нежелательны (побочные эффекты). Для многих ЛВ известны места их преимущественного действия в организме, т.е. локализация действия. Одни ЛВ преимущественно действуют на определенные структуры головного мозга (противопаркинсонические, антипсихотические средства), другие в основном действуют на сердце (сердечные гликозиды). Благодаря современным методическим приемам можно определить локализацию действия ЛВ не только на системном и органном, но и на клеточном и молекулярном уровнях. Например, сердечные гликозиды действуют на сердце (органный уровень), на кардиомиоциты (клеточный уровень), на Na+-,K+-АТФазу мембран кардиомиоцитов (молекулярный уровень). Глава 2. Фармакодинамика 105 Одни и те же фармакологические эффекты могут быть вызваны различными способами. Так, ЛВ могут вызывать снижение артериального давления, уменьшая синтез ангиотензина II (ингибиторы ангиотензинпревращающего фермента) или блокируя поступление Са2+ в гладкомышечные клетки (блокаторы потенциал-зависимых кальциевых каналов), либо уменьшая выделение норадреналина из окончаний симпатических нервов (симпатолитики). Способы, с помощью которых ЛВ вызывают фармакологические эффекты, определяются как механизмы действия. Большинство ЛВ вызывает фармакологические эффекты, действуя на определенные молекулярные субстраты — так называемые мишени. К основным молекулярным «мишеням» для ЛВ относят: ● рецепторы; ● ионные каналы; ● ферменты; ● транспортные системы. 2.1.1. Рецепторы Свойства и виды рецепторов. Взаимодействие рецепторов с ферментами и ионными каналами Рецепторы представляют собой функционально активные макромолекулы или их фрагменты (в основном это белковые молекулы — липопротеины, гликопротеины, нуклеопротеины и др.), обеспечивающие ответ клетки на воздействие медиаторов или иных веществ. При воздействии веществ (лигандов) на рецепторы происходит изменение конформации белковой молекулы рецептора, что приводит к возникновению в клетке цепи биохимических реакций и дальнейшему развитию определенных эффектов на тканевом, органном и системном уровнях (фармакологических эффектов, если лигандом является ЛВ). Рецепторы служат мишенями как для эндогенных лигандов (нейромедиаторов, гормонов, цитокинов и других эндогенных биологически активных веществ), так и для экзогенных биологически активных веществ (в том числе для ЛВ). И в том, и другом случаях рецепторы взаимодействуют только с веществами, имеющими определенную химическую структуру и пространственную ориентацию, т.е. обладают избирательностью, поэтому их называют специфическими рецепторами. Большое значение для специфического взаимодействия вещества и рецептора имеют присутствие в структуре вещества определенных функциональных групп, расстояние между ними, размер и пространственная 106 Часть I. Общая фармакология ориентация молекулы вещества. Влияние ориентации молекулы вещества в пространстве по отношению к рецептору определяет стереоспецифичность взаимодействия вещества и рецептора. Из двух стереоизомеров, которые имеют противоположную пространственную ориентацию (т.е. являются зеркальными отображениями друг друга), только L-изомер ориентирован в пространстве таким образом, что может эффективно взаимодействовать с рецептором и, следовательно, обладает активностью. Алкалоид гиосциамин, являющийся активным L-изомером, в процессе выделения из растения частично превращается в неактивный D-изомер. Поэтому полученная при экстракции смесь двух изомеров (атропин) в 2 раза менее активна, чем гиосциамин. Рецепторы могут находиться в мембране клеток (мембранные рецепторы) или внутри клеток — в цитоплазме или ядре (внутриклеточные рецепторы) (рис. 2.1). Мембранные рецепторы Эти рецепторы состоят из липофильного участка (домена), пронизывающего мембрану, и гидрофильных доменов, находящихся снаружи и внутри клеток (внеклеточный и внутриклеточный домены). На внеклеточном домене имеются места связывания для лигандов (веществ, взаимодействующих с рецепторами). Внутриклеточные домены взаимодействуют 1 2 3 4 Рис. 2.1. Виды рецепторов: 1 — внутриклеточные рецепторы (регулируют транскрипцию генов); 2 — рецепторы, непосредственно сопряженные с ферментами; 3 — рецепторы, непосредственно сопряженные с ионными каналами; 4 — рецепторы, взаимодействующие с G-белками; Р — рецепторы; — лиганды рецепторов; ТК — тирозинкиназа; G — G-белок; ЭБ — эффекторный белок (фермент или ионный канал); S — субстрат; S-P — фосфорилированный субстрат Глава 2. Фармакодинамика 107 с эффекторными белками (ферментами или ионными каналами) либо сами обладают ферментативной активностью (см. рис. 2.1). При связывании определенных лигандов с внеклеточным доменом изменяется конформация внутриклеточного домена, что оказывает влияние на его взаимодействие с эффекторными белками. Это может вызвать как повышение, так и понижение ферментативной активности эффекторного белка и/или открытие ионного канала, что сопровождается возникновением внутриклеточных реакций, в дальнейшем реализуемых в виде эффектов на тканевом, органном и системном уровнях. Известны 3 вида мембранных рецепторов, различающихся по механизму передачи сигнала от вне- и внутриклеточного домена к эффекторным белкам. 1. Рецепторы, непосредственно сопряженные с ферментами. Поскольку внутриклеточный домен этих рецепторов проявляет ферментативную активность, их называют также рецепторами-ферментами, или каталитическими рецепторами. Большинство рецепторов этой группы обладает тирозинкиназной активностью. При связывании рецептора с веществом происходит активация тирозинкиназы, которая фосфорилирует внутриклеточные белки и таким образом изменяет их активность. К этим рецепторам относят рецепторы для инсулина, некоторых факторов роста и цитокинов. Наиболее хорошо изученный рецептор такого типа — рецептор для инсулина. Этот рецептор состоит из двух внеклеточных α-субъединиц, каждая из которых ковалентно связана с β-субъединицей, пронизывающей мембрану и обладающей тирозинкиназной активностью (см. рис. 2.1). Связывание инсулина с α-субъединицами вызывает изменение конформации соседних β-субъединиц, что приводит к их более тесному сближению и способствует трансфосфорилированию β-субъединиц по остаткам тирозина в реакции, когда одна субъединица фосфорилирует другую («аутофосфорилирование»). Фосфорилированные остатки тирозина затем вовлекают в этот процесс другие цитозольные белки, известные как субстраты инсулинового рецептора. К этому виду рецепторов относят также рецепторы, непосредственно связанные с гуанилатциклазой. Связывание с лигандом (натрийуретическими пептидами) стимулирует внутреннюю гуанилатциклазную активность рецептора, в результате чего происходит превращение гуанозинтрифосфата (ГТФ) в циклический гуанозинмонофосфат (цГМФ). Это самое небольшое семейство трансмембранных рецепторов. Предсердный натрийуретический пептид В-типа, секретируемый желудочками в ответ на чрезмерное увеличение объема циркулирующей крови, действует 108 Часть I. Общая фармакология подобным образом. Рекомбинантный препарат этого пептида — незиритид — одобрен для лечения декомпенсированной сердечной недостаточности. 2. Рецепторы, непосредственно сопряженные с ионными каналами, состоят из нескольких субъединиц, пронизывающих клеточную мембрану и формирующих ионный канал. При связывании вещества с внеклеточным доменом рецептора ионные каналы (так называемые рецептор-управляемые ионные каналы) открываются, в результате изменяется проницаемость клеточных мембран для различных ионов. К таким рецепторам относят никотин-чувствительные холинорецепторы (Н-холинорецепторы), рецепторы к гамма-аминомасляной кислоте (ГАМКА-рецепторы), глициновые рецепторы, глутаматные рецепторы. ● Н-холинорецептор состоит из 5 субъединиц, пронизывающих цитоплазматическую мембрану и окружающих центральный ионный (натриевый) канал. Две из них обозначены как α-субъединицы, каждая из которых имеет свой внеклеточный участок связывания для ацетилхолина (рис. 2.2). Когда рецептор не связан с лигандом, боковая аминокислотная цепь закрывает канал и таким образом препятствует вхождению ионов. Связывание двух молекул ацетилхолина с α-субъединицами рецептора вызывает конформационные изменения рецептора, что приводит к открытию ионного канала, проникновению ионов натрия в клетку и, как следствие, к деполяризации клеточной мембраны. Это вызывает сокращение и повыNa+ А АЦХ В АЦХ Na+ С Рис. 2.2. Рецептор, непосредственно сопряженный с ионным (натриевым) каналом (Н-холинорецептор): А — рецептор состоит из пяти субъединиц, которые пронизывают мембрану; В — в отсутствии лиганда натриевый канал рецептора закрыт; С — при взаимодействии рецептора с двумя молекулами ацетилхолина натриевый канал рецептора находится в открытом состоянии Глава 2. Фармакодинамика 109 шение тонуса скелетных мышц. Н-холинорецепторы опосредуют действие ацетилхолина в нервно-мышечных синапсах при применении ингибиторов ацетилхолинэстеразы, повышающих концентрацию ацетилхолина. ● ГАМКА-рецепторы непосредственно сопряжены с хлорными каналами. При взаимодействии этих рецепторов с ГАМК хлорные каналы открываются, и ионы хлора поступают в клетку, вызывая гиперполяризацию клеточной мембраны (это приводит к усилению тормозных процессов в ЦНС). Таким же образом функционируют глициновые рецепторы. К ЛВ, действие которых связано с изменением проницаемости хлорных каналов, относятся бензодиазепины, угнетающие передачу нервных импульсов в ЦНС. Бензодиазепины потенцируют способность ГАМК повышать проникновение ионов хлора через мембраны нейронов. Рецепторы, непосредственно сопряженные с ионными каналами, опосредуют действие ЛВ очень быстро (в течение нескольких миллисекунд). 3. Рецепторы, взаимодействующие с G-белками, пронизывают цитоплазматическую мембрану семь раз и заканчиваются внутриклеточным доменом, который активирует уникальный класс сигнальных молекул, называемых G-белками (рис. 2.3). Эти белки имеют такое название, потому что они связывают гуаниновые нуклеотиды, гуанозинтрифосфат (ГТФ) и гуанозиндифосфат (ГДФ). G-белки выполняют роль посредников между рецепторами и эффекторными белками (ферментами и ионными каналами) клеток. В состоянии покоя (при отсутствии стимуляции рецептора лигандом) внутриклеточный домен рецептора нековалентно соединен с G-белком, состоящим из α- и βγ-субъединиц. При действии вещества на рецептор ГДФ, связанный с α-субъединицей G-белка, заменяется на ГТФ, этот комплекс отделяется от рецептора и вступает во взаимодействие с эффекторными белками. К эффекторам относят ферменты (аденилатциклаза, фосфолипаза С), различные ионные каналы и некоторые другие белки. Передача сигнала, опосредованная G-белками, завершается, когда ГТФ гидролизуется с образованием ГДФ под каталитическим действием α-субъединицы, обладающей внутренней ГТФ-азной активностью. Как правило, один рецептор сопряжен с несколькими G-белками, а каждый G-белок может одновременно взаимодействовать с несколькими молекулами ферментов или несколькими ионными каналами. В результате такого взаимодействия происходит амплификация (усиление) эффекта вещества. 110 Часть I. Общая фармакология Рецептор А ГТФ ГДФ Агонист ГДФ Эффектор ГТФ В С ГТФ Хорошо изучено взаимодействие G-белков с аденилатциклазой и фосфолипазой С. Аденилатциклаза — мембраносвязанный фермент, гидролизующий АТФ. В результате гидролиза АТФ образуется циклический аденозинмонофосфат (цАМФ), который активирует цАМФ-зависимые протеинкиназы, фосфорилирующие клеточные белки. При этом изменяется активность белков и регулируемых ими процессов. По влиянию на активность аденилатциклазы G-белки подразделяются на Gs-белки, стимулирующие аденилатциклазу, и Gi-белки, ингибирующие этот фермент. Примером рецепторов, взаимодействующих с G s-белками, являются β1-адренорецепторы (опосредуют стимулирующее влияние на сердце симпатической иннервации), а примером рецепторов, взаимодействующих с Gi-белками, — М2-холинорецепторы (опосредуют тормозное влияние на сердце парасимпатической иннервации). Эти рецепторы локализованы в мембране кардиомиоцитов. При стимуляции β1-адренорецепторов повышается активность адениРис. 2.3. Рецептор, взаимодействующий с G-белками: А — рецептор находится в состоянии покоя (не взаимодействует с веществом), α-субъединица G-белка связана с ГДФ; В — при взаимодействии рецептора с веществом (агонистом) α-субъединица G-белка связывается с ГТФ; С — комплекс α-субъединицы с ГТФ отсоединяется от рецептора и вступает во взаимодействие с эффектором (ферментом и/или ионным каналом) Глава 2. Фармакодинамика 111 латциклазы и увеличивается содержание цАМФ в кардиомиоцитах. В результате активируется протеинкиназа, фосфорилирующая кальциевые каналы мембран кардиомиоцитов. Через эти каналы ионы кальция поступают в клетку. Вход Са2+ в клетку увеличивается, что приводит к повышению автоматизма синусного узла и увеличению частоты сердечных сокращений. Внутриклеточные эффекты противоположной направленности развиваются при стимуляции М2-холинорецепторов кардиомиоцитов, в результате чего происходит снижение автоматизма синусного узла и частоты сердечных сокращений. С фосфолипазой С взаимодействуют Gq-белки, вызывая ее активацию. Примером рецепторов, сопряженных с Gq-белками, являются α1-адренорецепторы гладкомышечных клеток сосудов (опосредующие влияние на сосуды симпатической иннервации). При стимуляции этих рецепторов повышается активность фосфолипазы С. Фосфолипаза С гидролизует фосфатидилинозитол-4,5-дифосфат клеточных мембран с образованием гидрофильного вещества инозитол-1,4,5-трифосфата, взаимодействующего с кальциевыми каналами саркоплазматического ретикулума клетки и вызывающего высвобождение Са2+ в цитоплазму. При повышении концентрации Са2+ в цитоплазме гладкомышечных клеток увеличивается скорость образования комплекса Са2+-кальмодулин, который активирует киназу легких цепей миозина. Этот фермент фосфорилирует легкие цепи миозина, в результате чего облегчается взаимодействие актина с миозином и происходит сокращение гладких мышц сосудов. Рецепторы, сопряженные с G-белками, — наиболее распространенный вид рецепторов в организме человека. К ним относятся также дофаминовые рецепторы, некоторые подтипы серотониновых рецепторов, опиоидные рецепторы, гистаминовые рецепторы, рецепторы для большинства пептидных гормонов и др. Внутриклеточные рецепторы Внутриклеточные рецепторы — растворимые цитозольные или ядерные белки, которые опосредуют регулирующее действие веществ на транскрипцию ДНК (см. рис. 2.2). Лигандами внутриклеточных рецепторов являются липофильные вещества (стероидные и тиреоидные гормоны, витамины А, D), беспрепятственно проникающие в клетки через цитоплазматические мембраны, а затем в клеточное ядро. Взаимодействие лиганда (например, глюкокортикоидов) с цитозольными рецепторами вызывает их конформационное изменение, в результате чего комплекс вещество–рецептор перемещается в ядро клетки, где связывается с определенными участками молекулы ДНК. 112 Часть I. Общая фармакология Происходит изменение (активация или репрессия) транскрипции генов, кодирующих синтез различных функционально активных белков (ферментов, цитокинов и т.д.). Увеличение (или уменьшение) синтеза ферментов и других белков приводит к изменению биохимических процессов в клетке и возникновению фармакологических эффектов. Так, глюкокортикоиды, активируя гены, ответственные за синтез ферментов глюконеогенеза, стимулируют синтез глюкозы, способствуя развитию гипергликемии. В результате репрессии генов, кодирующих синтез цитокинов, молекул межклеточной адгезии, циклооксигеназы глюкокортикоиды оказывают иммуносупрессивное и противовоспалительное действия. Фармакологические эффекты веществ при их взаимодействии с внутриклеточными рецепторами развиваются медленно (в течение нескольких часов и даже суток). Взаимодействие с ядерными рецепторами характерно для тиреоидных гормонов, витаминов А (ретиноидов) и D. Рецепторы, активируемые пролифераторами пероксисом (подтип ядерных рецепторов), участвуют в регуляции липидного обмена и других метаболических процессов и являются мишенями для клофибрата (гиполипидемический препарат). Связывание вещества с рецептором. Понятие об аффинитете Для того чтобы ЛВ подействовало на рецептор, оно должно с ним связаться. В результате образуется комплекс «вещество–рецептор». Образование подобного комплекса осуществляется с помощью межмолекулярных связей. Существует несколько видов таких связей. ● Ковалентные связи — самый прочный вид межмолекулярных связей, образующихся между двумя атомами двух молекул за счет общей пары электронов. Ковалентные связи наиболее часто обеспечивают необратимое связывание веществ, однако такой вид связывания не характерен для взаимодействия ЛВ с рецепторами. ● Ионные связи возникают между группировками, несущими разноименные заряды (электростатическое взаимодействие). Ионные связи сильнее, чем водородные, но менее прочны, чем ковалентные. ● Ион-дипольные и диполь-дипольные связи близки по характеру к ионным связям. В электронейтральных молекулах ЛВ, попадающих в электрическое поле клеточных мембран или находящихся в окружении ионов, происходит образование индуцированных диполей. Ионные и дипольные связи характерны для взаимодействия ЛВ с рецепторами. ● Водородные связи — результат взаимодействия между положительно (атомы водорода, присоединенные к азоту или кислороду) и отри- Глава 2. Фармакодинамика 113 цательно заряженными атомами (кислород, азот или сера). Водородные связи слабее, чем ионные, для их образования необходимо, чтобы молекулы находились друг от друга на расстоянии не более 0,3 нм. Эти связи играют весьма существенную роль во взаимодействии ЛВ с рецепторами. ● Ван-дер-ваальсовы связи возникают вследствие появления в молекуле наведенной полярности, вызванной сдвигом электронов. Они образуются между двумя любыми атомами, находящимися на расстоянии не более 0,2 нм, и обеспечивают слабое взаимодействие между веществом и рецептором. При увеличении расстояния эти связи ослабевают. Такая индуцированная полярность весьма распространена при молекулярных взаимодействиях. ● Гидрофобные связи образуются при взаимодействии неполярных молекул в водной среде. Связывание ЛВ с рецепторами — результат множественных химических взаимодействий между этими двумя молекулами, что обеспечивает достаточную силу связывания, необходимую для образования комплекса «вещество–рецептор». Как правило, связывание веществ с рецепторами происходит при участии нескольких слабых связей: например, десяти и более Ван-дер-ваальсовых связей и нескольких водородных связей; ионные взаимодействия встречаются реже. Ковалентные связи между ЛВ и рецепторами возникают достаточно редко и представляют собой особый случай. Образование ковалентных необратимых связей приводит к формированию длительно существующего комплекса «вещество–рецептор». В результате для восстановления первоначальной активности должны быть синтезированы новые рецепторы, а поскольку молекулы ЛВ оказываются необратимо связанными и не могут быть вытеснены из комплекса с рецептором, его действие не может быть быстро прекращено. Примером участия ковалентных связей в образовании комплекса «вещество–рецептор» может служить необратимое связывание неизбирательного α-адреноблокатора феноксибензамина с α1- и α2-аренорецепторами, вследствие чего продолжительность его действия составляет 48 ч (см. стр 253). Поскольку большинство ЛВ связываются с рецепторами посредством слабых связей, т.е. обратимо, комплекс «вещество-рецептор» [ВР] через какой-то промежуток времени распадается с образованием свободной молекулы вещества [В] и незанятого рецептора [Р]: k1 [B] [P] [ВР]. k2 114 Часть I. Общая фармакология При установлении равновесия между процессом образования комплекса и процессом его диссоциации, согласно закону действующих масс, в каждый момент времени скорость образования комплекса равна скорости его распада: k1 [В] [Р] = k 2 [ВР], где [ВР] — количество комплексов «вещество–рецептор», [В] — концентрация свободного вещества, [Р] — количество свободных рецепторов, k1 и k2 — константы скорости образования и распада комплексов соответственно. Для характеристики обратимого связывания вещества с рецептором используют термин «aффинитет». Аффинитет (от лат. affinis — родственный) — способность вещества связываться с рецептором, в результате чего образуется комплекс «вещество– рецептор». Кроме того, термин «аффинитет» используют для характеристики прочности связывания вещества с рецептором (чем выше прочность связывания, тем дольше не распадается комплекс «вещество–рецептор» и тем выше аффинитет). Количественная мера аффинитета, отражающая прочность связывания вещества с рецептором, — константа диссоциации (Кd), численно равная отношению константы распада комплекса «вещество–рецептор» (k2) к константе образования этого комплекса (k1). При равновесии между процессами образования и диссоциации комплекса «вещество–рецептор» согласно предыдущему уравнению: Кd = k 2 / k1 = [В] [Р] / [ВР]. Из данного уравнения видно, что, если [ВР] = [Р], т.е. если половина рецепторов связана с веществом, а половина — нет, то Кd = [В] (концентрации вещества). Следовательно, константа диссоциации численно равна концентрации вещества, при которой половина рецепторов в данной системе связана с веществом. Выражается этот показатель в молях/л (М). Между аффинитетом и константой диссоциации существует обратно пропорциональное соотношение: чем меньше Кd, тем выше аффинитет. Например, если Кd вещества А равна 10–3 М, а Кd вещества В равна 10–10 М, аффинитет вещества В выше, чем аффинитет вещества А. Фракция рецепторов (f), связанных с веществом, по отношению к общему количеству рецепторов может быть выражена уравнением: f = [ВР] / ([Р] + [ВР]), Глава 2. Фармакодинамика 115 где [ВР] — количество рецепторов, связанных с веществом, [Р] — количество свободных рецепторов. Это уравнение может быть преобразовано с использованием Кd следующим образом: Внутренняя активность лекарственных веществ. Понятие об агонистах и антагонистах рецепторов Вещества, обладающие аффинитетом, могут проявлять внутреннюю активность. Внутренняя активность — способность вещества при взаимодействии с рецептором стимулировать его и таким образом вызывать определенные эффекты. 1,0 A B 0,5 0 KdAKdB Lg C Рис. 2.4. График полулогарифмической зависимости степени связывания рецепторов с веществами А и В от концентрации этих веществ. Вещество А связано с 0,5 от общего количества рецепторов в меньшей концентрации, чем вещество В (Кd вещества А меньше, чем Кd вещества В) 1,0 Эффект (Е) Отсюда следует, что если Кd = [В], т.е. концентрации свободного вещества, то f = 1/2 от общего количества рецепторов. Это уравнение может быть выражено графически (рис. 2.4). Константа диссоциации при определенных условиях коррелирует с активностью вещества, которая характеризуется концентрацией вещества (ЕС50), вызывающей полумаксимальный эффект (0,5 от максимального эффекта). ЕС50 может быть определена на графике зависимости эффекта от концентрации вещества (рис. 2.5). Чем меньше эта концентрация, тем выше активность вещества, что может быть следствием его более высокого аффинитета (и, следовательно, меньшего значения Кd). Степень связывания с рецептором f = [В] / ([В] + Кd). A B 0,5 0 EC50(A) EC50(B) Lg C Рис. 2.5. График полулогарифмической зависимости эффекта, вызываемого веществами А и В, от концентрации этих веществ. Вещество А вызывает полумаксимальный эффект в меньшей концентрации, чем вещество В (ЕС50 вещества А меньше, чем ЕС50 вещества В) 116 Часть I. Общая фармакология В зависимости от наличия внутренней активности ЛВ подразделяют на агонисты и антагонисты рецепторов. Агонисты (от греч. agonistes — соперник, agon — борьба), или миметики рецепторов, — вещества, обладающие аффинитетом и внутренней активностью. При взаимодействии со специфическими рецепторами они стимулируют их, т.е. вызывают изменения конформации рецепторов, в результате чего внутри клеток возникает цепь биохимических реакций, приводящих к развитию определенных фармакологических эффектов. Полные агонисты, взаимодействуя с рецепторами, стимулируют их в максимально возможной степени и вызывают максимальный эффект (обладают максимальной внутренней активностью). Например, ацетилхолин, связываясь с никотиновыми холинорецепторами, вызывает конформационные изменения в сопряженных с рецепторами ионных каналах, переводя их из закрытого в полностью открытое состояние. Частичные агонисты при взаимодействии с рецепторами вызывают эффект, меньший максимального (не обладают максимальной внутренней активностью). Многие ЛВ относятся к частичным агонистам рецепторов. Среди них — блокаторы β-адренорецепторов с внутренней симпатомиметической активностью, частичный агонист μ-опиоидных рецепторов бупренорфин и др. Существуют также обратные (инверсированные) агонисты, переводящие конститутивно (спонтанно) активные рецепторы в неактивное состояние. Антагонисты (от греч. antagonisma — соперничество, anti —против, agon — борьба) — вещества, обладающие аффинитетом, но лишенные внутренней активности. Связываясь с рецепторами, они препятствуют действию на них как эндогенных лигандов (нейромедиаторов, гормонов), которые являются полными агонистами этих рецепторов, так и экзогенных агонистов (в том числе ЛВ). Поэтому антагонисты также называют блокаторами рецепторов. Фармакологические эффекты антагонистов обусловлены устранением или ослаблением действия эндогенных лигандов данных рецепторов (нейромедиаторов, гормонов и др.) и поэтому противоположны эффектам агонистов. Так, ацетилхолин вызывает брадикардию, а антагонист М-холинорецепторов атропин, устраняя действие ацетилхолина на сердце, повышает частоту сердечных сокращений. Если антагонисты занимают те же места связывания, что и агонисты, они могут вытеснять друг друга из связи с рецепторами. Подобный вид антагонизма обозначают как конкурентный антагонизм, а антагонисты называют конкурентными. Конкурентный антагонизм зависит от сравнительного аффинитета конкурирующих веществ к данному рецепто- Глава 2. Фармакодинамика 117 ру и их концентрации. В достаточно высоких концентрациях вещество с низким аффинитетом может вытеснить вещество с более высоким аффинитетом из связи с рецептором. Поэтому при конкурентном антагонизме эффект агониста может быть полностью восстановлен при увеличении его концентрации в среде (рис. 2.6а). Конкурентный антагонизм часто используют для устранения токсических эффектов ЛВ. Так, при отравлении атропином его действие может быть устранено с помощью физостигмина (антихолинэстеразное средство), повышающего уровень ацетилхолина в синаптической щели, в результате чего ацетилхолин конкурентно вытесняет атропин из связи с М-холинорецепторами. Частичные агонисты также могут конкурировать с полными агонистами за места связывания. Вытесняя полные агонисты из связи с рецепторами, частичные агонисты уменьшают их эффекты и поэтому в клинической практике могут быть использованы вместо антагонистов. Неконкурентный антагонизм возникает, когда антагонист занимает так называемые аллостерические места связывания на рецепторах (участки макромолекулы, не являющиеся местами связывания агониста, но регулирующие активность рецепторов). При связывании с этими участками неконкурентные антагонисты изменяют конформацию рецепторов таким образом, что они теряют способность взаимодействовать с агонистами. При этом увеличение концентрации агониста не может привести к полному восстановлению его эффекта (см. рис. 2.6 б). Кроме того, неконкурентный антагонизм имеет место при необратимом (ковалентном) связывании вещества с рецептором. 100 А А+B 50 а Lg дозы Эффект Эффект 100 А А+C 50 б Lg дозы Рис. 2.6. Виды фармакологического антагонизма (полулогарифмические графики зависимости эффекта от дозы). а: вещество А — агонист; вещество В — конкурентный антагонист; при повышении концентрации агониста (А) он вытесняет антагонист (В) из связи с рецептором и его эффект полностью восстанавливается; б: вещество А — агонист, вещество С — неконкурентный антагонист; при повышении концентрации агониста (А) его эффект полностью не восстанавливается 118 Часть I. Общая фармакология Некоторые ЛВ сочетают способность стимулировать один подтип рецепторов и блокировать другой. Такие вещества обозначают как агонисты-антагонисты (например, буторфанол — антагонист μ- и агонист κ-опиоидных рецепторов). Как правило, агонисты-антагонисты обладают меньшими побочными эффектами, чем агонисты всех подтипов рецепторов. В частности, буторфанол в меньшей степени угнетает дыхание, чем морфин. Рецепторы не являются стабильными, постоянно существующими структурами клеток. Количество их может увеличиваться вследствие преобладания синтеза рецепторных белков или уменьшаться вследствие превалирования процесса их деградации. Кроме того, рецепторы могут терять свою функциональную активность (десенситизация рецепторов), вследствие чего при взаимодействии рецептора с агонистом не возникают биохимические реакции, приводящие к фармакологическому эффекту. Изменение количества и чувствительности рецепторов регулируется природой лиганда (агонист или антагонист), концентрацией лиганда и длительностью его воздействия на рецепторы. При длительном воздействии агониста развивается десенситизация рецепторов и/или снижается их количество (down-регуляция), и, наоборот, отсутствие лиганда (а также снижение его концентрации) или длительное воздействие антагониста приводит к увеличению количества рецепторов (up-регуляция). Десенситизации подвергаются многие рецепторы. Например, клеточный ответ на повторную стимуляцию β-адренорецепторов адреналином постоянно снижается с течением времени. Десенситизация β-адренорецепторов происходит вследствие фосфорилирования цитоплазматического участка молекулы рецептора, взаимодействующего с G-белком (рис. 2.7). Фосфорилирование способствует связыванию рецептора с β-аррестином, что угнетает способность рецептора стимулировать G-белки (Gs-белки), а также облегчает интернализацию рецепторов внутрь клеток. Вследствие уменьшения количества активированных Gsбелков аденилатциклазная активность снижается, в связи с чем синтезируется меньше молекул цАМФ. Это приводит к уменьшению эффектов на клеточном, тканевом и органном уровнях. Длительным воздействием веществ может быть вызвано также изменение количества рецепторов на поверхности или внутри клеток. Длительная стимуляция рецепторов агонистом способствует эндоцитозу и интернализации рецепторов клеточными везикулами, а также их деградации в лизосомах, что приводит к уменьшению количества рецепторов (down-регуляция). Когда вещество перестает контактировать Глава 2. Фармакодинамика 119 Агонист P ATФ AДФ я ци а из ал r -APK P P P ar Gs OH OH OH н ер - OH OH G s OH т Ин Дег рад аци я P P эндосома P P P лизосома Рис. 2.7. Десенситизация β-адренорецепторов и уменьшение их количества (downрегуляция) при длительном воздействии агониста (пояснения в тексте): β-АРК — киназа β-адренорецептора с рецепторами, рецепторы снова могут быть восстановлены на поверхности клеток и приведены в состояние функциональной активности (см. рис. 2.7). Кроме того, клетки обладают также способностью изменять интенсивность синтеза рецепторов и таким образом регулировать количество рецепторов, связывающихся с ЛВ. Интенсивность синтеза может повышаться (up-регуляция) при длительном воздействии антагониста или снижаться (down-регуляция) при длительном контакте рецептора с агонистом. Интернализация рецепторов и изменение их синтеза происходят медленнее и продолжаются дольше, чем процесс фосфорилирования рецепторов (десенситизация). Процессы десенситизации и down-регуляции лежат в основе развития толерантности к действию ЛВ при его длительном контакте с рецептором. Примером может служить развитие толерантности к анальгетическому и другим эффектам морфина при его повторном применении вследствие десенситизации и down-регуляции опиоидных рецепторов. Пример upрегуляции рецепторов в результате длительного воздействия антагонистов — развитие «синдрома отмены» при внезапном прекращении продолжительного приема β-адреноблокаторов, который может выражаться в резком подъеме артериального давления или других осложнениях, связанных с действием медиатора норадреналина на большее количество 120 Часть I. Общая фармакология β-адренорецепторов. Для предупреждения таких осложнений рекомендуют постепенную отмену препарата. 2.1.2. Другие «мишени» для лекарственных веществ К другим «мишеням» для ЛВ относят ионные каналы, ферменты, транспортные белки. Ионные каналы Одна из основных «мишеней» для ЛВ — потенциалозависимые ионные каналы, избирательно проводящие Nа+, Са2+, К+ и другие ионы через клеточную мембрану. В отличие от рецептор-управляемых ионных каналов, открываемых при взаимодействии вещества с рецептором, эти каналы регулируются потенциалом действия (открываются при деполяризации клеточной мембраны). ЛВ могут блокировать потенциалозависимые ионные каналы и таким образом нарушать поступление через них ионов или активировать их, способствуя прохождению ионных токов. Большинство ЛВ блокируют ионные каналы. Местные анестетики блокируют потенциалозависимые Nа+-каналы. К числу блокаторов Nа+-каналов относят и многие противоаритмические средства (хинидин, лидокаин, прокаинамид). Некоторые противоэпилептические средства (фенитоин, карбамазепин) также блокируют потенциалозависимые Nа+-каналы, с чем связана их противосудорожная активность. Блокаторы натриевых каналов нарушают вхождение в клетку Nа+, препятствуя деполяризации клеточной мембраны. Весьма эффективными при лечении многих сердечно-сосудистых заболеваний (гипертонической болезни, сердечных аритмий, стенокардии) оказались блокаторы Са2+-каналов (нифедипин, верапамил и др.). Ионы кальция принимают участие во многих физиологических процессах: сокращении гладких мышц, генерации импульсов в синусо-предсердном узле и проведении возбуждения по предсердно-желудочковому узлу, агрегации тромбоцитов и др. Блокаторы медленных кальциевых каналов препятствуют вхождению ионов кальция внутрь клетки через потенциалозависимые каналы и вызывают расслабление гладких мышц сосудов, уменьшение частоты сокращений сердца и атриовентрикулярной проводимости, нарушают агрегацию тромбоцитов. В качестве ЛВ используют как активаторы, так и блокаторы калиевых каналов. Активаторы калиевых каналов (миноксидил, диазоксид) нашли применение в качестве антигипертензивных средств. Они способствуют Глава 2. Фармакодинамика 121 выходу ионов калия из клетки, что приводит к гиперполяризации клеточной мембраны и уменьшению тонуса гладких мышц сосудов. В результате происходит снижение АД. Ферменты Многие ЛВ являются ингибиторами ферментов. Ингибиторы МАО нарушают метаболизм (окислительное дезаминирование) катехоламинов (норадреналина, дофамина, серотонина) и повышают их содержание в ЦНС. На этом принципе основано действие антидепрессантов — ингибиторов МАО (например, ниаламида). Механизм действия нестероидных противовоспалительных средств связан с ингибированием циклооксигеназы, в результате чего снижается биосинтез простагландинов Е2 и I2 и развивается прововоспалительное действие. Ингибиторы ацетилхолинэстеразы (антихолинэстеразные средства) препятствуют гидролизу ацетилхолина и повышают его содержание в синаптической щели. Препараты этой группы применяют для повышения тонуса гладкомышечных органов (ЖКТ, мочевого пузыря) и скелетных мышц. Транспортные системы ЛВ могут действовать на транспортные системы (транспортные белки), переносящие молекулы некоторых веществ или ионы через мембраны клеток. Например, трициклические антидепрессанты блокируют транспортные белки, переносящие норадреналин и серотонин через пресинаптическую мембрану нервного окончания (блокируют обратный нейрональный захват норадреналина и серотонина). Сердечные гликозиды блокируют Nа+–К+-АТФазу мембран кардиомиоцитов, осуществляющую транспорт Nа+ из клетки в обмен на К+. Перспективная «мишень» для ЛВ — гены. С помощью избирательно действующих ЛВ возможно оказывать прямое влияние на функцию определенных генов. В ряде случаев встречаются и другие «мишени», на которые действуют ЛВ. Структурные белки, такие как тубулин, служат мишенями для противоопухолевых веществ, способных проникать через цитоплазматические мембраны клеток злокачественных опухолей. Алкалоиды барвинка, обладающие антимитотическим действием, связываются с мономерами тубулина и препятствуют полимеризации этих молекул в микротрубочки. Антацидные средства нейтрализуют соляную кислоту желудка, их применяют при повышенной кислотности желудочного сока (гиперацидный гастрит, язвенная болезнь желудка). Некоторые ЛВ оказывают действие, 122 Часть I. Общая фармакология не имея никаких «мишеней»: диуретик маннитол повышает осмотическое давление почечного фильтрата, препятствуя реабсорбции воды и удерживая ее в почечных канальцах. 2.2. ВИДЫ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ Различают следующие виды действия ЛВ: местное и резорбтивное, рефлекторное, прямое и косвенное, основное и побочное и некоторые другие. Местное действие ЛВ оказывает при контакте с тканями в месте его нанесения (обычно это кожа или слизистые оболочки). Например, при поверхностной анестезии местный анестетик действует на окончания чувствительных нервов только в месте нанесения на слизистую оболочку. Для оказания местного действия ЛВ назначают в форме мазей, примочек, полосканий, пластырей. При назначении некоторых ЛВ в виде глазных или ушных капель также рассчитывают на их местное действие. Однако какое-то количество ЛВ обычно всасывается с места нанесения в кровь и оказывает общее (резорбтивное) действие. При местном применении ЛВ возможно также рефлекторное действие. Резорбтивное действие (от лат. resorbeo — поглощаю) ЛВ вызывается пocлe их всасывания в кровь или непосредственного введения в кровеносный сосуд и распределения в организме. При резорбтивном действии, так же как и при местном, вещество может возбуждать чувствительные рецепторы и вызывать рефлекторные реакции. Рефлекторное действие. Некоторые ЛВ способны возбуждать окончания чувствительных нервов кожи, слизистых оболочек (экстерорецепторы), хеморецепторы сосудов (интерорецепторы) и вызывать рефлекторные реакции со стороны органов, расположенных в удалении от места непосредственного контакта вещества с чувствительными рецепторами. Примером возбуждения экстерорецепторов кожи эфирным горчичным маслом является действие горчичников. Лобелин при внутривенном введении возбуждает хеморецепторы сосудов, приводя к рефлекторной стимуляции дыхательного и сосудодвигательного центров. Прямое (первичное) действие на сердце, сосуды, кишечник и другие органы развивается при непосредственном воздействии ЛВ на эти органы. Например, сердечные гликозиды вызывают кардиотонический эффект (усиление сокращений миокарда) вследствие их непосредственного влияния на кардиомиоциты. Вызываемое сердечными гликозидами повышение диуреза у больных с сердечной недостаточностью обусловлено Глава 2. Фармакодинамика 123 увеличением сердечного выброса и улучшением гемодинамики. Действие ЛВ, изменяющее функцию одних органов, воздействуя на другие органы, обозначают как косвенное (вторичное) действие. Основное действие — действие, ради которого применяют ЛВ при лечении данного заболевания. Например, фенитоин обладает противосудорожными и антиаритмическими свойствами. У больного эпилепсией основное действие фенитоина — противосудорожное, а у больного с сердечной аритмией, вызванной передозировкой сердечных гликозидов, — антиаритмическое. Все остальные (кроме основного) действия ЛВ, возникающие при его приеме в терапевтических дозах, расценивают как побочные действия (эффекты). Эти эффекты часто бывают неблагоприятными (отрицательными) (см. главу «Побочное и токсическое действие лекарственных веществ»). Например, ацетилсалициловая кислота может вызвать изъязвление слизистой оболочки желудка, антибиотики из группы аминогликозидов (канамицин, гентамицин и др.) — нарушение слуха. Отрицательное побочное действие часто служит причиной ограничения применения того или иного ЛВ и даже исключения его из списка лекарственных препаратов. Избирательное действие ЛВ направлено преимущественно на один орган или систему организма. Так, сердечные гликозиды обладают избирательным действием на миокард, окситоцин — на матку, снотворные средства — на ЦНС. Центральное действие развивается вследствие прямого влияния ЛВ на ЦНС. Центральное действие характерно для веществ, проникающих через ГЭБ. Для снотворных средств, антидепрессантов, анксиолитиков, средств для наркоза — это основное действие. В то же время центральное действие может быть побочным (нежелательным). Так, многие антигистаминные средства вследствие центрального действия вызывают сонливость. Периферическое действие обусловлено влиянием ЛВ на периферический отдел нервной системы или на органы и ткани. Курареподобные средства (миорелаксанты периферического действия) расслабляют скелетные мышцы, блокируя передачу возбуждения в нервно-мышечных синапсах. Некоторые периферические вазодилататоры расширяют кровеносные сосуды, действуя непосредственно на гладкомышечные клетки. Для веществ с основным центральным действием периферические эффекты обычно являются побочными. Например, антипсихотическое средство хлорпромазин вызывает расширение сосудов и снижение АД (нежелательное действие), блокируя периферические α-адренорецепторы. 124 Часть I. Общая фармакология Обратимое действие — следствие обратимого связывания ЛВ с «мишенями» (рецепторами, ферментами). Действие такого вещества можно прекратить путем его вытеснения другим ЛВ из связи c «мишенью». Необратимое действие возникает, как правило, в результате прочного (ковалентного) связывания ЛВ с «мишенями». Например, ацетилсалициловая кислота необратимо блокирует циклооксигеназу, поэтому действие препарата прекращается лишь после синтеза нового фермента. Вопросы и задания для самоконтроля 1. Аффинитет: а) зависит от прочности связывания вещества с рецептором; б) зависит от способности вещества вызывать определенный эффект; в) характеризуется константой ионизации; г) характеризуется константой диссоциации; д) коррелирует с внутренней активностью вещества. 2. Антагонисты рецепторов: а) способны специфически связываться с рецепторами; б) обладают внутренней активностью; в) вызывают изменение конформации рецептора; г) могут вытеснить агонист из связи с рецептором; д) изменяют активность ферментов клетки. 3. Конкурентные антагонисты: а) ковалентно связываются с местами связывания агонистов на рецепторе; б) вызывают биохимические реакции в клетке; в) оказывают фармакологические эффекты, вытесняя эндогенные агонисты из связи с рецепторами; г) не могут быть вытеснены агонистами из связи с рецепторами. 4. Уменьшение количества рецепторов на постсинаптической мембране: а) может быть следствием длительной стимуляции рецепторов агонистом; б) происходит вследствие длительного воздействия антагониста; в) может быть при снижении или прекращении выделения медиатора; г) может привести к развитию толерантности к действию вещества; д) является результатом экзоцитоза и деградации рецепторов внутри клетки. Глава 2. Фармакодинамика 125 5. Побочные эффекты лекарственных веществ неаллергической природы: а) не зависят от дозы; б) возникают при применении ЛВ в терапевтических дозах; в) возникают только при повторном приеме ЛВ; г) могут быть вызваны теми же механизмами, что и терапевтические эффекты. Глава 3 ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ФАРМАКОДИНАМИКУ И ФАРМАКОКИНЕТИКУ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ К факторам, влияющим на действие ЛС, относят свойства вещества (химическая структура, физико-химические свойства, дозы и концентрации), лекарственную форму и особенности ее технологии, состояние организма и его индивидуальные особенности (пол, возраст, генетические факторы и др.), а также режим назначения и условия применения (повторное введение, комбинированное применение, время суток, время года, температура воздуха, атмосферное давление, экологические факторы и т.д.). 3.1. СВОЙСТВА ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ. ЛЕКАРСТВЕННЫЕ ФОРМЫ К факторам, влияющим на действие ЛВ, относят: ● химическую структуру; ● физико-химические свойства; ● дозы (концентрации) ЛВ. Химическая структура ЛВ определяет характер их действия (фармакологические эффекты) и фармакокинетические особенности. Вещества, близкие по химической структуре и относящиеся к одной химической группе (например, бензодиазепины), как правило, вызывают одинаковые фармакологические эффекты. Связано это с тем, что взаимодействие веществ с «мишенями» определяется химическим строением, наличием определенных функционально активных групп, пространственной ориентацией и размером молекул ЛВ. Так, для того чтобы вещество подействовало на рецептор, оно должно не только иметь Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 127 соответствующую химическую структуру, но и пространственно соответствововать данному рецептору, т.е. быть ему комплементарным. Примером влияния комплементарности на действие ЛВ служит различие в действии стереоизомеров, молекулы которых имеют «зеркальную» пространственную ориентацию. Гиосциамин —L-изомер — в 2 раза активнее атропина, смеси активного L- и малоактивного D-изомеров. Значение имеет также расстояние между функционально активными группировками веществ. Зная зависимость действия ЛВ от их химической структуры, можно синтезировать ЛВ с определенными фармакологическими свойствами. Фармакокинетику и фармакодинамику ЛВ определяют также их физико-химические свойства: липофильность, гидрофильность, полярность, степень ионизации. Так, липофильность веществ определяет их способность проникать через ГЭБ и оказывать действие на ЦНС. Действие ЛВ (скорость развития фармакологического эффекта, его выраженность, продолжительность и даже характер) зависит от дозы. Доза (от греч. dosis — порция) — количество ЛВ на один прием (обозначается как разовая доза). Дозы приводят в весовых или объемных единицах. Дозы можно выражать в виде количества вещества на 1 кг массы тела или на 1 м2 поверхности тела (например, 1 мг/кг, 1 мг/м2), что позволяет более точно дозировать препарат. Жидкие лекарственные препараты дозируют столовыми, десертными или чайными ложками, а также каплями. Дозы некоторых антибиотиков и гормонов выражают в единицах действия (ЕД). При увеличении дозы ЛВ его фармакологический эффект, как правило, усиливается и через определенное время достигает максимальной (постоянной) величины (Еmax). Поэтому по арифметической шкале доз зависимость «доза–эффект» имеет гиперболический характер (градуальная зависимость). По логарифмической шкале доз эта зависимость выражается S-образной кривой (рис. 3.1). По величине дозы, вызывающей эффект определенной величины, судят об активности вещества. Обычно для этих целей на графике зависимости «доза–эффект» определяют дозу, вызывающую эффект, равный 50% от максимального, и обозначают ее как ЭД50 (ED50). Такие дозы ЛВ используют для сравнения их активности. Чем меньше ЭД50, тем выше активность вещества (если ЭД 50 вещества А в 10 раз меньше, чем ЭД50 вещества В, вещество А в 10 раз активнее). Помимо активности ЛВ сравнивают по эффективности (определяется величиной максимального эффекта, Еmax). Если максимальный эффект вещества А в 2 раза больше максимального эффекта вещества В, вещество А в 2 раза эффективнее. 128 Часть I. Общая фармакология Эффект (Е) Эффект (Е) А Б Еmax Еmax 1/2Еmax 1/2Еmax ЭD50 Доза D ЭD50 log D Рис. 3.1. Зависимость «доза–эффект» (пояснения в тексте) Различают терапевтические, токсические и летальные дозы. Выделяют следующие терапевтические дозы: ● минимальные действующие; ● средние терапевтические; ● высшие терапевтические. Минимальные действующие дозы (пороговые дозы) вызывают минимальный терапевтический эффект. Обычно они в 2–3 раза меньше средней терапевтической дозы. Средние терапевтические дозы оказывают у большинства больных необходимое фармакотерапевтическое действие. Разовая доза (pro dosi) — количество ЛВ на один прием, суточная доза (pro die) — количество ЛВ, которое больной принимает в течение суток. Поскольку индивидуальная чувствительность больных и тяжесть заболеваний могут варьировать, средние терапевтические дозы обычно выражают в виде диапазона доз (например, разовая доза диклофенака составляет 0,025–0,05 г). Ударная доза — доза, превышающая среднюю терапевтическую дозу. С нее обычно начинают лечение противомикробными средствами (антибиотиками, сульфаниламидами) для быстрого создания высокой концентрации вещества в крови. После достижения определенного терапевтического эффекта назначают поддерживающие дозы. При длительном применении ЛВ указывается его доза на курс лечения (курсовая доза). Высшие терапевтические дозы — предельные дозы, превышение которых может привести к развитию токсических эффектов. Их назначают, если применение средних доз не оказывает желаемого действия. Для ядовитых и сильнодействующих веществ в законодательном порядке Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 129 установлены высшие разовые и высшие суточные дозы. К назначению ЛС в высших терапевтических дозах нужно относиться с большой осторожностью, поскольку это связано с риском возникновения серьезных побочных эффектов. Провизор не имеет права отпускать ЛС с превышением высшей разовой и высшей суточной доз, если в рецепте нет специального указания врача. Токсические дозы — дозы, оказывающие токсическое действие на организм. Летальные дозы (от лат. Letum — смерть) — дозы, вызывающие смертельный исход (определяется на стадии доклинических испытаний). Диапазон доз от минимальной действующей до высшей терапевтической определяется как широта терапевтического действия. Чем она больше, тем безопаснее применение ЛС. При включении ЛВ в различные лекарственные формы должен сохраняться характер его действия. Вместе с тем лекарственная форма и технологический процесс ее изготовления влияют на скорость выделения действующего вещества, место и скорость его всасывания, а следовательно, на скорость наступления эффекта и его продолжительность. С помощью различных технологических процессов могут быть созданы готовые лекарственные формы пролонгированного действия с регулируемой скоростью высвобождения ЛВ. В лекарственную форму включают не только формообразующие вещества, но и различные добавки, улучшающие вкус, уменьшающие местное раздражающее действие и др. Таким образом, фармацевтические технологии используют для создания оптимальных условий применения ЛВ. 3.2. СВОЙСТВА ОРГАНИЗМА К факторам, влияющим на действие ЛВ, относят пол, возраст, массу тела, состояние организма, генетические особенности пациента. 3.2.1. Пол Проблема зависимости фармакологического действия ЛВ от пола исследована недостаточно. Эксперименты на животных и клинические наблюдения свидетельствуют о некоторых половых различиях в метаболизме ЛВ и чувствительности к определенным фармакологическим воздействиям. Так, мужские половые гормоны стимулируют синтез микросомальных ферментов печени, поэтому элиминация некоторых ЛВ (парацетамола, верапамила, бензодиазепинов, пропранолола) происхо- 130 Часть I. Общая фармакология дит быстрее у мужчин. Существующие половые различия в метаболизме этанола связаны с большей активностью алкогольдегидрогеназы у мужчин. Имеются клинические данные о большей чувствительности женщин к действию некоторых ЛВ. Аритмогенный эффект противоаритмических средств (желудочковые аритмии типа «пируэт») чаще возникает у женщин, болеутоляющее действие морфина возникает у женщин в меньших дозах, чем у мужчин. Средства, угнетающие ЦНС (морфин и барбитураты), могут вызывать состояние возбуждения у женщин и не оказывать подобного действия у мужчин. 3.2.2. Возраст Изменения действия ЛВ, связанные с возрастом, особенно проявляются у новорожденных и пациентов старше 60 лет. Особенности действия ЛВ на детский организм изучает педиатрическая фармакология. Отдельно рассматривают действие ЛВ на новорожденных (до 4 нед жизни) и плод в последний триместр беременности (перинатальная фармакология). Новорожденные имеют более высокую чувствительность к ЛВ. По скорости всасывания, распределения, метаболизма и выведения веществ они существенно отличаются от взрослых. Это связано с несовершенством метаболизма ЛВ (вследствие недостаточности ферментов), со сниженной функцией почек, с повышенной проницаемостью ГЭБ, недоразвитием эндокринной, нервной и других систем организма. Так, у новорожденных снижена активность ферментов, участвующих в конъюгации хлорамфеникола, что усиливает его токсическое действие. Более чувствительны новорожденные к морфину, неостигмину. Поэтому детям ЛВ назначают в меньших дозах, чем взрослым (а некоторые ЛВ противопоказаны). Уменьшение дозы препаратов у детей связано также с меньшей массой их тела. При назначении детям ядовитых и сильнодействующих ЛВ руководствуются специальными таблицами, приведенными в Государственной фармакопее. В них указаны дозы ЛВ для детей разного возраста. Каждый лекарственный препарат следует использовать в дозах, рекомендуемых для определенного возраста. В пожилом и старческом возрасте фармакокинетические процессы протекают медленнее. Изменение скорости всасывания связано с изменением кислотности желудочного сока, уменьшением кровотока в кишечнике, угнетением систем активного всасывания и др. Распределение ЛВ у пожилых людей может варьироваться вследствие изменения связывания с белками плазмы крови (уменьшение содержания Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 131 альбуминов в плазме увеличивает Vd препарата), снижения кровотока в органах и тканях. Замедление с возрастом метаболизма ЛВ связано с меньшей активностью ферментов печени и уменьшением печеночного кровотока. Снижение функции почек приводит к замедленному выведению ЛВ. Поэтому больным старше 60 лет дозы веществ, угнетающих ЦНС (снотворные, препараты группы морфина), сердечных гликозидов, мочегонных средств следует уменьшать на Ѕ, а дозы других сильнодействующих и ядовитых ЛВ — до ⅔ от доз, рекомендуемых для лиц среднего возраста. Изучением особенностей действия и применения ЛС у лиц пожилого и старческого возраста занимается гериатрическая фармакология. 3.2.3. Масса тела Концентрация ЛВ в плазме крови, в органах и тканях, а следовательно, их действие зависят от массы тела. Как правило, с увеличением массы тела назначаемую дозу ЛВ также следует увеличить. Поэтому при необходимости более точного дозирования дозы некоторых веществ приводят в расчете на 1 кг массы тела. 3.2.4. Состояние организма Различные патологические состояния могут вызвать изменение фармакокинетики и фармакодинамики ЛВ. При заболеваниях ЖКТ может происходить снижение скорости и степени всасывания ЛВ. Некоторые заболевания легких и сердечно-сосудистой системы приводят к существенным изменениям гемодинамики, отражающимся на характере распределения ЛВ. При нарушении функции почек удлиняется действие веществ, выводимых почками в неизмененном виде (например, при нормальной функции почек tЅ ампициллина составляет 1,3 ч, а при почечной недостаточности — 13–20 ч). Аналогичные изменения с действием ЛВ, метаболизируемых в печени, происходят при нарушениях ее функций. При повторных введениях ЛВ может происходить его накопление (кумуляция) в организме, повышающее опасность развития токсического действия при обычных схемах дозирования. В очаге воспаления резко ослабляется действие местноанестезирующих средств, а действие сульфаниламидов снижается в гнойных ранах. Действие некоторых ЛВ проявляется только при патологических состояниях. Например, кардиотоническое действие сердечных гликозидов 132 Часть I. Общая фармакология проявляется при сердечной недостаточности, ацетилсалициловая кислота снижает повышенную температуру тела. Патологические процессы, как правило, изменяют чувствительность и реактивность организма на ЛВ. Фармакологические эффекты зависят от функционального состояния организма. Существенное значение имеют также физическое развитие и питание больного. Физически крепкие лица меньше реагируют на ЛС, чем ослабленные. Истощенным и обезвоженным больным дозы большинства препаратов необходимо уменьшать в 1,5–2 раза. С особой осторожностью следует назначать ЛВ беременным и кормящим женщинам. Необходимо учитывать не только измененную чувствительность организма, но и возможность проникновения ЛВ через плацентарный барьер, выделения его с молоком и вредного влияния на плод и ребенка. 3.2.5. Генетические факторы Существуют значительные различия в индивидуальной чувствительности людей к ЛВ, определяемые генетическими факторами. Раздел фармакологии — фармакогенетика — изучает роль генетических факторов в изменении действия ЛВ. Очень часто индивидуальные различия в действии ЛВ обусловлены различиями в их метаболизме. Это происходит вследствие изменения активности ферментов, метаболизирующих ЛВ, часто связанное с мутацией генов, контролирующих синтез данных ферментов. Нарушение структуры и функции фермента называют энзимопатией (ферментопатией). При энзимопатиях активность фермента может быть снижена (замедление метаболизма может привести к усилению действия ЛВ и появлению токсических эффектов). При генетической недостаточности некоторых ферментов могут возникать атипичные реакции на вещества (идиосинкразия). Типичным примером идиосинкразии служит гемолитическое действие некоторых противомалярийных средств (хинина, примахина, хлорохина) при врожденной недостаточности в эритроцитах глюкозо-6-фосфатдегидрогеназы, в результате чего образуется хинон, вызывающий гемолиз эритроцитов. 3.3. РЕЖИМ НАЗНАЧЕНИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ На действие ЛВ оказывают влияние повторность введения и комбинированное (совместное, одновременное) применение ЛВ. Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 133 3.3.1. Повторное применение лекарственных веществ Повторные введения одного и того же ЛВ могут приводить к количественному (увеличение или уменьшение) и качественному изменению фармакологических эффектов. Среди явлений, наблюдаемых при повторных введениях ЛС, различают: ● кумуляцию; ● сенсибилизацию; ● привыкание (толерантность); ● лекарственную зависимость. Кумуляция (от лат. cumulatio — увеличение, скопление) — накопление в организме ЛВ или вызываемых им эффектов. ● Материальная кумуляция — увеличение в крови и/или тканях содержания ЛВ после каждого последующего введения по сравнению с предыдущим. При повторных введениях могут накапливаться ЛВ, медленно инактивируемые и выводимые из организма, а также ЛВ, прочно связывающиеся с белками плазмы крови или депонирующиеся в тканях, например некоторые снотворные средства из группы барбитуратов, препараты наперстянки. Материальная кумуляция может быть причиной токсических эффектов, что нужно учитывать при дозировании подобных препаратов. ● Функциональная кумуляция — усиление эффекта ЛВ при повторных введениях в отсутствии повышения его концентрации в крови и/ или тканях. Этот вид кумуляции возникает при повторных приемах алкоголя. При развитии алкогольного психоза («белая горячка») у восприимчивых лиц бред и галлюцинации развиваются в то время, когда этиловый спирт уже метаболизировался и не определяется в организме. Функциональная кумуляция также характерна для ингибиторов МАО. Сенсибилизация. Многие ЛВ образуют комплексы с белками плазмы крови, приобретающие при определенных условиях антигенные свойства. Это сопровождается образованием антител и сенсибилизацией организма. Повторное введение тех же ЛВ вызывает аллергические реакции. Часто такие реакции возникают при повторных введениях пенициллинов, прокаина, водорастворимых витаминов, сульфаниламидов и др. Привыкание (толерантность, от лат. tolerantia — терпение) — уменьшение фармакологического эффекта ЛВ при его повторных введениях в той же дозе. При развитии привыкания для достижения прежнего эффекта необходимо увеличивать дозу ЛВ. Толерантность развивается 134 Часть I. Общая фармакология как к терапевтическим, так и токсическим эффектам ЛВ. Например, при длительном применении морфина возникает толерантность не только к его анальгетическому действию, но и к угнетающему влиянию на дыхательный центр. Привыкание к ЛВ, стимулирующим рецепторы (агонистам рецепторов), может быть обусловлено снижением чувствительности (десенситизацией) рецепторов и/или уменьшением их плотности (количества). Привыкание к некоторым ЛВ связано с изменением их фармакокинетики (уменьшением всасывания, увеличением скорости метаболизма и/ или выведения). Так, основной причиной привыкания к фенобарбиталу считают активацию его метаболизма вследствие индукции ферментов печени, вызываемой самим фенобарбиталом. Привыкание к ЛВ может развиться в течение нескольких дней или месяцев. В случае развития привыкания делают перерыв в применении данного вещества, а при необходимости продолжения лечения назначают препараты с аналогичным действием, но из другой химической группы. При замене одного вещества на другое независимо от его химической структуры может возникнуть перекрестное привыкание (если эти вещества взаимодействуют с одними и теми же рецепторами или ферментами). Частный случай привыкания — тахифилаксия (от греч. tachys — быстрый, phylaxis — защита) — быстрое развитие привыкания при повторных введениях препарата через короткие промежутки времени (10–15 мин). Хорошо известна тахифилаксия к эфедрину, обусловленная истощением запасов норадреналина в окончаниях симпатических нервных волокон. С каждым последующим введением эфедрина количество выделяющегося в синаптическую щель норадреналина уменьшается, и гипертензивный эффект препарата (повышение АД) ослабляется. Другой частный случай привыкания — митридатизм — постепенное развитие нечувствительности к действию ЛС и ядов, возникающее при длительном их применении вначале в очень малых, а затем в возрастающих дозах. Согласно древнегреческой легенде, царь Митридат приобрел таким образом нечувствительность ко многим ядам. При повторном приеме некоторых веществ, вызывающих чрезвычайно приятные ощущения (эйфорию), у предрасположенных лиц развивается лекарственная зависимость. Лекарственная зависимость — настоятельная потребность (непреодолимое стремление) в постоянном или периодически возобновляемом приеме определенного ЛВ или группы веществ. Вначале вещество принимают для достижения состояния эйфории, благополучия и комфорта, устранения тягостных переживаний, испытания новых ощущений. Однако Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 135 через определенное время потребность в повторном приеме становится непреодолимой, что усугубляется синдромом отмены: возникновением при прекращении приема данного вещества тяжелого состояния, связанного с психическими и соматическими нарушениями (нарушениями функции органов и систем организма). Такое состояние обозначают термином «абстиненция» (от лат. abstinentia — воздержание). Различают психическую и физическую лекарственную зависимость. ● Психическая лекарственная зависимость характеризуется резким ухудшением настроения и эмоциональным дискомфортом, ощущением усталости при лишении препарата. Она возникает при применении кокаина и других психостимуляторов (амфетамина), галлюциногенов (диэтиламид лизергиновой кислоты, LSD-25), никотина, индийской конопли (анаша, гашиш, план, марихуана). ● Физическая лекарственная зависимость характеризуется не только эмоциональным дискомфортом, но и возникновением синдрома абстиненции — состояния, включающего объективные и субъективные нарушения. Физическая лекарственная зависимость развивается к опиоидам (героину, морфину), барбитуратам, бензодиазепинам, алкоголю (этиловому спирту). Лекарственная зависимость часто сочетается с привыканием, при этом для получения эйфории требуются все более высокие дозы вещества. Наиболее тяжело лекарственная зависимость протекает в случае сочетания психической зависимости, физической зависимости и привыкания. Токсикомания — использование веществ с целью получения одурманивающего действия. Наркомания — частный случай токсикомании, когда в качестве одурманивающего средства используют вещество, отнесенное к перечню веществ, вызывающих лекарственную зависимость (наркотических веществ) и подлежащих контролю. 3.3.2. Комбинированное применение и взаимодействие лекарственных веществ При комбинированном применении ЛВ их действие может усиливаться (синергизм) или ослабляться (антагонизм). Синергизм (от греч. syn — вместе, erg — работа) — однонаправленное действие двух или нескольких ЛВ, при котором развивается фармакологический эффект, превышающий эффекты каждого вещества в отдельности. 136 Часть I. Общая фармакология Если эффект комбинированного применения ЛВ равен сумме эффектов отдельных веществ, входящих в комбинацию, действие определяют как суммирование, или аддитивное действие. Суммирование возникает при введении в организм ЛВ, влияющих на одни и те же субстраты (рецепторы, клетки и др.). Например, суммируются сосудосуживающие и гипертензивные эффекты норэпинефрина и фенилэфрина, стимулирующих α-адренорецепторы периферических сосудов; суммируются эффекты средств для ингаляционного наркоза. Если одно вещество значительно усиливает фармакологический эффект другого вещества, такое взаимодействие называют потенцированием. При потенцировании общий эффект комбинации двух веществ превышает сумму их эффектов. Например, хлорпромазин (антипсихотическое средство) потенцирует действие средств для наркоза, что позволяет снизить концентрацию последних. ЛВ могут действовать на один и тот же субстрат (прямой синергизм) или иметь разную локализацию действия (косвенный синергизм). Явление синергизма часто применяют в медицинской практике, так как оно позволяет получить желаемый фармакологический эффект при назначении нескольких ЛС в меньших дозах. При этом риск появления побочных эффектов уменьшается. Антагонизм (от греч. anti — против, agon — борьба) — уменьшение или полное устранение фармакологического эффекта одного ЛВ другим при их совместном применении. Явление антагонизма используют при лечении отравлений и для устранения нежелательных реакций на ЛС. Различают следующие виды антагонизма: прямой функциональный антагонизм, косвенный функциональный антагонизм, физический антагонизм, химический антагонизм. ● Прямой функциональный антагонизм развивается, когда ЛВ оказывают противоположное (разнонаправленное) действие на одни и те же функциональные элементы (рецепторы, ферменты, транспортные системы и др.). Например, к функциональным антагонистам относятся стимуляторы и блокаторы β-адренорецепторов, стимуляторы и блокаторы М-холинорецепторов. Частный случай прямого антагонизма — конкурентный антагонизм, возникающий у ЛВ, имеющих близкую химическую структуру и конкурирующих за связь с рецептором. Так, в качестве конкурентного антагониста морфина и других наркотических анальгетиков применяют налоксон. Некоторые ЛВ имеют сходную химическую структуру с метаболитами микроорганизмов или опухолевых клеток и вступают с ними в конкуренцию за участие в одном из звеньев биохимического процесса. Такие ве- Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 137 щества получили название антиметаболиты. Замещая один из элементов в цепи биохимических реакций, антиметаболиты нарушают размножение микроорганизмов, опухолевых клеток. Например, сульфаниламиды — конкурентные антагонисты парааминобензойной кислоты, необходимой для развития некоторых микроорганизмов, метотрексат — конкурентный антагонист дигидрофолатредуктазы в опухолевых клетках. ● Косвенный функциональный антагонизм развивается в тех случаях, когда ЛВ оказывают противоположное влияние на работу какоголибо органа, и при этом в основе их действия лежат разные механизмы. Например, к косвенным антагонистам в отношении действия на гладкомышечные органы относят ацеклидин (повышает тонус гладкомышечных органов, стимулируя М-холинорецепторы) и папаверин (снижает тонус гладкомышечных органов вследствие прямого миотропного действия). ● Физический антагонизм возникает в результате физического взаимодействия ЛВ: адсорбции одного ЛВ на поверхности другого, в результате чего образуются неактивные или плохо всасывающиеся комплексы (например, адсорбция ЛВ и токсинов на поверхности активированного угля). Явление физического антагонизма применяют при лечении отравлений. ● Химический антагонизм возникает в результате химической реакции между веществами, в результате которой образуются неактивные соединения или комплексы. Антагонисты, действующие подобным образом, получили название антидотов. Например, при отравлении соединениями мышьяка, ртути, свинца применяют натрия тиосульфат, в результате химической реакции с которым образуются неядовитые сульфиты. При передозировке или отравлении сердечными гликозидами применяют дигибайнд — моноклональное антитело, образующее с ними неактивные комплексные соединения. При передозировке гепарина вводят протамина сульфат, катионные группы которого связываются с анионными центрами гепарина, нейтрализуя его антикоагулянтное действие. При комбинированном назначении ЛС следует убедиться в отсутствии между ними антагонизма. Одновременное назначение нескольких лекарственных препаратов (полипрагмазия) может привести к изменению скорости возникновения фармакологических эффектов, их выраженности и продолжительности. Если в результате комбинированного применения ЛВ достигнут более выраженный терапевтический эффект, ослаблены или предупрежде- 138 Часть I. Общая фармакология ны отрицательные реакции, такое сочетание лекарственных препаратов считают рациональным и терапевтически целесообразным. Например, для предупреждения нейротоксического действия изониазида назначают витамин В6, для профилактики кандидоза как осложнения при лечении антибиотиками широкого спектра действия — нистатин или леворин, для устранения гипокалиемии при лечении салуретиками — калия хлорид. Если в результате одновременного применения нескольких ЛС ослаблен, предупрежден или извращен терапевтический эффект либо развиваются нежелательные эффекты, такие сочетания считают нерациональными, терапевтически нецелесообразными (несовместимость ЛС). Взаимодействие ЛС можно разделить на 2 группы: ● фармацевтическое взаимодействие; ● фармакологическое взаимодействие. Фармацевтическое взаимодействие возникает до введения ЛС в организм, т.е. на этапах изготовления, хранения или введения препаратов в одном шприце. Фармацевтическое взаимодействие, приводящее к невозможности дальнейшего использования препаратов, получило название фармацевтическая несовместимость. Ее причинами могут быть химическое взаимодействие ЛВ с образованием неактивных или токсических соединений, ухудшение растворимости ЛВ, коагуляция коллоидных систем, расслоение эмульсий, отсыревание и расплавление порошков, адсорбция одного ЛВ на поверхности другого и др. В результате образуется осадок, изменяются цвет, запах и консистенция ЛВ. Подобные нерациональные рецептурные прописи известны провизорам. Поэтому ЛВ по таким прописям либо не изготавливают, либо осуществляют их фармацевтическую коррекцию, в результате чего больной получает качественно приготовленное лекарство. Фармакологическое взаимодействие ЛВ происходит после введения их в организм больного. В результате такого взаимодействия могут изменяться фармакологические эффекты препаратов, введенных одновременно. Различают 2 вида фармакологического взаимодействия: ● фармакокинетический; ● фармакодинамический. Фармакокинетический вид взаимодействия лекарственных веществ Фармакокинетический вид взаимодействия возникает на этапах всасывания, распределения, метаболизма и выведения ЛВ. Возможно изменение одного или нескольких фармакокинетических параметров. Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 139 В результате фармакокинетического взаимодействия обычно изменяется концентрация активной формы ЛВ в крови и тканях и, как следствие, — конечный фармакологический эффект. Взаимодействие лекарственных веществ при всасывании из желудочнокишечного тракта При одновременном нахождении нескольких ЛВ в просвете желудка и тонкого кишечника могут изменяться степень и скорость всасывания или оба показателя одновременно. Причины таких изменений следующие. ● Изменение рН среды. Большинство ЛВ всасывается из ЖКТ в кровь с помощью пассивной диффузии. Таким путем всасываются липофильные неполярные молекулы ЛВ. Многие ЛВ являются слабыми основаниями или слабыми кислотами, и степень их ионизации зависит от рН среды. Изменяя рН среды желудка или кишечника, можно изменить степень ионизации молекул, а следовательно, и их всасывание. Например, антацидные средства, повышая рН желудочного содержимого, замедляют всасывание слабокислых соединений (дигоксина, барбитуратов, ацетилсалициловой кислоты), ослабляя тем самым их фармакологический эффект. ● Образование трудновсасывающихся комплексов. ЛС, обладающие адсорбционными свойствами (активированный уголь), анионообменные смолы (колестирамин), ионы кальция, магния, алюминия образуют со многими ЛВ комплексы, плохо всасывающиеся из ЖКТ. ● Изменение перистальтики ЖКТ. Стимуляция перистальтики кишечника под влиянием холиномиметических, антихолинэстеразных средств, слабительных средств уменьшает всасывание дигоксина. Блокатор М-холинорецепторов атропин, напротив, замедляет опорожнение желудка и улучшает всасывание дигоксина. Взаимодействие лекарственных веществ на этапе связывания с белками плазмы крови После поступления в системный кровоток многие ЛВ обратимо связываются с белками плазмы крови. На этом этапе может происходить лекарственное взаимодействие в результате конкурентного вытеснения одного ЛВ другим из комплекса с белком. Так, ацетилсалициловая кислота вытесняет толбутамид из комплекса с белками, в результате чего концентрация свободных молекул последнего возрастает, и его гипогликемический эффект усиливается. Таким же действием обладают сульфаниламиды. 140 Часть I. Общая фармакология Взаимодействие лекарственных веществ в процессе метаболизма Метаболизм многих ЛВ осуществляется с участием микросомальных ферментов печени. ЛВ способны изменять их активность. Активность ферментов печени индуцируют (повышают) фенобарбитал, рифампицин, фенитоин и др. Это приводит к ускорению инактивации и ослаблению терапевтического эффекта других ЛВ (например, дигоксина, теофиллина, хинидина). Активность ферментов печени ингибируют верапамил, циметидин, ципрофлоксацин. В этом случае метаболизм таких веществ, как фенитоин, теофиллин, кофеин, снижается, их действие усиливается, и могут проявиться токсические эффекты. Некоторые ЛВ влияют на активность немикросомальных ферментов. Так, аллопуринол ингибирует ксантиноксидазу, участвующую в метаболизме меркаптопурина, и его токсическое действие на систему кроветворения резко возрастает. Взаимодействие лекарственных веществ в процессе выведения из организма Основной орган выведения ЛВ и их метаболитов — почки, поэтому рН мочи имеет большое значение для экскреции многих ЛВ. Этот показатель регулирует степень ионизации слабокислых и слабощелочных соединений, определяющую их реабсорбцию в почечных канальцах. При низких значениях рН мочи (в кислой среде) увеличивается выведение слабощелочных веществ (например, кофеина, морфина, диазепама), поэтому их действие ослабляется и укорачивается. При значениях рН мочи, соответствующих щелочной среде, ускоряется выведение слабых кислот (барбитуратов, салицилатов, сульфаниламидов), и их эффекты снижаются. Таким образом, вещества, изменяющие рН мочи, могут влиять на скорость выведения из организма слабокислых и слабощелочных ЛВ. Некоторые вещества, такие как натрия гидрокарбонат и аммония хлорид, применяют для ускорения выведения из организма слабых кислот и слабых оснований соответственно. Фармакодинамический вид взаимодействия лекарственных веществ Фармакодинамический вид взаимодействия ЛВ возникает в процессе реализации их фармакологического действия. Наиболее часто такое взаимодействие отмечают при одновременном применении агонистов и антагонистов одних и тех же рецепторов. Например, блокаторы адренорецепторов (фентоламин, пропранолол) снижают или полностью устраняют действие стимуляторов адренорецепторов (норэпинефрина и изопреналина соответственно). Симпатолитики (резерпин, гуанетидин) Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 141 ослабляют действие симпатомиметика эфедрина, поскольку оказывают противоположное влияние на выделение норадреналина из окончаний адренергических нервов. К ослаблению эффектов приводит одновременное применение ЛС, действующих противоположным образом на системы организма, например угнетающих (снотворные, седативные средства) и стимулирующих (психостимуляторы) ЦНС. К усилению действия приводит одновременное применение ЛС, оказывающих однонаправленное действие. Имея четкие представления о видах взаимодействия ЛС, провизор может дать следующие рекомендации для предупреждения нежелательных последствий для больного комбинированного приема лекарственных препаратов: ● принимать лекарственные препараты не одновременно, а с интервалами в 30, 40, 60 мин; ● заменить один из лекарственных препаратов на другой; ● изменить режим дозирования (дозу и интервал между введениями) препаратов; ● отменить один из препаратов (если первые три действия не устраняют отрицательных последствий взаимодействия назначенной комбинации препаратов). 3.4. ХРОНОФАРМАКОЛОГИЯ Фармакокинетика и фармакодинамика ЛВ зависят от того, в какое время их принимает пациент, что связано с периодическими (циклическими) изменениями активности эндогенных биологически активных веществ (ферментов и др.), а также с другими ритмическими процессами в организме. Изучением ритмических процессов в живой природе и роли фактора времени в биологических процессах занимается хронобиология (от греч. сhronos — время) — направление в биологии, сформировавшееся в 60-е годы прошлого столетия. Один из разделов хронобиологии — хронофармакология, изучающая периодические изменения активности ЛВ в зависимости от времени введения и влияния ЛВ на биологические ритмы организма. Биологические ритмы — периодически повторяющиеся изменения характера и интенсивности биологических процессов (рис. 3.2): ● акрофаза — время, когда исследуемая функция или процесс достигает своих максимальных значений; 142 Часть I. Общая фармакология ● батифаза — время, когда исследуемые функция или процесс достигают своих минимальных значений; ● амплитуда — степень отклонения исследуемого показателя в обе стороны от средней; мезор (от лат. мesos — средний) — среднесуточное значение ритма, т.е. среднее значение исследуемого показателя в течение суток. Периоды биологических ритмов приурочены к определенному времени, например: ● циркадианные (околосуточные, от лат. сirca — около, dies — день) — с периодом 20–28 ч; ● околочасовые — с периодом 3–20 ч; ● инфрадианные — с периодом 28–96 ч; ● околонедельные — с периодом 4–10 сут; ● околомесячные — с периодом 25–35 сут. В хронофармакологии приняты следующие термины: хронофармакокинетика (хронокинетика), хронестезия и хронергия. Хронофармакокинетика (хронокинетика) включает ритмические изменения всасывания, распределения, метаболизма и выведения ЛВ. Хронестезия — ритмические изменения чувствительности организма к ЛВ в течение суток. Хронергия — совокупное влияние хронокинетики и хронестезии на величину фармакологического эффекта ЛВ. Эффект (сила и длительность) Рис. 3.2. Основные характеристики колебаний биологических ритмов Глава 3. Влияние различных факторов на фармакодинамику и фармакокинетику… 143 от применения одной и той же дозы вещества различается в зависимости от времени суток. Так, нитроглицерин эффективнее устраняет приступ стенокардии при приеме в утренние часы, чем во второй половине дня. Глюкокортикоиды наиболее активны при приеме в 8 ч утра, а морфин — в 16 ч. Для некоторых ЛВ известны изменения фармакокинетических параметров в зависимости от времени суток. Так, противогрибковый препарат гризеофульвин лучше всасывается при приеме приблизительно в 12 ч дня, амфетамин лучше выводится почками ранним утром. Данные об изменении концентрации некоторых ЛВ в течение суток приведены в табл. 3.1. Таблица 3.1. Хронофармакокинетика (A. Reinberg, M. Smolensky) Лекарственный препарат Время введения, ч Показатели исследования Суточные изменения фармакокинетических параметров Пик концентрации и площадь под фармакокинетической кривой наибольшие в 6 ч утра, наименьшие — в 23 ч Высокий пик концентрации в 8 ч, самое быстрое исчезновение ЛВ — в 19 ч Высокий пик концентрации в7ч Ацетилсалициловая 6; 10; 18; 20 кислота (1,5 г однократно) Концентрация в плазме крови Индометацин (100 мг однократно) Теофиллин (4 мг/кг) 7; 11; 15; 19 Концентрация в плазме крови 7; 13; 19 Концентрация в плазме крови и в слюне Концентрация Пик концентрации и площадь в плазме крови под фармакокинетической кривой меньше после введения препарата в 14 ч по сравнению с 8, 20 и 2 ч ночи Концентрация Пик концентрации в плазме крови наибольший — в 11 ч 30 мин, Smax — в 12 ч Экскреция Пик концентрации и площадь с мочой под фармакокинетической и креатин мочи кривой наибольшие в 6 ч, нефротоксичность минимальна в 18 ч Пропранолол (80 мг однократно) 2; 8; 14; 20 Эритромицин (250 мг) 2; 8; 14; 20 Цисплатин (60 мг/м² внутривенно) 6; 18 Хронофармакологический подход к назначению ЛС имеет значение для их рационального дозирования в зависимости от времени их приема. При традиционной терапии назначают фиксированные дозы (например, 144 Часть I. Общая фармакология по 1 таблетке 3 раза в день), а при хронотерапии применяют динамические дозы с учетом циркадианных колебаний чувствительности и реактивности организма и ритма фармакокинетических процессов. Цель хронотерапии — достижение максимального лечебного эффекта при наименьших дозах ЛВ и, соответственно, уменьшение побочных эффектов. На основе экспериментальных и клинических исследований, выполненных на кафедре фармакологии фармацевтического факультета ММА им. И.М. Сеченова и ВМА им. С.М. Кирова, были выявлены хронофармакологические особенности в действии ряда ЛВ. Так, наибольшая чувствительность к действию внутривенно введенного стрихнина проявлялась в 16 ч, наибольшая устойчивость — в 10 ч утра. Установлена также роль сезонных факторов в действии некоторых ЛВ. Адаптогенное действие женьшеня, элеутерококка корневищ и корней, родиолы розовой корневищ и корней, аралии маньчжурской корней у больных с хирургическими и неврологическими болезнями наиболее выражено в период января–марта, а в летние месяцы значительно снижается. Кроме того, в весенне-летний период антигипоксический эффект женьшеня и элеутерококка отсутствует в широком диапазоне исследованных доз. Вопросы и задания для самоконтроля Накопление ЛВ в организме при повторных введениях называется: а) привыканием; б) тахифилаксией; в) физической зависимостью; г) материальной кумуляцией; д) абстиненцией. Глава 4 ПОНЯТИЕ О ФАРМАКОПРОФИЛАКТИКЕ И ФАРМАКОТЕРАПИИ. ВИДЫ ЛЕКАРСТВЕННОЙ ТЕРАПИИ Фармакопрофилактика — предупреждение заболеваний с помощью ЛС. С профилактической целью для предупреждения распространения инфекционных заболеваний применяют антисептические и дезинфицирующие ЛС, для профилактики гиповитаминозов — витаминные препараты, для профилактики эндемического зоба — препараты йода и др. Фармакотерапия (лекарственная терапия) — лечение заболеваний с помощью ЛС. Для будущих провизоров фармакотерапия соответствует учебной дисциплине «клиническая фармакология» и является следующей после общей и частной фармакологии ступенью в освоении науки о взаимодействии ЛС с живыми организмами. Применение ЛС для предупреждения и лечения основано на знаниях причин и условий возникновения (этиологии), механизмов развития (патогенеза) и внешних проявлений (симптомов) заболеваний. Различают следующие виды лекарственной терапии. ● Этиотропная терапия (от греч. aethia — причина, tropos — направление) направлена на устранение или ослабление причины заболевания. ЛС, устраняющие причину заболевания, называют этиотропными. К ним относят химиотерапевтические средства, подавляющие жизнедеятельность патогенных микроорганизмов, вывзывающих инфекционные заболевания, антидоты, связывающие токсические вещества, вызывающие отравления. ● Патогенетическая терапия (от греч. pathos — болезнь, genesis — происхождение) направлена на ограничение или устранение механизмов развития заболевания. ЛС, применяемые с этой целью, называют патогенетическими. Так, антигистаминные средства устраняют действие гистамина, выделяющегося при аллергической реакции, но не прекращают контакта организма с аллергеном и не устраняют причины развития аллергической реакции. Сердечные гликозиды 146 Часть I. Общая фармакология повышают сократимость миокарда при сердечной недостаточности, но не устраняют причин, вызвавших ее. ● Заместительная терапия направлена на восполнение дефицита эндогенных веществ в организме. С этой целью применяют соляную кислоту и ферментные препараты при недостаточной функции пищеварительных желез, гормональные препараты — при гипофункции эндокринных желез, витаминные препараты при гиповитаминозах. Препараты заместительной терапии не устраняют причины заболевания, но ослабляют или устраняют проявления дефицита того или иного вещества, необходимого для жизнедеятельности организма. Как правило, такие препараты применяют длительно. ● Симптоматическая терапия направлена на ограничение или устранение симптомов заболевания. ЛС, применяемые с этой целью, называют симптоматическими. Они не оказывают влияния на причину и механизмы развития заболевания. Например, болеутоляющие и жаропонижающие средства уменьшают выраженность таких симптомов различных заболеваний, как боль и повышенная температура тела. Вопросы и задания для самоконтроля 1. Применение антиконгестантов (средств, уменьшающих отек слизистой оболочки носа) при остром респираторном заболевании относится к: а) этиотропной терапии, потому что устраняет причину неприятных ощущений; б) патогенетической терапии, потому что уменьшает выраженность патологического процесса в слизистой оболочке носа; в) симптоматической, потому что при этом устраняется лишь симптом заболевания без влияния на возбудителя болезни и вызванный им патологический процесс. 2. Введение инсулина при 1 типе сахарного диабета является: а) заместительной терапией, потому что это заболевание характеризуется абсолютной недостаточностью инсулина; б) этиотропной терапией, потому что устраняет главную причину развития заболевания; в) симптоматической терапией, потому что введение инсулина приводит к устранению симптомов сахарного диабета. Глава 5 ПОБОЧНЫЕ И ТОКСИЧЕСКИЕ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ 5.1. ПОБОЧНЫЕ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ ЛВ используют для получения определенного лечебного эффекта. Фармакологический эффект, являющийся целью применения лекарственного препарата, получил название основного (главного) эффекта. Основной эффект может быть профилактическим или лечебным. Например, антибиотики могут предупреждать инфекционные осложнения после хирургической операции, анальгетики назначают для устранения боли и т.д. Побочный эффект — любой непреднамеренный эффект фармацевтического продукта, развивающийся при использовании в обычных (терапевтических) дозах и обусловленный его фармакологическим действием. Побочные эффекты вызывают практически все известные ЛВ. Побочные эффекты ЛВ в большинстве случаев известны и ожидаемы и обычно исчезают после прекращения приема или снижения дозы (или скорости введения) препарата. Однако в ряде случаев с побочными эффектами может быть связано такое серьезное осложнение, как «лекарственная болезнь». Точное соблюдение предписаний врача и требований инструкции к препарату во многих случаях позволяют избежать серьезных проявлений побочного действия. Осложнения лекарственной терапии могут развиться через несколько секунд и минут (особенно при внутривенном и ингаляционном введении препарата), через несколько часов (при приеме внутрь) и даже через несколько дней и месяцев. Осложнения могут быть кратковременными (несколько минут и часов), средней продолжительности (несколько дней) и длительными (несколько недель, месяцев, лет). Степень тяжести осложнений может колебаться от кратковременного дискомфорта до серьезных нарушений функций вплоть до летального исхода. 148 Часть I. Общая фармакология При выявлении опасных для жизни побочных эффектов, не указанных в аннотации на препарат, повлекших временную потерю трудоспособности или госпитализацию пациента, а также любых побочных эффектов, связанных с приемом нового ЛС (при его клинических испытаниях), провизор обязан оказать неотложную доврачебную помощь, заполнить «Карту-извещение регистрации побочных реакций» установленной формы и известить руководство аптечного учреждения и лечащего врача; в других случаях достаточно рекомендовать обратиться к врачу. 5.2. ТОКСИЧЕСКИЕ ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ ЛС, назначаемые женщинам во время беременности, могут оказывать отрицательное влияние на развитие эмбриона или плода. ● Эмбриотоксическое действие (от греч. embryon — зародыш) ЛС развивается в первые 12 нед беременности. На ранних стадиях беременности (1–3 нед) оно является следствием действия ЛВ на зиготу и бластоцист, находящиеся в просвете фаллопиевых труб и матки (до имплантации), а также на процесс имплантации зародыша в матку; как правило, проявляется нарушением развития эмбриона. Эмбриотоксическое действие могут оказывать антиметаболиты (меркаптопурин, фторурацил), антимитотические средства (колхицин и др.). ● Тератогенное действие (от греч. teras — урод) развивается в период 4–8 нед беременности. В это время происходят формирование скелета и закладка внутренних органов. На этой стадии беременности органы и ткани наиболее чувствительны к действию повреждающих факторов, поэтому применение ЛВ может привести к аномалиям развития скелета и внутренних органов. Примером тератогенного действия ЛВ может служить так называемая талидомидная катастрофа: возникновение врожденных уродств у новорожденных в Западной Европе, Америке и Австралии, обусловленное приемом женщинами в ранние сроки беременности снотворного препарата талидомида. При его приеме наиболее часто возникали аномалии развития конечностей: фокомелия (от греч. phoke — тюлень, melos — конечность). Руки и ноги ребенка имели форму ласт. Известно, что тератогенное действие могут оказывать противоопухолевые средства — антагонисты фолиевой кислоты (метотрексат), этиловый спирт (принимаемый женщиной при зачатии). Глава 5. Побочные и токсические действия лекарственных веществ 149 ● Фетотоксическое действие (от греч. fetus — плод) — следствие влияния ЛВ на плод в период, когда уже сформированы внутренние органы и физиологические системы. Например, антикоагулянты могут снижать свертываемость крови и провоцировать кровотечения у новорожденного, производные сульфонилмочевины — вызывать гипогликемию; алкоголь, наркотики, снотворные и транквилизаторы — угнетение ЦНС. Поскольку ЛС способны вызывать гибель эмбриона и аномалии развития плода, применение их беременными должно находиться под строгим контролем. ● Мутагенное действие (от лат. mutatio — изменение и греч. genos — род) — способность ЛВ вызывать изменение генетического аппарата в женских и мужских половых клетках на стадии их формирования и в клетках эмбриона. Канцерогенное действие (от лат. cancer — рак) — способность ЛВ вызывать развитие злокачественных новообразований. При создании лекарственных препаратов и внедрении их в медицинскую практику эмбриотоксическое, тератогенное, фетотоксическое, мутагенное и канцерогенное действия у них должны быть исключены. Дисбиоз — нарушение естественного состава микрофлоры слизистых оболочек (ЖКТ, полости рта, влагалища и др.). Это состояние развивается в результате гибели полезной микрофлоры под влиянием некоторых противомикробных средств, в основном антибиотиков широкого спектра действия. На фоне угнетения полезной микрофлоры и иммунитета размножаются грибы рода Candida. Для профилактики и лечения кандидозов одновременно с антибиотиками применяют противогрибковые средства (нистатин), а также препараты, содержащие лиофилизированные бифидум- и лактобактерии (бифидумбактерин♠, бифидум♠ и др.). Аллергические реакции В основе аллергических реакций (от греч. allos — другой, ergon — действие), возникающих при повторном введении ЛВ, лежат иммунные механизмы. ЛВ в этом случае выступают как аллергены. Аллергические реакции не зависят от дозы вводимого вещества и разнообразны по своему характеру и тяжести: от легких кожных проявлений до анафилактического шока. Выделяют 4 типа аллергических реакций (по классификации Джелла–Кумбса). ● Немедленная типаллергическая реакция (анафилаксия) развивается в первые часы после введения в сенсибилизированный организм разрешающей дозы ЛВ (может быть очень маленькой). Ведущую 150 Часть I. Общая фармакология роль в развитии этого типа реакций играют иммуноглобулины типа E (IgE), антитела, связывающиеся с антигенами на поверхности тучных клеток и вызывающие их дегрануляцию и выделение гистамина. Клинические проявления — зудящие сыпи на коже (крапивница), отеки, насморк, слезотечение, а в тяжелых случаях — бронхоспазм, отек гортани, анафилактический шок. Такой тип реакции возможен при применении, например, антибиотиков пенициллинового ряда. ● Цитолитическая типреакция. При этом типе реакции IgG и IgМ, активируя систему комплемента, взаимодействуют с антигеном на поверхности форменных элементов крови и вызывают их лизис. Например, метилдопа может вызывать гемолитическую анемию, хинидин — тромбоцитопеническую пурпуру, метамизол натрия — агранулоцитоз. ● Иммунокомплексная типреакция. В этом случае IgG, IgM и IgE образуют комплексы с антигеном и комплементом, взаимодействующие с эндотелием сосудов и повреждающие его. В результате развивается сывороточная болезнь, проявляющаяся крапивницей, артралгией, артритом, лимфаденопатией, лихорадкой, зудом. Сывороточную болезнь могут вызывать, в частности, пенициллины, сульфаниламиды. ● Замедленная типаллергическая реакция возникает в виде контактного дерматита при нанесении ЛВ на кожу. В этот тип реакций вовлечены клеточные механизмы иммунитета, включающие сенсибилизированные Т-лимфоциты и макрофаги. Токсическое действие ЛВ развивается, как правило, при поступлении в организм токсических доз вещества (при передозировке). При абсолютной передозировке (введение ЛВ с абсолютным превышением разовых, суточных и курсовых доз) в крови и тканях создаются чрезмерно высокие его концентрации. Токсическое действие возникает также при относительной передозировке ЛВ, возникающей при назначении средних терапевтических доз больным со сниженными дезинтоксикационной функцией печени или выделительной функцией почек, при длительном лечении ЛВ, способными к кумуляции. В этих случаях ЛВ может оказывать токсическое действие на определенные органы или физиологические системы, например на жизненно важные центры продолговатого мозга (остановка дыхания при действии опиатов); на сердце — нарушение проведения возбуждения по проводящей системе сердца (при действии сердечных гликозидов); на печень — нарушение функции и некроз Глава 5. Побочные и токсические действия лекарственных веществ 151 (при применении парацетамола); на систему кроветворения — лейкопения, агранулоцитоз (при применении метамизола натрия). Классификация нежелательных побочных реакций В настоящее время выделяют следующие типы нежелательных побочных реакций (НПР). ● Тип А — предсказуемые НПР — частые реакции, связанные с фармакологической активностью ЛС. Эти реакции хорошо описаны, возникновение их обусловлено фармакологическим действием ЛС (например, изъязвление слизистой оболочки желудка на фоне приема ацетилсалициловой кислоты). ● Тип В — непредсказуемые НПР — независимые от дозы, не связанные с фармакологическим действием ЛС. В основе патогенеза НПР лежит индивидуальная чувствительность человека — лекарственная непереносимость, идиосинкразия, аллергические реакции. ● Тип С — возникновение НПР при длительном приеме ЛС. Возможно развитие толерантности, синдрома отмены, лекарственной зависимости, кумулятивных эффектов, эффектов подавления выработки гормонов. ● Тип D — НПР с отсроченными эффектами. Канцерогенные, мутагенные, тератогенные реакции, эмбриотоксическое действие. Диагностировать данные НПР очень сложно из-за длительного временного промежутка отделяющего прием ЛС и развитие опухоли или хромосомных и геномных мутаций. По тяжести клинического течения НПР подразделяют на следующие: ● легкие — отсутствует необходимость в отмене препарата и специальном лечении, клинические проявления исчезают самостоятельно с течением времени; ● умеренные — требующие отмены препарата и проведения специального лечения, увеличения сроков госпитализации; ● тяжелые — с угрозой для жизни больного и высоким риском развития инвалидизации, требующие увеличения сроков госпитализации; ● смертельные. Вопросы и задания для самоконтроля 1. В период с 1956 по 1962 г. беременным женщинам назначали талидомид в качестве седативного и противорвотного средства. Это привело к рождению детей с врожденным недоразвитием конечностей (фокомелия, от греч. phoke — тюлень и mйlos — часть тела) в результате: 152 Часть I. Общая фармакология а) эмбриотоксического действия; б) тератогенного действия; в) мутагенного действия; г) фетотоксического действия. 2. К какому типу нежелательных реакций относится действие талидомида: а) А; б) В; в) С; г) D? 3. Сыпь на теле после первого приема препарата, скорее всего, возникла в результате: а) аллергии; б) идиосинкразии; в) кумуляции. Часть II ЧАСТНАЯ ФАРМАКОЛОГИЯ НЕЙРОТРОПНЫЕ СРЕДСТВА В главах 6–16 рассмотрены нейротропные средства — ЛС, оказывающие действие на нервную регуляцию функций организма. Выделяют ЛС, преимущественно действующие на периферическую нервную систему, и ЛС с преимущественным действием на центральную нервную систему (головной и спинной мозг). В главах 6–9 рассмотрены ЛС, влияющие на периферическую нервную систему. Средства, влияющие на периферическую нервную систему Периферическая нервная система включает афферентную иннервацию (нервные волокна, по которым возбуждение от органов и тканей поступает в ЦНС) и эфферентную иннервацию (нервные волокна которой проводят возбуждение от ЦНС к органам и тканям). В главах 6–7 рассмотрены ЛС, действующие на афферентную иннервацию. В главах 8–9 рассмотрены ЛС, действующие на эфферентную иннервацию. СРЕДСТВА, ДЕЙСТВУЮЩИЕ НА АФФЕРЕНТНУЮ ИННЕРВАЦИЮ Афферентная иннервация представлена чувствительными нервными окончаниями (окончаниями чувствительных нервных волокон) и первичными афферентными нервными клетками, волокна которых входят в спинной мозг через задние рога. Чувствительные нервные окончания (чувствительные рецепторы) расположны в тканях и органах и способны воспринимать раздражения, в ответ на которые происходит генерация импульсов, распространяющихся по чувствительным нервным волокнам в ЦНС. Периферические окончания первичных афферентных соматических и висцеральных волокон отвечают на термические, механические и химические стимулы. На термические воздействия реагируют два типа чувствительных нейронов: первый тип нейронов возбуждается при температуре менее 10 °С, второй — при более 42 °С. Болевое воздействие связано с температурной активацией катионных каналов ванилоидных рецепторов. Ванилоидные рецепторы (TRPV1 и TRPV2) представляют собой доменные белки молекулярной массой около 95 кд, состоят из шести доменных белков и небольшого гидрофобного фрагмента (между пятым и шестым доменами), формирующего ионный канал. Лиганд этого рецептора — содержащееся в остром перце вещество капсаицин (от греч. capsicum — кусать, щипать), алкиламидамид гомованилиновой кислоты, откуда и возникло название «ванилоиды». Ванилоидные рецепторы активируются при ацидозе (один из факторов возникновения боли при ишемии и воспалении) и продуктами метаболизма арахидоновой кислоты. Тонкие демиелинизованные нервные окончания, ноцицепторы содержат также ионные каналы, чувствительные к ацидозу (ASIC), пуринергические рецепторы к АТФ (P2X и P2Y), кининовые рецепторы В1 и В2, чувствительные к цитокинам и бактериальным липополисахаридам. По волокнам первичных афферентных волокон возбуждение поступает в задние рога спинного мозга. Глава 6 СРЕДСТВА, УГНЕТАЮЩИЕ АФФЕРЕНТНУЮ ИННЕРВАЦИЮ ЛВ, угнетающие афферентную иннервацию, могут понижать чувствительность окончаний афферентных нервных волокон и/или угнетать проведение возбуждения по чувствительным нервным волокнам (местноанестезирующие средства). Также используют ЛВ, препятствующие воздействию раздражающих факторов (в том числе раздражающих веществ) на чувствительные нервные окончания (вяжущие, обволакивающие и адсорбирующие средства). 6.1. МЕСТНОАНЕСТЕЗИРУЮЩИЕ СРЕДСТВА (МЕСТНЫЕ АНЕСТЕТИКИ) Местноанестезирующие средства (от aesthesiа — ощущение и an — приставка отрицания) понижают чувствительность окончаний афферентных нервных волокон или угнетают проведение возбуждения по нервным волокнам. Афферентные волокна более чувствительны к местным анестетикам, чем эфферентные, поэтому вначале нарушается чувствительность и лишь позднее — двигательная активность. Местные анестетики менее эффективно блокируют миелинизированные волокна (миелин предотвращает проникновение местных анестетиков в клетку). Тонкие волокна более чувствительны, чем толстые. Местные анестетики в первую очередь устраняют болевую чувствительность, а затем температурную и другие виды чувствительности (в последнюю очередь — тактильную). В связи с эффективным угнетающим действием местных анестетиков на болевые рецепторы и чувствительные нервные волокна их применяют для местного обезболивания (местной анестезии). Механизм действия местных анестетиков связан с блокадой потенциалозависимых натриевых каналов клеточных мембран чувствительных нервных волокон. Местные анестетики (слабые основания) в неионизированной форме (рН крови 7,4) проникают через клеточную мембрану 156 Часть II. Частная фармакология внутрь аксона и там ионизируются. Ионизированные молекулы вещества взаимодействуют со специфическими местами связывания натриевых каналов с внутренней стороны мембраны и, блокируя натриевые каналы, препятствуют входу Nа+ в клетку и деполяризации мембраны. В результате нарушаются генерация потенциала действия и распространение импульсов по нервному волокну. Местные анестетики имеют сродство к натриевому каналу, находящемуся в инактивированном состоянии (потенциалозависимые натриевые каналы имеют три состояния: покоя — закрыт, активации — открыт и инактивированное — закрыт). Таким образом, местные анестетики блокируют переход этих каналов в открытое состояние. Действие местных анестетиков обратимо (после инактивации вещества функция чувствительных нервных окончаний и нервных волокон полностью восстанавливается). Среди одинаковых по диаметру волокон быстрее блокируются те, активность которых выше. Чувствительные волокна возбуждаются чаще, поэтому блокируются быстрее. Это объясняется тем, что при более частом возбуждении вероятность связывания анестетика с каналом в инактивированном состоянии выше (use-dependent block). Поскольку местные анестетики — слабые основания, степень их проникновения через мембрану зависит от рН среды (чем ниже рН, тем большая часть вещества находится в ионизированной форме и не проникает внутрь аксона). Поэтому эффективность местных анестетиков снижается в кислой среде (с пониженным рН), в частности при воспалении тканей. Большинство местных анестетиков имеет в основе ароматическую структуру (липофильный фрагмент), соединенную посредством эфирных или амидных связей (промежуточная цепочка) с аминогруппой (гидрофильный фрагмент). Для проявления местноанестезирующего действия необходимо оптимальное соотношение между липофильным и гидрофильным фрагментами молекулы. Характер промежуточной алифатической цепочки имеет значение для продолжительности действия вещества. Поскольку эфирные связи легче гидролизуются, сложные эфиры (прокаин) оказывают более короткое действие, чем амиды (лидокаин). В зависимости от способа применения местных анестетиков различают следующие основные виды местной анестезии. ● Поверхностная (терминальная) анестезия. При нанесении на поверхность слизистой оболочки вещество блокирует чувствительные нервные окончания (терминали), расположенные в слизистой оболочке, в результате чего она теряет чувствительность. Такое же действие местные анестетики могут оказывать при нанесении на раневую и язвенную поверхности. Для терминальной анестезии Глава 6. Средства, угнетающие афферентную иннервацию 157 используют вещества, легко проникающие через эпителий слизистых оболочек и, следовательно, достигающие чувствительных нервных окончаний. При терминальной анестезии сначала утрачивается болевая чувствительность, а затем температурная и тактильная чувствительности. Терминальную анестезию применяют в глазной практике для обезболивания конъюнктивы и роговицы глаза при диагностических или оперативных вмешательствах, в отоларингологии — при операциях в полости носа, в зеве, гортани, а также при интубации трахеи, бронхоскопии, цистоскопии и т.д. К этому методу анестезии прибегают также для устранения болей при ожогах, язвенной болезни желудка. Местные анестетики могут частично всасываться со слизистых оболочек и оказывать резорбтивное токсическое действие. Для уменьшения всасывания веществ в кровь, а следовательно, для уменьшения опасности возникновения резорбтивных эффектов и продления местноанестезирующего действия в растворы местных анестетиков добавляют сосудосуживающие вещества (эпинефрин). ● Проводниковая анестезия. При введении местного анестетика в ткани, окружающие нерв, возникает блок проведения возбуждения по чувствительным нервным волокнам. В результате происходит потеря чувствительности (в первую очередь болевой) в области, иннервируемой этими нервными волокнами. При воздействии на смешанный нерв блокируется проведение импульсов сначала по чувствительным, а затем и по двигательным волокнам нерва. Двигательные волокна имеют больший диаметр, поэтому местные анестетики диффундируют внутрь волокна этих нервов медленнее. Таким образом, двигательные волокна более устойчивы к воздействию местноанестезирующих веществ. Проводниковую анестезию используют для обезболивания при хирургических операциях, в том числе в стоматологической практике. Чем проксимальнее (т.е. ближе к месту выхода нерва из ЦНС) расположено место введения местного анестетика, тем обширнее область анестезии. Максимальная область анестезии возможна при воздействии местноанестезирующего вещества на корешки спинного мозга. Проводниковую анестезию подразделяют на эпидуральную (перидуральную) и спинномозговую (по действию ЛВ на передние или задние корешки спинного мозга). —При эпидуральной анестезии местный анестетик вводят в пространство над твердой оболочкой спинного мозга. —Спинномозговую анестезию осуществляют путем введения раствора местноанестезирующего вещества в спинномозговую жидкость 158 Часть II. Частная фармакология на уровне поясничного отдела спинного мозга. При этом происходит блокада проведения импульсов по чувствительным волокнам, поступающим в пояснично-крестцовый отдел спинного мозга, что приводит к развитию анестезии нижних конечностей и нижней части туловища (в том числе и внутренних органов). Спинномозговую анестезию используют для обезболивания при хирургических операциях (обычно на органах малого таза и нижних конечностях). ● Инфильтрационная анестезия — широко распространенный метод местной анестезии, проводимый путем послойного пропитывания тканей в области операции раствором местноанестезирующего вещества. При этом вещество действует и на чувствительные нервные окончания, и на чувствительные нервные волокна, расположенные в инфильтрируемых тканях. Для инфильтрационной анестезии используют растворы местных анестетиков низкой концентрации (0,25–0,5%) в больших количествах (200–500 мл), которые вводят в ткани (кожу, подкожную клетчатку, мышцы, ткани внутренних органов) под давлением. Инфильтрационную анестезию используют при операциях на внутренних органах и многих других видах хирургических вмешательств. Растворяют анестетики в гипотоническом (0,6%) или изотоническом (0,9%) растворе натрия хлорида. Поскольку местные анестетики при их введении в ткани могут всасываться в кровь и попадать в системный кровоток, при проведении проводниковой и инфильтрационной анестезии следует использовать малотоксичные вещества. Для уменьшения резорбтивного действия и удлинения эффекта местных анестетиков в их растворы добавляют сосудосуживающие вещества (например, эпинефрин). Для проводниковой, спинномозговой и инфильтрационной анестезии используют только стерильные растворы местных анестетиков. Поэтому для этих видов обезболивания пригодны только такие местноанестезирующие вещества, которые достаточно хорошо растворимы в воде и не разрушаются при стерилизации. Для повышения растворимости и стабильности местные анестетики выпускают в виде солей (гидрохлоридов). В настоящее время в медицинской практике используют местноанестезирующие вещества с различной степенью активности и разной продолжительностью действия. По применению в клинической практике местные анестетики подразделяют на: ● средства, применяемые только для поверхностной анестезии: кокаин, тетракаин (дикаин♠), бензокаин (анестезин♠), бумекаин (пиромекаин♠), прамокаин (прамоксин♠); Глава 6. Средства, угнетающие афферентную иннервацию 159 инфильтрационной и про● средства, применяемые преимущественно для ♠ водниковой анестезии: прокаин (новокаин ), тримекаин, бупивакаин (маркаин♠), мепивакаин (изокаин♠), артикаин (ультракаин♠); применяемые для всех видов анестезии: лидокаин (кси● средства, каин♠). По химическому строению местноанестезирующие вещества можно разделить на 2 группы: ● сложные эфиры (кокаин, тетракаин, бензокаин, прокаин); ● замещенные амиды кислот (лидокаин, тримекаин, бупивакаин, мепивакаин, бумекаин, артикаин). Амиды не гидролизуются под влиянием эстераз плазмы крови и тканей, поэтому вещества этой группы оказывают более продолжительное местноанестезирующее действие, чем сложные эфиры. 6.1.1. Средства, применяемые только для поверхностной анестезии Только для поверхностной анестезии применяют вещества, обладающие довольно высокой токсичностью, вследствие чего их нельзя применять при других видах анестезии (кокаин, тетракаин), а также вещества, плохо растворимые в воде (L-бензокаин). Кокаин — алкалоид кустарника Erythroxylon coca, произрастающего в Южной Америке. По химической структуре представляет сложный эфир бензойной кислоты и метилэкгонина. Препарат получают из растительного сырья, а также полусинтетическим путем из экгонина, используют в виде гидрохлорида. Кокаин обладает высокой местноанестезирующей активностью. Однако он быстро всасывается со слизистых оболочек и оказывает резорбтивное действие. Побочные и токсические эффекты, возникающие при этом, ограничивают применение препарата. Растворы кокаина иногда применяют для поверхностной анестезии в глазной практике (закапывают в полость конъюнктивы). Анестезия продолжается около 1 ч. Кокаин обладает сосудосуживающим действием (суживает сосуды склеры), расширяет зрачки. Внутриглазное давление обычно снижается, однако у некоторых людей возникает его резкое повышение. При длительном применении кокаин может вызвать изъязвление эпителия роговицы. После всасывания в кровь кокаин оказывает стимулирующее действие на ЦНС. Вначале он вызывает эйфорию, психомоторное возбуждение, уменьшает ощущение утомления, чувство голода и состояние беспокойства. Кокаин стимулирует дыхательный, сосудодвигательный и рвотный центры, может вызвать судороги. В достаточно высоких дозах кокаин 160 Часть II. Частная фармакология вызывает угнетение ЦНС и остановку дыхания (вследствие угнетения дыхательного центра). Кокаин вызывает тахикардию и суживает сосуды, в результате чего повышается АД. Это действие кокаина связано как со стимулирующим влиянием на сосудодвигательный центр, так и с усилением эффектов возбуждения адренергической иннервации. При отравлении кокаином проводят мероприятия по его удалению со слизистых оболочек (смывают изотоническим раствором натрия хлорида), из желудка (промывают 0,05–0,1% раствором калия перманганата, назначают адсорбирующие средства и солевые слабительные). При введении препарата в ткани для уменьшения всасывания накладывают жгут проксимальнее от места введения. Для купирования возбуждения внутривенно вводят диазепам. При необходимости применяют искусственное дыхание. Вследствие того, что кокаин вызывает состояние эйфории (повышение настроения, усиление положительных и устранение неприятных эмоций), повышает работоспособность, снимает ощущение усталости, при его хроническом применении (в основном это вдыхание через нос порошка кокаина, жевание листьев коки, иногда внутривенное введение) развивается лекарственная зависимость — кокаинизм. При этом резкое прекращение приема кокаина сопровождают депрессия, состояние усталости, неприятные ощущения, тягостное психическое состояние, что определяют как возникновение психической зависимости. Тетракаин (дикаин♠) представляет собой 2-диметиламиноэтиловый эфир пара-бутил-аминобензойной кислоты, применяют в виде гидрохлорида (рис. 6.1). Легко растворим в воде, спирте. Эффективное местноанестезирующее средство, значительно превосходящее по активности кокаин (примерно в 10 раз), он также превосходит кокаин по токсичности (в 2–5 раз), поэтому имеет ограниченное применение; используют для поверхностной анестезии. Относят к списку А. Применяют тетракаин в глазной практике в виде 0,1% раствора при измерении внутриглазного давления, в виде 0,25–1% или 2% раствора при удалении инородных тел и оперативных вмешательствах. Через 1–2 мин развивается выраженная анестезия. Тетракаин в отличие от кокаина не влияет на внутриглазное давление и не расширяет зрачки. Растворы, содержащие свыше 2% тетракаина, могут вызвать повреждение эпителия роговицы и значительное расширение сосудов конъюнктивы. Обычно для анестезии при глазных хирургических вмешательствах достаточно 0,5% раствора. При необходимости длительной анестезии используют глазные пленки с тетракаином (0,75 мг), изготовленные на ос- Глава 6. Средства, угнетающие афферентную иннервацию Сложные эфиры 161 Замещенные амиды кислот Рис. 6.1. Химические структуры некоторых местноанестезирующих средств нове биорастворимого полимера. Для анестезии слизистых оболочек носа и носоглотки используют 1–2% растворы тетракаина. Поскольку тетракаин легко всасывается через слизистые оболочки, он может вызвать резорбтивные токсические эффекты в виде возбуждения ЦНС, сменяющегося ее угнетением. При тяжелых отравлениях наступает смерть от паралича дыхательного центра. Для уменьшения всасывания тетракаина к его растворам добавляют эпинефрин, что приводит также к удлинению и усилению местноанестезирующего эффекта тетракаина. Бумекаин (пиромекаин♠) близок по химической структуре к тримекаину, применяют для поверхностной анестезии в стоматологии в виде 0,5–2% растворов и 5% мази. Бензокаин (анестезин♠) представляет собой этиловый эфир парааминобензойной кислоты; белый кристаллический порошок без запаха, слабогорького вкуса; вызывает на языке чувство онемения. В отличие от других местных анестетиков малорастворим в воде, хорошо растворим в спирте, жирных маслах. В связи с малой растворимостью в воде препарат применяют для поверхностной анестезии в составе мазей, паст, присыпок при крапивни- 162 Часть II. Частная фармакология це, заболеваниях кожи, сопровождающихся зудом, а также для обезболивания раневой и язвенной поверхностей. Используют также готовые лекарственные препараты «Меновазин»♠ и «Ампровизоль»♠. Аэрозоль «Ампровизоль»♠ содержит бензокаин, ментол♠, раствор эргокальциферола (витамин D2♠) в спирте, глицерин♠, прополис и этанол. Применяют для местного обезболивания при солнечных и термических ожогах I–II степени. Для анестезии слизистых оболочек применяют 5–20% масляные растворы бензокаина. Кроме того, бензокаин применяют в ректальных суппозиториях при заболеваниях прямой кишки (трещины, зуд, геморрой), он входит в состав комбинированных свечей «Анестезол»♠. Внутрь принимают в порошках, таблетках и слизистых микстурах для обезболивания слизистых оболочек при спазмах и болях в желудке, повышенной чувствительности пищевода, при рвоте, морской и воздушной болезни. Входит в состав комбинированных таблеток «Белластезин»♠. Прамокаин (прамоксин♠) — средство для поверхностной анестезии, не относящееся к эфирам аминобензойной кислоты. Обладает достаточно высокой активностью в отношении слизистых оболочек и кожи, однако его раздражающее действие слишком выражено для анестезии слизистой оболочки носа и глаз. Применяют при аллергии на производные аминобензойной кислоты. 6.2.2. Средства, применяемые преимущественно для инфильтрационной и проводниковой анестезии Прокаин (новокаин♠) представляет собой β-диэтиламиноэтиловый эфир парааминобензойной кислоты, выпускают в виде гидрохлорида, хорошо растворим в воде (1:1) и спирте (1:8). При введении в ткани прокаин вызывает выраженную анестезию продолжительностью 30–60 мин. Обладает относительно низкой токсичностью. Прокаин используют для инфильтрационной и проводниковой анестезии. Для инфильтрационной анестезии применяют 0,25–0,5% растворы; для анестезии по методу А.В. Вишневского (тугая ползучая инфильтрация) — 0,125–0,25% растворы; для проводниковой анестезии — 1–2% растворы. Иногда прокаин используют для спинномозговой анестезии (5% раствор). Поскольку прокаин плохо проникает через слизистые оболочки, для поверхностной анестезии его можно применять только в достаточно высоких концентрациях — в виде 10% раствора. В организме прокаин относительно быстро гидролизуется холинэстеразой плазмы крови и эстеразами тканей с образованием параамино- Глава 6. Средства, угнетающие афферентную иннервацию 163 бензойной кислоты и диэтиламиноэтанола. Парааминобензойная кислота по химическому строению близка к сульфаниламидам и является их конкурентным антагонистом. Именно поэтому при одновременном применении прокаина с сульфаниламидами их антимикробное действие ослабляется. Диэтиламиноэтанол обладает умеренными сосудорасширяющими свойствами, что может способствовать всасыванию прокаина в кровь. Для предупреждения всасывания к растворам прокаина добавляют 0,1% раствор эпинефрина. Причем это не только снижает возможность резорбтивного действия прокаина, но и усиливает и удлиняет его местноанестезирующее действие. При всасывании в кровь прокаин оказывает преимущественно угнетающее действие на нервную систему: ● угнетает висцеральные рефлексы и некоторые полисинаптические спинальные рефлексы; ● уменьшает выделение ацетилхолина из преганглионарных волокон (блокирует вегетативные ганглии) и окончаний двигательных волокон (в больших дозах нарушает нервно-мышечную передачу); ● уменьшает спазм гладких мышц; ● оказывает гипотензивное и кратковременное антиаритмическое действия (понижает автоматизм и возбудимость кардиомиоцитов, увеличивает эффективный рефрактерный период). Тримекаин по химической структуре близок к лидокаину (относится к замещенным амидам), применяют в виде гидрохлорида. Тримекаин в 2–3 раза активнее прокаина и действует более продолжительно (2–4 ч). По токсичности он также превышает прокаин. Применяют в основном для инфильтрационной (0,125–0,5% растворы) и проводниковой (1–2% растворы) анестезии. Для спинномозговой анестезии используют более высокие концентрации (5% раствор). При поверхностной анестезии он уступает многим местным анестетикам (эффективен только при применении 2–5% растворов). При резорбтивном действии тримекаин оказывает угнетающее влияние на ЦНС. Вызывает седативный и снотворный эффекты. При интоксикации возможны клонические судороги. Для уменьшения резорбтивных токсических эффектов и удлинения местноанестезирующего действия в растворы тримекаина добавляют эпинефрин. Бупивакаин по химической структуре близок к лидокаину (относят к замещенным амидам), используют в виде гидрохлорида. Бупивакаин — один из наиболее активных и длительно действующих местных анестетиков. Применяют для инфильтрационной (0,25%), проводниковой (0,25–0,5%), эпидуральной (0,75%), спинномозговой (0,25–0,5%) 164 Часть II. Частная фармакология анестезии; ретробульбарной блокады (0,75%). В акушерской и гинекологической практике используют только 0,25–0,5% растворы. Препарат обеспечивает выраженную и длительную (от 3 до 10 ч и более) анестезию. При интоксикации возможны судороги, угнетение сердечной деятельности (вплоть до остановки cepдца). Мепивакаин представляет собой метильный аналог бупивакаина. Применяют в виде 1–3% растворов. Действует быстро и относительно длительно (около 3 ч). Артикаин (ультракаин♠) — метиловый эфир 4-метил-3-[2-(пропиламино)-пропионамидо]-2-тиофенкарбоновой кислоты, используют в виде гидрохлорида. Оказывает быстрое и относительно длительное (1–3 ч) местноанестезирующее действие при инфильтрационной, проводниковой и спинномозговой анестезии. Применяемый в акушерской практике артикаин (2% раствор) считают препаратом выбора, так как он в значительно меньшей степени, чем другие местноанестезирующие средства, проникает через плацентарный барьер, вследствие чего не оказывает вредного влияния на плод. Применяют артикаин в стоматологической практике (вызывает анестезию даже при воспалительных заболеваниях полости рта). При использовании артикаина возможны побочные и токсические эффекты: головная боль, помутнение в глазах, диплопия, подергивания мышц; тошнота, рвота, в редких случаях — нарушение сознания (вплоть до полной его потери). Возможны также аллергические реакции: отек и покраснение кожи в месте инъекции, ангионевротический отек и др. Препарат противопоказан при повышенной чувствительности к самому артикаину и содержащемуся в его готовых лекарственных формах консерванту, при выраженных нарушениях ритма сердца, закрытоугольной глаукоме. Препарат не следует вводить внутривенно. Для снижения резорбтивных токсических эффектов и удлинения действия артикаина в растворы анестетика добавляют эпинефрин. 6.2.3. Средства, применяемые для всех видов анестезии Лидокаин (ксикаин♠) — 2-диэтиламино-2’,6’-ацетоксилидид, или α-диэтиламино-2,6-диметилацетанилид моногидрат, выпускают в виде гидрохлорида, хорошо растворим в воде, растворим в cпирте. Лидокаин — замещенный амид, поэтому он медленнее метаболизируется в организме и действует более продолжительно, чем прокаин. Лидокаин — эффективное местноанестезирующее средство, использующееся при всех видах местной анестезии: поверхностной, инфильтраци- Глава 6. Средства, угнетающие афферентную иннервацию 165 онной, проводниковой. По сравнению с прокаином он обладает большей местноанестезирующей активностью (примерно в 2,5 раза). Кроме того, лидокаин действует быстрее и в 2 раза продолжительнее, чем прокаин. Относительная токсичность лидокаина зависит от концентрации раствора. В малых концентрациях (0,5%) он существенно не отличается по токсичности от прокаина; с увеличением концентрации (1–2%) токсичность повышается. Лидокаин применяют: для инфильтрационной анестезии при хирургических вмешательствах; для проводниковой анестезии в стоматологии, хирургии конечностей; для блокады нервных сплетений; для эпидуральной и спинномозговой анестезии при операциях на органах малого таза, нижних конечностях; для терминальной анестезии слизистых оболочек в урологии, офтальмологии, стоматологии, при ожогах, при бронхоскопии и др. Для анестезии растворы лидокаина применяют парентерально и местно. Количество раствора и его концентрация зависят от вида анестезии и характера оперативного вмешательства. При интоксикации лидокаином возможны головная боль, головокружение, сонливость, беспокойство, шум в ушах, онемение языка и слизистой оболочки рта, нарушение зрения, судорожные подергивания, тремор, брадикардия. В тяжелых случаях возможно угнетение дыхания. Для снижения резорбтивных токсических эффектов и удлинения местноанестезирующего действия в раствор лидокаина добавляют 0,1% раствор эпинефрина. Лидокаин показан при непереносимости прокаина. В связи с тем, что при метаболизме лидокаина не происходит образования парааминобензойной кислоты, он не снижает антимикробного действия сульфаниламидов. Наряду с местноанестезирующей активностью лидокаин обладает выраженными антиаритмическими свойствами, поэтому его применяют также в качестве противоаритмического средства (см. главу «Антиаритмические средства»). 6.2. ВЯЖУЩИЕ, ОБВОЛАКИВАЮЩИЕ И АДСОРБИРУЮЩИЕ СРЕДСТВА Вяжущие, обволакивающие и адсорбирующие средства предохраняют окончания чувствительных нервов от воздействия на них различных раздражающих факторов, в том числе раздражающих веществ. 166 Часть II. Частная фармакология 6.2.1. Вяжущие средства Вяжущие средства вызывают частичную денатурацию белков слизи или раневого экссудата. Образовавшаяся белковая пленка защищает чувствительные нервные окончания от действия раздражающих веществ, снижая болевые ощущения. Кроме того, происходит местное сужение сосудов, снижаются их проницаемость и выделение экссудата, что способствует уменьшению воспалительной реакции. Такое действие оказывают многие вещества растительного происхождения (органические вяжущие средства), а также слабые растворы солей некоторых металлов (неорганические вяжущие средства). К органическим вяжущим средствам относят танин♠, отвар коры дуба, настои травы зверобоя, листьев шалфея, цветков ромашки, плодов черники и др. К неорганическим вяжущим средствам относят висмута субнитрат, дерматол♠, ксероформ♠, свинца ацетат, квасцы♠ — KAl (SO4)2, цинка сульфат, меди сульфат и др. Помимо вяжущего они оказывают некоторое противомикробное (антисептическое) действие, вызывая коагуляцию белков микробных клеток. Вяжущие средства применяют внутрь и местно. Показания к применению вяжущих средств: ● острые воспалительные заболевания ЖКТ (назначают вяжущие средства растительного происхождения, которые в случае инфекционной природы заболевания сочетают с антибактериальными препаратами); ● язвенная болезнь желудка и двенадцатиперстной кишки, хронические гастриты, дуодениты — применяют препараты растительного происхождения, а также висмута субнитрат (входит в состав комплексных таблеток «Викаир»♠ и «Викалин»♠); ● острые и хронические воспалительные заболевания полости рта, горла (назначают препараты растительного происхождения). Кроме того, вяжущие средства применяют в виде глазных капель при конъюнктивитах; в виде промываний при уретритах, вагинитах — цинка сульфат; в виде промываний и примочек — свинца ацетат, квасцы♠. В качестве вяжущего средства применяют галлодубильную кислоту, известную под названием танин♠. Танин♠ получают из чернильных орешков (Gallae turcicae), наростов на молодых побегах малоазиатского дуба, или из некоторых растений семейства сумаховых. Танин♠ используют при отравлении алкалоидами и солями тяжелых металлов, с которыми танин образует малорастворимые соединения (вво- Глава 6. Средства, угнетающие афферентную иннервацию 167 дят 0,5% водный раствор танина♠ для промывания желудка с последующим удалением промывных вод). C некоторыми алкалоидами (морфин, кокаин, атропин, никотин) танин♠ образует нестойкие соединения, требующие их быстрого удаления из желудка. Танин♠ применяют для полоскания рта и горла в виде 1–2% раствора, при ожогах и язвах — наружно в виде 3–10% растворов и мазей. Внутрь при диарее применяют соединения танина♠ с белком (танальбин♠, теальбин♠) для избежания нежелательного осаждения танина на слизистой оболочке желудка. При гастритах, энтеритах применяют внутрь, при колитах внутрь и в виде клизм настоев и отваров растений, содержащих танин♠ (травы зверобоя, листьев шалфея, цветков ромашки; корневища змеевика, кровохлебки, лапчатки; ягод черники и черемухи, коры дуба). При воспалительных заболеваниях кожи и слизистых оболочек (дерматиты, язвы, экземы) в виде присыпок и мазей применяют висмута субнитрат, а также препараты дерматол♠ (висмута субгаллат), ксероформ♠ (трибромфенолят висмута основной), оказывающие вяжущее и антисептическое действия. Эффект вяжущих средств непродолжителен и обратим, поэтому их применяют повторно. Обычно препараты этой группы хорошо переносимы. Побочные действия (тошнота, рвота, диспептические явления и др.) могут проявиться лишь при длительном применении препаратов, содержащих металлы, либо в случае индивидуальной их непереносимости. В больших концентрациях соли тяжелых металлов могут оказывать прижигающее действие. Растворы солей алюминия, серебра, меди, цинка используют как прижигающие (при обработке слизистых оболочек и кожи, для удаления избыточных грануляций) или кровоостанавливающие средства (при неглубоких порезах). 6.2.2. Обволакивающие средства Обволакивающие средства образуют с водой коллоидные растворы, покрывающие слизистые оболочки и препятствующие действию на них раздражающих веществ, защищая чувствительные нервные окончания. К обволакивающим средствам относят крахмальную слизь, слизь из семян льна и др. Растворы обволакивающих веществ образуют коллоидную пленку на воспаленных участках, язвах и таким образом предохраняют ткани и находящиеся в них окончания чувствительных нервов от раздражения. 168 Часть II. Частная фармакология Именно поэтому обволакивающие средства оказывают местное противовоспалительное и болеутоляющее действия. Они находят применение при воспалительных и язвенных поражениях слизистой оболочки желудка (гастрит, язвенная болезнь) и кишечника (энтероколит). Крахмальную и льняную слизи назначают совместно или непосредственно перед приемом ЛВ, обладающих раздражающими свойствами. Обволакивающие средства назначают продолжительное время (2–3 нед) при отравлениях кислотами и щелочами с целью защиты воспаленной и изъязвленной поверхности. 6.2.3. Адсорбирующие средства Адсорбирующие средства — тонко измельченные порошкообразные вещества, имеющие большую адсорбционную поверхность. Адсорбирующие вещества не должны растворяться в воде, оказывать раздражающее действие и вступать во взаимодействие с другими веществами. Покрывая кожу или слизистые оболочки, эти вещества адсорбируют на своей поверхности различные химические соединения и защищают чувствительные нервные окончания от их раздражающего действия. В качестве адсорбирующего средства можно применять тальк (4SiO2 3MgO H2O), адсорбирующий при нанесении на кожу выделения потовых желез и предохраняющий кожу от механического раздражения. Тальк входит в состав паст, обладающих подсушивающим действием. Одно из основных адсорбирующих средств в медицинской практике — уголь активированный. Это уголь растительного или животного происхождения, тонко измельченный и поэтому имеющий большую адсорбирующую поверхность. Препарат назначают внутрь при отравлении алкалоидами, солями тяжелых металлов, а также при пищевых интоксикациях. Применяют в виде взвеси в воде. При назначении внутрь уголь активированный адсорбирует токсичные вещества и препятствует их всасыванию в ЖКТ. Кроме того, таблетки активированного угля назначают при метеоризме (избыточном скоплении газов в кишечнике). Вопросы и задания для самоконтроля 1. Для всех видов анестезии применяют: а) прокаин; б) тетракаин; в) лидокаин; г) кокаин; д) бензокаин. Глава 6. Средства, угнетающие афферентную иннервацию 169 2. Эффективность блокады проведения по нерву зависит от: а) толщины нерва; б) миелинизации нерва; в) рН среды; г) состояния натриевых каналов; д) частоты импульсов, проводимых по нерву. 3. Местные анестетики связываются с потенциалозависимыми натриевыми каналами: а) в состоянии покоя; б) в активированном состоянии; в) в неактивированном состоянии; г) ковалентно; д) только афферентных нервов. 4. Вяжущие средства: а) раздражают рецепторы и вызывают локальное увеличение кровоснабжения; б) используются при миозитах; в) денатурируют белки раневой поверности; г) используются при ожогах и язвах; д) танин используют при отравлении морфином и солями тяжелых металлов. 5. Обволакивающие средства: а) обволакивают токсические вещества в ЖКТ и способствуют их выведению; б) применяют при воспалительных и язвенных поражениях ЖКТ; в) предохраняют нервные окончания пораженной области от раздражения; г) сорбируют токсические вещества; д) используются при отравлениях солями тяжелых металлов. Глава 7 СРЕДСТВА, СТИМУЛИРУЮЩИЕ ОКОНЧАНИЯ АФФЕРЕНТНЫХ НЕРВОВ B медицинской практике используют вещества, возбуждающие окончания чувствительных нервных волокон (чувствительные рецепторы) кожи и слизистых оболочек и не повреждающие расположенные рядом ткани. Некоторые вещества избирательно стимулируют определенные группы чувствительных рецепторов. К ним относят горечи (избирательно возбуждают вкусовые рецепторы), рвотные и отхаркивающие средства рефлекторного действия (избирательно возбуждают рецепторы желудка), слабительные средства (избирательно возбуждают рецепторы кишечника). (Подробнее об этих препаратах см. разделы «Средства, влияющие на функции органов пищеварения» и «Средства, влияющие на функции органов дыхания»). B медицинской практике используют также вещества, не избирательно стимулирующие чувствительные рецепторы кожи и слизистых оболочек, — раздражающие средства. Раздражающие средства стимулируют окончания чувствительных нервов кожи и слизистых оболочек. В качестве раздражающих средств используют горчичное эфирное масло, этанол (спирт этиловый♠) (20–40%), скипидар живичный, перцовый пластырь♠, аммиак (раствор аммиака 10%♠), рацементол (ментол♠) и др. Раздражающие вещества используют при воспалительных заболеваниях дыхательных путей, мышечных и суставных болях (миозитах, невритах, артритах и т.д.). В этом случае при воздействии на здоровые участки кожи, имеющие сопряженную иннервацию с пораженными органами или тканями, раздражающие вещества оказывают отвлекающее действие, уменьшая болевые ощущения. Отвлекающий эффект объясняют взаимодействием возбуждения, поступающего в ЦНС с пораженных органов, и возбуждения, поступающего с чувствительных рецепторов кожи, расположенных над этими органами (сегментарное строение тела), при действии на них Глава 7. Средства, стимулирующие окончания афферентных нервов 171 раздражающих веществ. При этом снижается восприятие афферентной импульсации с патологически измененных органов и тканей. При использовании раздражающих препаратов происходит также улучшение трофики органов и тканей, вовлеченных в патологический процесс. Трофическое действие раздражающих веществ объясняют тем, что при возбуждении чувствительных рецепторов кожи активируется симпатическая иннервация пораженных органов и тканей. Полагают, что возбуждение распространяется от рецепторов кожи к пораженным органам через разветвления постганглионарных симпатических волокон по типу аксон-рефлекса (минуя ЦНС). Трофическое действие может осуществляться и путем обычного кожно-висцерального рефлекса (через ЦНС). При раздражении кожи положительное влияние может оказывать высвобождение биологически активных веществ (гистамина, брадикинина и др.). Отвлекающее и трофическое действия оказывает горчичное эфирное масло, которое выделяется при применении горчичников. Горчичники — горчичная бумага, покрытая тонким слоем порошка из семян горчицы, содержащих гликозид синигрин и фермент мирозин. Перед употреблением горчичники помещают на короткое время в теплую воду (примерно 38 °С). Эта температура является оптимальной для ферментативной реакции, в результате которой под влиянием мирозина происходит расщепление синигрина с образованием действующего раздражающего вещества горчицы — эфирного горчичного масла (аллилизотиоцианата). Применяют горчичники при заболеваниях органов дыхания, стенокардии, невралгиях, миалгиях. В результате уменьшаются болевые ощущения, улучшается трофика соответствующих органов и тканей. Для получения отвлекающего эффекта используют и другие раздражающие средства. Спиртовые компрессы. Для компрессов применяют этанол (спирт этиловый 40%♠), так как именно в этой концентрации спирт оказывает выраженное раздражающее действие (в детской практике используют спирт этиловый 20%♠). Плоды перца стручкового, содержащие основной раздражающий гликозид капсаицин, используют для приготовления препаратов: настойки перца стручкового, пластыря перцового♠, кремов и мазей («Никофлекс»♠, «Эфкамон»♠). Скипидар живичный (масло терпентинное очищенное♠) — продукт перегонки живицы из сосны обыкновенной. Содержит липофильное вещество терпеновой структуры — α-пинен, проникающее через эпидермис и раздражающее чувствительные нервные окончания. Оказывает отвле- 172 Часть II. Частная фармакология кающее и антисептическое действия. Входит в состав мази скипидарной. Отвлекающим действием обладает мазь «Финалгон»♠. Эти препараты применяют как отвлекающие средства при радикулитах, миозитах, артритах. Раздражающие вещества, возбуждая чувствительные рецепторы слизистых оболочек, оказывают рефлекторное действие (возбуждение с чувствительных рецепторов передается по афферентным волокнам в ЦНС, в результате чего изменяется состояние соответствующих нервных центров и иннервируемых ими органов). Рефлекторное действие раздражающих веществ используют при применении аммиака (раствора аммиака♠), рацементола (ментола♠). Аммиак (раствор аммиака♠, нашатырный спирт, NH4OH) используют для рефлекторной стимуляции дыхательного центра при обморочных состояниях. Для этого вату, смоченную раствором аммиака♠, подносят к носу больного. Вдыхание паров аммиака♠ приводит к возбуждению окончаний чувствительных нервов верхних дыхательных путей. В результате рефлекторно возбуждается дыхательный центр, и больной приходит в сознание. Однако вдыхание больших количеств паров аммиака♠ может вызвать резкое снижение частоты сердечных сокращений, остановку дыхания. Рацементол (ментол♠) — основной компонент эфирного масла мяты перечной (спирт терпенового ряда). Оказывает избирательное возбуждающее влияние на холодовые рецепторы, вызывает ощущение холода, сменяемое местной анестезией. Раздражение ментолом♠ холодовых рецепторов полости рта сопровождается рефлекторным расширением спазмированных коронарных сосудов. На основе ментола выпускают препарат валидол♠ (25% раствор ментола в ментиловом эфире изовалериановой кислоты). Применяют рацементол (ментол♠) при воспалительных заболеваниях верхних дыхательных путей в виде капель, ингаляций и т.д. Рацементол (ментол♠) как отвлекающее средство входит в состав многих комбинированных препаратов для наружного применения (меновазин♠, бороментол♠, эфкамон♠ и др.). Вопросы и задания для самоконтроля 1. Укажите группы ЛВ, стимулирующих окончания афферентных нервов: а) вяжущие средства; б) горечи; в) спирт этиловый; г) отхаркивающие средства; д) обволакивающие средства. Глава 7. Средства, стимулирующие окончания афферентных нервов 2. Эффективность горчичников обусловлена: а) антибактериальным действием; б) повышением температуры кожи под горчичником; в) стимуляцией аксон-рефлекса. 173 СРЕДСТВА, ДЕЙСТВУЮЩИЕ НА ЭФФЕРЕНТНУЮ ИННЕРВАЦИЮ Эфферентная иннервация органов и тканей осуществляется из ЦНС и представлена двигательными нервными волокнами, иннервирующими скелетные мышцы (соматическая иннервация), и вегетативными нервными волокнами, иннервирующими внутренние органы, кровеносные сосуды, железы и др. (вегетативная иннервация). Соматическая нервная система включает центральную часть — двигательные нервные клетки (мотонейроны), тела которых расположены в головном и спинном мозге, и периферическую часть — длинные отростки мотонейронов (аксоны). Аксоны представляют собой двигательные нервные волокна, образующие контакты (синапсы) со скелетными мышцами, так называемые нервно-мышечные синапсы. В отличие от соматической вегетативная иннервация обеспечивается двумя последовательно расположенными нейронами. Тела первых нейронов находятся в ЦНС. Аксоны этих нейронов выходят из ЦНС и заканчиваются в вегетативных ганглиях, поэтому их называют преганглионарными волокнами. В ганглиях эти волокна образуют синаптические контакты со вторыми, ганглионарными, нейронами. Аксоны ганглионарных нейронов, называемые постганглионарными волокнами, оканчиваются на клетках иннервируемых органов (эффекторных органов). Исключение составляет вегетативная иннервация хромаффинных клеток мозгового вещества надпочечников, эмбриогенетически родственных нейронам симпатических ганглиев. Эти клетки иннервируются только преганглионарными нервными волокнами (схема 1). Вегетативная нервная система состоит из симпатического и парасимпатического отделов, имеющих анатомические и физиологические различия (схема 2). В симпатической системе тела преганглионарных нейронов находятся в боковых рогах торако-люмбального (грудного и поясничного) отдела спинного мозга, а в парасимпатической системе — в среднем и продолговатом мозге и сакральном отделе (крестцовой части) спинного мозга. Ганглии симпатической нервной системы локализованы вне иннервируемых органов — в двух симпатических стволах, расположенных по обе стороны позвоночника (вертебральные ганглии), а также в сим- Средства, действующие на эфферентную иннервацию 175 Схема 1. Схема эфферентной иннервации: АЦХ — ацетилхолин; Адр — адреналин; НА — норадреналин; М-ХР — М-холинорецепторы; Н-ХР — Н-холинорецепторы; А, Б, В, Г, Д — холинергические синапсы; Г — нервно-мышечный синапс; Е — адренергический синапс; 1 — ганглий парасимпатической системы; 2 — ганглий симпатической системы патических узлах (верхний и средний шейные узлы, чревное сплетение, верхний и нижний брыжеечные узлы). Парасимпатические ганглии локализованы в непосредственной близости или внутри эффекторных органов (интрамуральные ганглии). Передача возбуждения с преганглионарных волокон на постганглионарные нейроны в симпатических и парасимпатических ганглиях осуществляется с помощью медиатора ацетилхолина. Ацетилхолин выделяется также из окончаний нервных волокон, иннервирующих хромаффинные клетки мозгового вещества надпочечников, эмбриогенетически Схема 2. Схема вегетативной нервной системы Парасимпатическая иннервация s) gu (n. va Симпатическая иннервация 176 Часть II. Частная фармакология Средства, действующие на эфферентную иннервацию 177 родственные нейронам симпатических ганглиев (эти клетки выделяют адреналин и норадреналин). Кроме того, ацетилхолин является медиатором, передающим возбуждение с постганглионарных парасимпатических нервных волокон на эффекторные органы (см. схему 1). Передача возбуждения, осуществляемая посредством ацетилхолина, называется холинергической. Холинергическими называют также и нервные волокна, выделяющие ацетилхолин. Передача возбуждения с постганглионарных симпатических нервных волокон на эффекторные органы осуществляется другим медиатором — норадреналином и называется адренергической. Нервные волокна, выделяющие норадреналин, также называются адренергическими. Исключение составляют постганглионарные симпатические волокна, иннервирующие большинство потовых желез, и пилоэректоры (эти симпатические волокна выделяют ацетилхолин и поэтому относятся к холинергическим). Основные эффекты возбуждения симпатической иннервации: ● расширение зрачков (мидриаз) вследствие сокращения радиальной мышцы радужки; ● увеличение силы и частоты сердечных сокращений (увеличение сердечного выброса); ● увеличение скорости проведения импульсов по атриовентрикулярному узлу (облегчение атриовентрикулярной проводимости); ● повышение автоматизма проводящей системы сердца; ● сужение кровеносных сосудов; ● повышение АД вследствие увеличения сердечного выброса и сужения сосудов. Основные эффекты возбуждения парасимпатической иннервации: ● сужение зрачков (миоз) вследствие сокращения круговой мышцы радужки; ● спазм аккомодации (зрение устанавливается на ближнюю точку видения) вследствие сокращения цилиарной (ресничной) мышцы; ● уменьшение ЧСС; ● уменьшение скорости проведения импульсов по атриовентрикулярному узлу (ухудшение атриовентрикулярной проводимости); ● повышение тонуса бронхов; ● повышение тонуса гладких мышц ЖКТ, мочевого пузыря (тонус сфинктеров снижается), миометрия; ● увеличение секреции бронхиальных желез, пищеварительных желез (слюнных желез, желез ЖКТ), слезных и носоглоточных желез. ЛВ, влияющие на эфферентную иннервацию, действуют в области синапсов, т.е. контактов между окончаниями нервных волокон и ганглио- 178 Часть II. Частная фармакология нарными клетками или клетками эффекторных органов. Синапсы (от греч. sinapsis — соединение, связь) состоят из трех основных элементов: пресинаптической мембраны (мембраны нервного окончания), синаптической щели и постсинаптической мембраны (часть мембраны иннервируемой клетки, которая непосредственно граничит с нервным окончанием). В нервных окончаниях происходит синтез медиатора и его депонирование в синаптических пузырьках (везикулах). При деполяризации пресинаптической мембраны, вызываемой нервными импульсами, происходят выделение (экзоцитоз) содержимого везикул и высвобождение медиатора в синаптическую щель. Медиатор диффундирует через синаптическую щель и возбуждает специфические рецепторы, находящиеся на постсинаптической мембране. Возбуждение рецепторов на постсинаптической мембране клеток эффекторных органов приводит к усилению или угнетению их функции. Синапсы, в которых передача возбуждения осуществляется медиатором ацетилхолином, называют холинергическими, а синапсы, медиатором которых является норадреналин, — адренергическими. Выделяют 2 основные группы веществ, действующих на эфферентную иннервацию: ● средства, действующие на холинергические синапсы; ● средства, действующие на адренергические синапсы. Глава 8 СРЕДСТВА, ДЕЙСТВУЮЩИЕ НА ХОЛИНЕРГИЧЕСКИЕ СИНАПСЫ Холинергические синапсы локализованы во внутренних органах, получающих постганглионарные парасимпатические волокна, в вегетативных ганглиях, мозговом слое надпочечников, каротидных клубочках, скелетных мышцах. Передача возбуждения в холинергических синапсах происходит с помощью ацетилхолина. Ацетилхолин синтезируется в цитоплазме окончаний холинергических нервов из ацетил-КоА и холина при участии фермента холинацетилтрансферазы (холинацетилазы) и депонируется в синаптических пузырьках (везикулах). Под влиянием нервных импульсов ацетилхолин высвобождается из везикул в синаптическую щель. Происходит это следующим образом. Распространяющийся по аксону импульс вызывает деполяризацию пресинаптической мембраны окончания холинергического нерва, в результате чего открываются потенциалозависимые кальциевые каналы, через которые ионы кальция проникают в нервное окончание. Концентрация Са2+ в цитоплазме нервного окончания повышается, способствуя слиянию мембраны везикул с пресинаптической мембраной (слияние происходит вследствие взаимодействия белков везикулярной и пресинаптической мембран) и выделению содержимого везикул в синаптическую щель (рис. 8.1). Этот процесс носит название экзоцитоза. Слиянию везикулярной и пресинаптической мембран и, следовательно, высвобождению ацетилхолина в синаптическую щель препятствует ботулинический токсин. Высвобождение ацетилхолина блокируют также вещества, снижающие поступление Са2+ в цитоплазму нервных окончаний, например аминогликозидные антибиотики, ионы магния. После высвобождения в синаптическую щель ацетилхолин стимулирует холинорецепторы, локализованные на постсинаптической и пресинаптической мембранах холинергических синапсов. В синаптической щели ацетилхолин очень быстро гидролизуется ферментом ацетилхолинэстеразой с образованием холина и уксусной кислоты. Холин захватывается нервными окончаниями (подвергается 180 Часть II. Частная фармакология холин АцКоА Гемихолиний Na+ Везамикол АцКоА+холин АцХ Ca++ АцХ Ca++ Ботулинический токсин АцХ холин М-холиномиметики М-холиноблокаторы ацетат Ганглиоблокаторы Курареподобные средства Антихолинэстеразные средства Рис. 8.1. Схема холинергического синапса. Локализация действия веществ, влияющих на холинергическую иннервацию: АцХ — ацетилхолин; АцКоА — ацетилкоэнзим А; Н-ХР — никотиновый холинорецептор; М-ХР — мускариновый холинорецептор; АХЭ — ацетилхолинэстераза; SNAPs — synaptosome-associated proteins; VAMPs — vesicle-associated membrane proteins обратному нейрональному захвату) и вновь включается в синтез ацетилхолина. В плазме крови, печени и других органах присутствует фермент бутирилхолинэстераза (псевдохолинэстераза, ложная холинэстераза), которая также может инактивировать ацетилхолин. На передачу возбуждения в холинергических синапсах могут воздействовать вещества, оказывающие влияние на следующие процессы: ● синтез ацетилхолина и его депонирование в везикулах; ● высвобождение ацетилхолина; ● взаимодействие ацетилхолина с холинорецепторами; ● гидролиз ацетилхолина в синаптической щели; ● обратный нейрональный захват холина. Депонирование ацетилхолина в везикулах уменьшает везамикол, блокирующий транспорт ацетилхолина из цитоплазмы в везикулы. Высвобождение ацетилхолина в синаптическую щель стимулирует аминопиридин (пимадин♠). Блокирует высвобождение ацетилхолина ботулинический токсин. Обратный нейрональный захват холина ингибирует Глава 8. Средства, действующие на холинергические синапсы 181 гемихолиний, блокирующий транспортные белки пресинаптической мембраны нервного окончания (см. рис. 8.1). Однако эти вещества (за исключением препаратов ботулинического токсина) не нашли применение в качестве ЛС. В медицинской практике в основном используют вещества, непосредственно взаимодействующие с холинорецепторами: холиномиметики (вещества, стимулирующие холинорецепторы) и холиноблокаторы (вещества, блокирующие холинорецепторы и препятствующие действию на них ацетилхолина). Применяют также вещества, ингибирующие гидролиз ацетилхолина, — ингибиторы ацетилхолинэстеразы (антихолинэстеразные средства). 8.1. СРЕДСТВА, СТИМУЛИРУЮЩИЕ ХОЛИНЕРГИЧЕСКИЕ СИНАПСЫ В этой группе выделяют: ● холиномиметики — вещества, подобно ацетилхолину непосредственно стимулирующие холинорецепторы; ● антихолинэстеразные средства, ингибирущие ацетилхолинэстеразу, повышающие концентрацию ацетилхолина в синаптической щели, усиливающие и пролонгирующие действие ацетилхолина. 8.1.1. Холиномиметики Холинорецепторы разных холинергических синапсов одинаково чувствительны к ацетилхолину, однако проявляют неодинаковую чувствительность к другим веществам. Холинорецепторы, локализованные в постсинаптической мембране клеток эффекторных органов у окончаний постганглионарных парасимпатических волокон, проявляют высокую чувствительность к мускарину (алкалоиду, выделенному из некоторых видов мухоморов). Такие рецепторы называют мускариночувствительными, или М-холинорецепторами. Холинорецепторы, расположенные в постсинаптической мембране нейронов симпатических и парасимпатических ганглиев, хромаффинных клеток мозгового вещества надпочечников, в каротидных клубочках (находящихся в месте деления общих сонных артерий) и на концевой пластинке скелетных мышц (в нервно-мышечных синапсах), наиболее чувствительны к никотину и поэтому называются никотиночувствительными рецепторами, или Н-холинорецепторами. Эти рецепторы подразделяются на Н-холинорецепторы нейронального типа (Нн) и Н-холинорецепторы 182 Часть II. Частная фармакология мышечного типа (Нм), различающиеся по локализации (табл. 8.1) и чувствительности к фармакологическим веществам. Таблица 8.1. Подтипы холинорецепторов и эффекты, вызываемые их стимуляцией Подтипы холинорецепторов Локализация рецепторов Эффекты, вызываемые стимуляцией холинорецепторов М-холинорецепторы Выделение гистамина, стимулиМ1 ЦНС: диффузно располорующего секрецию хлористоводоженные холинергические родной кислоты париетальными синапсы в коре головного мозга, среднем мозге, ство- клетками желудка ле и спинном мозге. Энтерохромаффиноподобные клетки желудка М2 Сердце Уменьшение ЧСС. Снижение сократительной активности предсердий. Угнетение проводимости и удлинение рефрактерного периода в атриовентрикулярном узле Пресинаптическая мембра- Снижение высвобождения ацена окончаний постганглио- тилхолина нарных парасимпатических волокон М3 Круговая мышца радужной Сокращение, сужение зрачков. Сокращение, спазм аккомодации (иннервируемые) оболочки. (глаз устанавливается на ближЦилиарная (ресничная) нюю точку видения) мышца глаза Повышение тонуса (за исключеГладкие мышцы бронхов, желудка, кишечника, желч- нием сфинктеров) и усиление моторики желудка, кишечника ного пузыря и желчных протоков, мочевого пузыря, и мочевого пузыря, повышение тонуса миометрия. матки. Повышение секреции Экзокринные железы (бронхиальные железы, железы желудка, кишечника, слюнные, слезные, носоглоточные и потовые железы) М3 (неиннерви- Эндотелиальные клетки Выделение эндотелиального реруемые) кровеносных сосудов лаксирующего фактора (NО), вызывающего расслабление гладких мышц сосудов Глава 8. Средства, действующие на холинергические синапсы 183 Окончание табл. 8.1 Подтипы холинорецепторов Нм Нн Локализация рецепторов Эффекты, вызываемые стимуляцией холинорецепторов Н-холинорецепторы Скелетные мышцы Сокращение Вегетативные ганглии. Возбуждение ганглионарных нейЭнтерохромаффинные ронов. клетки мозгового вещества Секреция адреналина и норадренадпочечников. налина. Каротидные клубочки Рефлекторное возбуждение дыхательного и сосудодвигательного центров Вещества, избирательно блокирующие Нн-холинорецепторы ганглиев мозгового вещества надпочечников и каротидных клубочков, называются ганглиоблокаторами, а вещества, преимущественно блокирующие Нм-холинорецепторы скелетных мышц, — курареподобными средствами. Среди холиномиметиков выделяют вещества, преимущественно стимулирующие М-холинорецепторы (М-холиномиметики), Н-холинорецепторы (Н-холиномиметики) или оба подтипа холинорецепторов одновременно (М-, Н-холиномиметики). Классификация холиномиметиков: , цевимелин; ● М-холиномиметики: мускарин, пилокарпин, ацеклидин ● Н-холиномиметики: никотин, цитизин (цититон♠), лобелин; ♠ ● М-, Н-холиномиметики: ацетилхолин, карбахол (карбахолин ). М-холиномиметики М-холиномиметики стимулируют М-холинорецепторы, расположенные в мембране клеток эффекторных органов и тканей, получающих парасимпатическую иннервацию (см. рис. 8.2). М-холинорецепторы подразделяют на несколько подтипов, проявляющих неодинаковую чувствительность к разным фармакологическим веществам. Известно 5 подтипов М-холинорецепторов (М1, М2, М3, М4, М5). Наиболее подробно изучены М1-, М2- и М3-холинорецепторы (см. табл. 8.1). Все М-холинорецепторы относятся к мембранным рецепторам, взаимодействующим с G-белками, а через них с ферментами или ионными каналами. Так, М2-холинорецепторы мембран кардиомиоцитов взаимодействуют с Gi-белками, угнетающими аденилатциклазу. При их стимуляции в клетках снижается синтез цАМФ и, как следствие, активность цАМФзависимых протеинкиназ, фосфорилирующих белки. Нарушается фос- 184 Часть II. Частная фармакология форилирование кальциевых каналов кардиомиоцитов, в результате чего во время деполяризации мембраны в кардиомиоциты поступает меньше Са2+. Это приводит к замедлению спонтанной диастолической деполяризации и, следовательно, к снижению автоматизма синоатриального узла и ЧСС. Кроме того, при стимуляции М2-холинорецепторов активируются калиевые каналы, усиливается выход калия из клетки и возникает гиперполяризация мембраны. Вход Са2+ через потенциалозависимые каналы в кардиомиоциты при этом снижается, что также способствует развитию тормозных эффектов. Уменьшаются также сократимость предсердий и атриовентрикулярная проводимость, рефрактерный период в атриовентрикулярном узле удлиняется. Напротив, в предсердиях рефрактерный период укорачивается, проведение импульсов по предсердиям ускоряется. Влияние парасимпатической иннервации на желудочки выражено в меньшей степени, поэтому при стимуляции М2-холинорецепторов сократимость желудочков по сравнению с предсердиями снижается незначительно. М2-холинорецепторы локализованы также на пресинаптической мембране окончаний постганглионарных парасимпатических волокон. При их возбуждении уменьшается выделение ацетилхолина в синаптическую щель. М3-холинорецепторы гладкомышечных клеток и клеток экзокринных желез взаимодействуют с Gq-белками, активирующими фосфолипазу С. При участии этого фермента из фосфолипидов клеточных мембран образуется инозитол-1,4,5-трифосфат, способствующий высвобождению Са2+ из саркоплазматического ретикулума (внутриклеточного депо кальция). В результате при стимуляции М3-холинорецепторов в цитоплазме увеличивается концентрация Са2+, что вызывает повышение тонуса гладких мышц внутренних органов и увеличение секреции экзокринных желез. В мембранах эндотелиальных клеток сосудов находятся неиннервируемые (внесинаптические) М3-холинорецепторы. При их стимуляции увеличиваются синтез и высвобождение из эндотелиальных клеток эндотелиального релаксирующего фактора (NO), вызывающего расслабление гладкомышечных клеток сосудов. Это приводит к снижению тонуса сосудов и уменьшению АД. М1-холинорецепторы также сопряжены с Gq-белками. Стимуляция М1-холинорецепторов энтерохромаффиноподобных клеток желудка приводит к повышению концентрации цитоплазматического Са2+ и увеличению секреции этими клетками гистамина. Гистамин, в свою очередь, действуя на париетальные клетки желудка, стимулирует секрецию хло- Глава 8. Средства, действующие на холинергические синапсы 185 ристоводородной кислоты. Подтипы М-холинорецепторов и эффекты, вызываемые их стимуляцией, представлены в табл. 8.1. Прототип М-холиномиметиков — алкалоид мускарин (рис. 8.2), содержащийся в ядовитых грибах (мухоморах). Мускарин вызывает эффекты, связанные со стимуляцией всех подтипов М-холинорецепторов, приведенных в табл. 8.1. Через ГЭБ мускарин не проникает и поэтому не оказывает существенного влияния на ЦНС. Мускарин не используют в качестве ЛС. При отравлении мухоморами, содержащими мускарин, проявляется его токсическое действие, связанное с возбуждением М-холинорецепторов. При этом отмечают сужение зрачков, спазм аккомодации, обильное слюнотечение и потоотделение, брадикардию и снижение АД, повышение тонуса бронхов и секреции бронхиальных желез (проявляется ощущением удушья), спастические боли в животе, диарею, тошноту и рвоту. Центральные эффекты при отравлении мухоморами вызваны содержащимися в них галлюциногенами (иботеновая кислота, мусцимол). При тяжелом отравлении развиваются гипертермия, миоклонус, судороги и кома. При отравлении мухоморами проводят промывание желудка, дают энтеросорбенты и солевые слабительные. Для устранения М-холиномиметического действия мускарина применяют М-холиноблокатор атропин. М-холиномиметическим действием обладает также алкалоид ареколин, содержащийся в семенах растения Areca catechu, произрастающей в субконтинентальной Индии и на прилегающих островах. Ареколин вызывает эйфорию, поэтому листья и семена (орехи Четвертичные аммониевые соединения Третичные аммониевые соединения Рис. 8.2. Химические структуры некоторых М-холиномиметиков 186 Часть II. Частная фармакология бетель) в смеси с другими компонентами (листья перечного растения Piper betle и др.) используются местным населением как легкое наркотическое средство под названием бетель. Пилокарпин — алкалоид листьев кустарника Pilocarpus pinnatifolius Jaborandi, произрастающего в Южной Америке. Пилокарпин, применяемый в медицинской практике, получают синтетическим путем. Пилокарпин оказывает прямое стимулирующее действие на М-холинорецепторы и вызывает все эффекты, характерные для препаратов этой группы (см. табл. 8.1). Особенно сильно пилокарпин повышает секрецию желез (слюнных, слезных, потовых), поэтому его иногда в небольших дозах (5–10 мг) назначают внутрь при ксеростомии (сухость слизистой оболочки полости рта), которая может быть результатом радиационного облучения области головы и шеи или болезни Шегрена. В ряде стран выпускают препарат пилокарпина для введения внутрь (салаген ). Но поскольку пилокарпин, будучи третичным амином (см. рис. 8.2), и, следовательно, неполярным липофильным соединением, проникает в ЦНС и обладает довольно высокой токсичностью, его в основном применяют местно в виде глазных лекарственных форм для снижения внутриглазного давления. Величина внутриглазного давления зависит от двух процессов: образования и оттока внутриглазной жидкости (водянистой влаги глаза). Внутриглазная жидкость продуцируется ресничным телом, а оттекает через дренажную систему угла передней камеры глаза (между радужкой и роговицей). Эта дренажная система включает трабекулярную сеть (гребешковую связку) и венозный синус склеры (шлеммов канал). Через щелевидные пространства между трабекулами (фонтановы пространства) трабекулярной сети жидкость фильтруется в шлеммов канал, а оттуда по коллекторным сосудам оттекает в поверхностные вены склеры (рис. 8.3). Снизить внутриглазное давление можно, уменьшив продукцию внутриглазной жидкости и/или увеличив ее отток. Отток внутриглазной жидкости во многом зависит от размера зрачка, величина которого регулируется двумя мышцами радужной оболочки: круговой (m. sphincter pupillae) и радиальной (m. dilatator pupillae). Круговая мышца иннервируется парасимпатическими волокнами (n. oculomotorius), а радиальная — симпатическими (n. sympaticus). При сокращении круговой мышцы зрачок сужается, а при сокращении радиальной мышцы — расширяется. Способность пилокарпина снижать внутриглазное давление используют при лечении глаукомы — заболевания, характеризующегося постоянным или периодическим повышением внутриглазного давления, что мо- Глава 8. Средства, действующие на холинергические синапсы 187 Рис. 8.3. Действие на глаз веществ, влияющих на холинергическую иннервацию (толщиной стрелки показана интенсивность оттока внутриглазной жидкости) жет привести к атрофии зрительного нерва и потере зрения. Глаукома бывает закрытоугольной и открытоугольной. Закрытоугольная форма развивается при нарушении доступа к углу передней камеры глаза, чаще всего при его частичном или полном закрытии корнем радужки. Внутриглазное давление при этом может повышаться до 60–80 мм рт.ст. (в норме внутриглазное давление составляет 16–26 мм рт.ст.). Открытоугольная форма глаукомы связана с нарушением дренажной системы угла передней камеры глаза, через которую осуществляется отток внутриглазной жидкости; сам угол при этом открыт. 188 Часть II. Частная фармакология Пилокарпин, как и все М-холиномиметики, вызывает сокращение круговой мышцы радужной оболочки и сужение зрачков (миоз), а также сокращение цилиарной (ресничной) мышцы. В связи со способностью сужать зрачки (миотическое действие) пилокарпин обладает высокой эффективностью при лечении закрытоугольной формы глаукомы, и в этом случае его используют в первую очередь (препарат выбора). При сужении зрачков радужная оболочка становится тоньше, способствуя раскрытию угла передней камеры глаза (между радужкой и роговицей) и оттоку внутриглазной жидкости через фонтановы пространства в шлеммов канал. Это приводит к снижению внутриглазного давления. При закрытоугольной форме глаукомы пилокарпин часто применяют перед оперативным вмешательством и для купирования острого приступа глаукомы. Назначают пилокарпин и при открытоугольной форме глаукомы, при которой имеет значение действие пилокарпина на цилиарную мышцу. Сокращение цилиарной мышцы вызывает натяжение трабекул гребешковой связки, вследствие чего фонтановы пространства увеличиваются в размерах, и улучшается отток внутриглазной жидкости. Пилокарпин применяют в виде 1–2% водных растворов (продолжительность действия 4–8 ч), растворов с добавлением полимерных соединений, оказывающих пролонгированное действие (8–12 ч), мазей и специальных глазных пленок из полимерного материала (глазные пленки с пилокарпином закладывают за нижнее веко 1–2 раза в сутки). Выпускают комбинированные препараты, содержащие пилокарпин с эпинефрином (глазные пленки пиларен♠) и пилокарпин с тимололом (глазные капли фотил♠). Вызываемое пилокарпином сокращение ресничной мышцы приводит к расслаблению цинновой связки, расстягивающей хрусталик. Кривизна хрусталика увеличивается, и он приобретает более выпуклую форму. При увеличении кривизны хрусталика повышается его преломляющая способность — глаз устанавливается на ближнюю точку видения (лучше видны предметы, находящиеся вблизи). Это явление, называемое спазмом аккомодации, относится к побочным эффектам пилокарпина. Кроме того, пилокарпин вызывает макропсию (предметы кажутся увеличенными и видны нечетко). При длительном применении пилокарпина возможны фиброзные изменения внутриглазных мышц, необратимый миоз, повышение проницаемости капилляров, появление отеков и кровоизлияний. В связи с этим один раз в год рекомендуется прерывать лечение пилокарпином на несколько месяцев с заменой его на тимолол. Пилокарпин и другие М-холиномиметики в глазных лекарственных формах противопоказаны при ирите и иридоциклите. Глава 8. Средства, действующие на холинергические синапсы 189 При закапывании в конъюнктивальный мешок пилокарпин практически не всасывается в кровь и не оказывает заметного резорбтивного действия. При системном введении пилокарпин вызывает все эффекты возбуждения парасимпатической системы, а проникая в ЦНС, может провоцировать приступы эпилепсии. В экспериментальных исследованиях на животных пилокарпин используется для создания модели эпилепсии. Ацеклидин — синтетическое соединение с прямым стимулирующим действием на М-холинорецепторы, вызывает все эффекты, связанные с возбуждением этих рецепторов (см. табл. 8.1). Ацеклидин можно применять местно (инстиллировать в конъюнктивальный мешок) для понижения внутриглазного давления при глаукоме. После однократной инстилляции снижение уровня внутриглазного давления продолжается до 6 ч. Однако растворы ацеклидина обладают местнораздражающим действием и могут вызвать раздражение конъюнктивы. В связи с меньшей по сравнению с пилокарпином токсичностью ацеклидин можно применять для резорбтивного действия при атонии кишечника и мочевого пузыря, пониженном тонусе матки и маточных кровотечениях в послеродовом периоде. Назначают внутрь и парентерально. Побочные эффекты: слюнотечение, диарея, спазмы гладкомышечных органов, снижение АД. Вследствие того, что ацеклидин повышает тонус гладких мышц бронхов, он противопоказан при бронхиальной астме и обструктивной болезни легких. Противопоказан ацеклидин при стенокардии, брадикардии, язвенной болезни желудка и гиперацидном гастрите (повышает секрецию НСl), при беременности (повышает тонус миометрия). Бетанехол — синтетический М-холиномиметик, применяемый при атонии кишечника и мочевого пузыря. Назначают внутрь и парентерально. Побочные эффекты и противопоказания такие же, как у ацеклидина. Цевимелин — агонист М1- и М3-холинорецепторов с преимущественным действием на М3-холинорецепторы, используется для повышения секреции слюнных желез при ксеростомии, вызванной болезнью Шегрена или радиационным облучением области головы и шеи. При передозировке М-холиномиметиков используют их антагонисты — М-холиноблокаторы (атропин и атропиноподобные вещества). Н-холиномиметики К этой группе относят алкалоиды никотин, лобелин, цитизин, действующие преимущественно на Н-холинорецепторы нейронального типа, локализованные на нейронах симпатических и парасимпатических ган- 190 Часть II. Частная фармакология глиев, хромаффинных клетках мозгового вещества надпочечников, в каротидных клубочках и ЦНС. На Н-холинорецепторы скелетных мышц эти вещества действуют в значительно больших дозах. Н-холинорецепторы относят к мембранным рецепторам, непосредственно связанным с ионными каналами (см. рис. 2.2). По структуре это гликопротеины, состоящие из нескольких субъединиц. Так, Н-холинорецептор нервно-мышечных синапсов включает 5 белковых субъединиц (α, α, β, γ, δ), окружающих ионный (натриевый) канал. При связывании двух молекул ацетилхолина с α-субъединицами происходит открытие Na+-канала. Ионы Na+ поступают в клетку, что приводит к деполяризации постсинаптической мембраны концевой пластинки скелетных мышц и мышечному сокращению. Н-холинорецепторы нейронального (ганглионарного) типа имеют такую же структуру, но в отличие от Нм-холинорецепторов состоят из α- и β-субъединиц различных подтипов. Различия α- и β-субъединиц этих рецепторов, по-видимому, определяют разнообразие эффектов, опосредуемых Н-холинорецепторами нейронального типа в ЦНС и вегетативных ганглиях. Никотин — алкалоид, содержащийся в листьях табака (Nicotiana tabacum, Nicotiana rustica). Никотин попадает в организм человека во время курения табака, примерно 3 мг за время курения одной сигареты (смертельная доза никотина — 60 мг). Никотин быстро всасывается со слизистых оболочек дыхательных путей (также хорошо проникает через неповрежденную кожу). Никотин стимулирует Н-холинорецепторы симпатических и парасимпатических ганглиев, хромаффинных клеток мозгового вещества надпочечников (повышает выделение адреналина и норадреналина) и каротидных клубочков (стимулирует дыхательный и сосудодвигательный центры). Стимуляция симпатических ганглиев, мозгового вещества надпочечников и каротидных клубочков приводит к наиболее характерным для никотина эффектам со стороны сердечно-сосудистой системы: увеличению ЧСС, сужению сосудов и повышению АД. Стимуляция парасимпатических ганглиев вызывает повышение тонуса и моторики кишечника и повышение Рис. 8.4. Химические структуры некоторых Н-холиномиметиков Глава 8. Средства, действующие на холинергические синапсы 191 секреции экзокринных желез (большие дозы никотина оказывают на эти процессы угнетающее влияние). Стимуляция Н-холинорецепторов парасимпатических ганглиев также является причиной брадикардии, которую можно наблюдать в начале действия никотина. В связи с тем, что никотин обладает высокой липофильностью (третичный амин, см. рис. 8.4), он быстро проникает через ГЭБ в ткани мозга. В ЦНС никотин вызывает высвобождение дофамина, некоторых других биогенных аминов и возбуждающих аминокислот, с чем связывают субъективные приятные ощущения, возникающие у курильщиков. В небольших дозах никотин стимулирует дыхательный центр, а в больших дозах вызывает его угнетение вплоть до остановки дыхания (паралич дыхательного центра). В больших дозах никотин вызывает тремор и судороги. Действуя на триггерную зону рвотного центра, никотин может вызвать тошноту и рвоту. Никотин метаболизируется в печени и выводится почками в неизмененном виде и в виде метаболитов. Таким образом, он быстро элиминируется из организма (t½ 1,5–2 ч). К действию никотина быстро развивается толерантность (привыкание). Острое отравление никотином может произойти при попадании растворов никотина на кожу или слизистые оболочки. При этом отмечают гиперсаливацию, тошноту, рвоту, диарею, брадикардию, сменяющуюся тахикардией, повышение АД, одышку, а затем угнетение дыхания, возможны судороги. Смерть наступает от паралича дыхательного центра. Основная мера помощи — искусственное дыхание. При курении табака возможно хроническое отравление никотином, а также другими токсичными веществами, содержащимися в табачном дыме и оказывающими раздражающее и канцерогенное действия. Для большинства курильщиков типичны воспалительные заболевания дыхательных путей, например хронический бронхит; чаще отмечают рак легких. Повышен риск сердечно-сосудистых заболеваний. К никотину развивается психическая зависимость, при прекращении курения у курильщиков возникает синдром отмены, связанный с возникновением тягостных ощущений, снижением работоспособности. Для уменьшения синдрома отмены рекомендуют в период отвыкания от курения использовать жевательную резинку, содержащую никотин (2 или 4 мг), или трансдермальную терапевтическую систему (специальный накожный пластырь, в течение 24 ч равномерно выделяющий небольшие количества никотина). В медицинской практике иногда используют Н-холиномиметики лобелин и цитизин. 192 Часть II. Частная фармакология Лобелин — алкалоид тропического растения Lobelia inflata, третичный амин (см. рис. 8.4). Стимулируя Н-холинорецепторы каротидных клубочков, лобелин рефлекторно возбуждает дыхательный и сосудодвигательный центры. Вначале лобелин, возбуждая центр блуждающего нерва в продолговатом мозге, вызывает брадикардию и снижение АД, но затем вследствие стимуляции Н-холинорецепторов симпатических ганглиев и мозгового вещества надпочечников АД повышается. Цитизин — алкалоид, содержащийся в растениях ракитник (Cytisus Laburnum) и термопсис (Thermopsis Lanceolata), вторичный амин. По действию сходен с лобелином, но несколько сильнее возбуждает дыхательный центр. Выпускается в виде 0,15% раствора под названием «Цититон»♠. В отличие от лобелина цитизин вызывает только прессорный эффект (повышает АД). Цитизин и лобелин входят в состав таблеток «Табекс»♠ и «Лобесил»♠, применяемых для облегчения отвыкания от курения. Препарат цититон♠ и раствор лобелина иногда вводят внутривенно для рефлекторной стимуляции дыхания. Действуют эти вещества кратковременно (в течение 2–5 мин). Однако эти препараты эффективны только при сохранении рефлекторной возбудимости дыхательного центра. Поэтому их не применяют при отравлении веществами, снижающими рефлекторную возбудимость дыхательного центра (снотворные средства, средства для наркоза). Применяют их редко при отравлении угарным газом и асфиксии новорожденных (в основном в тех случаях, когда невозможно провести искусственную вентиляцию легких). При передозировке эти вещества могут вызвать рвоту, тонико-клонические судороги и остановку сердца. Противопоказаны при артериальной гипертензии, атеросклерозе, отеке легких. М-, Н-холиномиметики Ацетилхолин — нейромедиатор, передающий возбуждение во всех холинергических синапсах, стимулирует как М-, так и Н-холинорецепторы. Ацетилхолин выпускают в виде лиофилизированного препарата ацетилхолин-хлорида♠. При введении ацетилхолина в организм преобладают его эффекты, связанные со стимуляцией М-холинорецепторов: брадикардия, расширение сосудов и понижение АД, повышение тонуса и усиление перистальтики ЖКТ, повышение тонуса гладких мышц бронхов, желчного и мочевого пузыря, матки, усиление секреции бронхиальных, пищеварительных и др. экзокринных желез. Действие ацетилхолина на частоту сердечных сокращений (ЧСС) может осложняться возникновением рефлекторных эффектов со стороны симпатической системы и зависит Глава 8. Средства, действующие на холинергические синапсы 193 от вводимой дозы: при внутривенной инфузии ацетилхолина со скоростью 20–50 мкг/мин снижение АД (вследствие расширения сосудов) сопровождается рефлекторным повышением ЧСС. При введении более высоких доз превалирует прямое действие ацетилхолина на М-холинорецепторы, и он, несмотря на снижение АД, вызывает брадикардию и угнетение атриовентрикулярной проводимости. Стимулирующее влияние ацетилхолина на периферические Н-холинорецепторы (никотиноподобное действие) проявляется при блокаде М-холинорецепторов (атропином). В результате на фоне атропина ацетилхолин вызывает тахикардию, сужение сосудов и, как следствие, повышение АД. Происходит это вследствие возбуждения симпатических ганглиев, повышения выделения адреналина хромаффинными клетками мозгового вещества надпочечников и стимуляции каротидных клубочков. В очень больших дозах ацетилхолин может вызвать стойкую деполяризацию постсинаптической мембраны и блокаду передачи возбуждения в холинергических синапсах. По химической структуре ацетилхолин — четвертичное аммониевое соединение (см. рис. 8.2), поэтому плохо проникает через ГЭБ и не оказывает существенного влияния на ЦНС. В организме ацетилхолин быстро разрушается холинэстеразой плазмы крови и синаптической ацетилхолинэстеразой, поэтому оказывает кратковременное действие, продолжающееся всего несколько минут. Ацетилхолин используется в экспериментальных исследованиях. Медицинское применение ацетилхолина весьма ограничено. Его сосудорасширяющее действие иногда используют при спазмах периферических сосудов (перемежающаяся хромота) и артерий сетчатки. Для этих целей используют отечественный препарат ацетилхолин-хлорид♠, вводят подкожно или внутримышечно. В сочетаниях с другими препаратами применяют местно для сужения зрачка при глазных хирургических операциях. Карбахол (карбахолин♠) — аналог ацетилхолина, но в отличие от него практически не разрушается ацетилхолинэстеразой и поэтому действует более продолжительно (в течение 1–1,5 ч). Вызывает такие же фармакологические эффекты. Раствор карбахола в виде глазных капель можно использовать при глаукоме и парентерально при атонии мочевого пузыря. 8.1.2. Антихолинэстеразные средства Антихолинэстеразные средства ингибируют ацетилхолинэстеразу — фермент, гидролизующий ацетилхолин в синаптической щели, и холинэстеразу плазмы крови (бутирилхолинэстераза, псевдохолинэстераза, 194 Часть II. Частная фармакология ложная холинэстераза). Ингибирование ацетилхолинэстеразы в холинергических синапсах приводит к повышению концентрации ацетилхолина в синаптической щели, вследствие чего значительно усиливается и удлиняется действие ацетилхолина. Таким образом, все эффекты антихолинэстеразных средств вызваны эндогенным ацетилхолином. При введении антихолинэстеразных средств стимулирующее действие ацетилхолина на М-холинорецепторы приводит к сужению зрачков, спазму аккомодации, брадикардии, снижению сердечного выброса, повышению тонуса гладких мышц бронхов, тонуса и моторики ЖКТ, мочевого пузыря, увеличению секреции экзокринных желез. Антихолинэстеразные средства практически не влияют на тонус сосудов. Связано это с тем, что в сосудах находятся в основном неиннервируемые (внесинаптические) М3-холинорецепторы. АД снижается гораздо в меньшей степени, чем при введении М-холиномиметиков (в основном за счет снижения сердечного выброса). При введении антихолинэстеразных средств из Н-холиномиметических эффектов ацетилхолина наиболее отчетливо проявляется его стимулирующее влияние на нервно-мышечную передачу, в результате чего повышается тонус скелетных мышц. Стимулирующее влияние ацетилхолина на вегетативные ганглии проявляется в меньшей степени. Однако при введении больших доз антихолинэстеразных средств стимуляция Н-холинорецепторов симпатических ганглиев, хромаффинных клеток мозгового вещества надпочечников и каротидных клубочков может привести к возникновению тахикардии и повышению АД. Антихолинэстеразные средства, проникающие через ГЭБ, оказывают возбуждающее действие на ЦНС. По характеру взаимодействия с ацетилхолинэстеразой различают антихолинэстеразные вещества обратимого и необратимого действия. Антихолинэстеразные средства обратимого действия К ним относят физостигмин (эзерин), неостигмина метилсульфат (прозерин♠), пиридостигмина бромид (калимин♠), ривастигмин (экселон♠), галантамина гидробромид (реминил♠, нивалин♠), донепезил (арисепт♠), эдрофоний (тензилон). Ацетилхолинэстераза имеет два активных центра: анионный и эстеразный. Положительно заряженный четвертичный атом азота в молекуле ацетилхолина связывается с анионным центром (с карбоксильной группой остатка глутаминовой кислоты), а углерод карбонильной группы — с эстеразным центром (с ОН-группой остатка серина). В результате происходит гидролиз ацетилхолина с образованием холина и ацетилиро- Глава 8. Средства, действующие на холинергические синапсы 195 ванного фермента (ковалентно связанного с ацетильной группой). Отщепление ацетильной группы и, как следствие, восстановление активности ацетилхолинэстеразы происходят очень быстро. Весь процесс гидролиза ацетилхолина занимает приблизительно 100–150 мкс. Многие антихолинэстеразные вещества (физостигмин, неостигмина метилсульфат, пиридостигмина бромид, ривастигмин и некоторые другие) являются эфирами карбаминовой кислоты — карбаматами (рис. 8.5). Эти вещества подобно ацетилхолину связываются как с анионным, так и с эстеразным центрами ацетилхолинэстеразы и подвергаются гидролизу, при этом ацетилхолинэстераза в своем эстеразном центре (через ОН-группу остатка серина) оказывается ковалентно связанной с карбамильной группой. Гидролиз этой более прочной связи происходит Четвертичные аммониевые соединения 2 Третичные аммониевые соединения 5 Рис. 8.5. Химические структуры некоторых антихолинэстеразных средств обратимого действия 196 Часть II. Частная фармакология медленнее — от 30 мин до нескольких часов. Это определяет продолжительность действия карбаматов. Препараты этой группы ингибируют ацетилхолинэстеразу в течение 3–6 ч. Другие обратимые ингибиторы ацетилхолинэстеразы (эдрофоний, галантамин, донепезил) связываются посредством нековалентных связей только с одним центром фермента и препятствуют его взаимодействию с ацетилхолином. Эдрофоний связывается с анионным центром ацетилхолинэстеразы при участии непрочных электростатических и водородных связей. Этот комплекс существует 5–10 мин, поэтому эдрофоний оказывает кратковременное антихолинэстеразное действие. Эдрофоний — полярное гидрофильное соединение (четвертичный амин), быстро выводится из организма. Донепезил и галантамин обладают большим сродством к ферменту и действуют намного продолжительнее. Эти вещества в отличие от эдрофония являются третичными аминами (см. рис 8.5) и проникают через ГЭБ в ткани мозга. Неостигмина метилсульфат (прозерин♠) — синтетическое соединение, содержащее четвертичный атом азота (см. рис. 8.5), плохо проникает через ГЭБ. Обладает выраженной антихолинэстеразной активностью, усиливая и удлиняя действие ацетилхолина преимущественно в периферических холинергических синапсах. При применении неостигмина метилсульфата преобладают эффекты, связанные с возбуждением парасимпатической иннервации. Фармакологические эффекты неостигмина. Неостигмин вызывает сужение зрачков (вследствие сокращения круговой мышцы радужки), что приводит к понижению внутриглазного давления (открывается угол передней камеры глаза и облегчается отток внутриглазной жидкости через фонтановы пространства в шлеммов канал). Одновременно развивается спазм аккомодации (вследствие сокращения цилиарной мышцы расслабляется циннова связка — хрусталик становится более выпуклым, и глаз устанавливается на ближнюю точку видения). Неостигмин вызывает брадикардию и замедление атриовентрикулярной проводимости, повышает тонус бронхов, тонус и моторику ЖКТ, тонус и сократительную активность мочевого пузыря, матки, секрецию экзокринных желез. Из эффектов неостигмина, вызываемых стимуляцией Н-холинорецепторов, отмечают действие на нервно-мышечные синапсы. Облегчение нервно-мышечной передачи и повышение тонуса скелетных мышц связано не только с ингибированием ацетилхолинэстеразы и увеличением концентрации ацетилхолина в синаптической щели, но и с прямым стимулирующим действием неостигмина на Нм-холинорецепторы скелетных мышц. Этот эффект в особенности выражен при пониженном тонусе скелетной му- Глава 8. Средства, действующие на холинергические синапсы 197 скулатуры, связанном с патологическими состояниями или введением курареподобных средств. В клинической практике в основном используют стимулирующее действие неостигмина на тонус скелетных мышц и тонус гладких мышц ЖКТ и мочевого пузыря. Основные показания к применению неостигмина: ● миастения (аутоиммунное заболевание, при котором образуются антитела к Н-холинорецепторам скелетных мышц, вследствие чего уменьшается их количество; проявляется мышечной слабостью и повышенной утомляемостью скелетных мышц, в тяжелых случаях возможно нарушение дыхания из-за ослабления сократимости дыхательных мышц); препарат назначают внутрь, под кожу и внутримышечно, при миастеническом кризе — внутривенно; ● послеоперационная атония кишечника и мочевого пузыря; препарат вводят внутрь, под кожу или внутримышечно; ● в качестве антагониста курареподобных средств антидеполяризующего конкурентного типа действия для снятия остаточного нервномышечного блока; препарат вводят внутривенно; ● редко — при закрытоугольной глаукоме. Неостигмин будучи полярным гидрофильным соединением после приема внутрь всасывается из ЖКТ не полностью (дозы для приема внутрь в 30 раз превышают дозы для парентерального введения), действует непродолжительно (2–4 ч). Побочные эффекты неостигмина связаны со стимуляцией М-холинорецепторов: тошнота, рвота, диарея, гиперсаливация, брадикардия, снижение АД, повышение тонуса бронхов. Для устранения этих симптомов можно использовать атропин. Стимуляция Нм-холинорецепторов высокими концентрациями ацетилхолина может вызвать подергивание скелетных мышц. Увеличение дозы неостигмина повышает и удлиняет действие ацетилхолина в нервно-мышечном синапсе, что может вызвать стойкую деполяризацию постсинаптической мембраны (деполяризационный блок) и угнетение нервно-мышечной передачи. Поэтому при передозировке препарата наряду с усилением симптомов, связанных со стимуляцией парасимпатической системы, возможно усиление симптомов миастении вследствие нарушения нервно-мышечной передачи (холинергический криз). Препарат противопоказан при эпилепсии, болезни Паркинсона, стенокардии, нарушениях проводящей системы сердца, бронхиальной астме, язвенной болезни желудка и двенадцатиперстной кишки и других заболеваниях, при которых усиление парасимпатических влияний на органы и ткани нежелательно. 198 Часть II. Частная фармакология Пиридостигмина бромид — четвертичное аммониевое соединение, не проникает через ГЭБ, действует подобно неостигмину, но более продолжительно (около 6 ч). В основном его применяют при лечении миастении, а также при атонии кишечника и мочевого пузыря, назначают внутрь и парентерально. По сравнению с неостигмином обладает менее выраженным мускариноподобным действием. Побочные эффекты и противопоказания аналогичны таковым для неостигмина метилсульфата. При передозировке пиридостигмина бромида возможен холинергический криз. Другой длительно действующий препарат, применяемый при миастении, — амбенония хлорид (оксазил♠) — оказывает эффект продолжительностью до 10 ч, принимают внутрь. Амбенония хлорид является четвертичным аммониевым соединением, не проникает через ГЭБ. Эдрофоний — короткодействующий препарат (продолжительность действия 5–15 мин), четвертичный амин, оказывает периферическое действие. Применяют для диагностики миастении, вводят внутривенно (эффект наступает через 30–60 с). Повышение тонуса скелетных мышц после введения препарата является признаком заболевания. Эдрофоний также используют для дифференциальной диагностики между холинергическим кризом, вызванным передозировкой антихолинэстеразных средств при лечении миастении, и обострением миастении. При передозировке антихолинэстеразных средств (неостигмина метилсульфата, пиридостигмина бромида) вместо ожидаемого улучшения состояния отмечают мышечную слабость, связанную со стойкой деполяризацией постсинаптической мембраны, что препятствует передаче возбуждения в нервно-мышечных синапсах (деполяризационный блок). В такой ситуации введение эдрофония не вызывает повышения тонуса скелетных мышц и может даже усилить мышечную слабость (благодаря небольшой продолжительности действия препарата этот эффект исчезает быстро). Эдрофоний применяют также в качестве антагониста курареподобных средств антидеполяризующего типа действия. Физостигмин — алкалоид калабарских бобов, семян древовидного кустарника Physostigma venenosum, произрастающего в Западной Африке, был первым антихолинэстеразным веществом, которое стали применять в медицинской практике — сначала как антидот при отравлении беленой, а затем при глаукоме. В больших концентрациях физостигмин может непосредственно стимулировать холинорецепторы. Поскольку физостигмин по структуре является третичным амином (см. рис. 8.5) и хорошо проникает через ГЭБ, его можно использовать Глава 8. Средства, действующие на холинергические синапсы 199 как антидот при отравлении холиноблокаторами, проникающими в ЦНС (например, атропином). Растворы физостигмина салицилата используют в глазной практике при закрытоугольной форме глаукомы (в основном при острых приступах) как миотическое средство, облегчающее отток внутриглазной жидкости. По эффективности физостигмин превосходит пилокарпин (в большей степени снижает внутриглазное давление), но вследствие сильного сокращения радужной оболочки при его применении возникают болевые ощущения в глазах и надбровных дугах. Кроме того, физостигмин послужил прототипом для создания ЛВ, используемых при лечении болезни Альцгеймера. Это заболевание характеризуется прогрессирующей потерей памяти и развитием слабоумия, что связывают с атрофией нейронов коры и подкорковых структур мозга, в том числе холинергических нейронов. При этом отмечают снижение концентрации ацетилхолина в тканях мозга, что явилось основанием для использования антихолинэстеразных средств. Сам физостигмин в настоящее время при болезни Альцгеймера не применяют по причине непродолжительного действия и выраженных побочных эффектов, связанных со стимуляцией периферических холинорецепторов. Препарат такрин (когнекс), ранее рекомендованный к применению при болезни Альцгеймера, имеет ограниченное использование, так как обладает многими побочными эффектами, наиболее серьезное из которых — нарушение функции печени. Галантамин, алкалоид, выделенный из луковиц и цветков растений рода Galanthus (в виде галантамина гидробромида под названием реминил)♠, ривастигмин (экселон♠) и донепезил (арисепт♠), которые в настоящее время применяются при болезни Альцгеймера и других видах деменции, имеют определенные преимущества. Эти вещества, особенно ривастигмин, в меньшей степени ингибируют ацетилхолинэстеразу периферических тканей (скелетных мышц, внутренних органов), чем ацетилхолинэстеразу мозга, и поэтому вызывают менее выраженные побочные эффекты, связанные со стимуляцией периферических холинорецепторов. Кроме того, они не обладают характерной для такрина♠ гепатотоксичностью. Препараты оказывают продолжительное антихолинэстеразное действие (донепезил назначают 1 раз, а галантамин и ривастигмин — 2 раза в сутки). Ривастигмин выпускают также в виде трансдермальных терапевтических систем для накожного применения (продолжительность действия — 24 ч). Курсовое применение этих препаратов способствует улучшению памяти, внимания, речи и других когнитивных функций, частично уменьшаются и другие проявления болезни Альцгеймера. 200 Часть II. Частная фармакология Антихолинэстеразные средства, проникающие в ЦНС, в небольших дозах оказывая благотворное стимулирующее действие (повышение внимания, укорочение рефлекторных реакций), в высоких дозах могут вызвать генерализованные судороги с последующей комой и угнетением дыхания. Среди побочных эффектов отмечают тошноту, рвоту, диарею, головокружение, головную боль и др. Галантамина гидробромид (нивалин♠), кроме того, назначают при параличах скелетных мышц, связанных с нарушениями ЦНС, например при остаточных явлениях после перенесенного полиомиелита, спастических формах церебрального паралича. Кроме того, препарат используют при атонии кишечника и мочевого пузыря, при миастении, вводят подкожно. Препарат также применяют как антагонист курареподобных средств антидеполяризующего действия, вводят внутривенно. Некоторые обратимые ингибиторы ацетилхолинэстеразы из группы карбаматов используются в сельском хозяйстве в качестве инсектицидов. Первым таким продуктом был карбосульфан, разработанный в США. В настоящее время эти вещества используются под разными названиями. Проникая через слизистые оболочки и кожу, карбаматы могут вызвать тяжелую интоксикацию, симптомы и методы лечения которой такие же, как при отравлении фосфорорганическими соединениями — ФОС. Но в отличие от необратимых ингибиторов ацетилхолинэстеразы карбаматы вызывают менее выраженные нарушения и действуют менее продолжительно (вследствие быстрого восстановления активности фермента), поэтому при интоксикации этими веществами нет необходимости использовать реактиваторы холинэстераз. Введение атропина в дозах, достаточных для конкурентного вытеснения ацетилхолина из связи с холинорецепторами, является неотложным мероприятием, необходимым для устранения симптомов как при отравлении инсектицидами, так и при передозировке антихолинэстеразных средств обратимого действия медицинского назначения. Ипидакрин (нейромидин♠) обладает слабой антихолинэстеразной активностью и непосредственно стимулирует проведение импульсов в холинергических синапсах, блокируя калиевые каналы мембран нервных клеток, способствует ее деполяризации. Действует как в периферической нервной системе, так и в ЦНС. Применяется по тем же показаниям, что и другие антихолинэстеразные средства: для лечения миастении и миастенического криза, при невритах, полиневритах, параличах и парезах центрального происхождения, деменции, включая болезнь Альцгеймера. Назначают также при атонии кишечника и слабости родовой деятельности. Побочные эффекты и противопоказания такие же, как у других антихолинэстеразных средств. Глава 8. Средства, действующие на холинергические синапсы 201 Антихолинэстеразные средства необратимого действия К этой группе относят ФОС, ингибирующие ацетилхолинэстеразу за счет образования ковалентных связей между фосфорильным остатком ФОС и ОН-группой серина в эстеразном центре фермента. Эти связи очень прочные и гидролизуются очень медленно (в течение сотен часов). Поэтому ФОС ингибируют ацетилхолинэстеразу практически необратимо. В медицинской практике ФОС применяют только местно, что связано с их высокой токсичностью. Препараты армин♠ и экотиопат могут быть использованы в качестве миотических средств для снижения внутриглазного давления при глаукоме. Экотиопат — гидрофильное полярное соединение, плохо проникает через конъюнктиву, поэтому при его применении меньше опасность возникновения системных побочных эффектов. Продолжительность действия около 4 сут. В отличие от других ФОС экотиопат устойчив в водном растворе. В основном ФОС, такие как карбофос (малатион), тиофос (паратион), используются с немедицинскими целями — в сельском хозяйстве в качестве инсектицидов для уничтожения насекомых, вредителей растений. В организме млекопитающих и более интенсивно у насекомых они превращаются в активные метаболиты — малаоксон и параоксон, в которых атом серы замещен на атом кислорода. Некоторые ФОС использовались как боевые отравляющие вещества (зоман). Поскольку ФОС обладают высокой липофильностью, они легко всасываются через неповрежденную кожу и слизистые оболочки, с поверхности легких, поэтому нередко бывают причиной отравлений. Попав в организм, значительная часть ФОС депонируется в жировой ткани, сорбируется на эндотелии сосудов и эритроцитах, проникает в липиды клеточных мембран и, выделяясь с желчью в просвет кишечника, подвергается энтерогепатической циркуляции. В результате продолжительного пребывания ФОС в организме могут возникать повторные интоксикации. Симптомы и принципы лечения острого отравления ФОС. При острых отравлениях ФОС наблюдаются эффекты возбуждения как М-, так и Н-холинорецепторов. Эти эффекты связаны со значительной концентрацией ацетилхолина в холинергических синапсах и проявляются в чрезмерной степени. М-холиномиметические эффекты проявляются в виде сильного миоза (сужение зрачков до размеров булавочной головки), слезотечения, профузного потоотделения, обильной саливации, спазма бронхов и повышения секреции бронхиальных желез, ощущения удушья. Возникают рвота, спастические боли в животе, диарея, непроизвольное мочеиспускание, брадикардия и снижение АД. Развитие та- 202 Часть II. Частная фармакология хикардии и повышение АД связаны с преимущественной стимуляцией Н-холинорецепторов симпатических ганглиев и мозгового вещества надпочечников, стимуляцией сосудодвигательного центра. Вследствие стимуляции Н-холинорецепторов скелетных мышц вначале возникают мышечные подергивания, затем — паралич скелетной мускулатуры вследствие стойкой деполяризации постсинаптической мембраны. Со стороны ЦНС отмечаются спутанность сознания, психомоторное возбуждение судороги, коматозное состояние. Смерть наступает от паралича дыхательного центра. При попадании ФОС на кожу и слизистые оболочки следует быстро вытереть кожу сухим ватным тампоном и промыть 5–6% раствором натрия гидрокарбоната и теплой водой с мылом, а при введении внутрь — промыть желудок и дать адсорбирующие и слабительные средства. Если вещество всосалось в кровь, для ускорения его выведения применяют форсированный диурез. Используют также гемодиализ, гемосорбцию, перитонеальный диализ. Поскольку основные симптомы острого отравления ФОС вызываются стимуляцией М-холинорецепторов, для их устранения применяют М-холиноблокаторы, чаще всего атропин, который вводят внутривенно в больших дозах (2–4 мл 0,1% раствора). Применяют также реактиваторы холинэстеразы — вещества, восстанавливающие активность фермента. Реактиваторы холинэстеразы содержат в молекуле оксимную группу (–NOH), обладающую высоким сродством к атому фосфора. Они взаимодействуют с фосфорильными остатками ФОС, связанными с ацетилхолинэстеразой, дефосфорилируют фермент и таким образом восстанавливают его активность. Реактиваторы холинэстеразы эффективны только в течение нескольких часов после отравления. Это связано с изменением химических связей между ацетилхолинэстеразой и остатками ФОС («старением» комплекса), в результате чего этот комплекс становится более устойчивым к действию реактиваторов. В качестве реактиваторов холинэстеразы применяют тримедоксима бромид (дипироксим♠), аллоксим♠ и изонитрозин♠. Тримедоксима бромид и аллоксим♠ относятся к четвертичным аммониевым соединениям, которые плохо проникают через ГЭБ. Изонитрозин♠ — третичный амин, хорошо проникает в ЦНС и устраняет не только периферические, но и центральные эффекты ФОС. Препараты используют в неотложной помощи в условиях стационара, вводят парентерально. Реактиваторы холинэстеразы не применяют при отравлениях антихолинэстеразными средствами обратимого действия. Глава 8. Средства, действующие на холинергические синапсы 203 8.2. СРЕДСТВА, БЛОКИРУЮЩИЕ ХОЛИНЕРГИЧЕСКИЕ СИНАПСЫ К препаратам этой группы относят вещества, блокирующие М-холинорецепторы (М-холиноблокаторы), Н-холинорецепторы вегетативных ганглиев (ганглиоблокаторы) и вещества, блокирующие Н-холинорецепторы скелетных мышц (курареподобные средства). 8.2.1. М-холиноблокаторы М-холиноблокаторы блокируют М-холинорецепторы, локализованные на мембране клеток эффекторных органов, препятствуя их взаимодействию с ацетилхолином. Поскольку М-холинорецепторы располагаются в органах и тканях, получающих парасимпатическую иннервацию, М-холиноблокаторы, устраняя ее влияние, вызывают эффекты, противоположные эффектам возбуждения парасимпатической нервной системы. М-холиноблокаторы вызывают: ● расширение зрачков (мидриаз); ● паралич аккомодации (глаз устанавливается на дальнюю точку видения); ● повышение ЧСС (тахикардию); ● повышение атриовентрикулярной проводимости; ● снижение тонуса гладких мышц бронхов; ● снижение тонуса и моторики ЖКТ и мочевого пузыря; ● уменьшение секреции бронхиальных и пищеварительных желез. Кроме того, М-холиноблокаторы устраняют влияние симпатической системы на секрецию потовых желез, получающих симпатическую холинергическую иннервацию, уменьшая их секрецию. Среди М-холиноблокаторов выделяют вещества растительного происхождения и синтетические соединения. К веществам растительного происхождения относятся алкалоиды тропанового ряда, полученные из растений семейства пасленовых (Solanaceae): красавки (Atropa belladonna), белены (Hyosciamus niger), дурмана (Datura stramonium) и скополии (Scopolia carniolica). Главный алкалоид этих растений — L-гиосциамин, который при выделении превращается в рацемическую смесь L- и D-гиосциамина — атропин. Атропин по химической структуре представляет собой сложный эфир тропина и D-, L-троповой кислоты (рис. 8.6), относится к третичным аминам (липофильным неполярным соединениям), получен синтетическим путем. В тех же растениях содержится другой алкалоид с М-холиноблоки- 204 Часть II. Частная фармакология рующей активностью — сложный эфир скопина и троповой кислоты — скополамин (l-гиосцин). Из крестовника широколистного (Senecio platyphyllus) выделен алкалоид платифиллин (производное метилпирролизидина). Атропин — наиболее известный М-холиноблокатор, поэтому другие препараты этой группы часто называют атропиноподобными средствами. Атропин блокирует М1-, М2- и М3-подтипы холинорецепторов и устраняет влияние парасимпатической иннервации на многие органы и ткани, поэтому обладает широким спектром фармакологического действия. Фармакологические эффекты атропина. Блокируя М3-холинорецепторы круговой мышцы радужной оболочки, атропин вызывает ее расслабление, вследствие чего происходит расширение зрачков (мидриаз). Он также вызывает расслабление ресничной (цилиарной) мышцы. Это приводит к натяжению цинновой связки, вследствие чего уменьшается кривизна хрусталика (снижается его преломляющая способность) — глаз устанавливается на дальнюю точку видения. Такое состояние называют параличом аккомодации. При расширении зрачков и расслаблении цилиарной мышцы нарушается отток внутриглазной жидкости, и у больных глаукомой может повыситься внутриглазное давление. В связи с этим атропин и другие М-холиноблокаторы противопоказаны при глаукоме. Атропин, блокируя М2-холинорецепторы сердца, устраняет тормозное влияние блуждающего нерва (вагуса) на синоатриальный узел и повышает его автоматизм — возникает тахикардия. Блокада тормозного влияния вагуса на атриовентрикулярный узел приводит к повышению атриовентрикулярной проводимости. В связи с тем, что атропин стимулирует центры блуждающего нерва в ЦНС, тахикардии может предшествовать кратковременная брадикардия, возникающая при применении низких доз атропина (этому также может способствовать усиление выделения ацетилхолина вследствие блокады пресинаптических М2-холинорецепторов). Блокируя М3-холинорецепторы гладкомышечных клеток, атропин устраняет стимулирующее влияние парасимпатической иннервации на гладкие мышцы бронхов, желудка, кишечника, мочевого пузыря, желчевыводящих протоков и снижает их тонус, моторику ЖКТ и сокращения детрузора мочевого пузыря. Тонус сфинктеров при этом повышается. Рис. 8.6. Химическая структура атропина Глава 8. Средства, действующие на холинергические синапсы 205 Атропин блокирует М3-холинорецепторы экзокринных желез (желез внешней секреции) и уменьшает секрецию бронхиальных, слюнных желез, желез желудка и поджелудочной железы, слезных, носоглоточных и потовых желез. Атропин блокирует М1-холинорецепторы энтерохромаффиноподобных клеток желудка, уменьшая выделение гистамина, стимулирующего секрецию хлористоводородной кислоты париетальными клетками желудка. В результате секреция хлористоводородной кислоты снижается. Атропин блокирует неиннервируемые М3-холинорецепторы эндотелия сосудов, но при этом не вызывает изменения тонуса сосудов. Однако он препятствует взаимодействию рецепторов с М-холиномиметическими веществами и устраняет их сосудорасширяющее действие. Многие из этих эффектов атропина (и других М-холиноблокаторов) используют в медицинской практике. Показания к применению атропина. Способность атропина вызывать расширение зрачков используют в офтальмологии для исследования глазного дна, а также для лечения воспалительных заболеваний (ириты, иридоциклиты) и травм глаза, так как при расширении зрачка снижается опасность образования спаек между радужкой и капсулой хрусталика. Вызываемый атропином паралич аккомодации (циклоплегия) позволяет использовать его для определения истинной рефракции глаза (определение преломляющей способности хрусталика). После инстилляции в глаз 0,5–1% раствора атропина максимальное расширение зрачка наблюдается через 30–40 мин, паралич аккомодации — через 1–3 ч. Действие атропина на величину зрачков и аккомодацию сохраняется в течение 10–14 дней. Продолжительное расширение зрачков — преимущество атропина при лечении воспалительных заболеваний глаза. При длительном применении атропина возможны местное раздражение и гиперемия конъюнктив, развитие конъюнктивита. Системные реакции при закапывании атропина в глаз (гипертермия, сухость во рту) чаще возникают у маленьких детей и лиц преклонного возраста. В связи со способностью повышать атриовентрикулярную проводимость атропин применяют при атриовентрикулярном блоке вагусного происхождения. Используют его также при синусовой брадикардии. В связи с тем, что атропин понижает тонус бронхов и уменьшает секрецию бронхиальных желез, его можно применять как бронхолитическое средство для купирования и предупреждения бронхоспазма. Однако в настоящее время с этой целью в основном используют другие М-холиноблокаторы — ипратропий♠, тиотропий♠ и тровентол♠, которые 206 Часть II. Частная фармакология вводят ингаляционно. Эти вещества — четвертичные аммониевые соединения, в отличие от атропина они плохо всасываются в кровь с поверхности легких (а также из кишечника при проглатывании) и практически не вызывают системных побочных эффектов. Эффект уменьшения секреции хлористоводородной кислоты париетальными клетками желудка позволяет использовать атропин и некоторые другие М-холиноблокаторы при язвенной болезни желудка и двенадцатиперстной кишки. Это действие атропина связано с блокадой нескольких подтипов М-холинорецепторов, в том числе М3-холинорецепторов париетальных клеток, секретирующих хлористоводородную кислоту, и М1-холинорецепторов энтерохромаффиноподобных клеток желудка, выделяющих гистамин (гистамин стимулирует секрецию хлористоводородной кислоты париетальными клетками). В настоящее время из препаратов этой группы при язвенной болезни желудка и двенадцатиперстной кишки в основном используют избирательный блокатор М1-холинорецепторов — пирензепин. Пирензепин снижает секрецию хлористоводородной кислоты в меньшей степени, чем атропин, но в отличие от атропина практически не вызывает побочных эффектов, связанных с блокадой М2- и М3-холинорецепторов. Атропин снижает тонус гладких мышц и обладает сильным спазмолитическим действием, поэтому он применяется при болезненных спазмах гладкомышечных органов (коликах): спазмах кишечника (кишечная колика), желчных протоков (печеночная колика); в меньшей степени атропин эффективен при почечной колике. Атропин уменьшает секрецию трипсиногена поджелудочной железой, поэтому его можно применять в лечении острого панкреатита (при остром панкреатите трипсиноген превращается в трипсин непосредственно в тканях поджелудочной железы, что приводит к ее разрушению). Атропин применяют в анестезиологии для премедикации перед хирургическими операциями, при этом атропин, блокируя М2-холинорецепторы сердца, предупреждает рефлекторную брадикардию и возможность рефлекторной остановки сердца. При этом полезной также является способность атропина уменьшать секрецию слюнных и бронхиальных желез, снижать риск развития рефлекторного ларингоспазма и оказывать противорвотное действие. Атропин используют как специфический антидот при отравлении М-холиномиметиками и антихолинэстеразными средствами. Кроме того, атропин, проникая через ГЭБ, блокирует М-холинорецепторы экстрапирамидной системы и может уменьшать проявления болезни Паркинсона: тремор, ригидность, гипокинезию. В настоящее Глава 8. Средства, действующие на холинергические синапсы 207 время в качестве противопаркинсонических средств используют центральные М-холиноблокаторы: тригексифенидил (циклодол♠), бипериден (акинетон♠), которые преимущественно блокируют М-холинорецепторы ЦНС и в отличие от атропина оказывают менее выраженные периферические побочные эффекты. Применяют атропин в виде атропина сульфата♠ внутрь, парентерально и местно в виде глазных лекарственных форм. Внутрь назначают за 30– 40 мин до еды. Атропин хорошо всасывается из кишечника и быстро проникает в ЦНС (в течение 30–60 мин). Метаболизируется в печени путем гидролиза с образованием тропина и троповой кислоты. Период полуэлиминации составляет около 2 ч. Выводится почками (около 30–50% в неизмененном виде). Продолжительность действия при введении внутрь составляет 4–6 ч. Побочные эффекты атропина. При применении атропина возникают следующие побочные эффекты, связанные с блокадой М-холинорецепторов в различных органах и тканях: ● сухость во рту вследствие снижения секреции слюнных желез; ● нарушение ближнего видения вследствие паралича аккомодации; ● повышение ЧСС (тахикардия); ● обстипация (запор) вследствие снижения тонуса и перистальтики ЖКТ и повышения тонуса сфинктеров; ● нарушение мочеиспускания вследствие снижения тонуса и моторики стенки мочевого пузыря и повышения тонуса сфинктера. Атропин вызывает повышение внутриглазного давления, поэтому он противопоказан при глаукоме. Вследствие задержки мочеиспускания атропин противопоказан при доброкачественной гиперплазии предстательной железы. Атропин противопоказан также при тахиаритмиях, атонии кишечника. Симптомы и принципы лечения острого отравления атропином. В больших дозах атропин вызывает эффекты, связанные со стимулирующим действием на ЦНС: двигательное и психическое возбуждение, сильное беспокойство, нарушение памяти, координации. Эти эффекты возникают в основном при отравлении атропином. Кроме того, для отравления атропином характерны: ● расширение зрачков и затуманенность зрения; ● фотофобия (светобоязнь); ● ощущение присутствия песка в глазах вследствие снижения секреции слезных желез; ● сухость слизистых оболочек полости рта, носоглотки, гортани, что может привести к нарушению глотания и речи; 208 Часть II. Частная фармакология ● жажда, сухость, покраснение кожи и повышение температуры тела (вследствие нарушения потоотделения и теплоотдачи; у детей температура может повышаться до 42 °С); ● тахикардия (160–190 в минуту), экстрасистолы, ишемия миокарда; ● головная боль, головокружение; ● задержка мочеиспускания; ● в тяжелых случаях возникают зрительные и слуховые галлюцинации, бред, возможны судороги, которые сменяются состоянием угнетения и комой. Смерть наступает от паралича дыхательного центра (летальная доза для взрослых 100 мг). Частой причиной отравления, особенно у детей, бывает употребление растений, содержащих атропин (красавка, дурман, белена, летальная доза для детей — 10 мг — содержится в 2–3 ягодах красавки). Появление токсических концентраций атропина в крови может быть при передозировке атропина, а также при его закапывании в глаз. Симптомы интоксикации могут вызвать и другие средства с М-холиноблокирующей активностью, например трициклические антидепрессанты имипрамин и амитриптилин, антипсихотическое средство аминазин♠. Для устранения эффектов атропина парентерально вводят антихолинэстеразные средства, проникающие в ЦНС (физостигмин, галантамин). Другие меры при отравлении атропином состоят в промывании желудка раствором перманганата калия и назначении солевых слабительных, энтеросорбентов (активированный уголь), танина♠ (можно крепкий чай); для удаления вещества из крови используют гемосорбцию, форсированный диурез. При сильном возбуждении применяют диазепам или барбитураты короткого действия. При необходимости проводится искусственное дыхание. Препараты красавки (белладонны) содержат атропин. Применяют в виде настойки и экстрактов (сухой и густой) в качестве спазмолитических средств при болезненных спазмах гладких мышц ЖКТ, желчевыводящих протоков и других гладкомышечных органов. Препараты красавки входят в состав таблеток «Бекарбон»♠, «Бесалол»♠, «Бепасал»♠, «Беллалгин»♠; свечей «Бетиол»♠, «Анузол»♠; «Капель Зеленина»♠ и др. Скополамин (l-гиосцин) — алкалоид, содержащийся в тех же растениях, что и атропин, по химической структуре близок к атропину (сложный эфир скопина и троповой кислоты). Выпускают в виде скополамина гидробромида (рис. 8.7). Скополамин вызывает эффекты, связанные с блокадой М-холинорецепторов в периферических органах и тканях и в ЦНС. Через ГЭБ ско- Глава 8. Средства, действующие на холинергические синапсы 209 Рис. 8.7. Химические структуры некоторых М-холиноблокаторов поламин (рКа=7,2) проникает в большей степени, чем атропин (рКа= 9,0), так как при рН плазмы крови большее его количество находится в неионизированном состоянии. Периферические эффекты скополамина сходны с эффектами атропина. В то же время центральные эффекты существенно различаются. Скополамин в отличие от атропина в терапевтических дозах оказывает выраженное угнетающее действие на ЦНС. Обычно это проявляется в виде общего успокоения, сонливости. Кроме того, скополамин вызывает выраженную амнезию (ухудшение памяти). В токсических дозах скополамин подобно атропину может вызвать возбуждение ЦНС и затем коматозное состояние. Применение скополамина в клинической практике во многом связано с особенностями его действия на ЦНС. Используют его способность угнетать вестибулярные центры и оказывать противорвотное действие при вестибулярных расстройствах, проявляющихся в виде головокружений, тошноты, рвоты, нарушения равновесия, а также для профилакти- 210 Часть II. Частная фармакология ки морской и воздушной болезни. Скополамин входит в состав таблеток «Аэрон»♠, которые принимают для предупреждения тошноты и рвоты перед полетом или морским путешествием, действие их продолжается 6 ч. Эти таблетки содержат камфорнокислые соли скополамина и гиосциамина (камфора, оказывая тонизирующее действие на дыхательный центр, устраняет угнетающий эффект скополамина на дыхание). Для обеспечения более длительного действия скополамина используют специальные трансдермальные терапевтические системы доставки, представляющие собой накожные пластыри (приклеивают на здоровую кожу за ухом), выделяющие скополамин в течение 48–72 ч. Скополамин (как и атропин) применяют для премедикации перед хирургическими операциями для предотвращения рефлекторной брадикардии и уменьшения секреции слюнных и бронхиальных желез. При этом полезным также оказывается успокаивающее действие скополамина. Скополамин, так же как и атропин, можно применять в качестве спазмолитика при болезненных спазмах гладкомышечных органов (коликах), в офтальмологии его применяют для расширения зрачков с диагностической целью, а также при иритах и иридоциклитах. В качестве спазмолитических средств при спазмах желудка, кишечника, желчевыводящих и мочевыводящих путей, а также в комплексной терапии синдрома раздраженного кишечника используют гиосциамина сульфат и гиосцина бутилбромид (бускопан♠). Эти препараты также применяют в комплексной терапии язвы желудка и двенадцатиперстной кишки. Платифиллин — алкалоид крестовника широколистного (Senecio platyphyllus), третичное аммониевое основание, хорошо всасывается в кишечнике и проникает через ГЭБ. По М-холиноблокирующему действию менее активен, чем атропин. Обладает прямым миотропным спазмолитическим действием (расслабляющее действие оказывает непосредственно на гладкие мышцы внутренних органов и кровеносных сосудов), вследствие чего расширяет сосуды и несколько снижает АД. Применяют платифиллин в виде платифиллина гидротартрата♠ при спазмах гладкомышечных органов (в частности, при почечной колике), язвенной болезни желудка и двенадцатиперстной кишки, спазмах сосудов головного мозга и периферических сосудов. Пирензепин (гастроцепин ♠) блокирует преимущественно М 1-холинорецепторы, угнетая выделение гистамина энтерохромаффиноподобными клетками желудка (см. рис. 8.9). При этом снижается вызываемая гистамином секреция хлористоводородной кислоты париетальными клетками желудка. Кроме того, пирензепин снижает стимулирующее влияние центра вагуса на желудочную секрецию (связано с блокадой Глава 8. Средства, действующие на холинергические синапсы 211 М1-холинорецепторов интрамуральных ганглиев). Препарат применяют в качестве антисекреторного средства при лечении язвенной болезни желудка и двенадцатиперстной кишки (см. главу «Средства, влияющие на функции органов пищеварения»). Поскольку в средних терапевтических дозах пирензепин не блокирует М2- и М3-холинорецепторы, он не оказывает выраженного влияния на ЧСС, атриовентрикулярную проводимость, величину зрачка, тонус гладких мышц и перистальтику кишечника. При применении пирензепина несколько уменьшается секреция слюнных желез, в связи с чем возникает сухость во рту, возможны незначительные нарушения ближнего зрения (паралич аккомодации), диарея, булимия, аллергические реакции. Пирензепин не проникает через ГЭБ и не оказывает влияния на ЦНС. Толтеродин (детрузитол♠) блокирует М2 и М3-холинорецепторы и оказывает преимущественное спазмолитическое действие на гладкие мышцы мочевого пузыря, расслабляет детрузор и уменьшает его спонтанные сокращения. В меньшей степени оказывает угнетающее действие на секрецию слюнных желез. Преимущественное действие на мочевой пузырь (М-холиноблокирующее и прямое миотропное действие) оказывает также оксибутинин (дриптан♠). Оба препарата применяют при учащенном мочеиспускании, связанном с гиперреактивностью мочевого пузыря. К М-холиноблокаторам, недавно разрешенным для использования при лечении гиперактивного мочевого пузыря, относят троспия хлорид (спазмекс♠), дарифенацин (энаблекс♠) и солифенацин (везикар♠), зарегистрированные в нашей стране. Троспия хлорид — неселективный антагонист М-холинорецепторов, обладающий также прямым спазмолитическим действием. Дарифенацин и солифенацин — конкурентные М-холиноблокаторы с преимущественным действием на М3-холинорецепторы. Ипратропия бромид (атровент♠, иправент♠) неизбирательно блокирует разные подтипы М-холинорецепторов (М1, М2, М3). Блокируя М3-холинорецепторы гладких мышц бронхов (преимущественно на уровне крупных и средних бронхов) и бронхиальных желез, препарат оказывает выраженное бронхорасширяющее действие и снижает секрецию желез. Последний эффект нежелателен, так как приводит к уменьшению объема и повышению вязкости мокроты, затрудняя ее отделение. Отсутствие угнетающего действия на мукоцилиарный транспорт является положительным свойством препарата (см. рис. 8.7). В связи с блокадой М2-холинорецепторов, расположенных на пресинаптической мембране холинергических нервных окончаний, усиливается выделение ацетилхолина, конкурентно вытесняющего ипратропий 212 Часть II. Частная фармакология из связи с М3-холинорецепторами бронхов. Это уменьшает как продолжительность действия препарата, так и выраженность его эффекта. Применяют ипратропия бромид ингаляционно при обструктивных заболеваниях дыхательных путей, бронхиальной астме. При хронической обструктивной болезни легких (ХОБЛ) М-холиноблокаторы являются препаратами выбора, так как в патогенезе этого заболевания большое значение имеет повышение холинергических влияний на тонус бронхов. Эффект ипратропия развивается через 15 мин после ингаляционного введения препарата и сохраняется и течение 6–8 ч. При ингаляционном введении только лишь 10% препарата достигает мелких бронхов и альвеол, остальная часть вещества оседает в глотке и полости рта и проглатывается. Ипратропий относится к четвертичным аммониевым соединениями и, будучи гидрофильным веществом, при ингаляционном введении плохо всасывается в кровь со слизистой оболочки дыхательных путей, а также из кишечника в случае его проглатывания, поэтому практически не оказывает системных побочных (атропиноподобных) эффектов. В связи с уменьшением секреции слюнных и бронхиальных желез отмечаются сухость во рту и повышение вязкости мокроты. Ипратропий также уменьшает секрецию желез слизистой оболочки полости носа, поэтому он может использоваться для уменьшения ринореи при вазомоторном рините. Еще одно применение ипратропия (препарат итроп♠) связано с его способностью повышать атриовентрикулярную проводимость и ЧСС. Применяется этот препарат внутривенно и внутрь при синусовой брадикардии и блокадах сердца, обусловленных влиянием вагуса. Тиотропия бромид (спирива♠) преимущественно блокирует М3-холинорецепторы дыхательных путей, а также М1-холинорецепторы. Препарат не блокирует М2-холинорецепторы, локализованные на пресинаптической мембране нервных окончаний, поэтому не повышает выделение ацетилхолина в синаптическую щель. Это благоприятно сказывается на продолжительности его действия (около 12 ч). Тиотропия бромид назначают ингаляционно 1 раз в сутки при ХОБЛ для предупреждения обострения заболевания. Эффект развивается медленнее, чем при применении ипратропия бромида. Тиотропия бромид — полярное гидрофильное соединение, плохо проникающее через мембраны клеток, поэтому при его применении системные побочные (атропиноподобные) эффекты выражены незначительно. В офтальмологической практике применяют гоматропин, циклопентолат и тропикамид, которые оказывают менее продолжительное действие на глаз, чем атропин. Глава 8. Средства, действующие на холинергические синапсы 213 Тропикамид (мидриацил♠), блокируя М-холинорецепторы круговой мышцы радужки и цилиарной мышцы, вызывает мидриаз и паралич аккомодации (циклоплегию) (см. рис. 8.7). Расширение зрачков наступает быстро — через 5–10 мин и продолжается до 6 ч. Паралич аккомодации отмечается через 20–40 мин и длится 1–2 ч. Применяют для исследования глазного дна. Непродолжительность циклоплегии в этом случае является преимуществом препарата. Тропикамид хорошо всасывается со слизистой оболочки слезного канала в кровь и может оказывать нежелательные системные эффекты: головную боль, тахикардию, фотофобию, сухость во рту, гипертермию (в особенности у детей). Циклопентолат (цикломед♠) вызывает мидриаз и паралич аккомодации продолжительностью 20–24 ч. Его применяют для исследования рефракции у маленьких детей и непродолжительных диагностических исследований. Гоматропин — синтетическое атропиноподобное вещество, сложный эфир тропина и миндальной кислоты, относится к третичным аминам. По фармакологическим свойствам близок к атропину, но отличается меньшей активностью и меньшей продолжительностью действия (15–20 ч). Его применяют в офтальмологической практике для расширения зрачков в виде гоматропина метилбромида. Все М-холиноблокаторы противопоказаны при глаукоме. 8.2.2. Ганглиоблокаторы Ганглиоблокаторы блокируют Н-холинорецепторы нейронов симпатических и парасимпатических ганглиев и таким образом нарушают передачу возбуждения с преганглионарных на постганглионарные волокна. В результате уменьшается или устраняется влияние как симпатической, так и парасимпатической иннервации на эффекторные органы и ткани. Кроме того, ганглиоблокаторы блокируют Н-холинорецепторы хромаффинных клеток мозгового вещества надпочечников (уменьшая выделение адреналина и норадреналина), а также Н-холинорецепторы каротидных клубочков (препятствуя рефлекторному возбуждению дыхательного и сосудодвигательного центров). Блокада ганглиев симпатической системы приводит к уменьшению стимулирующего влияния симпатической системы на сердце и тонус сосудов (артерий и вен), при этом снижается ударный объем, расширяются артерии и вены — в результате снижается артериальное и венозное давление. Уменьшение выделения адреналина и норадреналина надпочеч- 214 Часть II. Частная фармакология никами под влиянием ганглиоблокаторов также способствует снижению артериального и венозного давления. Блокада ганглиев парасимпатической системы приводит к нарушению аккомодации (паралич аккомодации), учащению сокращений сердца (тахикардия), понижению тонуса гладких мышц ЖКТ и мочевого пузыря, угнетению секреции слюнных, бронхиальных желез, желез желудка и кишечника. В отличие от М-холиноблокаторов ганглиоблокаторы в меньшей степени расширяют зрачки и не оказывают существенного влияния на тонус бронхов. Показания к применению ганглиоблокаторов. В медицинской практике используют в основном гипотензивное действие ганглиоблокаторов. Препараты этой группы применяют для купирования гипертензивных кризов (быстрого снижения АД), при отеке легких на фоне повышенного АД, при спазме периферических артерий. Ганглиоблокаторы короткого действия можно использовать для управляемой гипотензии при хирургических операциях. Препараты вводят внутривенно капельно, при этом снижение АД способствует уменьшению кровотечения из сосудов операционного поля, а при нейрохирургических операциях препятствует развитию отека мозга. Кроме того, таким образом можно уменьшить нежелательные рефлекторные реакции на сердце и сосуды, которые возникают во время операции. Побочные эффекты ганглиоблокаторов связаны с блокадой симпатических и парасимпатических ганглиев. Так, расширение венозных сосудов, связанное с блокадой симпатических ганглиев, может стать причиной ортостатической гипотензии (резкого снижения давления крови при перемене положения тела из горизонтального в вертикальное). В результате может возникнуть обморок. Для предупреждения этого побочного эффекта после введения ганглиоблокатора больным рекомендуется лежать не менее 1,5–2 ч. К побочным эффектам ганглиоблокаторов, связанным с блокадой парасимпатических ганглиев, относят мидриаз, паралич аккомодации, сухость во рту, тахикардию, снижение моторики кишечника и тонуса мочевого пузыря. Снижение моторики кишечника приводит к обстипации и даже может быть причиной паралитического илеуса (непроходимости кишечника), а снижение тонуса мочевого пузыря приводит к задержке мочеиспускания. При таких осложнениях вводят М-холиномиметические (ацеклидин) или антихолинэстеразные (неостигмина бромид) средства. В связи с выраженными побочными эффектами ганглиоблокаторы не используют длительно. При передозировке ганглиоблокаторов развивается гипотензия, для устранения которой применяют α-адреномиметики, повышающие Глава 8. Средства, действующие на холинергические синапсы 215 АД. Показано применение аналептических средств, восстанавливающих дыхание. Ганглиоблокаторы противопоказаны при выраженной гипотензии, гиповолемии и шоке, остром инфаркте миокарда, гиперплазии предстательной железы, почечной и печеночной недостаточности. Ганглиоблокаторы нельзя применять у больных с закрытоугольной глаукомой, так как в связи с расширением зрачков происходит ухудшение оттока жидкости из передней камеры глаза, что может привести к повышению внутриглазного давления. В связи с замедлением тока крови ганглиоблокаторы противопоказаны при повышенной склонности к тромбообразованию. По химической структуре ганглиоблокаторы подразделяют на: ● бис-четвертичные аммониевые соединения: ♠ —гексаметония бензосульфонат (бензогексоний ); —азаметония бромид (пентамин♠); —трепирия йодид (гигроний♠); (арфонад♠); ● сульфониевое соединение: триметафана камсилат ♠ ● третичные амины: пемпидина тозилат (пирилен ), в настоящее время не используется. Бис-четвертичные аммониевые соединения и сульфониевое соединение — гидрофильные полярные вещества, поэтому плохо всасываются из ЖКТ и не проникают через ГЭБ. Они различаются по длительности действия. Гексаметония бензосульфонат и азаметония бромид при внутримышечном и подкожном введении действуют 2–3 ч и в основном применяются для купирования гипертензивных кризов. Триметафан и гигроний ♠ вызывают кратковременный эффект продолжительностью 10–20 мин. Поэтому эти препараты вводят внутривенно капельно для управляемой гипотензии. В настоящее время в связи с появлением более эффективных и безопасных гипотензивных средств ганглиоблокаторы используют редко. Мекамиламин — неполярное липофильное соединение (вторичный амин), легко проникает в ЦНС. Препарат может быть использован для облегчения отвыкания от курения (мекамиламинустраняет вызываемую никотином эйфорию, вытесняя его из связи с Н-холинорецепторами мозга). 8.2.3. Средства, блокирующие нервно-мышечные синапсы Средства, блокирующие нервно-мышечные синапсы, вызывают расслабление скелетных мышц (миорелаксацию) вследствие блокады передачи нервных импульсов с двигательных нервов на мышцы. Препараты этой группы называют также миорелаксантами периферического действия, 216 Часть II. Частная фармакология в отличие от веществ, которые расслабляют скелетные мышцы, действуя на ЦНС (миорелаксанты центрального действия, см. бензодиазепины). В зависимости от механизма нервно-мышечного блока выделяют миорелаксанты антидеполяризующего (недеполяризующего) действия и миорелаксанты деполяризующего действия. Миорелаксанты антидеполяризующего действия Вещества этой группы блокируют Н-холинорецепторы, локализованные на концевой пластинке скелетных мышц, препятствуя их взаимодействию с ацетилхолином, в результате чего ацетилхолин не вызывает деполяризацию мембраны мышечных волокон — мышцы не сокращаются. Такое состояние называется нервно-мышечным блоком. Однако при повышении концентрации ацетилхолина в синаптической щели (например, при применении антихолинэстеразных средств) ацетилхолин конкурентно вытесняет миорелаксант из связи с Н-холинорецептором и вызывает деполяризацию постсинаптической мембраны — происходит восстановление нервно-мышечной передачи. Вещества, действующие подобным образом, называют миорелаксантами антидеполяризующего конкурентного действия. Первым препаратом этой группы был алкалоид тубокурарин♠ — основное действующее вещество стрельного яда кураре. В состав этого яда входят экстракты южноамериканских растений видов Strychnos и Chondodendron. Индейцы Южной Америки использовали кураре во время охоты на животных, смазывая им наконечники стрел. Кураре, попав в организм животного, вызывало паралич скелетных мышц, и животное теряло способность двигаться, но его мясо было пригодно к употреблению в пищу. Впоследствии было установлено, что по химической структуре тубокурарин♠ — четвертичное аммониевое соединение, имеющее положительные заряды при двух атомах азота (рис. 8.8), поэтому, являясь гидрофильным полярным веществом и имея молекулу больших размеров, практически не всасывается из ЖКТ. Вещества, близкие тубокурарину♠ по действию, стали называть курареподобными средствами. Большинство курареподобных средств, так же как тубокурарин♠, относят к четвертичным аммониевым соединениям. В молекуле большинства веществ есть два положительно заряженных атома азота (катионные центры), которые и взаимодействуют с анионными структурами Н-холинорецепторов скелетных мышц, вызывая нервномышечный блок. Антидеполяризующие миорелаксанты относят к двум химическим группам: Глава 8. Средства, действующие на холинергические синапсы 217 Рис. 8.8. Химические структуры некоторых миорелаксантов антидеполяризующего действия безилат (атракурий), ● бензилизохинолины: тубокурарин♠, атракурия ♠ цисатракурия безилат (цисатракурий , нимбекс ), мивакурия хлорид (мивакурий, мивакрон♠); бромид (панкуроний), пипекурония ● аминостероиды: панкурония бромид (пипекуроний , ардуан♠), векурония бромид (векуроний, норкурон♠), рокурония бромид (рокуроний, рокуроний Каби♠). В зависимости от продолжительности вызываемого ими нервно-мышечного блока выделяют препараты: действия (30–60 мин и более) — тубокурарин♠, панку● длительного роний , пипекуроний (действует около 2 ч); действия (20–40 мин) — атракурий, ● средней продолжительности векуроний , рокуроний ; ● короткого действия (10–15 мин) — мивакурий. Антидеполяризующие миорелаксанты действуют медленнее, чем деполяризующие; исключение составляет рокуроний, вызывающий наиболее быстрый миопаралитический эффект (через 60–90 с). 218 Часть II. Частная фармакология Продолжительность действия курареподобных средств определяется характером их элиминации. Наиболее продолжительно действуют вещества, выделяющиеся почками (пипекуроний). Вещества средней продолжительности действия в большей степени выделяются с желчью в неизмененном виде и в виде метаболитов (векуроний, рокуроний) или подвергаются спонтанному неферментативному гидролизу (элиминация Хоффмана) в плазме крови (атракурий). Короткое действие мивакурия связано с тем, что он быстро разрушается холинэстеразой плазмы крови (псевдохолинэстераза). В связи с особым характером элиминации продолжительность действия атракурия не зависит от функционального состояния печени и почек (препарат может быть использован у больных с почечной и печеночной недостаточностью). Показания к применению антидеполяризующих миорелаксантов. Курареподобные средства используют для расслабления скелетных мышц при хирургических операциях. Под действием курареподобных средств мышцы расслабляются в следующей последовательности: сначала мышцы лица, гортани, шеи, затем мышцы конечностей, туловища и в последнюю очередь дыхательные мышцы — наступает остановка дыхания. При выключении дыхания больного переводят на искусственную вентиляцию легких. Кроме того, курареподобные средства применяют для устранения тонических судорог при столбняке и при отравлении стрихнином. При этом расслабление скелетных мышц способствует устранению судорог. Побочные эффекты. Побочные эффекты большинства курареподобных средств из группы бензилизохинолинов (тубокурарин♠, атракурий, мивакурий) связаны с их способностью высвобождать гистамин. Это может быть причиной гипотензии, бронхоспазма, покраснения кожи, а также других анафилактоидных реакций. В большей степени высвобождению гистамина способствует тубокурарин♠. Способность высвобождать гистамин практически отсутствует у препаратов из группы аминостероидов, но некоторые из них (панкуроний, рокуроний) оказывают умеренное М-холиноблокирующее действие и поэтому могут вызвать тахикардию. Поскольку тубокурарин♠ обладает выраженным гистаминогенным действием и, кроме того, в некоторой степени блокирует Н-холинорецепторы симпатических ганглиев (вследствие чего при его применении возможно существенное снижение АД и существует опасность анафилактоидных реакций), в настоящее время его применяют редко. Антагонисты миорелаксантов антидеполяризующего действия — антихолинэстеразные средства. Угнетая активность ацетилхолинэстеразы, они предотвращают гидролиз ацетилхолина, увеличивая его концентрацию в синаптической щели. Ацетилхолин вытесняет препарат из связи Глава 8. Средства, действующие на холинергические синапсы 219 с Н-холинорецепторами, восстанавливая нервно-мышечную передачу. Антихолинэстеразные средства (в частности, неостигмина бромид) применяют для прерывания нервно-мышечного блока или устранения остаточных явлений после введения антидеполяризующих мышечных релаксантов. Для предотвращения эффектов неостигмина бромида, вызываемых стимуляцией М-холинорецепторов, за 10 мин до неостигмина вводят атропин. Для устранения нервно-мышечного блока, вызванного аминостероидами, такими как векуроний и рокуроний, может быть использован новый препарат с принципиально другим механизмом действия — сугаммадекс (брайдан♠). Это модифицированный гамма-циклодекстрин, который состоит из восьми молекул глюкозы, образующих кольцеобразную структуру. Сугаммадекс формирует комплекс с упомянутыми аминостероидами, инкапсулируя их внутри своей структуры, и таким образом препятствует их взаимодействию с Н-холинорецепторами скелетных мышц. Препарат вводят внутривенно болюсно. От антихолинэстеразных средств сугаммадекс выгодно отличается отсутствием М-холиномиметических эффектов, в связи с чем при его применении нет необходимости вводить атропин. Миорелаксанты деполяризующего действия К этой группе относят суксаметоний (сукцинилхолин), который выпускается в виде суксаметония хлорида (листенон♠), суксаметония йодида (дитилин♠), суксаметония бромида. По химической структуре суксаметоний представляет собой удвоенную молекулу ацетилхолина (рис. 8.9). Суксаметоний взаимодействует с Нм-холинорецепторами, локализованными на концевой пластинке скелетных мышц, стимулируя их, и подобно ацетилхолину вызывает деполяризацию постсинаптической мембраны. При этом мышечные волокна сокращаются, что проявляется в виде отдельных подергиваний скелетных мышц — фасцикуляций. Однако в отличие от ацетилхолина суксаметоний обладает устойчивостью к ацетилхолинэстеразе (он гидролизуется только холинэстеразой плазмы крови) и поэтому практически не разрушается в синаптической щели. В результате суксаметоний вызывает стойкую деполяризацию постсинаптической мембраны кон- Рис. 8.9. Химическая структура суксаметония йодида 220 Часть II. Частная фармакология цевой пластинки скелетных мышц (деполяризационный блок). Это приводит к нарушению нервно-мышечной передачи и расслаблению скелетных мышц. При этом выделяющийся в синаптическую щель ацетилхолин лишь усиливает деполяризацию мембраны и углубляет нервно-мышечный блок. По этой причине антихолинэстеразные средства не устраняют действие суксаметония. Более того, подавляя активность ацетилхолинэстеразы, антихолинэстеразные средства повышают концентрацию ацетилхолина в синаптической щели, что поддерживает стойкую деполяризацию постсинаптической мембраны. Кроме того, ингибируя холинэстеразу плазмы крови, они препятствуют разрушению суксаметония, вследствие чего также усиливают и удлиняют его действие. Суксаметоний применяют при интубации трахеи, эндоскопических процедурах (бронхо-, эзофаго-, цистоскопии), кратковременных операциях (наложение швов на брюшную стенку, вправление вывихов, репозиция костных отломков), для устранения тонических судорог при столбняке. После внутривенного введения суксаметония его миопаралитическое действие начинается через 30–60 с и продолжается до 10 мин. Такое кратковременное действие препарата связано с его быстрым разрушением псевдохолинэстеразой (бутирилхолинэстеразой) плазмы крови (образуются холин и янтарная кислота). При генетической недостаточности этого фермента действие суксаметония может продолжаться до 2–6 ч. Миорелаксирующее действие препарата можно прекратить переливанием свежей цитратной крови, которая содержит активную псевдохолинэстеразу. При частых повторных введениях суксаметония возможно развитие десенситизации Нм-холинорецепторов концевой пластинки скелетных мышц, вследствие чего в ответ на введение препарата уже не возникает деполяризация мембраны, и деполяризационный блок сменяется на антидеполяризационный. При таком характере нервно-мышечного блока антихолинэстеразные средства могут ослабить миопаралитическое действие суксаметония. Побочные эффекты суксаметония: ● послеоперационные мышечные боли, что объясняется микротравмами мышц во время их фасцикуляций; ● угнетение дыхания (апноэ); ● гиперкалиемия вследствие выхода ионов калия из скелетных мышц при стойкой деполяризации постсинаптической мембраны и связанные с этим аритмии и возможная остановка сердца; ● гипертензия, вызываемая стимуляцией Н-холинорецепторов симпатических ганглиев и мозгового вещества надпочечников при повторных введениях препарата; Глава 8. Средства, действующие на холинергические синапсы 221 ● брадикардия и повышение секреции слюнных желез, связанные с наличием у суксаметония М-холиномиметического действия; ● повышение внутриглазного давления вследствие тонического сокращения под действием суксаметония экстраокулярных мышц, что затрудняет отток внутриглазной жидкости. Кроме того, возможны тяжелые осложнения — рабдомиолиз и миоглобинемия, а также злокачественная гипертермия, проявляющаяся быстрым повышением температуры тела (до 41–42 °С) и тоническим сокращением скелетных мышц, что связывают с повышенным выбросом ионов кальция из саркоплазматического ретикулума (для устранения этого эффекта вводят дантролен). Развитию злокачественной гипертермии при введении суксаметония способствует одновременное применение таких средств для наркоза, как галотан или изофлуран. Суксаметоний противопоказан при глаукоме, нарушении функции печени, анемии, беременности, в младенческом возрасте. Сравнительные характеристики миорелаксантов антидеполяризующего и деполяризующего действия приведены в табл. 8.2. Таблица 8.2. Сравнительные характеристики миорелаксантов антидеполяризующего и деполяризующего действия Показатели Механизм развития нервно-мышечного блока Фазы действия Влияние антихолиэстеразных средств Антидеполяризующие миорелаксанты Блокада H-холинорецепторов концевой пластинки скелетных мышц, устранение вызываемой ацетилхолином деполяризации постсинаптической мембраны Фаза миорелаксации Деполяризующие миорелаксанты Стимуляция H-холинорецепторов концевой пластинки скелетных мышц, стойкая деполяризация постсинаптической мембраны Фаза мышечной фасцикуляции. Фаза миорелаксации Устранение нервно-мышечно- Усиление и удлинение го блока нервно-мышечного блока 8.2.4. Средства, уменьшающие выделение ацетилхолина Нейротоксин, вырабатываемый анаэробной бактерией Clostridium botulinum, так называемый ботулинический (или ботулиновый) токсин (ботулотоксин) вызывает паралич скелетных мышц, препятствуя выделению ацетилхолина из окончаний холинергических нервных волокон в нервно- 222 Часть II. Частная фармакология мышечных синапсах. В токсических дозах, измеряемых в нанограммах (около 1 нг/кг массы тела), ботулотоксин вызывает паралич дыхательной мускулатуры со смертельным исходом и довольно часто является причиной пищевых отравлений (если продукты контаминированы спорами этих бактерий и предварительно не подвергаются обработке). В значительно меньших дозах ботулотоксин снимает спазм скелетных мышц, существенно не влияя на двигательную активность. Известны 7 иммунологически различающихся типов нейротоксинов (А, B, C, D, E, F, G), из них серотип А превосходит по активности серотипы В и F и используется в качестве медицинского препарата. Ботулинический токсин А Ботулинический токсин состоит из двух пептидных цепей (тяжелой и легкой, различающихся молекулярной массой — 100 и 50 кД соответственно), объединенных дисульфидным мостиком. Тяжелая цепь ботулинического токсина обладает способностью связываться со специфическими рецепторами мембран нервных клеток. После связывания с пресинаптической мембраной нервного окончания ботулинический токсин путем эндоцитоза проникает внутрь нейрона. По некоторым данным, легкая цепь токсина обладает протеазной активностью, вызывая протеолиз белков пресинаптической мембраны (SNAP-25, синтаксин) и везикулярной мембраны (синаптобревин). При взаимодействии этих белков происходит процесс слияния мембран. В результате энзиматического расщепления белков не происходит слияния мембраны везикул с пресинаптической мембраной и нарушается высвобождение ацетилхолина в синаптическую щель (см. рис. 8.1). Вследствие уменьшения выделения ацетилхолина в нервно-мышечных синапсах развивается паралич скелетных мышц. При этом не нарушается выделение из везикул веществ, выполняющих трофические функции, поэтому даже повторные инъекции ботулинического токсина не вызывают полной атрофии скелетных мышц. Кроме того, нарушается передача нервных импульсов в других холинергических синапсах, в том числе с симпатических холинергических волокон, иннервирующих потовые железы. Ботулинический токсин типа А получают из культур Clostridium botulinum и после ферментации, очистки и кристаллизации выпускают в виде комплекса с гемагглютинином (предохраняющим токсин от разрушения и ограничивающим его распределение в другие ткани) в форме лиофилизированного порошка для инъекций (препараты ботокс♠, диспорт♠). Глава 8. Средства, действующие на холинергические синапсы 223 В связи с тем, что препараты ботулинического токсина различаются по составу, их активность оценивают методом биологической стандартизации на мышах и выражают в мышиных единицах. При этом 1 ЕД эквивалентна количеству токсина, вызывающему при внутрибрюшинном введении летальный исход у 50% мышей определенной линии и массы тела в течение 3 дней. Показания к применению препаратов ботулинического токсина. Препараты применяют при спастических заболеваниях скелетных мышц, в том числе в офтальмологии при блефароспазме, для лечения косоглазия, а также для лечения спастичности, являющейся следствием инсульта, травмы спинного или головного мозга, при рассеянном склерозе, детском церебральном параличе, нейродегенеративных заболеваниях, спастической кривошее, лицевом гемиспазме, спастической дистонии и других спастических состояниях. Вводят внутримышечно и реже подкожно. Действие продолжается 4–6 мес. Длительное действие препаратов объясняется тем, что мышечные сокращения восстанавливаются только вследствие процесса реиннервации (появления боковых отростков нервных окончаний). Вследствие того, что ботулинический токсин препятствует выделению ацетилхолина окончаниями симпатических холинергических волокон, иннервирующих потовые железы, препараты применяют при гипергидрозе для уменьшения секреции апокринных потовых желез (подмышечные впадины, ладони, стопы). Вводят внутрикожно, эффект продолжается 6–8 мес. Препараты ботулинического токсина применяют в косметологии. Ботулинический токсин концентрируется в месте инъекции в течение некоторого времени, а затем попадает в системный кровоток, не проникает через ГЭБ и быстро метаболизируется. Побочные эффекты препаратов ботулинического токсина. В качестве побочных эффектов отмечаются боль и микрогематомы в месте инъекции, незначительная общая слабость в течение 1 нед (при применении больших доз), в зависимости от места введения возможны птоз, слезотечение или дисфагия. У некоторых пациентов при применении препаратов появляются антитела к комплексу «гемагглютинин–ботулинический токсин». Этому способствует введение препаратов в высоких дозах. Препараты ботулинического токсина противопоказаны при миастении, беременности, грудном вскармливании, их нельзя сочетать с препаратами, вызывающими релаксацию скелетных мышц, например с аминогликозидными антибиотиками. 224 Часть II. Частная фармакология Вопросы и задания для самоконтроля 1. Снижение АД при внутривенном введении ацетилхолина связано со стимуляцией: а) Н-холинорецепторов симпатических ганглиев; б) М3-холинорецепторов эндотелиальных клеток сосудов; в) М3-холинорецепторов гладкомышечных клеток сосудов; г) Н-холинорецепторов энтерохромаффинных клеток надпочечников; д) М2-холинорецепторов синоатриального узла. 2. При денервации круговой мышцы глаза сужение зрачка вызывают: а) физостигмин; б) пилокарпин; в) армин; г) неостигмин; д) карбахолин. 3. Антихолинэстеразные средства и М-холиномиметики различаются по действию на: а) гладкие мышцы кишечника; б) круговую мышцу радужки; в) скелетные мышцы; г) экзокринные железы; д) эндотелиальные клетки сосудов; е) кардиомиоциты. 4. При болезни Альцгеймера применяют: а) донепезил; б) неостигмин; в) ривастигмин; г) пиридостигмин; д) галантамин; е) эдрофоний. 5. Какой эффект неостигмина не устраняется атропином: а) спазм гладких мышц бронхов; б) брадикардия; в) повышение тонуса скелетных мышц; г) сужение зрачков; д) повышение секреции хлористоводородной кислоты? Глава 8. Средства, действующие на холинергические синапсы 225 6. На фоне атропина ацетилхолин повышает АД, действуя на: а) Н-холинорецепторы симпатических ганглиев; б) М-холинорецепторы эндотелиальных клеток; в) Н-холинорецепторы парасимпатических ганглиев; г) М-холинорецепторы гладких мышц сосудов; д) Н-холинорецепторы мозгового вещества надпочечников. 7. Пирензепин в отличие от атропина: а) в большей степени угнетает секрецию хлористоводородной кислоты; б) в основном угнетает гистамин-индуцированную секрецию HCl; в) оказывает более выраженные побочные эффекты; г) избирательно блокирует М1-холинорецепторы; д) повышает выделение гистамина из энтерохромаффиноподобных клеток желудка. 8. Что является общими характеристиками ипратропия и тиотропия: а) способность проникать через мембраны клеток путем пассивной диффузии; б) низкая выраженность системных побочных эффектов при ингаляционном введении; в) избирательная блокада М3-холинорецепторов; г) выраженное угнетение мукоцилиарного транспорта; д) эффективность при ХОБЛ? 9. Тиотропий в отличие от ипратропия: а) избирательно блокирует М1- и М3-холинорецепторы; б) проникает через ГЭБ; в) повышает выделение ацетилхолина из окончаний парасимпатических нервов; г) действует продолжительнее. 10. Какой эффект скополамина является основным при болезни движения: а) противорвотный; б) антидиарейный; в) угнетение мочеиспускания; г) антисекреторный; д) амнестический? 11. Какой эффект ганглиоблокаторов связан с блокадой ганглиев симпатической системы: 226 Часть II. Частная фармакология а) тахикардия; б) мидриаз; в) гипотензия; г) снижение перистальтики кишечника; д) снижение секреции экзокринных желез? 12. В отличие от мивакурия♠ суксаметоний♠: а) не разрушается холинэстеразой плазмы крови; б) вызывает стойкую деполяризацию постсинаптической мембраны нервно-мышечного синапса; в) блокирует Нм-холинорецепторы скелетных мышц; г) стимулирует Нм-холинорецепторы скелетных мышц; д) вызывает паралич скелетных мышц, который, как правило, не устраняется неостигмином. Глава 9 СРЕДСТВА, ДЕЙСТВУЮЩИЕ НА АДРЕНЕРГИЧЕСКИЕ СИНАПСЫ Адренергические синапсы, локализованные в органах, получающих симпатическую иннервацию, образованы окончаниями постганглионарных симпатических (адренергических) волокон и эффекторными клетками. Аксоны ганглионарных клеток симпатической системы имеют анатомическую особенность — в непосредственной близости от эффекторных органов они разветвляются с образованием сети адренергических волокон с множеством варикозных утолщений. Именно варикозные утолщения участвуют в образовании синаптических контактов с клетками эффекторных органов. В варикозных утолщениях есть везикулы (пузырьки), содержащие норадреналин. Норадреналин является медиатором адренергических синапсов. Норадреналин синтезируется из аминокислоты тирозина, проникающей в варикозные утолщения путем активного транспорта (тирозин синтезируется в печени из аминокислоты фенилаланина). В цитоплазме адренергических нейронов тирозин подвергается ряду последовательных превращений: сначала из тирозина при участии фермента тирозингидроксилазы образуется ДОФА, из которого под действием ДОФА-декарбоксилазы образуется дофамин. Дофамин путем активного транспорта поступает в везикулы и внутри везикул превращается в норадреналин (рис. 9.1). Под влиянием нервного импульса происходит деполяризация пресинаптической мембраны, открываются потенциалозависимые кальциевые каналы, через которые ионы кальция проникают в варикозные утолщения: концентрация Са2+ в цитоплазме варикозного утолщения увеличивается. Это приводит к экзоцитозу везикул и выделению норадреналина в синаптическую щель. После высвобождения в синаптическую щель норадреналин стимулирует адренорецепторы, локализованные на постсинаптической мембране эффекторных клеток. Действует норадреналин непродолжительно, большая его часть (около 80%) захватывается нервными окончаниями с помощью специальных транспортных систем (обратный нейрональный захват). 228 Часть II. Частная фармакология РЕЗЕРПИН Рис. 9.1. Схема адренергического синапса: Адр — адреналин; НА — норадреналин; ДОФА — диоксифенилаланин; МАО — моноаминоксидаза; КОМТ — катехол-Ометилтрансфераза; 1 — транспортная система мембран везикул; 2 — система обратного нейронального захвата В цитоплазме варикозного утолщения часть норадреналина подвергается окислительному дезаминированию под действием МАО, локализованной на внешней мембране митохондрий, но большее количество норадреналина захватывается везикулами. Небольшая часть норадреналина захватывается эффекторными клетками (например, гладкомышечными клетками). Этот процесс называется экстранейрональным захватом. В эффекторных клетках норадреналин метаболизируется цитоплазматическим ферментом катехол-орто-метилтрансферазой (КОМТ). Под действием КОМТ происходит О-метилирование норадреналина. Вещества, воздействующие на передачу возбуждения в адренергических синапсах, могут оказывать влияние на разные процессы. Превращение тирозина в ДОФА в цитоплазме нервных окончаний угнетает ингибитор тирозингидроксилазы альфа-метил-п-тирозин (метирозин), а превращение ДОФА в дофамин — ингибиторы ДОФА-декарбоксилазы карбидопа и бенсеразид (см. гл. «Противопаркинсонические средства»). Симпатолитик резерпин блокирует транспортные системы мембран везикул, в результате уменьшается захват везикулами дофамина и нарушается синтез норадреналина в везикулах. Высвобождение норадре- Глава 9. Средства, действующие на адренергические синапсы 229 налина из пресинаптических окончаний повышают симпатомиметики: тирамин♠, содержащийся в некоторых пищевых продуктах (например, в сыре), эфедрин и амфетамин. Некоторые вещества оказывают прямое стимулирующее действие на адренорецепторы (адреномиметики) или блокируют адренорецепторы (адреноблокаторы). Обратный нейрональный захват норадреналина ингибируют кокаин и трициклические антидепрессанты (имипрамин, амитриптилин), при этом повышается концентрация норадреналина в синаптической щели. Разрушению норадреналина в цитоплазме нервных окончаний препятствуют вещества, ингибирующие МАО (например, неизбирательный ингибитор МАО ниаламид). Проникновение норадреналина через мембрану везикул нарушает резерпин, блокирующий транспортные системы мембран везикул, при этом нарушается депонирование норадреналина в везикулах (см. рис. 9.1). Многие из этих веществ — трициклические антидепрессанты, ингибиторы МАО, некоторые симпатомиметики и симпатолитики используют в качестве лекарственных препаратов. Наибольшее применение в медицинской практике нашли вещества, непосредственно воздействующие на адренорецепторы (адреномиметики и адреноблокаторы). Адренорецепторы различают по чувствительности к одним и тем же веществам. Выделяют α-адренорецепторы и β-адренорецепторы. α-Адренорецепторы подразделяют на α 1 -адренорецепторы и α 2 адренорецепторы, а среди β-адренорецепторов различают β1-, β2- и β3адренорецепторы. На постсинаптической мембране эффекторных клеток локализованы α1- и β1-адренорецепторы (постсинаптические рецепторы). Эти рецепторы стимулируются норадреналином, высвобождаемым из окончаний адренергических волокон. α2- и β2-Адренорецепторы могут быть внесинаптическими и пресинаптическими. Внесинаптические α2- и β2-адренорецепторы, локализованные вне синапсов на мембране эффекторных клеток, не получают симпатическую иннервацию (неиннервируемые рецепторы). Они возбуждаются циркулирующим в крови адреналином, выделяющимся из хромаффинных клеток мозгового вещества надпочечников, α2-адренорецепторы, кроме того, могут возбуждаться циркулирующим в крови норадреналином. Находящиеся на пресинаптической мембране (пресинаптические) α2-адренорецепторы регулируют высвобождение норадреналина по принципу отрицательной обратной связи. Стимуляция этих рецепторов норадреналином или другими веществами с α2-адреномиметической активностью тормозит высвобождение норадреналина из варикозных утолщений. 230 Часть II. Частная фармакология В отличие от пресинаптических α2-адренорецепторов стимуляция пресинаптических β-адренорецепторов приводит к повышению выделения норадреналина (см. рис. 9.1). Основные эффекты, вызываемые стимуляцией адренорецепторов, представлены в табл. 9.1. Таблица 9.1. Подтипы адренорецепторов и основные эффекты, вызываемые их стимуляцией Подтипы адренорецепторов Эффекты, вызываемые стимуляцией адренорецепторов Сокращение гладких мышц сосудов (сужение кровеносных сосудов) Сокращение радиальной мышцы радужки (расширение зрачков) Сокращение гладких мышц сосудов (сужение кровеносных α2 сосудов) α2 Пресинаптические Снижение выделения норадреналина окончаниями адренергических волокон Увеличение силы сердечных сокращений, ЧСС, β1 атриовентрикулярной проводимости Секреция ренина юкстагломерулярными клетками почек Расслабление гладких мышц сосудов, бронхов, матки. β2 Расширение кровеносных сосудов. Расширение бронхов. Снижение тонуса и сократительной активности миометрия. Активация гликогенолиза α1 Средства, действующие на адренергические синапсы, подразделяют на: ● средства, стимулирующие адренергические синапсы; ● средства, блокирующие адренергические синапсы. 9.1. СРЕДСТВА, СТИМУЛИРУЮЩИЕ АДРЕНЕРГИЧЕСКИЕ СИНАПСЫ Средства, стимулирующие адренергические синапсы, подразделяют на 2 группы: ● адреномиметики — средства, непосредственно стимулирующие адренорецепторы; ● симпатомиметики (адреномиметики непрямого действия) — средства, повышающие выделение медиатора. Глава 9. Средства, действующие на адренергические синапсы 231 9.1.1. Адреномиметики По преимущественному влиянию на α- или β-адренорецепторы адреномиметики подразделяют на: ● α-адреномиметики (средства, преимущественно стимулирующие α-адренорецепторы); ● β-адреномиметики (средства, преимущественно стимулирующие β-адренорецепторы); ● α-, β-адреномиметики (средства, стимулирующие α- и β-адренорецепторы). α-Адреномиметики По преимущественному влиянию на α1- или α2-адренорецепторы выделяют α1-адреномиметики и α2-адреномиметики. α1-Адреномиметики (стимуляторы α1-адренорецепторов) α1-Адренорецепторы локализованы на постсинаптической мембране эффекторных клеток, получающих симпатическую иннервацию: гладкомышечных клеток сосудов (артерий внутренних органов, кожи, слизистых оболочек, головного мозга, коронарных артерий, вен), радиальной мышцы радужки, сфинктера мочевого пузыря, простатической части уретры, предстательной железы, миометрия, сфинктеров ЖКТ, капсулы селезенки. Стимуляция α1-адренорецепторов (связанных с Gq-белками) вызывает сокращение гладких мышц. Сокращение гладких мышц сосудов приводит к их сужению, увеличению общего периферического сопротивления и повышению АД. При сокращении капсулы селезенки происходит выброс крови в циркуляторное русло. Сокращение радиальной мышцы глаза вызывает расширение зрачка, а сокращение сфинктера мочевого пузыря и уретры приводит к задержке мочеиспускания. Сокращение сфинктеров ЖКТ при снижении моторики и тонуса гладких мышц желудка и кишечника (связано с активацией Са2+-зависимых К+-каналов и гиперполяризацией мембран гладкомышечных клеток) приводит к задержке продвижения содержимого по ЖКТ. Стимуляция α1-адренорецепторов матки приводит к сокращению миометрия. К α1-адреномиметикам относится фенилэфрин (применяют в виде фенилэфрина гидрохлорида♠, препарата мезатон♠). В отличие от адреналина♠ и норадреналина♠ фенилэфрин не является катехоламином (содержит только одну гидроксильную группу в ароматическом кольце) и практиче- 232 Часть II. Частная фармакология ски не инактивируется КОМТ (рис. 9.2). В связи с этим он оказывает более продолжительное действие и эффективен при приеме внутрь. Стимулируя α1-адренорецепторы сосудов, фенилэфрин вызывает сужение сосудов и, как следствие, повышение АД (прессорное действие). При повышении АД происходит стимуляция барорецепторов дуги аорты и возникает рефлекторная брадикардия. Рефлекторный эффект является доминирующим: рефлекторно повышается активность центра блуждающего нерва, вследствие чего усиливаются его тормозные влияния на ЧСС и, кроме того, рефлекторно понижается активность сосудодвигательного центра и уменьшаются симпатические влияния на сердце. Препарат вызывает расширение зрачков вследствие сокращения радиальной мышцы радужки и не влияет на аккомодацию (цилиарная мышца глаза имеет преимущественную парасимпатическую иннервацию), понижает внутриглазное давление при открытоугольной глаукоме. Показания к применению фенилэфрина. Применяют фенилэфрин для повышения АД при гипотензии (внутривенно, подкожно и внутримышечно). Прессорный эффект продолжается 20 мин при внутривенном введении и 40–50 мин при подкожном введении. Фенилэфрин в виде глазных капель (ирифрин♠) применяют в офтальмологии для расширения зрачков. Продолжительность мидриатического эффекта составляет 4–6 ч, в отличие от М-холиноблокаторов не вызывает паралич аккомодации. Фенилэфрин также используют для снижения внутриглазного давления при открытоугольной глаукоме. Фенилэфрин в комбинации с блокатором Н1-гистаминовых рецепторов диметинденом (препарат Виброцил♠) используют местно при рините (вызывает сужение сосудов и уменьшает отек слизистой оболочки носовой полости), а также добавляют в растворы местных анестетиков (сужение сосудов пролонгирует местное и снижает резорбтивное действие местных анестетиков). Основные побочные эффекты фенилэфрина — чрезмерное повышение АД, головная боль, головокружение, рефлекторная брадикардия, ишемия тканей вследствие сужения периферических сосудов, нарушение мочеиспускания. Рис. 9.2. Химическая структура фенилэфрина гидрохлорида Глава 9. Средства, действующие на адренергические синапсы 233 Фенилэфрин противопоказан при гипертонической болезни, спазмах сосудов (в том числе коронарных), гипертиреозе, закрытоугольной глаукоме. α2-Адреномиметики (стимуляторы α2-адренорецепторов) α2-Адренорецепторы находятся в сосудах (артериолах кожи и слизистых оболочек, коронарных артериолах, венах) в основном вне синапсов, также они обнаружены на постсинаптических мембранах и на пресинаптических мембранах варикозных утолщений окончаний адренергических нервов. Стимуляция α2-адренорецепторов сосудов (сопряженных с Gi/0белками, угнетающими аденилатциклазу) уменьшает уровень цАМФ и активность цАМФ-зависимой протеинкиназы А в гладкомышечных клетках. В результате повышается активность киназы, фосфорилирующей легкие цепи миозина. Фосфорилирование легких цепей миозина облегчает его взаимодействие с актином, что приводит к сокращению гладких мышц кровеносных сосудов. При снижении активности протеинкиназы А повышается также активность фосфоламбана, угнетающего Са2+-АТФазу саркоплазматического ретикулума (место депонирования внутриклеточного Са2+). В результате Са2+-АТФаза не переносит Са2+ из цитоплазмы в саркоплазматический ретикулум, и концентрация Са2+ в цитоплазме клетки повышается. Это также способствует сокращению гладких мышц сосудов. Стимуляция пресинаптических α 2 -адренорецепторов вызывает уменьшение выделения норадреналина из варикозных утолщений (по принципу отрицательной обратной связи). Этот эффект (связанный с активацией Gi/0-белков) возникает как в результате снижения активности протеинкиназы А, так и в результате открытия калиевых каналов в мембранах нервных окончаний, что приводит к выходу из них ионов К+ и гиперполяризации клеточных мембран. В результате стимуляции α2-адренорецепторов нарушается открытие кальциевых каналов и поступление ионов Са2+ в нервные окончания, что уменьшает экзоцитоз содержимого везикул в синаптическую щель. α2-Адренорецепторы находятся также на пресинаптических мембранах окончаний холинергических нервов, где они таким же образом регулируют выделение ацетилхолина. Внесинаптические α2-адренорецепторы находятся в тромбоцитах, где они опосредуют агрегацию тромбоцитов. Тонус и моторика желудка и кишечника, а также секреция пищеварительных желез снижаются при стимуляции α2-адренорецепторов. В β-клетках поджелудочной железы стимуляция α2-адренорецепторов снижает секрецию инсулина. 234 Часть II. Частная фармакология В ЦНС с возбуждением α2-адренорецепторов связывают гипотензивный, анальгетический, седативный, гипотермический и некоторые другие эффекты агонистов этих рецепторов. К α2-адреномиметикам относят нафазолин (нафтизин♠, санорин♠), оксиметазолин (назол♠), ксилометазолин (галазолин♠, ксилен♠), тетризолин (тизин♠, октилия♠), клонидин (клофелин♠, гемитон♠), гуанфацин (эстулик♠), метилдопа (допегит♠), тизанидин (сирдалуд♠). По химической структуре нафазолин, оксиметазолин, тетризолин и ксилометазолин — производные имидазолина (рис. 9.3). Стимулируя α2-адренорецепторы, они оказывают сосудосуживающее действие, вызывая более длительное сужение периферических сосудов, чем фенилэфрин. Применяют местно при ринитах в виде капель и спрея, нафазолин также в виде эмульсии (санорин♠). При интраназальном введении препаратов происходит сужение сосудов слизистой оболочки носовой полости, что уменьшает ее отечность и улучшает отток из околоносовых пазух. В результате снижается приток крови к венозным синусам и облегчается носовое дыхание. В зависимости от лекарственной формы и входящих в нее других компонентов действие продолжается от 6–8 ч до 10–12 ч. Не следует применять эти препарата более 3 раз в сутки. При длительном применении (более 5 сут) сосудосуживающий эффект препаратов снижается вследствие быстрого развития привыкания (тахифилаксии). После отмены препаратов возникает эффект последействия (заложенность носа вследствие рикошетной вазодилатации), наиболее выраженный у ксилометазолина и оксиметазолина. Тетризолин в виде глазных капель (препарат октилия♠) применяют при раздражении глаз, вызванном аллергическими, химическими или физическими факторами (дым, пыль, сильное освещение). Эти препараты, несмотря на местное применение, могут частично всасываться в системный кровоток и повышать АД, противопоказаны при гипертонической болезни, выраженном атеросклерозе. Клонидин и гуанфацин стимулируют α2-адренорецепторы в ЦНС, в частности α2-адренорецепторы, локализованные на нейронах ядер солитарного тракта в продолговатом мозге, а клонидин, кроме того, стимулирует имидазолиновые I1рецепторы той же локализации. Стимуляция нейронов ядер солитарного тракта Рис. 9.3. Химическая структура приводит к повышению активности нафазолина нитрата центра блуждающего нерва (вагуса) и уг- Глава 9. Средства, действующие на адренергические синапсы 235 нетению активности сосудодвигательного центра. Вследствие увеличения влияния вагуса на сердце снижается ЧСС. Угнетение активности сосудодвигательного центра сопровождается уменьшением симпатических влияний на сердце и сосуды: снижаются сила и ЧСС (уменьшается сердечный выброс) и расширяются кровеносные сосуды. Снижение симпатических влияний приводит также к уменьшению секреции ренина юкстагломерулярными клетками почек (уменьшается образование ангиотензина II). В результате происходит снижение АД. Кроме того, клонидин и гуанфацин, стимулируя пресинаптические α2-адренорецепторы на окончаниях адренергических волокон, уменьшают выделение норадреналина, что также снижает влияние симпатической иннервации на сердце и сосуды и приводит к понижению АД. Показания к применению клонидина и гуанфацина. Гипотензивный эффект этих препаратов используют при лечении артериальной гипертензии (см. главу «Гипотензивные средства»). Клонидин применяют при гипертензивных кризах. При его быстром внутривенном введении возможно кратковременное повышение АД из-за стимуляции α2-адренорецепторов сосудов, поэтому препарат необходимо вводить медленно. Гуанфацин отличается от клонидина большей продолжительностью действия. Клонидин оказывает болеутоляющее действие и применяется в качестве анальгетика (см. главу «Анальгезирующие средства»), кроме того, он может быть использован для уменьшения абстинентного синдрома при зависимости к опиоидам. Клонидин вызывает седативный эффект и потенцирует действие алкоголя. Анальгетический и седативный эффекты связаны со стимуляцией α2-адренорецепторов в ЦНС. Клонидин уменьшает продукцию внутриглазной жидкости (суживает сосуды ресничного тела и уменьшает фильтрацию жидкости), вследствие чего снижает внутриглазное давление. Это свойство клонидина используют при открытоугольной глаукоме (во время операции по поводу глаукомы, при неэффективности β-адреноблокаторов). Клонидин и гуанфацин вызывают следующие побочные эффекты: сухость во рту, брадикардию, сонливость и заторможенность, снижение скорости реакции (не рекомендуется вождение автомобиля), задержку жидкости в организме, констипацию, возможно развитие синдрома отмены. Метилдопа является пролекарством и в организме превращается в α-метилнорадреналин, стимулирующий α2-адренорецепторы, в том числе в продолговатом мозге. Применяют как гипотензивное средство при артериальной гипертензии. Тизанидин, как и клонидин, — производное имидазолина. Стимулируя пресинаптические α2-адренорецепторы в нейронах спинного мозга, 236 Часть II. Частная фармакология уменьшает высвобождение возбуждающих аминокислот и угнетает передачу возбуждения в синапсах. Это вызывает снижение тонуса скелетных мышц (препарат относят к миорелаксантам центрального действия). Применяют внутрь при мышечной боли, связанной со спазмом скелетных мышц (оказывает болеутоляющее действие). Препарат понижает АД, вызывает головокружение, слабость, сонливость, заторможенность. Как и при применении клонидина, гуанфацина и метилдопы, следует избегать видов деятельности, требующих высокой концентрации внимания (например, вождения автомобиля). β-Адреномиметики Выделяют вещества с преимущественным действием на β1-адренорецепторы (β 1-адреномиметики), с преимущественным действием на β2-адренорецепторы (β2-адреномиметики) и вещества неизбирательного действия (β1-, β2-адреномиметики). β1-Адреномиметики (стимуляторы β1-адренорецепторов) β1-Адренорецепторы преимущественно локализованы в сердце, в мембране кардиомиоцитов. Стимуляция β1-адренорецепторов (связанных с Gs-белками, активирующими аденилатциклазу) приводит к увеличению уровня цАМФ, повышению активности протеинкиназы А, которая, фосфорилируя кальциевые каналы, способствует их открытию и поступлению Са2+ в кардиомиоциты, происходит также мобилизация Са2+ из саркоплазматического ретикулума — в результате концентрация цитоплазматического кальция повышается. Увеличение поступления Са2+ в кардиомиоциты повышает автоматизм синоатриального узла и, следовательно, ЧСС. В атриовентрикулярном узле увеличение входа Са2+ приводит к облегчению атриовентрикулярной проводимости и повышению автоматизма, повышается также автоматизм волокон Пуркинье. В клетках рабочего миокарда кальций, в большом количестве высвобождающийся из саркоплазматического ретикулума в цитоплазму, связывается с тропонином С — одним из компонентов тропонин-тропомиозинового комплекса. При этом происходит изменение конформации комплекса и устраняется его тормозное влияние на сократительные белки миокарда, в результате облегчается взаимодействие актина с миозином, что приводит к увеличению силы сердечных сокращений. При стимуляции β1-адренорецепторов юкстагломерулярных клеток почек увеличивается секреция ренина, вследствие чего повышается образование ангиотензина II. Глава 9. Средства, действующие на адренергические синапсы 237 К препаратам, стимулирующим преимущественно β1-адренорецепторы сердца, относится добутамин (добутрекс♠). Добутамин — рацемическая смесь двух стереоизомеров, которые будучи агонистами β1-адренорецепторов, разнонаправленно действуют на α1-адренорецепторы: (–) изомер является агонистом, а (+) изомер — антагонистом α1-рецепторов (рис. 9.4). В итоге рацемическая смесь этих стереоизомеров, используемая в клинической практике, практически обладает только β1-адреномиметической активностью; причем в отношении β1-адренорецепторов (+) изомер приблизительно в 10 раз активнее (–) изомера. Добутамин увеличивает силу сокращений сердца (оказывает положительное инотропное действие) и в меньшей степени повышает ЧСС и атриовентрикулярную проводимость. В терапевтических дозах препарат практически не изменяет общее периферическое сопротивление сосудов. Применяется добутамин как кардиотоническое средство при острой сердечной недостаточности (см. главу «Кардиотонические средства»). Поскольку добутамин разрушается в ЖКТ, его вводят внутривенно (инфузионно). Начинает действовать через 1–2 мин, максимальный эффект отмечается через 10 мин. Метаболизируется в печени КОМТ. Выводится почками в виде конъюгатов t½ — 2 мин). Из побочных эффектов отмечают тахикардию, аритмии, повышение работы сердца и потребления миокардом кислорода (вследствие этого может увеличиваться зона инфаркта), загрудинные боли, гипертензию. К действию добутамина в течение нескольких дней развивается толерантность. Стимулирующим действием на β1-адренорецепторы сердца обладает препарат дофамина допамин, также применяемый как кардиотоническое средство при острой сердечной недостаточности (повышает силу сокращений миокарда и, следовательно, сердечный выброс). Кроме того, в невысоких дозах допамин, стимулируя дофаминовые D1-рецепторы, вызывает расширение сосудов внутренних органов и почек (это препятствует развитию ишемии внутренних органов при кардиогенном шоке). Этот эффект связан со стимулирующим действием дофамина на активность аденилатциклазы и повышением уровня цАМФ в ангиомиоцитах. Рис. 9.4. Химическая структура добутамина 238 Часть II. Частная фармакология Повышая почечный кровоток и скорость клубочковой фильтрации, допамин увеличивает диурез, повышается экскреция натрия. В высоких дозах допамин стимулирует α1-адренорецепторы и вызывает сужение сосудов. Так же как и при введении добутамина, повышается ЧСС, возможны аритмии, возникновение ангинальной боли, повышение АД. В связи с тем, что дофамин разрушается МАО и КОМТ, его препарат применяется парентерально (внутривенно инфузионно). Добутамин и допамин не применяют для длительного лечения хронической сердечной недостаточности. β2-Адреномиметики (стимуляторы β2-адренорецепторов) β2-Адренорецепторы находятся в мембранах гладкомышечных клеток бронхов, матки, ЖКТ, детрузора мочевого пузыря, кровеносных сосудов (сосудов скелетных мышц, легких, коронарных сосудов). При стимуляции этих рецепторов происходит расслабление гладких мышц бронхов, снижаются тонус и сократительная активность миометрия, мочевого пузыря, желчного пузыря и желчных протоков, моторика и тонус желудка и кишечника, расширяются кровеносные сосуды. Расслабление гладких мышц при стимуляции β2-адренорецепторов (сопряженных с Gs-белками, стимулирующими аденилатциклазу) связано с повышением уровня цАМФ и активацией цАМФ-зависимой протеинкиназы в гладкомышечных клетках. цАМФ-зависимая протеинкиназа А угнетает киназу легких цепей миозина, в результате нарушается фосфорилирование легких цепей миозина и не происходит его взаимодействия с актином. Кроме того, цАМФ-зависимая протеинкиназа А угнетает фосфоламбан (ингибитор Са2+-АТФазы), в результате в гладкомышечных клетках повышается активность Са2+-АТФазы, транспортирующей Са2+ из цитоплазмы в саркоплазматический ретикулум, и уменьшается концентрация цитоплазматического Са2+. Все это приводит к снижению тонуса и сократительной активности гладких мышц. β2-Адренорецепторы локализованы также на мембранах кардиомиоцитов, но в меньшем количестве, чем β1-адренорецепторы, где они также, но в меньшей степени опосредуют процессы стимуляции функций сердца под воздействием агонистов. β2-Адренорецепторы контролируют процесс гликогенолиза в печени и скелетных мышцах и секрецию инсулина в поджелудочной железе. При стимуляции этих рецепторов активируется фосфорилаза и усиливается распад гликогена, в результате повышается уровень глюкозы в крови. Секреция инсулина при стимуляции β2-адренорецепторов увеличивается. Кроме того, стимуляция β2-адренорецепторов вызывает сокращение ске- Глава 9. Средства, действующие на адренергические синапсы 239 летных мышц и захват К+ клетками, вследствие чего снижается концентрация калия в крови. К препаратам, стимулирующим преимущественно β2-адренорецепторы, относят: сальбутамол (саламол♠, сальтос♠, вентолин♠, сальгим♠), фенотерол (беротек♠), тербуталин (бриканил♠), гексопреналин (гинипрал♠), салметерол (серевент♠), формотерол (форадил♠, атимос♠, оксис турбухалер♠), кленбутерол. β2-Адреномиметики снижают тонус бронхов, понижают тонус и сократительную активность миометрия и расширяют кровеносные сосуды (скелетных мышц, печени, коронарные сосуды). Применяют указанные препараты в качестве бронхорасширяющих средств при бронхиальной астме, хроническом обструктивном бронхите и других бронхообструктивных заболеваниях. При ингаляционном пути введения, который является в этих случаях основным, β2-адреномиметики преимущественно расширяют мелкие и средние бронхи (где преобладают β2-адренорецепторы), уменьшают выделение медиаторов аллергии и воспаления из тучных клеток, стимулируют мукоцилиарный транспорт и таким образом облегчают отделение мокроты. Поскольку селективность этих веществ в отношении β2-адренорецепторов не является абсолютной, они в высоких концентрациях стимулируют также β1-адренорецепторы сердца. По химической структуре эти вещества отличаются от катехоламинов наличием гидроксильных групп в положениях 3 и 5 вместо положений 3 и 4 (фенотерол, тербуталин) или модификациями в положении 3 (сальбутамол, салметерол, формотерол) ароматического кольца (рис. 9.5), что делает их малочувствительными к действию катехол-о-метилтрансферазы. Показания к применению β2-адреномиметиков. Сальбутамол и фенотерол рекомендуют применять для купирования приступов бронхиальной астмы (бронхоспазма). С этой целью препараты вводят в основном ингаляционно с помощью специальных дозирующих устройств. При ингаляционном введении эффект наступает быстро, в течение 1–3 мин. Продолжительность бронхорасширяющего действия составляет от 3 до 6 ч (табл. 9.2). При необходимости эти препараты можно использовать для предупреждения бронхоспазма, вводят ингаляционно, а сальбутамол и внутрь (за 30– 40 мин до еды). Сальбутамол, кроме того, выпускается в лекарственных формах для парентерального введения, его вводят подкожно, внутримышечно и внутривенно. Для предупреждения бронхоспазма рекомендуют применять препараты более длительного действия — салметерол, формотерол, которые вводят только ингаляционно. Салметерол и формотерол отличаются от β2-адреномиметиков короткого действия высокой липофильностью, 240 Часть II. Частная фармакология Рис. 9.5. Химические структуры некоторых β2-адреномиметиков благодаря которой они не только хорошо проникают в мембраны гладкомышечных клеток бронхов, но и задерживаются в липидном слое мембран, создавая своеобразное депо вещества в непосредственной близости от рецептора. Формотерол, постепенно высвобождаясь из мембраны в интерстициальную жидкость, вызывает быструю и продолжительную стимуляцию β2-адренорецепторов. Салметерол по сравнению с формотеролом характеризуется более высокой липофильностью, что мешает его высвобождению из липидных слоев мембраны и задерживает активацию β2-адренорецепторов (по-видимому, небольшая скорость диффузии через липидные слои мембраны определяет отсроченное начало действия вещества на рецептор и является причиной медленного развития эффекта препарата). Поскольку эффект салметерола наступает не сразу (приблизительно через 30 мин после ингаляции), для купирования бронхоспазма его не применяют. Продолжительность бронхорасширяющего действия салметерола и формотерола составляет более 12 ч, в связи с чем их можно применять Глава 9. Средства, действующие на адренергические синапсы 241 для предупреждения ночных приступов бронхиальной астмы. Учитывая, что формотерол начинает действовать уже через 1–3 мин (табл. 9.2), он при необходимости может быть использован при острых приступах удушья, как в виде монотерапии, так и в комбинации с глюкокортикоидами (препарат симбикорт♠). Таблица 9.2. Время наступления и продолжительность бронхорасширяющего действия β2-адреномиметиков в зависимости от пути введения Препараты, путь введения Фенотерол Ингаляционно Сальбутамол Ингаляционно Перорально Подкожно Салметерол Ингаляционно Формотерол Ингаляционно Начало эффекта Максимальный эффект Продолжительность эффекта 1–3 мин 30–60 мин 4–6 ч 1–3 мин 15–30 мин 15 мин 30 мин 2–3 ч 30–60 мин 3–6 ч До 8 ч 1,5–4 ч 15–30 мин 2–4 ч 12 ч 1–3 мин 2ч 12 ч При лечении ХОБЛ рациональными считают комбинации β2-адреномиметиков с М-холиноблокаторами, позволяющими снизить дозу β2-адреномиметика при усилении его бронхорасширяющего действия. Таким комбинированным препаратом является беродуал♠, в состав которого входят фенотерола гидробромид и ипратропия бромид. Препарат вводят ингаляционно, эффект наступает через 5–10 мин и продолжается 3–4 ч. Агонисты β2-адренорецепторов — одна из основных групп бронхорасширяющих средств, применяемых при лечении бронхообструктивных заболеваний. Сведения об этих препаратах приводятся также в разделе «Средства, применяемые при бронхиальной астме». Поскольку β2-адреномиметики снижают тонус и сократительную активность миометрия, их применяют при угрозе выкидышей, для предупреждения преждевременных родов и при чрезмерной родовой деятельности (токолитические средства, см. раздел «Средства, влияющие на миометрий»). С этой целью используют сальбутамол (сальгим♠), фенотерол (партусистен♠) и гексопреналин (гинипрал♠). В качестве токолитических средств можно применять также тербуталин и ритодрин. При угрозе выкидышей препараты, как правило, назначают внутрь в виде 242 Часть II. Частная фармакология таблеток, в экстренных случаях вводят внутривенно. β2-Адреномиметики, проникая через плаценту, могут вызвать тахикардию и опасность аритмий у плода (салбутамол). Менее опасны в этом отношении фенотерол и гексопреналин. Побочные эффекты при применении β2-адреномиметиков связаны в основном со стимуляцией β2-адренорецепторов: ● расширение периферических сосудов и снижение диастолического давления; ● тахикардия (при ингаляционном введении незначительная), рефлекторно возникающая в ответ на снижение АД, а также связанная с прямым возбуждением β2-адренорецепторов сердца (плотность которых в предсердиях составляет 25%); некоторое значение имеет также частичная стимуляция β1-адренорецепторов (так как эти препараты не обладают абсолютной селективностью по отношению к β2-адренорецепторам); ● тремор вследствие стимуляции β2-адренорецепторов скелетных мышц; ● повышение уровня глюкозы в крови вследствие усиления гликогенолиза; ● гипокалиемия вследствие захвата ионов К+ клетками. Отмечают также беспокойство, повышенную потливость, головокружение, задержку мочеиспускания, атонию кишечника. Изопреналин (изадрин♠, изопротеренол) по химической структуре является N-изопропилнорадреналином (рис. 9.6). Оказывает прямое стимулирующее влияние на β1- и β2-адренорецепторы. Основные эффекты препарата связаны с влиянием на сердце и гладкие мышцы. Стимулируя β1-адренорецепторы сердца, он увеличивает силу и частоту сердечных сокращений, облегчает атриовентрикулярную проводимость, повышает автоматизм кардиомиоцитов. В связи с увеличением силы и ЧСС повышается потребление миокардом кислорода. Стимулируя β2-адренорецепторы, изопреналин расширяет сосуды, вследствие чего уменьшает общее периферическое сопротивление сосудов (ОПСС) и снижает диастолическое давление. Систолическое давление повышается вследствие увеличения сердечного выброса, среднее давление снижается. Применяют препарат для повышения атриовентрикулярной проводимости при атриовентрикулярном блоке в виде таблеток под язык (сублингвально). Учитывая, что изопреналин снижает тонус бронхов (вследствие стимуляции β2-адренорецепторов), его можно использовать в качестве бронхорасширяющего средства. Для этих целей используют водные растворы для ингаляций и таблетки для рассасывания Глава 9. Средства, действующие на адренергические синапсы 243 Рис. 9.6. Химическая структура изопреналина гидрохлорида в полости рта. Стимулируя β2-адренорецепторы, изопреналин вызывает те же побочные эффекты, что и сальбутамол. Побочные эффекты изопреналина, связанные со стимуляцией β1-адренорецепторов, выражены в большей степени, чем у препаратов, преимущественно стимулирующих β2-адренорецепторы: он вызывает более выраженную тахикардию, в большей степени повышает потребность миокарда в кислороде, высока опасность возникновения аритмий. Поэтому в качестве бронхорасширяющего средства изопреналин уступает по значению препаратам β2-адреномиметиков. При длительном применении β-адреномиметиков (как селективных, так и неселективных) развивается привыкание (толерантность), в результате чего со временем снижается эффект препаратов, назначаемых в той же дозе. Развитие толерантности связано со снижением чувствительности рецепторов к β-адреномиметикам (десенситизацией) и уменьшением их количества. Сначала возникает толерантность к побочным эффектам, таким как тремор, тахикардия, и только позже к бронхолитическому действию β2-адреномиметиков. α-, β-Адреномиметики (стимуляторы α- и β-адренорецепторов) Норэпинефрин (норадреналин♠) по химической структуре (рис. 9.7) соответствует медиатору симпатической нервной системы норадреналину. В медицинской практике применяется в виде норэпинефрина (норадреналина) гидротартрата♠. Действие норадреналина связано с прямым стимулирующим влиянием на α1- и α2-адренорецепторы сосудов и β1-адренорецепторы сердца. β2-Адренорецепторы малочувствительны к норадреналину. Основной эффект норадреналина — выраженное, но непродолжительное (в течение нескольких минут) повышение АД, связанное с его влиянием на α1- и α2-адренорецепторы сосудов. В отличие от адреналина♠ при введении норадреналина♠ не наблюдается последующего снижения АД, так как норадреналин♠ практически не стимулирует β2-адренорецепторы сосудов. Вследствие повышения АД возникает рефлекторная брадикардия: 244 Часть II. Частная фармакология Рис. 9.7. Химические структуры эпинефрина и норэпинефрина рефлекс с барорецепторов дуги аорты в ответ на повышение АД приводит к стимуляции центра блуждающего нерва и усилению его тормозного влияния на ЧСС. Поэтому вызываемую норадреналином♠ брадикардию можно предотвратить введением атропина. Стимуляция норадреналином♠ β1-адренорецепторов сердца приводит к повышению силы сердечных сокращений, ударный объем при этом увеличивается, но вследствие рефлекторного снижения ЧСС не происходит увеличения минутного объема (сердечного выброса). При пероральном введении норадреналин♠ разрушается, а при подкожном и внутримышечном введениях вызывает спазм сосудов на месте инъекции и может вызвать некроз ткани, поэтому его вводят внутривенно. При однократном введении действует непродолжительно, несколько минут (быстро инактивируется МАО и катехол-орто-метилтрансферазой и захватывается адренергическими нервными окончаниями), поэтому обычно растворы норадреналина♠ вводят внутривенно капельно. Метаболиты и незначительная часть неизмененного норадреналина♠ выводятся почками. Норадреналин♠ применяют при состояниях, сопровождающихся острым снижением АД. При его применении возможны нарушение дыхания, головная боль, нарушения ритма сердца. Препарат противопоказан при сердечной слабости, выраженном атеросклерозе, атриовентрикулярном блоке, галотановом наркозе (повышается опасность аритмий). Эпинефрин (адреналин♠) по химической структуре (см. рис. 9.7) соответствует биогенному катехоламину адреналину, образующемуся главным образом в хромаффинных клетках мозгового вещества надпочечников (был впервые выделен из мозгового вещества надпочечников). Для медицинских целей адреналин получают синтетическим путем или выделяют из надпочечников убойного скота. Выпускают в виде эпинефрина (адреналина♠) гидрохлорида и эпинефрина (адреналина♠) гидротартрата. По действию эти препараты не отличаются друг от друга, но из-за разной молекулярной массы гидротартрат применяют в большей дозе. Фармакологические эффекты адреналина. Действие адреналина ♠ при введении в организм связано с прямым стимулирующим влияни- Глава 9. Средства, действующие на адренергические синапсы 245 ем на β1-, β1-, α1- и α2-адренорецепторы различных органов и тканей. β-Адренорецепторы проявляют большую чувствительность к адреналину♠, чем α-адренорецепторы, поэтому в небольших дозах адреналин♠ преимущественно стимулирует β-адренорецепторы. Вследствие стимуляции α 1- и α 2-адренорецепторов адреналин ♠ оказывает сосудосуживающее действие. Стимуляция адреналином ♠ β 2-адренорецепторов приводит к расширению сосудов. Поскольку β2-адренорецепторы более чувствительны к адреналину♠, при введении небольших доз препарата происходит расширение сосудов (скелетных мышц), в которых преобладают β2-адренорецепторы, и ОПСС снижается, в результате понижается диастолическое давление. Из-за повышения систолического давления (стимуляция β1-адренорецепторов сердца) среднее давление изменяется незначительно. При введении более высоких доз адреналина♠ сначала проявляется его действие на α-адренорецепторы, при этом происходит сужение сосудов кожи, слизистых оболочек, органов брюшной полости и повышение ОПСС. Стимулируя β1-адренорецепторы миокарда, адреналин♠ повышает силу и ЧСС. Ударный объем сердца и сердечный выброс (минутный объем) при этом увеличиваются. Вследствие увеличения сердечного выброса и ОПСС адреналин♠ вызывает повышение АД. Прессорный эффект наиболее выражен при быстром внутривенном введении адреналина. Изменение АД при таком введении состоит из нескольких фаз (рис. 9.8). Резкое повышение АД вследствие увеличения сердечного выброса (1-я фаза) может вызвать кратковременную рефлекторную брадикардию, что сопровождается кратковременным незначительным снижением АД (2-я фаза), давление затем повышается (3-я фаза) вследствие вазоконстрикторного действия адреналина♠ (действие на α-адренорецепторы сосудов) и стимуляции секреции ренина (действие на β1-адренорецепторы юкстагломерулярных клеток почек). Прессорное действие адреналина♠ обычно продолжается несколько минут (концентрация адреналина быстро снижается вследствие его захвата тканями и инактивации КОМТ и МАО) и сменяется небольшой, но более продолжительной гипотензией (4-я фаза), связанной с возбуждением β2-адренорецепторов сосудов, более чувствительных к адреналину♠. Вследствие увеличения силы и ЧСС адреналин♠ стимулирует работу сердца. Потребление миокардом кислорода при этом возрастает, чему также способствует увеличение в миокарде окисления свободных жирных кислот, концентрация которых повышается под действием адреналина♠ (стимуляция липолиза). 246 Часть II. Частная фармакология Рис. 9.8. Влияние адреналина на артериальное давление (пояснения в тексте) Адреналин♠ облегчает атриовентрикулярную проводимость и повышает автоматизм проводящей системы сердца и кардиомиоцитов желудочков, что может привести к образованию эктопических очагов возбуждения и нарушениям сердечного ритма (аритмиям). Адреналин♠ расширяет зрачки (вследствие сокращения радиальной мышцы радужной оболочки) и снижает внутриглазное давление, что связывают как с уменьшением продукции внутриглазной жидкости (снижением фильтрации вследствие сужения сосудов), так и с увеличением ее оттока. Адреналин♠ стимулирует β2-адренорецепторы гладких мышц бронхов, вызывает их расслабление и устраняет бронхоспазм. Под влиянием адреналина♠ тонус и моторика ЖКТ преимущественно снижаются (действие на α- и β-адренорецепторы), тонус сфинктеров при этом повышается (действие на α-адренорецепторы). Повышается также тонус сфинктера мочевого пузыря, гладких мышц уретры и предстательной железы (связано со стимуляцией α1А-адренорецепторов), вследствие стимуляции β2-адренорецепторов происходит расслабление детрузора мочевого пузыря, все это приводит к задержке мочеиспускания. Действие адреналина♠ на матку зависит от многих факторов (от фазы цикла, периода беременности, дозы). Адреналин♠, стимулируя α1-адренорецепторы, вызывает сокращение, а стимулируя β2-адренорецепторы, способствует расслаблению миометрия. Стимулируя β2-адренорецепторы, адреналин♠ повышает расщепление гликогена в печени и скелетных мышцах (гликогенолиз), при этом в крови повышается концентрация глюкозы (возникает гипергликемия). Захват Глава 9. Средства, действующие на адренергические синапсы 247 скелетными мышцами ионов К+ вызывает гипокалиемию. Стимуляция адреналином♠ β3-адренорецепторов адипоцитов активирует триглицеридлипазу, и вследствие ускорения распада триглицеридов в плазме крови повышается содержание свободных жирных кислот. Адреналин♠ улучшает функциональное состояние скелетных мышц, особенно на фоне их утомления. Адреналин♠ разрушается при введении внутрь. Поэтому его вводят парентерально (подкожно, внутримышечно и внутривенно) и местно. В организме адреналин♠ быстро метаболизируется КОМТ и в меньшей степени МАО и подвергается нейрональному захвату, поэтому действует кратковременно (при внутривенном введении около 5 мин, при подкожном — до 30 мин). Метаболиты и небольшие количества неизмененного адреналина♠ выводятся почками. Показания к применению адреналина. В связи с тем, что адреналин♠ может повышать АД, расширять бронхи и стимулировать сердечную деятельность, его применяют при анафилактическом шоке (аллергическая реакция немедленного типа, возникающая в ответ на введение некоторых ЛВ, например бензилпенициллина, употребление пищевых продуктов, укус насекомых и сопровождающаяся падением АД и спазмом бронхов) и других острых аллергических реакциях. Препарат вводят внутривенно. При анафилактическом шоке адреналин♠ является препаратом выбора. Адреналин♠ применяют для купирования приступов бронхиальной астмы. Препарат вводят под кожу, эффект длится около 1 ч. При таком пути введения адреналина♠ меньше опасность повышения АД (так как в более низких концентрациях адреналин♠ мало влияет на α-адренорецепторы сосудов). Адреналин♠ применяют при остановке сердца. Препарат вводят длинной иглой через грудную клетку в полость левого желудочка. Адреналин♠ добавляют в растворы местных анестетиков с целью уменьшения их резорбтивного действия, так как сужение сосудов под действием адреналина♠ препятствует всасыванию местных анестетиков в кровь. Это также способствует удлинению их местноанестезирующего действия. Тампоны, смоченные раствором адреналина♠, применяют для остановки кровотечения. В офтальмологии адреналин♠ применяют для снижения внутриглазного давления при открытоугольной форме глаукомы. При закапывании в глаз адреналин♠ может вызвать выраженные местные (жжение и покраснение глаз, пигментация переднего отдела глаза и век) и системные (головная боль, тахикардия) побочные эффекты. При закрытоугольной форме глаукомы адреналин♠ противопоказан. 248 Часть II. Частная фармакология В связи со способностью адреналина♠ повышать уровень глюкозы в крови его можно использовать при гипогликемической коме, вызванной гипогликемическими средствами (например, при передозировке инсулина). Побочные эффекты адреналина. Наиболее частый побочный эффект при применении адреналина♠ — повышение АД, при его резком повышении возможно кровоизлияние в мозг. В высоких дозах адреналин♠ может вызывать нарушения сердечного ритма. Адреналин♠, проникая через ГЭБ в ткани мозга, оказывает умеренное возбуждающее действие на ЦНС. Центральное действие адреналина♠ проявляется в виде беспокойства, страха, головокружения, головной боли, тремора, тошноты, рвоты (эти эффекты в основном появляются при передозировке адреналина♠). Адреналин♠ противопоказан при гипертонической болезни, тиреотоксикозе, выраженном атеросклерозе, закрытоугольной форме глаукомы, сахарном диабете, беременности. Вследствие повышения потребности миокарда в кислороде адреналин♠ противопоказан при стенокардии. Нельзя применять адреналин♠ в сочетании с некоторыми средствами для наркоза (галотан), что повышает опасность возникновения аритмий. 9.1.2. Симпатомиметические средства (симпатомиметики, адреномиметики непрямого действия) Эфедрин — алкалоид, содержащийся в различных видах эфедры (Ephedra L.). По химической структуре (рис. 9.9) близок к адреналину, но в отличие от него не содержит гидроксильных групп в ароматическом кольце. Эфедрин, получаемый из растительного сырья, — левовращающий изомер, а эфедрин, получаемый синтетическим путем, представляет собой рацемическую смесь левовращающего и правовращающего изомеров и обладает меньшей активностью. В медицинской практике используется в виде эфедрина гидрохлорида♠. Эфедрин способствует высвобождению медиатора норадреналина из варикозных утолщений симпатических нервных волокон, а также непосредственно стимулирует адренорецепторы, это действие выражено в меньшей степени, поэтому эфедрин Рис. 9.9. Химическая структура относят к адреномиметикам непрямого действия. Кроме того, эфедрин снижает эфедрина гидрохлорида Глава 9. Средства, действующие на адренергические синапсы 249 активность МАО и катехол-о-метилтрансферазы. Эффективность эфедрина зависит от запасов медиатора в окончаниях симпатических нервов. При действии эфедрина возбуждаются те же подтипы α- и β-адренорецепторов, что и при действии адреналина (но в меньшей степени), поэтому в основном эфедрин вызывает фармакологические эффекты, характерные для адреналина♠. Эфедрин увеличивает силу и ЧСС и суживает сосуды, вследствие чего повышает АД. Сосудосуживающее действие эфедрина проявляется и при его местном применении, при нанесении на слизистые оболочки. Эфедрин расширяет бронхи, уменьшает перистальтику кишечника, расширяет зрачки (на аккомодацию не влияет), повышает содержание глюкозы в крови, повышает тонус скелетных мышц. Эфедрин оказывает стимулирующее действие на ЦНС: повышает активность дыхательного и сосудодвигательного центров, оказывает умеренное психостимулирующее действие: уменьшает ощущение усталости, потребность во сне, повышает работоспособность. По психостимулирующему действию эфедрин уступает амфетамину. Применяют эфедрин как бронхорасширяющее средство. Для купирования приступов бронхиальной астмы препарат вводят подкожно, а для предупреждения — внутрь (он входит в состав комбинированных препаратов «Теофедрин»♠, «Солутан»♠, «Бронхолитин»♠). Иногда эфедрин применяют для повышения АД. В частности, препарат используется для предупреждения сосудистого коллапса при спинномозговой и эпидуральной анестезии, при острой артериальной гипотензии, вызванной инфекционными заболеваниями или травмой). Эфедрин уступает адреналину♠ по активности (для достижения одинакового прессорного эффекта доза эфедрина должна быть в 50 раз выше, чем доза адреналина). Прессорный эффект эфедрина выражен в меньшей степени, чем у адреналина♠, но продолжается значительно дольше (1– 1,5 ч). При повторных введениях эфедрина через небольшие промежутки времени (10–30 мин) его прессорное действие снижается, развивается привыкание (тахифилаксия). Этот эффект обусловлен быстрым истощением запасов норадреналина в окончаниях адренергических волокон. Кроме того, эфедрин эффективен при аллергических заболеваниях (сенная лихорадка, сывороточная болезнь), его можно применять местно при насморке (сужение сосудов слизистой оболочки полости носа и уменьшение их проницаемости приводит к уменьшению воспалительной реакции). Внутривенное введение эфедрина эффективно при анафилактическом шоке. При атриовентрикулярном блоке эфедрин повышает атриовентрикулярную проводимость. 250 Часть II. Частная фармакология Благодаря стимулирующему действию на ЦНС эфедрин может применяться при отравлении снотворными и наркотическими средствами, при нарколепсии (патологическая сонливость), при ночном энурезе (эфедрин уменьшает глубину сна и повышает тонус сфинктера мочевого пузыря). Эфедрин вводят внутрь, подкожно, внутримышечно. Внутрь назначают до еды. Эфедрин быстро и полностью всасывается из ЖКТ в кровь, при этом он практически не метаболизируется МАО, t½ составляет 3–6 ч. Выводится почками. Эфедрин применяют только по назначению врача, поскольку он вызывает ряд серьезных побочных эффектов: сердцебиение, повышение АД, нервное возбуждение, бессонницу, расстройство кровообращения, дрожание конечностей, потерю аппетита, рвоту, усиленное потоотделение, задержку мочеиспускания. Препарат противопоказан при бессоннице, некомпенсированной гипертензии, атеросклерозе, феохромоцитоме, гипертиреозе, фибрилляции желудочков. Из-за характерного действия на ЦНС эфедрин имеет ограниченное применение, в частности он относится к допинговым препаратам и запрещен к приему у спортсменов. 9.2. СРЕДСТВА, БЛОКИРУЮЩИЕ АДРЕНЕРГИЧЕСКИЕ СИНАПСЫ Средства, блокирующие адренергические синапсы, нарушают передачу возбуждения с окончаний постганглионарных симпатических (адренергических) волокон на эффекторные органы и ткани. Нарушение передачи возбуждения в адренергических синапсах осуществляется путем блокады адренорецепторов, локализованных на постсинаптической мембране (адреноблокаторы), или путем уменьшения выделения медиатора норадреналина адренергическими нервными окончаниями (симпатолитики). 9.2.1. Адреноблокаторы Адреноблокаторы блокируют адренорецепторы и препятствуют действию на них медиатора норадреналина и циркулирующего в крови адреналина. В зависимости от преобладающего влияния на α- или β-адренорецепторы выделяют α-адреноблокаторы и β-адреноблокаторы, а также вещества, действующие на оба типа адренорецепторов: α-, β-адреноблокаторы. Глава 9. Средства, действующие на адренергические синапсы 251 Блокаторы α-адренорецепторов (α-адреноблокаторы) Препараты этой группы подразделяются на неселективные α1-, α2адреноблокаторы и α1-адреноблокаторы. Блокаторы α1-, α2-адренорецепторов (α1-, α2-адреноблокаторы) Среди препаратов этой группы выделяют: ● синтетические α-адреноблокаторы: фентоламин (регитин♠), феноксибензамин; алкалоидов спорыньи: дигидроэрготамин (дигидер● производные гот♠), ницерголин (сермион♠). К основным синтетическим препаратам, блокирующим α1- и α2ад рено рецепторы, относят производное имидазолина фентоламин (рис. 9.10). Фентоламин действует на постсинаптические α1-адренорецепторы, а также на постсинаптические, внесинаптические и пресинаптические α2-адренорецепторы. Поскольку фентоламин блокирует α1- и α2адренорецепторы сосудов, он оказывает выраженное сосудорасширяющее действие, в результате снижается артериальное и венозное давление. Вследствие снижения АД может возникнуть рефлекторная тахикардия. Блокада пресинаптических α2-адренорецепторов приводит к повышению выделения норадреналина окончаниями постганглионарных симпатических волокон и, как следствие, усилению стимулирующего действия норадреналина на β1-адренорецепторы сердца, что является еще одной причиной тахикардии, вызываемой фентоламином. Повышается также потребность миокарда в кислороде и секреция ренина. Кроме того, выделяющийся в больших количествах норадреналин препятствует блоку α1-адренорецепторов сосудов, вытесняя фентоламин из связи с рецепторами. Все это уменьшает и укорачивает сосудорасширяющее действие фентоламина (при внутривенном введении действие препарата продолжается 10–15 мин). Показания к применению фентоламина. Поскольку фентоламин блокирует как α1-, так и α2-адренорецепторы сосудов, он оказывает выраженное гипотензивное действие при феохромоцитоме (опухоли мозгового вещества надпочечников). Опухоль выделяет в кровь большое количество адреналина и норадреналина, что приводит к повышению АД, тахикардии и аритмиям. Фентоламин уменьшает Рис. 9.10. Химическая прессорное действие норадреналина, а также структура фентоламина 252 Часть II. Частная фармакология уменьшает и извращает прессорное действие адреналина (на фоне α1-, α2-адреноблокаторов адреналин не повышает, а снижает АД). Этот пародоксальный эффект связан с тем, что при блокаде α-адренорецепторов проявляется стимулирующее действие адреналина на β2-адренорецепторы сосудов, что приводит к расширению сосудов и снижению АД. Вследствие повышения концентрации адреналина в крови фентоламин при феохромоцитоме оказывает более выраженное гипотензивное действие, что может быть использовано для диагностики феохромоцитомы. Однако в основном с этой целью проводят определение циркулирующих в крови катехоламинов (адреналина и норадреналина) или определяют их метаболиты в моче. Обычно феохромоцитому удаляют хирургическим путем. Фентоламин может быть использован перед и/или во время операции для предупреждения и купирования гипертензивных кризов во время хирургического вмешательства, а также для снижения АД при неоперабельных опухолях (для предупреждения тахикардии после фентоламина вводят β1-адреноблокаторы). Фентоламин применяют для устранения гипертензивного криза, который может возникнуть при резкой отмене клонидина или при употреблении тирамин-содержащих продуктов во время приема неизбирательных ингибиторов МАО. Кроме того, при нарушении периферического кровообращения (болезни Рейно, облитерирующем эндартериите), трофических язвах конечностей, отморожениях используется способность фентоламина расширять периферические сосуды, особенно артериолы и прекапилляры, и таким образом улучшать кровоснабжение мышц, кожи, слизистых оболочек и устранять гипоксию тканей. Побочные эффекты фентоламина. Побочные эффекты, связанные с влиянием на сердце и сосуды: ● ортостатическая (постуральная) гипотензия связана с расширением вен и угнетением веноконстрикторных реакций, АД резко падает при перемене положения тела из горизонтального в вертикальное, уменьшается приток крви к мозгу, что может привести к потере сознания (обмороку); для предупреждения этого осложнения после введения препарата следует лежать в течение 1–2 ч; ● тахикардия связана как с рефлекторной стимуляцией сосудодвигательного центра, так и с увеличением секреции норадреналина вследствие блокады пресинаптических α2-адренорецепторов; ● заложенность носа вследствие расширения сосудов и отека слизистой оболочки носовой полости. Такие побочные эффекты, как диарея и повышение секреции слюнных желез и желез желудка, очасти связаны с усилением выделения аце- Глава 9. Средства, действующие на адренергические синапсы 253 тилхолина окончаниями постганглионарных парасимпатических нервов при блокаде пресинаптических α2-адренорецепторов. Из-за расслабления гладких мышц семявыносящего протока возможно нарушение эякуляции, а снижение тонуса сфинктера мочевого пузыря приводит к учащению мочеиспускания. Противопоказан фентоламин при артериальной гипотензии, стенокардии, после перенесенного инфаркта миокарда. Фентоламин действует непродолжительно (таблетки фентоламина назначают 3–4 раза в день). Поэтому при феохромоцитоме для длительного применения при неоперабельных опухолях, а также перед операциями для предупреждения гипертензивных кризов рекомендуют применять α1-, α2-адреноблокатор длительного действия — феноксибензамин. Феноксибензамин блокирует α-адренорецепторы необратимо (вследствие образования прочных ковалентных связей с рецептором) и поэтому вызывает гипотензивный эффект продолжительностью до 48 ч и более. Кроме блокады α-адренорецепторов феноксибензамин угнетает обратный нейрональный захват норадреналина. Побочные эффекты и противопоказания такие же, как у фентоламина. α1-, α2-Адреноблокирующими свойствами обладает группа дигидрированных производных алкалоидов спорыньи (дигидроэрготамин, дигидроэрготоксин, дигидроэргокриптин), основой химической структуры которых является тетрациклическое соединение — D-лизергиновая кислота (6-метилэрголин) (рис. 9.11). Алкалоиды спорыньи выделены из маточных рожков дикорастущей спорыньи, которые представляют собой покоящуюся стадию гриба Claviceps purpurea, паразитирующего на ржи. Эти алкалоиды наряду с адреноблокирующей активностью обладают способностью повышать тонус миометрия и оказывают прямое сосудосуживающее действие. Дигидрированные алкалоиды спорыньи отличаются от природных алкалоидов отсутствием стимулирующего влияния на матку, меньшим сосудосуживающим действием и большей α-адреноблокирующей активностью. Дигидроэрготамин, блокируя α 1 - и α 2 адренорецепторы, вызывает расширение периферических сосудов и снижает АД. Кроме того, дигидроэрготамин, будучи агонистом серотониновых рецепторов, оказывает стимулирующее влияние на тонус сосудов мозга. Поэтому Рис. 9.11. Химическая в основном его применяют для купирования структура D-лизергиноострых приступов мигрени. Вводят интрана- вой кислоты 254 Часть II. Частная фармакология зально, в виде аэрозоля, эффект наступает через 20–30 мин. Дигидроэрготамин применяют также при вазоспастической стенокардии. В связи с тем, что препарат тонизирует вены, возможно его применение при ортостатической гипотензии. Выпускается в виде метансульфоната (мезилата) под названием дигидергот♠ для интраназального и парентерального введения, а также в виде капель для приема внутрь. Вазобрал♠— комбинированный препарат, содержащий дигидроэргокриптина мезилат и кофеин (триметилксантин). Дигидроэргокриптин по фармакологическим свойствам близок к дигидроэрготамину: блокирует α-адренорецепторы, расширяет сосуды. Его применяют при заболеваниях сосудов мозга, в восстановительный период после инсульта, при лечении кохлеовестибулярных расстройств, а также при нарушениях периферического кровообращения. Ницерголин — аналог алколоидов спорыньи, в структуре которого имеется эрголиновое ядро и бромзамещенный остаток никотиновой кислоты. Кроме α-адреноблокирующих свойств препарат обладает миотропной спазмолитической активностью, особенно выраженной в отношении сосудов мозга и периферических сосудов, что связывают с наличием в молекуле ницерголина остатка никотиновой кислоты. Мало влияет на АД. Ницерголин угнетает агрегацию тромбоцитов. Оказывает ноотропное действие, улучшая память и внимание. Препарат показан при хронических нарушениях мозгового кровообращения, мигрени, расстройствах периферического кровообращения. Таблетки ницерголина принимают до еды. Действие препарата развивается постепенно, и поэтому его применяют длительно, иногда в течение нескольких месяцев. Пророксан (пирроксан♠) блокирует центральные и периферические α1- и α2-адренорецепторы, в том числе блокирует α-адренорецепторы в структурах гипоталамуса. Расширяет периферические сосуды, в большей степени артериолы и прекапилляры, снижает АД. Оказывает седативное и антиабстинентное действия. Уменьшает психическое напряжение и тревогу. Применяют при гипертензивных кризах, симпатоадреналовых кризах с высоким АД при гипоталамическом синдроме, при морфинной и алкогольной абстиненции, рекомендован при болезни Меньера, при укачивании. Кроме того, пророксан обладает противозудным действием и может применяться при аллергическом дерматозе. Блокаторы α1-адренорецепторов (α1-адреноблокаторы) К этой группе препаратов относят празозин, доксазозин (артезин♠, кардура♠, тонокардин♠), теразозин (корнам♠, сетегис♠), тамсулозин (омник♠), альфузозин (дальфаз♠). Глава 9. Средства, действующие на адренергические синапсы 255 Празозин (рис. 9.12), доксазозин, теразозин блокируют α 1 -адренорецепторы гладкомышечных клеток сосудов и устраняют сосудосуживающее влияние медиатора норадреналина и циркулирующего в крови адреналина. В результате происходит расширение артериальных и веноз- Рис. 9.12. Химическая структура ных сосудов — уменьшаются ОПСС празозина и венозный возврат крови к сердцу, снижается артериальное и венозное давление. Вследствие снижения АД развивается умеренная рефлекторная тахикардия. Вследствие расширения венозных сосудов возможна ортостатическая гипотензия. Поскольку эти препараты не блокируют пресинаптические α2-адренорецепторы, они не повышают высвобождение норадреналина из окончаний адренергических волокон (вследствие чего не происходит чрезмерной стимуляции β1-адренорецепторов сердца и не нарушается блок α1-адренорецепторов сосудов). Показания к применению α1-адреноблокаторов. Учитывая, что празозин, теразозин и доксазозин снижают АД, их применяют при артериальной гипертензии; назначают внутрь. Препараты различаются по продолжительности действия: празозин действует 6–8 ч, теразозин — 24 ч, а доксазозин — до 36 ч. Помимо артериальной гипертензии, длительно действующие α1адреномиметики (доксазозин, теразозин) применяют при нарушении мочеиспускания, связанном с доброкачественной гиперплазией предстательной железы. При этом заболевании вследствие увеличения размера предстательной железы происходит сужение просвета простатической части уретры, что приводит к задержке мочеиспускания. Действие препаратов связывают с блокадой α1А-адренорецепторов гладких мышц предстательной железы, простатической части уретры и шейки мочевого пузыря. Блокада этих рецепторов приводит к расслаблению гладких мышц и расширению просвета уретры, что улучшает отток мочи из мочевого пузыря. Празозин используют также при спазмах периферических сосудов (синдром Рейно). При применении празозина, теразозина и доксазозина отмечается уменьшение уровня атерогенных липопротеинов низкой плотности (ЛПНП) и повышение антиатерогенных липопротеинов высокой плотности (ЛПВП). 256 Часть II. Частная фармакология Побочные эффекты α1-адреноблокаторов. При применении препаратов этой группы возможны: ● умеренная рефлекторная тахикардия (наиболее выражена при приеме празозина); ● ортостатическая гипотензия, в особенности выраженная при первом приеме препарата («феномен первой дозы»), наблюдается при приеме празозина (рекомендуют принимать перед сном); ● учащенное мочеиспускание (вследствие снижения тонуса шейки мочевого пузыря и уретры); ● заложенность носа (вследствие расширения сосудов слизистой оболочки носовой полости); ● периферические отеки. Тамсулозин преимущественно блокирует α1А-адренорецепторы гладких мышц предстательной железы, уретры и шейки мочевого пузыря и мало влияет на тонус сосудов и АД. Препарат применяют при доброкачественной гиперплазии предстательной железы. Назначают внутрь утром после еды. Вследствие того, что препарат оказывает незначительное влияние на тонус сосудов, многие побочные эффекты, характерные для празозина, выражены в гораздо меньшей степени. При доброкачественной гиперплазии предстательной железы применяют также альфузозин. Блокаторы β-адренорецепторов (β-адреноблокаторы) Препараты этой группы подразделяют на β1-, β2-адреноблокаторы и β1-адреноблокаторы. Блокада β1-адренорецепторов сердца приводит к: ● ослаблению силы сокращений сердца; ● уменьшению ЧСС (вследствие снижения автоматизма синусного узла); ● угнетению атриовентрикулярной проводимости; ● снижению автоматизма атриовентрикулярного узла и волокон Пуркинье. Вследствие уменьшения силы и ЧСС происходит снижение сердечного выброса (минутного объема), работы сердца и, как следствие, потребности миокарда в кислороде. Блокада β1-адренорецепторов юкстагломерулярных клеток почек приводит к уменьшению секреции ренина, что нарушает образование ангиотензина II. Блокада β2-адренорецепторов вызывает следующие эффекты: ● сужение кровеносных сосудов (скелетных мышц, коронарных сосудов); ● повышение тонуса бронхов; Глава 9. Средства, действующие на адренергические синапсы 257 ● повышение сократительной активности миометрия; ● снижение гипергликемического действия адреналина (β-адреноблокаторы подавляют гликогенолиз: снижается распад гликогена в печени и скелетных мышцах и уменьшается уровень глюкозы в крови); ● снижение секреции инсулина β-клетками поджелудочной железы. Блокада как β1-, так и β2-адренорецепторов вызывает повышение моторики кишечника. Вследствие блокады β1-адренорецепторов β-адреноблокаторы оказывают антигипертензивное, антиангинальное и антиаритмическое действия. Кроме того, они снижают внутриглазное давление при открытоугольной форме глаукомы, что связывают с уменьшением продукции внутриглазной жидкости вследствие блокады β-адренорецепторов ресничного эпителия. В табл. 9.3 приведены механизмы развития терапевтических эффектов β-адреноблокаторов и основные показания к их применению. Блокаторы β1- и β2-адренорецепторов (неселективные β1-, β2-адреноблокаторы) К этой группе препаратов относят (рис. 9.13) пропранолол (анаприлин♠, обзидан♠, индерал♠), надолол (коргард♠), тимолол (оптимол♠, арутимол♠), пиндолол (вискен♠), окспренолол (тразикор♠). Пропранолол (см. рис. 9.13) вызывает эффекты, связанные с блокадой β1-адренорецепторов (уменьшение силы и ЧСС, угнетение атриовентрикулярной проводимости, снижение автоматизма атриовентрикулярного узла и волокон Пуркинье, угнетение секреции ренина) и β2-адренорецепторов (сужение кровеносных сосудов, повышение тонуса бронхов, повышение сократительной активности миометрия, снижение уровня глюкозы в крови). Блокируя β-адренорецепторы адипоцитов, пропранолол угнетает липолиз и снижает содержание в крови свободных жирных кислот. Показания к применению пропранолола. Пропранолол оказывает гипотензивное, антиангинальное и антиаритмическое действие и поэтому применяется при гипертонической болезни, стенокардии и тахиаритмиях. Гипертоническая болезнь. При однократном применении пропранолол снижает АД, в основном за счет уменьшения сердечного выброса (уменьшения силы и частоты сокращений сердца); однако тонус сосудов и ОПСС повышаются вследствие блокады β2-адренорецепторов гладких мышц сосудов и устранения действия на них адреналина (стимулирующее действие адреналина на α1-адренорецепторы сохраняет- 258 Часть II. Частная фармакология Рис. 9.13. Химические структуры некоторых неселективных β-адреноблокаторов ся). Кроме того, повышение тонуса артерий может возникнуть в ответ на уменьшение возбуждения барорецепторов дуги аорты вследствие снижения сердечного выброса (при этом компенсаторно усиливаются симпатические влияния на α1-адренорецепторы сосудов). Однако при длительном применении препарата в течение 1–2 нед происходит расширение сосудов и уменьшение ОПСС, АД при этом существенно и стабильно снижается. Стабильное снижение тонуса сосудов и АД при систематическом применении пропранолола объясняют несколькими причинами: ● уменьшением (на 60%) выделения ренина юкстагломерулярными клетками почек, в результате снижается синтез ангиотензина II, обладающего выраженными сосудосуживающими свойствами (ренин вызывает превращение ангиотензиногена в ангиотензин I, который затем превращается в ангиотензин II); ● восстановлением барорецепторного депрессорного рефлекса (при гипертонической болезни этот рефлекс подавлен вследствие снижения чувствительности барорецепторов дуги аорты); Глава 9. Средства, действующие на адренергические синапсы 259 ● уменьшением выделения норадреналина окончаниями адренергических волокон (вследствие блокады пресинаптических β2-адренорецепторов); ● угнетением центральных звеньев симпатической регуляции сосудистого тонуса. Таблица 9.3. Основные терапевтические эффекты β-адреноблокаторов Основные эффекты Основные механизмы развития терапевтических эффектов Антигипертензивный Снижение сердечного выброса, восстановление барорецепторного депрессорного рефлекса, уменьшение секреции ренина (уменьшается синтез ангиотензина II) Антиангинальный Снижение частоты и силы сердечных сокращений — уменьшение работы сердца, в результате — снижение потребности миокарда в кислороде Антиаритмический Угнетение автоматизма синусного узла, проводимости атриовентрикулярного узла Угнетение автоматизма эктопических очагов Снижение внутриУменьшение образования внуглазного давления триглазной жидкости ресничным эпителием Показания к применению Гипертоническая болезнь Стенокардия напряжения Наджелудочковые тахиаритмии Экстрасистолии Открытоугольная форма глаукомы (тимолол, бетаксолол) Стенокардия напряжения. При стенокардии напряжения происходит снижение доставки кислорода к сердцу из-за сужения просвета коронарных сосудов вследствие образования атеросклеротических бляшек. Уменьшая силу и ЧСС, пропранолол уменьшает работу сердца, в результате снижается потребность миокарда в кислороде (см. раздел «Антиангинальные средства»). Кроме того, пропранолол угнетает липолиз в жировой ткани, при этом в сердце уменьшается концентрация и, следовательно, окисление свободных жирных кислот (этот путь окисления требует значительного потребления кислорода). Это также снижает потребление миокардом кислорода. Вызывая сужение коронарных артерий (блокада β2-адренорецепторов), пропранолол снижает общий коронарный кровоток. При стабильной стенокардии в большей степени суживаются здоровые (интактные) 260 Часть II. Частная фармакология сосуды (в участках миокарда с гипоксией сосуды расширены из-за локального повышения концентрации ионов водорода, образования аденозина и других факторов), что может привести к перераспределению коронарного кровотока в зону ишемии. В то же время сосудосуживающее действие пропранолола не позволяет использовать его при вазоспастической (вариантной) стенокардии, так как при этом заболевании доставка кислорода к сердцу снижается из-за спазма коронарных сосудов. Пропранолол может ухудшить состояние больных с этой формой стенокардии, вызывая приступ загрудинных болей. Аритмии сердца. Пропранолол снижает автоматизм синоатриального узла (ЧСС), автоматизм и проводимость атриовентрикулярного узла, а также уменьшает автоматизм волокон Пуркинье и кардиомиоцитов желудочков, поэтому он эффективен при наджелудочковых тахикардиях и тахиаритмиях (уменьшает атриовентрикулярную проводимость при тахисистолической форме мерцания предсердий) и наджелудочковых и желудочковых экстрасистолиях (подавляет возникновение эктопических очагов). Инфаркт миокарда. Пропранолол назначают после острого периода инфаркта миокарда для предупреждения повторных инфарктов. При инфаркте миокарда пропранолол уменьшает зону некроза, снижает опасность фибрилляций желудочков и других осложнений, связанных со стимуляцией симпатической системы, уменьшает (на 20–50%) смертность больных. Тиреотоксикоз. При тиреотоксикозе пропранолол уменьшает симптомы заболевания (снижает ЧСС, пальпитацию, предотвращает развитие аритмий) и уменьшает превращение тироксина в трийодтиронин. Мигрень. Пропранолол применяют для профилактики приступов мигрени (тонизирует сосуды мозга и предупреждает их болезненную пульсацию), а также для уменьшения эссенциального тремора (блокирует β2-адренорецепторы скелетных мышц). В связи с тем, что пропранолол оказывает угнетающее влияние на ЦНС, его можно назначать при состояниях, сопровождающихся чувством страха и тревоги (панические атаки). Открытоугольная форма глаукомы. β-Адреноблокаторы снижают внутриглазное давление, уменьшая секрецию внутриглазной жидкости. В связи с тем, что пропранолол обладает местноанестезирующей активностью, его при глаукоме не применяют. С этой целью в офтальмологии используют препараты тимолола, бетаксолола и проксодолола. Пропранолол назначают внутрь и внутривенно. Пропранолол обладает высокой липофильностью, поэтому прием препарата во время или сразу после еды способствует его более полному всасыванию; метаболизиру- Глава 9. Средства, действующие на адренергические синапсы 261 ется в печени. В связи с интенсивным метаболизмом при первом прохождении через печень, биодоступность пропранолола невелика (порядка 30%). В процессе биотрансформации пропранолола образуется активный метаболит 4-гидроксипропранолол. Пропранолол и его метаболиты выводятся почками. Препарат более чем на 90% связывается с белками плазмы крови; будучи липофильным соединением, хорошо проникает через ГЭБ и плацентарный барьер; t1/2 пропранолола составляет 2–4 ч. При длительном применении период полуэлиминации может удлиняться, так как, уменьшая печеночный кровоток, пропранолол снижает скорость своего метаболизма. Побочные эффекты пропранолола. Вследствие блокады β1-адренорецепторов пропранолол может вызвать: ● чрезмерное снижение сердечного выброса и развитие сердечной недостаточности; ● выраженную брадикардию; ● угнетение атриовентрикулярной проводимости (вплоть до атриовентрикулярного блока); ● артериальную гипотензию. Вследствие блокады β2-адренорецепторов пропранолол может вызвать: ● повышение тонуса бронхов (может вызвать бронхоспазм у больных бронхиальной астмой); ● повышение тонуса периферических сосудов (вследствие нарушения кровотока в конечностях возникает ощущение холода и повышается риск ишемии тканей); ● угнетение гликогенолиза в печени, продление и усиление гипогликемии, вызванной ЛС; при этом может маскировать симптомы гипогликемии (тахикардию, тремор, повышенное потоотделение); ● снижение утилизации глюкозы в скелетных мышцах (вследствие сужения сосудов), уменьшение выделения инсулина вследствие блокады β2-адренорецепторов β-клеток поджелудочной железы; ● усиление сокращений беременной матки (создать угрозу выкидыша); ● усиление перистальтики кишечника (возможна диарея). Пропранолол может вызвать эректильную дисфункцию. Вследствие снижения активности эндотелиальной липопротеинлипазы пропранолол увеличивает концентрацию триглицеридов и уровень атерогенных липопротеинов очень низкой плотности (ЛПОНП), а также уменьшает уровень антиатерогенных липопротеинов (ЛПВП) в плазме крови. Пропранолол влияет также на уровень тиреоидных гормонов, препятствуя превращению тироксина в более активный трийодтиронин. 262 Часть II. Частная фармакология Пропранолол в значительной степени проникает в ЦНС и может вызвать побочные эффекты, связанные с угнетением ЦНС: вялость, сонливость, нарушение сна, депрессию, тошноту, рвоту. При резкой отмене препарата после его длительного применения могут возникнуть приступы стенокардии, обострение артериальной гипертензии, повышается риск инфаркта миокарда (синдром отмены). Эти эффекты вызываются увеличением синтеза β-адренорецепторов (up-регуляция) при продолжительном воздействии антагониста, вследствие чего после отмены препарата норадреналин и адреналин связываются с большим количеством рецепторов. В результате увеличивается секреция ренина, повышаются тонус сосудов, ЧСС, сердечный выброс, потребность миокарда в кислороде. Синдром отмены может возникнуть после регулярного приема препарата в течение 3 мес и более, наиболее выраженные последствия отмечаются в течение 7 дней после прекращения приема. Для предупреждения синдрома отмены дозу препарата следует снижать постепенно в течение 2 нед. Пропранолол противопоказан при бронхиальной астме, нарушении атриовентрикулярной проводимости и выраженной брадикардии, острой и тяжелой хронической сердечной недостаточности, артериальной гипотензии, облитерирующих заболеваниях периферических сосудов, вазоспастической стенокардии, беременности (вызывает нежелательные эффекты у плода и может спровоцировать преждевременные роды). При назначении пропранолола больным сахарным диабетом, принимающим гипогликемические средства, может развиться выраженная гипогликемия, симптомы которой (тахикардия, тремор, повышенное потоотделение) маскируются вследствие блокады β-адренорецепторов. Этот эффект пропранолола наиболее выражен при инсулинзависимом диабете (1-го типа). Пропранолол повышает резистентность тканей к инсулину при диабете 2-го типа. Кроме того, неселективные β-адреноблокаторы снижают секрецию инсулина, что может спровоцировать развитие диабета у лиц, предрасположенных к этому заболеванию. Поэтому применение пропранолола и других неселективных β-адреноблокаторов при сахарном диабете ограничено. В таких случаях при необходимости использования β-адреноблокаторов предпочтение отдается кардиоселективным препаратам. Надолол (см. рис. 9.13) относят к неселективным β-адреноблокаторам длительного действия (t½ составляет 20–24 ч). В связи с этим препарат назначают 1 раз в сут внутрь (независимо от приема пищи). Применяют при артериальной гипертензии, стенокардии напряжения, аритмиях, инфаркте миокарда, тиреотоксикозе. В отличие от пропранолола обладает Глава 9. Средства, действующие на адренергические синапсы 263 низкой липофильностью, не полностью всасывается из ЖКТ (из-за чего биодоступность составляет 30–35%) и плохо проникает в ЦНС (не оказывает депрессивного действия), в основном выводится почками в неизмененном виде. Побочные эффекты и противопоказания такие же, как у пропранолола (в меньшей степени выражены центральные побочные эффекты). Тимолол (см. рис. 9.13) применяют главным образом в офтальмологии при лечении открытоугольной формы глаукомы. Снижение внутриглазного давления связывают с уменьшением секреции внутриглазной жидкости. На величину зрачка препарат не влияет. Тимолол выпускается в виде глазных капель (арутимол♠, офтан тимолол♠). При их инстилляции в глаз действие наступает через 10–30 мин, достигает максимума через 1–2 ч и продолжается до 24 ч. Местные побочные эффекты: уменьшение секреции слезной жидкости, конъюнктивит, аллергические реакции. Тимолол (липофильное вещество) может всасываться через слизистые оболочки глаза и оказывать резорбтивное действие. Возможны брадикардия, артериальная гипотензия, слабость и быстрая утомляемость, у больных бронхиальной астмой — опасность бронхоспазма. Тимолол также назначают внутрь для лечения стенокардии и артериальной гипертензии. β-Адреноблокаторы с внутренней симпатомиметической активностью Пиндолол (см. рис. 9.13), окспренолол, бопиндолол относят к β1-, β2-адреноблокаторам с внутренней симпатомиметической активностью, оказывающим слабое стимулирующее действие на β1- и β2-адренорецепторы, так как они по существу являются не блокаторами, а частичными агонистами этих рецепторов (т.е. стимулируют их, но в меньшей степени, чем адреналин♠ и норадреналин♠). В качестве частичных агонистов эти вещества устраняют действие адреналина и норадреналина (полных агонистов). Поэтому на фоне повышенного влияния симпатической иннервации они действуют подобно истинным β-адреноблокаторам, снижают силу и ЧСС, но в сравнительно меньшей степени, в результате в меньшей степени снижается сердечный выброс. На фоне пониженного симпатического тонуса эти препараты не оказывают такого действия. β-Адреноблокаторы с внутренней симпатомиметической активностью применяют при гипертензии, стенокардии. Эти препараты не вызывают выраженной брадикардии, практически не снижают ЧСС в покое. В сравнении с другими неселективными β-адреноблокаторами они в меньшей степени влияют на тонус бронхов, периферических сосудов и действие гипогликемических средств. Бопиндолол отличается от окспренолола и пиндолола большей продолжительностью действия (24 ч). 264 Часть II. Частная фармакология Блокаторы β1-адренорецепторов (кардиоселективные) К этой группе препаратов относят метопролол (беталок♠, эгилок♠, вазокардин♠), талинолол (корданум♠), атенолол (тенормин♠), бетаксолол (локрен♠), эсмолол (бревиблок♠), бисопролол (конкор♠), небиволол (небилет♠). Селективные β 1-адреноблокаторы преимущественно блокируют β1-адренорецепторы (в частности, β1-адренорецепторы сердца), действуя гораздо в меньшей степени на β2-адренорецепторы, поэтому их называют кардиоселективными β-адреноблокаторами (рис. 9.14). Для характеристики β-адреноблокаторов используется индекс кардиоселективности, отражающий относительную блокаду β1/β2-адренорецепторов. Для метопролола и бетаксолола этот индекс составляет 25/1, для наиболее кардиоселективного препарата небиволола — 290/1 (для неселективного препарата пропранолола — 1/1,8). В сравнении с неселективными β-адреноблокаторами кардиоселективные препараты в меньшей степени повышают тонус бронхов и периферических сосудов, меньше влияют на действие гипогликемических средств и в меньшей степени повышают резистентность тканей к инсулину. Поскольку при бронхиальной астме эти препараты могут несколько повышать тонус бронхов, при этом заболевании они противопоказаны. Кардиоселективность β1-адреноблокаторов существенно снижается при применении препаратов в высоких дозах. Кардиоселективные β-адреноблокаторы применяются в основном по тем же показаниям, что и неселективные препараты: при артериальной гипертензии, стенокардии напряжения, инфаркте миокарда, тахиаритмиях. Положительным свойством этих препаратов при ишемической болезни сердца является, в частности, то, что они практически не повышают общее периферическое сопротивление сосудов и постнагрузку на сердце. Несмотря на то, что β-адреноблокаторы снижают сердечный выброс, некоторые кардиоселективные препараты (метопролол, бисопролол, Рис. 9.14. Химические структуры некоторых кардиоселективных β-адреноблокаторов Глава 9. Средства, действующие на адренергические синапсы 265 небиволол) применяют при хронической сердечной недостаточности (ХСН). При этом заболевании компенсаторно повышается активность симпатоадреналовой и ренин-ангиотензиновой систем. В результате их чрезмерной активации увеличивается выделение норадреналина и синтез ангиотензина II, повышается ЧСС, пред- и постнагрузка на сердце, увеличивается потребность миокарда в кислороде, возникает опасность желудочковых аритмий. Развиваются гипертрофия и фиброз миокарда, происходит усиление гибели кардиомиоцитов, что еще больше снижает сократительную функцию миокарда. β-Адреноблокаторы, устраняя действие норадреналина на β1-адренорецепторы, уменьшают ЧСС и удлиняют диастолу, снижают потребность миокарда в кислороде уменьшают выделение ренина и синтез ангиотензина II, препятствуют гипертрофии и фиброзным изменениям миокарда, устраняют опасность аритмий, оказывают кардиопротективное действие. При ХСН лечение β-адреноблокаторами начинают с назначения малых доз, постепенно их увеличивая. Сочетают с ингибиторами ангиотензинпревращающего фермента. Положительный эффект достигается через 2–3 мес после начала приема препаратов. Метопролол является липофильным соединением и всасывается из кишечника более чем на 90%, но из-за интенсивного пресистемного метаболизма в печени, биодоступность его невелика (40–50%). Вводят внутрь и внутривенно. При введении внутрь назначают 2–3 раза в сутки во время еды. Метаболизм метопролола имеет межиндивидуальные различия, что связано с генетическим полиморфизмом метаболизирующих его ферментов (CYP2D6), поэтому период полуэлиминации препарата может варьировать от 3 до 7–8 ч. У лиц с низкой активностью ферментов метаболизма риск побочных эффектов метопролола увеличивается приблизительно в 5 раз. В основном метопролол применяют по тем же показаниям, что и пропранолол. Препараты метопролола пролонгированного действия (Беталок зок♠, Эгилок ретард♠, Эмзок♠) применяют в комплексной терапии хронической сердечной недостаточности (легкой и средней тяжести). Считается, что высокая эффективность метопролола обусловлена его свойствами обратного агониста, способного угнетать спонтанную активацию β-адренорецепторов. Атенолол, обладая низкой липофильностью, всасывается из ЖКТ не полностью, по этой причине его биодоступность составляет около 50%, плохо проникает через ГЭБ, поэтому в отличие от пропранолола оказывает незначительное действие на ЦНС. Применяют атенолол при артериальной гипертензии, стенокардии напряжения, аритмиях, вводят внутрь и внутривенно. Внутрь назначают 2 раза в сутки за 30– 266 Часть II. Частная фармакология 40 мин до еды. Препарат действует менее продолжительно, чем надолол (t½ составляет 6–7 ч), но несколько продолжительнее, чем метопролол. Эффект от однократного применения препарата сохраняется до 24 ч. Кардиоселективность атенолола не очень высока (индекс селективности, по разным данным, составляет 14–35), поэтому хотя и в меньшей степени, но он вызывает побочные эффекты, характерные для неселективных препаратов. Бетаксолол действует более продолжительно, до 36 ч (t1/2 составляет 14–22 ч). Бетаксолол липофилен, практически полностью всасывается в ЖКТ (всасывание не зависит от приема пищи), практически не метаболизируется при первом прохождении через печень и имеет высокую биодоступность (порядка 80–90%). Бетаксолол элиминируется в основном (на 85%) путем метаболизма в печени с образованием неактивных метаболитов, которые экскретируются почками. Период полуэлиминации удлиняется при нарушении функции печени и почек, что требует коррекции назначаемых доз. Препарат применяют при артериальной гипертензии, стенокардии напряжения, тахиаритмиях, назначают внутрь 1 раз в сутки. Кроме того, бетаксолол в виде глазных капель (бетоптик♠) используется для снижения внутрилазного давления при открытоугольной форме глаукомы. После однократной инстилляции действие препарата продолжается 12 ч. Может вызвать местные побочные реакции: жжение, боль, зуд в глазах, гиперемию конъюнктивы, аллергический конъюнктивит. При местном применении возможны проявления резорбтивного действия бетаксолола: брадикардия, гипотензия, у больных бронхиальной астмой — бронхоспазм. Эсмолол — препарат ультракороткого действия (из-за наличия эфирных связей он быстро метаболизируется в крови эстеразами эритроцитов с образованием неактивных метаболитов). t½ составляет около 10 мин. Вводят внутривенно для быстрого прекращения приступов пароксизмальной суправентрикулярной тахикардии, для устранения аритмий при тиреотоксическом кризе, во время или после хирургических операций и при других острых состояниях. Бисопролол обладает высокой кардиоселективностью (индекс селективности — 75, в 75 раз сильнее действует на β1-, чем на β2-адренорецепторы). Кардиоселективность понижается при увеличении дозы. При приеме внутрь бисопролол хорошо всасывается (липофильное вещество) и практически не метаболизируется при первом прохождении через печень, биодоступность препарата составляет около 90%. Бисопролол элиминируется в равной степени путем метаболизма в печени и путем по- Глава 9. Средства, действующие на адренергические синапсы 267 чечной экскреции в неизмененном виде (50%). Неактивные метаболиты также экскретируются почками. Период полуэлиминации бисопролола около 10–12 ч. Применяют при артериальной гипертензии, стенокардии напряжения, назначают 1 раз в сутки независимо от приема пищи. Показан при умеренно выраженной хронической сердечной недостаточности (в комплексной терапии). Вызывает центральные побочные эффекты, характерные для липофильных β-адреноблокаторов. Небиволол отличается наиболее высокой кардиоселективностью (приблизительно в 300 раз больше действует на β1-, чем на β2-адренорецепторы), кроме β1-адреноблокирующей активности обладает сосудорасширяющими свойствами, так как стимулирует синтез оксида азота (NO) в эндотелиальных клетках сосудов, повышает также продукцию простациклина. Небиволол представляет собой рацемическую смесь L- и d-изомеров, из которых d-изомер обладает β-адреноблокирующей активностью, а L-изомер вызывает высвобождение NO. Небиволол обладает и другими положительными эффектами: оказывает антиоксидантное и антитромботическое действия (снижает адгезию и агрегацию тромбоцитов), в отличие от пропранолола уменьшает уровень триглицеридов и увеличивает концентрацию ЛПВП, повышает утилизацию глюкозы тканями и снижает инсулинорезистентность при сахарном диабете 2-го типа. Применяют препарат внутрь при артериальной гипертензии, стабильной стенокардии (в отличие от пропранолола расширяет коронарные сосуды вследствие выделения NO), назначают 1 раз в сутки. Показан при умеренной и легкой формах хронической сердечной недостаточности (в комплексной терапии). Побочные эффекты, характерные для неселективных β-адреноблокаторов (в том числе влияние на бронхи), проявляются в наименьшей степени. Кардиоселективность небиволола снижается при увеличении его дозы (с 5 мг до 10 мг) и существенно ниже у лиц с медленным метаболизмом препарата. Для небиволола характерно межиндивидуальное различие в скорости метаболизма, связанное с генетическим полиморфизмом метаболизирующих ферементов (CYP2D6). У лиц с быстрым метаболизмом (высокой активностью фермента) биодоступность небиволола вследствие интенсивного пресистемного метаболизма составляет 12%, а период полуэлиминации равен приблизительно 10 ч, у лиц с медленным метаболизмом биодоступность практически максимальная (96%), а период полуэлиминации удлиняется до 30–50 ч. Большое значение для фармакокинетики небиволола имеет также его одновременное применение с ЛВ, изменяющими активность CYP2D6. 268 Часть II. Частная фармакология Блокаторы α-, β-адренорецепторов (α-, β-адреноблокаторы) Карведилол представляет собой смесь двух энантиомеров, один из которых (S-энантиомер) блокирует β1-, β2- и α1-адренорецепторы, а другой (R-энантиомер) — только α 1-адренорецепторы. При этом β1- и β2-адренорецепторы карведилол блокирует в большей степени, чем α1-адренорецепторы. В результате блокады α1-адренорецепторов происходят расширение периферических сосудов и снижение ОПСС. В результате блокады β1-адренорецепторов сердца снижаются частота и сила сердечных сокращений (и, следовательно, сердечный выброс), уменьшается секреция ренина. Блокада пресинаптических β2-адренорецепторов приводит к уменьшению секреции норадреналина окончаниями симпатических нервов. Таким образом, карведилол в отличие от других β-адреноблокаторов снижает АД, не вызывая первоначального увеличения ОПСС, а в отличие от α-адреноблокаторов не вызывает тахикардии. Препарат оказывает длительный антигипертензивный эффект, снижает пред- и постнагрузку на сердце, кроме того, обладает антиоксидантными свойствами. В отличие от пропранолола карведилол понижает резистентность тканей к инсулину, уменьшает уровень триглицеридов, ЛПНП и повышает уровень ЛПВП в плазме крови, препятствуя развитию атеросклероза. Положительным свойством карведилола является также то, что в отличие от большинства β-адреноблокаторов при его длительном применении в сердце не увеличивается количество β1-адренорецепторов. Применяют карведилол при гипертонической болезни, стенокардии, а также в комплексном лечении хронической сердечной недостаточности умеренной степени тяжести. Назначают внутрь 1–2 раза в сутки. При лечении ХСН карведилол препятствует гибели кардиомиоцитов, гипертрофии и ремоделированию миокарда, оказывает кардиопротективное действие. Уменьшение секреции норадреналина вследствие блокады пресинаптических β2-адренорецепторов рассматривается как преимущество карведилола в сравнении с селективными β1-адреноблокаторами (метопрололом и бисопрололом) при лечении ХСН. Карведилол быстро и практически полностью всасывается при приеме внутрь, но из-за интенсивной пресистемной элиминации (быстрее метаболизируется S-энантиомер), его биодоступность составляет 25%. Карведилол, обладая высокой липофильностью, быстро распределяется по организму, проникая в жидкости и ткани, метаболизируется в печени при участии CYP2D6, CYP2С9 и некоторых других изоформ цитохрома Р-450 с образованием трех активных метаболитов, проявляющих в основном β-адреноблокирующую активность. Генетический полиморфизм Глава 9. Средства, действующие на адренергические синапсы 269 CYP2D6 (в большей степени влияет на метаболизм R-энантиомера) может быть причиной некоторых межиндивидуальных различий фармакокинетики карведилола. Карведилол и его метаболиты в основном выводятся с желчью; период полуэлиминации составляет 7–10 ч. Побочные эффекты карведилола: ● ортостатическая гипотензия вследствие блокады α 1-адренорецепторов сопряжена с опасностью потери сознания (рекомендуется лежать после приема первой дозы); ● брадикардия вследствие блокады β1-адренорецепторов сердца (менее выражена, чем при применении других β-адреноблокаторов). Блокада β2-адренорецепторов может привести к повышению тонуса бронхов у больных бронхиальной астмой, а также тонуса и перистальтики кишечника (возможна диарея). Поскольку карведилол проникает в ЦНС (липофильное соединение), он может вызывать расстройство сна, депрессию и другие центральные побочные эффекты. Проксодолол♠ — отечественный препарат с неселективной β-адреноблокирующей и слабой α-адреноблокирующей активностью. Блокируя β-адренорецепторы, угнетает продукцию водянистой влаги глаза. В виде глазных капель проксодолол♠ применяют для снижения внутриглазного давления при открытоугольной форме глаукомы. После однократной инстилляции действие препарата сохраняется 8–12 ч. Возможны системные побочные эффекты: брадикардия, гипотензия, повышение тонуса миометрия и тонуса бронхов. Проксодолол♠ выпускается также в лекарственных формах для приема внутрь (таблетки) и в виде раствора для парентерального введения, может использоваться для длительного лечения как антигипертензивное и антиангинальное средство и внутривенно для снижения АД при гипертензивном кризе. Противопоказан при бронхиальной астме. Сравнительная характеристика β-адреноблокаторов В настоящее время в медицинской практике используется большое количество β-адреноблокаторов, различающихся не только по селективности в отношении β1- и β2-адренорецепторов, но и по степени липофильности, а также по наличию дополнительных свойств, таких как внутренняя симпатомиметическая активность, мембраностабилизирующее действие (блокада Na+-каналов мембран), вазодилатирующие свойства (вследствие блокады α1-адренорецепторов и/или кальциевых каналов или выделения NО) и по продолжительности действия. Данные о некоторых широко используемых β-адреноблокаторах представлены в табл. 9.4. 270 Часть II. Частная фармакология Таблица 9.4. Классификация β-адреноблокаторов по их основным свойствам β-Адреноблокаторы Кардио- Внутренняя Мембрано- Блокада ВыделеЛипоселектив- симпатоми- стабилизи- α-адрено- ние NO фильность ность метическая рующее рецепактивность действие торов I поколение: неселективные β1, β2-адреноблокаторы Пропранолол – – ++ – – +++ Надолол – – – – – – Пиндолол – ++ + – – +** II поколение: селективные β1-адреноблокаторы Метопролол + – +* – – ++ Атенолол + – – – – – Бисопролол ++ – – – – +** Бетаксолол + – + – – ++ III поколение: β-адреноблокаторы с вазодилатирующими свойствами Небиволол +++ – – ++ ++ – ++ Карведилол – – ++ + (α1) – + (α2) + +** Целипролол + + (β2) Примечание: * — в высоких дозах; ** — обладают липофильными и гидрофильными (амфифильными) свойствами. Соотношение липофильных и гидрофильных свойств β-адреноблокаторов оказывает влияние на особенности их фармакокинетики. Наибольшей липофильностью обладает пропранолол. К липофильным β-адреноблокаторам относятся также небиволол, бетаксолол, тимолол, окспренолол, карведилол, метопролол, бисопролол, пиндолол. Липофильные вещества хорошо всасываются в кишечнике, но некоторые из них (пропранолол, метопролол, карведилол), подвергаясь интенсивному пресистемному метаболизму, имеют низкую биодоступность (около 25–50%). Для многих липофильных β-адреноблокаторов характерно связывание с белками плазмы крови. В связи с тем, что липофильные вещества в основном метаболизируются в печени (имеют высокий печеночный клиренс), необходимо снижать их дозы при назначении больным с заболеваниями печени или низким печеночным кровотоком (например, в старческом возрасте или при ХСН). Кроме того, некоторые липофильные β-адреноблокаторы (метопролол, небиволол, карведилол) метаболизируются в печени при участии изофермента CYP2D6, активность которого в значительной степени снижена при носительстве Глава 9. Средства, действующие на адренергические синапсы 271 дефектных вариантов гена, кодирующих синтез этого фермента. У таких лиц метаболизм этих β-адреноблокаторов происходит значительно медленнее, что сопровождается изменением фармакокинетических параметров (биодоступности, периода полуэлиминации) и, как правило, развитием более выраженных побочных эффектов (требуется корректировка дозы). Гидрофильные β-адреноблокаторы (атенолол, надолол) не полностью всасываются в ЖКТ и по этой причине имеют низкую биодоступность (порядка 30–50%), практически не метаболизируются при первом прохождении через печень и мало связываются с белками плазмы крови. Эти вещества в основном выводятся почками (имеют высокий почечный клиренс) в неизмененном виде и в меньшей степени в виде метаболитов. При почечной недостаточности и снижении клубочковой фильтрации (например, в старческом возрасте) гидрофильные β-адреноблокаторы накапливаются в организме, что требует корректировки их дозы. В отличие от липофильных препаратов фармакокинетика гидрофильных β-адреноблокаторов и выраженность их действия практически не зависят от функционального состояния печени и генетического полиморфизма ферментов метаболизма. Поскольку гидрофильные β-адреноблокаторы практически не проникают через ГЭБ, они в меньшей степени оказывают такие побочные эффекты, характерные для липофильных препаратов, как депрессия, нарушения сна и кошмарные сновидения, спутанность сознания. Некоторые липофильные β-адреноблокаторы (бисопролол, пиндолол, целипролол) достаточно хорошо растворяются не только в липидах, но и в воде. Эти препараты практически в равной степени метаболизируются в печени и выводятся почками в неизмененном виде. Продолжительность действия β-адреноблокаторов во многом зависит от скорости их элиминации. Препараты длительного действия, такие как надолол, бетаксолол, бисопролол, атенолол, карведилол, имеют период полуэлиминации от 7 до 24 ч, их можно назначать 1–2 раза в сутки. Препараты средней продолжительности действия: пропранолол, тимолол, метопролол, пиндолол — с периодом полуэлиминации от 2 до 5 ч требуют более частого назначения. Эффекты, вызванные блокадой β-адренорецепторов, могут быть более продолжительными, в особенности при повышении дозы препарата. Кроме того, при систематическом применении β-адреноблокаторов их элиминация может замедляться (так как сами β-адреноблокаторы снижают как почечный, так и печеночный кровоток) и продолжительность действия увеличивается. 272 Часть II. Частная фармакология Эсмолол — β-адреноблокатор короткого действия (период полуэлиминации — около 10 мин), применяют для быстрого купирования острых состояний. Для длительной медикаментозной терапии, в особенности при артериальной гипертензии и хронической сердечной недостаточности, предпочтительнее использовать препараты длительного действия, которые при приеме 1–2 раза в сутки могут поддерживать АД на постоянном адекватном уровне. β-Адреноблокаторы с вазодилатирующими свойствами, такие как небиволол, способствующий синтезу и выделению NО, или карведилол, блокирующий α1-адренорецепторы, имеют определенные преимущества: они вызывают расширение сосудов и снижают ОПСС и постнагрузку на сердце непосредственно после введения, не повышают резиситентность тканей к инсулину при диабете 2-го типа, снижают уровень триглицеридов, повышают уровень ЛПНП. В соответствии с международной классификацией, учитывающей селективность по отношению к β1-адренорецепторам и наличие сосудорасширяющих свойств, β-адреноблокаторы принято разделять по поколениям (см. табл. 9.4). 9.2.2. Симпатолитики Симпатолитики тормозят передачу возбуждения с окончаний постганглионарных адренергических волокон на эффекторные органы, уменьшая количества медиатора норадреналина в варикозных утолщениях. При этом уменьшается выделение норадреналина адренергическими нервными окончаниями. В результате устраняется влияние симпатической иннервации на сердце и кровеносные сосуды — уменьшается частота и сила сердечных сокращений (уменьшается сердечный выброс), сосуды расширяются, АД снижается. Устранение симпатических влияний приводит к тому, что начинает преобладать влияние парасимпатической иннервации. В результате происходит усиление моторики ЖКТ (возможна диарея), повышение секреции пищеварительных желез. Эти явления устраняются атропином. При уменьшении выделения медиатора адренергическими нервными окончаниями компенсаторно увеличивается количество адренорецепторов на постсинаптической мембране эффекторных клеток. Поэтому на фоне симпатолитиков препараты адреномиметиков оказывают более сильное и продолжительное действие. Таким образом, симпатолитики Глава 9. Средства, действующие на адренергические синапсы 273 не устраняют, а усиливают эффекты введенных извне (экзогенных) адреномиметиков. Выраженными симпатолитическими свойствами обладает резерпин — алкалоид раувольфии (Rauwolfia serpentina Benth) — растения, произрастающего в Индии. Корни этого растения с давних времен применяли в народной индийской медицине. По химической структуре резерпин — производное индола. Резерпин нарушает процесс депонирования норадреналина и дофамина в везикулах варикозных утолщений (окончаний адренергических волокон). Он накапливается в мембране везикул и препятствует захвату везикулами дофамина (при этом уменьшается синтез норадреналина) и обратному захвату везикулами норадреналина (см. рис. 9.1). В цитоплазме нервных окончаний норадреналин подвергается окислительному дезаминированию под влиянием МАО (инактивируется). В результате истощаются запасы норадреналина в окончаниях адренергических волокон, меньше медиатора выделяется в синаптическую щель и нарушается передача возбуждения в адренергических синапсах. Кроме того, резерпин истощает запасы адреналина и норадреналина в надпочечниках. Таким образом, резерпин ослабляет влияние симпатической иннервации на сердце и кровеносные сосуды. Вследствие расширения сосудов и уменьшения сердечного выброса АД снижается. Основной терапевтический эффект резерпина — гипотензивный. Резерпин проникает через ГЭБ и уменьшает содержание норадреналина, дофамина и серотонина в ЦНС. В связи со снижением уровня дофамина в ЦНС резерпин оказывает слабый антипсихотический эффект. Однако в настоящее время резерпин в качестве антипсихотического средства не используют. Применяют резерпин при гипертонической болезни. Назначают внутрь. АД при введении резерпина снижается постепенно, и максимальный эффект наблюдается через 1–2 нед. После отмены препарата его эффект сохраняется до 2 нед (резерпин действует необратимо, и для устранения его действия необходимо образование новых везикул). Резерпин назначают в сочетании с тиазидными диуретиками и другими антигипертензивными веществами. Резерпин входит в состав комплексных препаратов, выпускаемых под названиями: Адельфан♠, Бринердин♠, Кристепин♠, Трирезид К♠ (см. раздел «Гипотензивные средства»). Побочные эффекты резерпина, связанные с повышением влияния парасимпатической иннервации: усиление секреции желез желудка (возможно обострение язвенной болезни желудка), диарея, брадикардия. 274 Часть II. Частная фармакология Вследствие расширения сосудов возможен отек слизистой оболочки носовой полости (заложенность носа). При применении резерпина (чаще в высоких дозах) могут возникать побочные эффекты, связанные с его угнетающим действием на ЦНС: вялость, нарушение внимания, сонливость, депрессия, редко возникают экстрапирамидные расстройства, связанные с истощением запасов дофамина в неостриатуме (лекарственный паркинсонизм). При появлении признаков депрессии препарат необходимо отменить. Антагонистами резерпина в отношении его угнетающего влияния на ЦНС являются ингибиторы МАО (ниаламид), которые восстанавливают баланс катехоламинов и серотонина в тканях мозга. Ингибиторы МАО назначают только после отмены резерпина (при одновременном приеме с ингибиторами МАО резерпин повышает АД и оказывает возбуждающее действие). Резерпин противопоказан при депрессии, болезни Паркинсона, язвенной болезни желудка и двенадцатиперстной кишки, феохромоцитоме. Гуанетидин отличается от резерпина по механизму симпатолитического действия: вытесняя норадреналин из систем обратного нейронального захвата, он вместо норадреналина захватывается окончаниями симпатических волокон. В цитоплазме нервных окончаний он проникает внутрь везикул и вытесняет из них норадреналин. Вытесненный из везикул норадреналин разрушается МАО. В везикулах гуанетидин препятствует синтезу норадреналина из дофамина. Кроме того, гуанетидин угнетает выделение норадреналина из нервных окончаний. В результате происходит резкое снижение (истощение) запасов норадреналина. Это приводит к уменьшению влияния симпатической иннервации на сердце и сосуды и снижению АД (вследствие расширения сосудов и уменьшения сердечного выброса). Гипотензивное действие гуанетидина развивается медленно и достигает максимума только на 7–8-й день лечения. Препарат действует длительно, после его отмены действие продолжается в течение 2 нед. В отличие от резерпина гуанетидин вызывает выраженную ортостатическую гипотензию. Кроме того, возникают такие же побочные эффекты, как и при применении резерпина: брадикардия, увеличение секреции пищеварительных желез, диарея, заложенность носа. В отличие от резерпина гуанетидин не оказывает влияния на ЦНС (не проникает через ГЭБ). Гуанетидин не влияет на выделение катехоламинов из надпочечников, так как в них нет механизма обратного нейронального захвата. Из-за выраженных побочных эффектов гуанетидин в настоящее время в основном используют в экспериментальных исследованиях. Глава 9. Средства, действующие на адренергические синапсы 275 Вопросы и задания для самоконтроля 1. Какие из перечисленных эффектов фенилэфрин вызывает рефлекторно: а) повышение АД; б) расширение зрачков; в) снижение ЧСС; г) сужение сосудов кожи; д) снижение моторики кишечника; е) повышение тонуса сфинктера мочевого пузыря? 2. Клонидин снижает АД вследствие стимуляции: а) α2-адренорецепторов нейронов ядер солитарного тракта; б) внесинаптических β2-адренорецепторов сосудов; в) пресинаптических α2-адренорецепторов; г) внесинаптических α2-адренорецепторов сосудов. 3. Сальбутамол вызывает тахикардию: а) увеличивая высвобождение адреналина из мозгового вещества надпочечников; б) увеличивая высвобождение норадреналина из окончаний симпатических нервов; в) рефлекторно вследствие снижения АД; г) вследствие стимуляции β2-адренорецепторов сердца; д) вследствие стимуляции пресинаптических β2-адренорецепторов. 4. Основное терапевтическое действие добутамина при острой сердечной недостаточности: а) повышение ЧСС; б) повышение атриовентрикулярной проводимости; в) увеличение силы сердечных сокращений; г) расширение сосудов. 5. Основные эффекты адреналина♠ при анафилактическом шоке: а) повышение АД вследствие стимуляции α-адренорецепторов сосудов; б) снижение АД вследствие стимуляции β2-адренорецепторов сосудов; в) устранение бронхоспазма вследствие стимуляции β2-адренорецепторов бронхов; г) повышение уровня глюкозы в крови вследствие стимуляции гликогенолиза. 276 Часть II. Частная фармакология 6. Блокада каких рецепторов не приводит к расширению сосудов: а) α1-адренорецепторов сосудов; б) β2-адренорецепторов сосудов; в) α2-адренорецепторов сосудов; г) пресинаптических β2-адренорецепторов; д) пресинаптических α2-адренорецепторов; е) β1-адренорецепторов юкстагломерулярных клеток? 7. В отличие от пропранолола празозин применяют при: а) стабильной стенокардии; б) наджелудочковых аритмиях; в) заболеваниях периферических артерий; г) мигрени; д) тиреотоксическом кризе; е) гипертензии. 8. Какой адреноблокатор можно назначить для лечения артериальной гипертензии больным с бронхиальной астмой: а) карведилол; б) пропранолол; в) доксазозин; г) тамсулозин; д) метопролол? СРЕДСТВА, ВЛИЯЮЩИЕ НА ЦЕНТРАЛЬНУЮ НЕРВНУЮ СИСТЕМУ ЛС, действующие на ЦНС, были известны с древних времен. Препаратам опия, мандрагоры, белладонны в Древнем Египте, средневековой Европе приписывали магические свойства; алкоголь использовали для снижения болевой чувствительности. Вместе с тем арсенал средств, влияющих на функции ЦНС, был весьма незначительным в течение многих веков. Большинство заболеваний головного и спинного мозга оставались неизлечимыми. Лишь в ХХ в. были достигнуты значительные успехи в этой области. Во многом развитие фармакологии ЦНС было обусловлено достижениями физиологии и биохимии. В ЦНС нейроны связаны между собой посредством синапсов, т.е. специальных контактов между отростками одних нейронов и телами или отростками других нейронов. Передачу возбуждения в синапсах от одного нейрона к другому осуществляют медиаторы (нейромедиаторы), выделяющиеся из пресинаптических окончаний под воздействием нервного импульса. Нейромедиаторы действуют на специфические рецепторы, расположенные на постсинаптической мембране и связанные с ионными каналами, ферментами. При этом изменяется функциональная активность нейронов. Нейромедиаторы могут действовать на рецепторы, расположенные на пресинаптической мембране, регулируя таким образом выделение нейромедиатора в синаптическую щель. К числу нейромедиаторов, участвующих в синаптической передаче в ЦНС, относят моноамины, ацетилхолин, аминокислоты, пептиды. МОНОАМИНЫ К моноаминам относят катехоламины (дофамин, норадреналин) и серотонин. Дофамин Основные дофаминергические структуры головного мозга расположены в черном веществе, неостриатуме, мезолимбической системе, ги- 278 Часть II. Частная фармакология поталамусе, пусковой зоне рвотного центра. Патологические изменения дофаминергических структур мозга играют роль в возникновении таких заболеваний, как паркинсонизм, шизофрения. Нарушения дофаминергической передачи возникают при лекарственной зависимости. В настоящее время выделено несколько подтипов дофаминовых рецепторов, которые объединены в два класса: D1 (подтипы D1 и D5) и D2 (подтипы D2, D3 и D4). Между этими классами рецепторов существуют определенные функциональные различия, обусловленные тем, что дофаминовые рецепторы класса D1 связаны с Gs-белками (активируют аденилатциклазу, в результате в клетках повышается содержание цАМФ), а рецепторы класса D2 — с Gi-белками (ингибируют аденилатциклазу и снижают количество цАМФ, а также активируют калиевые каналы). Норадреналин Значительная часть норадренергических нейронов расположена в голубоватом месте (locus caeruleus) серого вещества моста, откуда аксоны нейронов проецируются в кору головного мозга, гиппокамп, гипоталамус, мозжечок, продолговатый и спинной мозг. В норадренергических синапсах ЦНС есть как α-, так и β-адренорецепторы. Серотонин (5-гидрокситриптамин) Серотонинергические пути начинаются из ядер шва, моста и ствола головного мозга. Волокна, входящие в эти пути, распределяются в головном мозге, контролируя многие функции ЦНС, — участвуют в регуляции аппетита, цикла сон–бодрствование, активности нейронов антиноцицептивной системы, рвотного центра, лимбической системы. Выделяют значительное число подтипов рецепторов, сгруппированных в подразделения 5-НТ1A-F, 5-НТ1A-C и т.д. При стимуляции различных подтипов рецепторов возникают как тормозные эффекты (5-НТ1A, 5-НТ1D), так и эффекты возбуждения (5-НТ1С, 5-НТ2, 5-НТ3 и 5-НТ4). Среди этих рецепторов только 5-НТ3-рецепторы ионотропны (непосредственно связаны с ионными каналами), остальные подтипы 5-НТ-рецепторов взаимодействуют с ионными каналами и ферментами через G-белки. АЦЕТИЛХОЛИН Холинергические нейроны локализованы в большинстве областей ЦНС. Холинергическая передача имеет большое функциональное значение в неостриатуме и коре головного мозга. Посредством холинерги- Средства, влияющие на центральную нервную систему 279 ческой передачи осуществляется регуляция как психических, так и моторных функций; установлена ее роль в процессах обучения и памяти. Н-холинорецепторы, сходные с Н-холинорецепторами вегетативных ганглиев, расположены на тормозных клетках Реншоу в спинном мозге, а М-холинорецепторы представлены широко и находятся в синапсах различных отделов головного мозга (в коре головного мозга, неостриатуме). АМИНОКИСЛОТЫ Тормозные аминокислоты ГАМК относят к монокарбоновым аминокислотам. ГАМК — основной тормозной медиатор в ЦНС. Среди ГАМК-рецепторов выделяют два основных подтипа: ГАМКА- и ГАМКB-рецепторы. ГАМКА-рецептор, состоящий из пяти субъединиц (выделяют α-, β-, γ-, δ-, ε-, π-, ρ-типы субъединиц рецептора), образует мембранный канал для Cl–, который открывается при возбуждении рецептора двумя молекулами ГАМК. При этом отрицательно заряженные ионы хлора поступают через канал внутрь клетки, что вызывает гиперполяризацию мембраны, т.е. тормозной эффект. В настоящее время существуют данные о гетерогенности субъединиц ГАМКА-рецепторов в различных отделах головного мозга, что объясняет различия в эффектах веществ угнетающего типа. ГАМКА-рецептор имеет несколько модулирующих мест связывания для бензодиазепинов, барбитуратов, нейростероидов, этанола, а также средств для наркоза, таких как изофлуран, энфлуран, пропофол, этомидат. В отличие от ГАМКА-рецепторов, ГАМКB-рецепторы оказывают длительное тормозное воздействие. Их действие связано с G-белками, регулирующими активность аденилатциклазы. При стимуляции ГАМКBрецепторов активируются калиевые каналы нейронов, а также в результате действия на кальциевые каналы уменьшается поступление в синапсы ионов Са2+, что приводит к уменьшению выделения медиаторов и развитию тормозных эффектов. ГАМКС-рецепторы, находящиеся в спинном и головном мозге, а также в сетчатке глаза, в настоящее время изучены недостаточно. Глицин, как и ГАМК, относят к монокарбоновым аминокислотам и, воздействуя на глициновые рецепторы, оказывает аналогичное тормозное влияние на нейроны (повышается проницаемость хлорных каналов, ионы Cl– поступают в клетку, возникает гиперполяризация мембраны). Наибольшая концентрация этого медиатора отмечена в сером веществе спинного мозга. 280 Часть II. Частная фармакология Возбуждающие аминокислоты L-глутамат — возбуждающий медиатор в ЦНС, обладает выраженным активирующим действием на нейроны; относится к дикарбоновым аминокислотам, присутствует во всех отделах головного и спинного мозга. Глутаматные рецепторы подразделяют на «метаботропные», связанные с G-белками, и «ионотропные», непосредственно взаимодействующие с ионными каналами. Ионотропные глутаматные рецепторы связаны с натриевыми каналами, открывающимися при стимуляции рецепторов. В результате ионы Na+ поступают в клетку, что вызывает деполяризацию мембраны, стимулируя возбуждающий эффект. Связанные с каналами рецепторы по чувствительности к химическим анализаторам подразделяют на AMPA-рецепторы (чувствительны к амино-3-гидрокси-5метил-4-изоксазолпропионовой кислоте), каинатные рецепторы (чувствительны к каиновой кислоте, выделенной из морских водорослей) и NMDA-рецепторы (чувствительны к N-метил-D-аспартату). Стимуляция каинатных и AMPA-рецепторов вызывает быструю деполяризацию в большинстве глутаматергических синапсов головного и спинного мозга. NMDA-рецепторы также вовлечены в синаптическую передачу, однако они в большей степени определяют пластичность синаптической передачи, что имеет большое значение для процессов обучения и памяти. Экспериментально было установлено, что блокада этих рецепторов предупреждает дегенерацию нейронов головного мозга при ишемии. АМРА и каинатные рецепторы выполняют роль «зажигания» для NMDAрецепторов. Они образуют комплекс с NMDA-рецепторами, легче активируются, и вызванная ими деполяризация мембраны способствует удалению иона магния из просвета катионного канала NMDA-зависимого комплекса канал-рецептор. Другая эндогенная возбуждающая аминокислота —L-аспартат — действует аналогично L-глутамату. ПЕПТИДЫ Роль пептидов в регуляции активности ЦНС установлена сравнительно недавно, поэтому уверенно говорить о пептидергической передаче можно лишь в отношении некоторых соединений. Так, энкефалины и эндорфины — агонисты опиоидных рецепторов мозга. Субстанция Р участвует в передаче болевых (ноцицептивных) импульсов в спинном мозге. Многие физиологически активные пептиды (холецистокинин, пептид дельта-сна, вазоинтестинальный пептид, нейропептид Y) имеют места связывания Средства, влияющие на центральную нервную систему 281 в ЦНС, но полностью их роль как нейромедиаторов пока не доказана. Предполагают, что эти вещества могут оказывать на синаптическую передачу регулирующее (нейромодуляторное) действие. Известны и другие вещества, которые, наряду с нейромедиаторной функцией (передачей возбуждения в синапсах), оказывают на синаптическую передачу в ЦНС регулирующее действие, т.е. играют роль нейромодуляторов. К таким веществам могут быть отнесены аденозин, АТФ, оксид азота, гистамин. В регуляции некоторых функций ЦНС принимают участие простагландины. Анализ нейромедиаторных систем головного мозга позволил найти возможные «мишени» действия для ЛВ. Большинство ЛВ, влияющих на ЦНС, воздействуют на синаптическую передачу в головном или спинном мозге. Вещества могут действовать на различных этапах синаптической передачи как на пресинаптическом, так и на постсинаптическом уровне. ЛВ могут оказывать влияние на синтез медиатора (леводопа), выделение медиатора в синаптическую щель (амфетамин). Эффекты многих ЛВ связаны со стимуляцией соответствующих рецепторов (опиоидные анальгетики, бензодиазепины) или с блокадой рецепторов (антипсихотические средства). Используют вещества, ингибирующие обратный нейрональный захват медиатора (трициклические антидепрессанты), нарушающие процесс депонирования медиатора в везикулах (резерпин) и процесс метаболической инактивации медиатора в цитоплазме нервной клетки (ингибиторы МАО). Кроме того, некоторые ЛВ оказывают влияние на ЦНС, непосредственно взаимодействуя с ионными каналами (противоэпилептические средства из группы блокаторов натриевых, кальциевых каналов) или ферментами (парацетамол — ингибитор ЦОГ). Известны вещества, оказывающие нормализующее действие на энергетический обмен в нервных клетках (ноотропные средства). Различают следующие группы ЛВ, действующих на ЦНС. ● Средства для наркоза. ● Снотворные средства. ● Противоэпилептические средства. ● Противопаркинсонические средства. ● Болеутоляющие средства (анальгетики). ● Аналептики. ● Психотропные средства: —нейролептики; —антидепрессанты; —соли лития; 282 Часть II. Частная фармакология —анксиолитики; —седативные средства; —психостимуляторы; —ноотропные средства. Средства для наркоза, снотворные наркотического типа действия оказывают неизбирательное (общее) угнетающее действие на ЦНС. Противоэпилептические и противопаркинсонические средства, анальгетики, нейролептики, анксиолитики оказывают относительно избирательное угнетающее влияние на определенные структуры и функции ЦНС. Аналептики стимулируют жизненно важные центры — дыхательный и сосудодвигательный. Психостимуляторы активируют высшую нервную деятельность. Глава 10 СРЕДСТВА ДЛЯ НАРКОЗА (ОБЩИЕ АНЕСТЕТИКИ) Наркоз (от греч. narkosis — оцепенение, оглушение) — обратимое угнетение функций ЦНС, сопровождающееся потерей сознания, утратой чувствительности, в том числе болевой, угнетением соматических и вегетативных рефлексов, снижением мышечного тонуса. Наркоз используют при проведении хирургических операций. Средства для наркоза (общие анестетики) — очень неоднородная по физико-химическим свойствам группа ЛВ. Так, в нормальных условиях ксенон — газ, пропофол — жидкость, тиопентал натрия — твердое вещество. Фармакологические свойства препаратов этой группы также отличаются. Так, например, пропофол вызывает потерю сознания с низким уровнем обезболивания, а динитроген оксид (азота закись♠) — снижение болевой чувствительности при сохраненном сознании. Множество различий позволяет охарактеризовать средства для наркоза лишь как средства, в низких концентрациях вызывающие обратимую потерю сознания. Попытки уменьшить боль во время хирургических операций предпринимали еще в Древнем Египте и Риме с помощью алкоголя или опия. Однако официальной датой открытия наркоза считают 16 октября 1846 г., когда Уильям Мортон во время операции использовал для наркоза диэтиловый эфир. Через год Джеймс Симпсон впервые применил для наркоза хлороформ. Большое значение для внедрения средств для наркоза в хирургическую практику имели работы выдающегося русского хирурга Н.И. Пирогова, который уже с 1847 г. стал использовать для обезболивания во время хирургических операций диэтиловый эфир. При применении средств для наркоза важны следующие основные характеристики: ● быстрое развитие наркоза без выраженного возбуждения; ● достаточная глубина наркоза, позволяющая проводить операцию в оптимальных условиях; ● хорошая управляемость глубиной наркоза; ● быстрый и без последствий выход из наркоза. 284 Часть II. Частная фармакология Средства для наркоза должны иметь достаточную широту наркотического действия (наркотическую широту) — диапазон между концентрацией вещества, в которой оно вызывает стадию глубокого хирургического наркоза, и минимальной токсической концентрацией, при которой наступает остановка дыхания вследствие угнетения дыхательного центра. Кроме того, средства для наркоза не должны вызывать раздражения тканей в месте введения, должны обладать минимальными побочными эффектами. Вещества этой группы не должны быть взрывоопасными. Однако в настоящее время нет препаратов, обладающих всеми указанными свойствами. В связи с этим в современной анестезиологической практике, как правило, применяют комбинации средств для наркоза, что позволяет уменьшать количество вводимых препаратов и, следовательно, их нежелательные эффекты. В зависимости от путей введения выделяют: ● средства для ингаляционного наркоза: ♠ —летучие жидкости: галотан (фторотан ), энфлуран (этран♠), изофлуран (форан♠), севофлуран, эфир диэтиловый; —газообразные вещества: динитрогена оксид (азота закись♠), ксенон; ● средства для неингаляционного наркоза: —тиопентал натрия, пропофол, кетамин, натрия оксибутират (натрия оксибат♠). 10.1. СРЕДСТВА ДЛЯ ИНГАЛЯЦИОННОГО НАРКОЗА Механизм действия средств для ингаляционного наркоза полностью не ясен. Известно, что препараты этой группы понижают спонтанную и вызванную активность нейронов различных областей головного мозга. Одной из концепций, объясняющих механизм их действия, считалась «липидная» теория. Средства для наркоза относят к высоколипофильным веществам. Эти соединения легко растворяются в липидном бислое мембран нейронов, что приводит к последующим конформационным изменениям ионных каналов и нарушению трансмембранного транспорта ионов. Препараты этой группы повышают проницаемость калиевых и уменьшают проницаемость быстрых натриевых каналов, что соответственно вызывает гиперполяризацию и нарушает процесс деполяризации мембран нейронов. В результате нарушается межнейронная передача возбуждения, и развиваются тормозные эффекты. Однако в настоящее время преобладает точка зрения, согласно которой средства для наркоза стимулируют или повышают чувствительность Глава 10. Средства для наркоза (общие анестетики) 285 к ГАМК и глицину соответствующих рецепторов в головном и спинном мозге, а также угнетают активность глутаматных рецепторов, в частности NMDA-рецепторов (NDMA — N-метил-D-аспартат). У большинства средств для наркоза определены места связывания с этими рецепторами. Наряду с этим полагают, что средства для ингаляционного наркоза могут уменьшать выделение некоторых медиаторов (ацетилхолина, дофамина, серотонина, норадреналина) в головном мозге. Чувствительность различных отделов мозга к средствам для наркоза неодинакова. Вначале угнетается синаптическая передача в ретикулярной формации и коре головного мозга, в последнюю очередь — в дыхательном и сосудодвигательном центрах. Это объясняет наличие определенных стадий в действии средств для наркоза. Так, в действии эталонного средства для наркоза — диэтилового эфира — выделяют 4 стадии. ● Cтадия аналгезии (от лат. an — отрицание и греч. algos — боль) характеризуется снижением болевой чувствительности, постепенным угнетением сознания (однако пациент еще находится в сознании). Частота дыхания, пульс и АД не изменены. К концу этой стадии развиваются выраженная аналгезия и амнезия (потеря памяти). ● Стадия возбуждения. У пациента утрачивается сознание, развивается речевое и двигательное возбуждение (характерны немотивированные движения). Дыхание нерегулярное, появляется тахикардия, зрачки расширены, усиливается кашлевой и рвотный рефлексы, вследствие чего возможно возникновение рвоты. Спинномозговые рефлексы и мышечный тонус повышены. Стадию возбуждения объясняют угнетением коры головного мозга, в связи с чем уменьшаются ее тормозные влияния на нижележащие центры, при этом происходит повышение активности подкорковых структур (в основном среднего мозга). ● Стадия хирургического наркоза. Начало этой стадии характеризуется нормализацией дыхания, отсутствием признаков возбуждения, значительным снижением мышечного тонуса и угнетением безусловных рефлексов. Сознание и болевая чувствительность отсутствуют. Зрачки сужены, дыхание регулярное, АД стабилизируется, в стадии глубокого хирургического наркоза происходит урежение пульса. При углублении наркоза частота пульса меняется, возможны сердечные аритмии и снижение АД. Происходит постепенное угнетение дыхания. В этой стадии выделяют 4 уровня: —1-й уровень (III1) — поверхностный наркоз; —2-й уровень (III2) — легкий наркоз; —3-й уровень (III3) — глубокий наркоз; —4-й уровень (III4) — сверхглубокий наркоз. 286 Часть II. Частная фармакология ● Стадия восстановления наступает после прекращении введения препарата. Постепенно происходит восстановление функций ЦНС в порядке, обратном их появлению. При передозировке средств для наркоза развивается агональная стадия, обусловленная угнетением дыхательного и сосудодвигательного центров. При применении других ингаляционных средств для наркоза стадия возбуждения выражена в меньшей степени, выраженность стадии аналгезии также может быть различной. Основным параметром, определяющим скорость развития наркоза и выхода из него, служит коэффициент распределения кровь/газ. Средства для ингаляционного наркоза, легко переходящие из воздуха альвеол в кровь (галотан, энфлуран, изофлуран, эфир диэтиловый), вызывают относительное медленное развитие наркоза и длительное пробуждение. Напротив, общие анестетики, хуже растворяющиеся в крови (азота закись♠, ксенон и севофлуран), вызывают быстрое вхождение в наркоз и быстрое восстановление. Как уже указывалось, существенный фактор в развитии наркоза — неодинаковая чувствительность различных отделов ЦНС к общим анестетикам. Так, высокая восприимчивость к ним нейронов студенистого вещества спинного мозга, участвующих в проведении болевых импульсов, служит причиной аналгезии на I стадии наркоза, когда сознание еще сохранено. Большая устойчивость нейронов подкорковых структур позволяет поддерживать основные параметры жизнедеятельности организма при угнетении коры головного мозга, отсутствии сознания во время стадии хирургического наркоза. К средствам для ингаляционного наркоза относят жидкие летучие вещества — галотан, энфлуран, изофлуран. Активность этих средств для ингаляционного наркоза очень высока, в связи с чем их введение проводят с помощью специальных наркозных аппаратов, позволяющих точно дозировать ингалируемые вещества. Пары летучих жидкостей поступают в дыхательные пути через интубационную трубку, введенную в трахею. Преимущество ингаляционного наркоза — высокая управляемость, поскольку препараты этой группы легко всасываются и быстро выделяются из организма через легкие. Галотан принадлежит к фторсодержащим алифатическим соединениям. Это бесцветная прозрачная, подвижная, легко летучая жидкость со специфическим запахом. В связи с тем, что галотан разлагается под действием света, препарат выпускают во флаконах из темного стекла (рис. 10.1). Галотан при смешивании с воздухом не горит и не взрывается, обладает высокой наркотической активностью. В смеси с кислородом или воздухом способен вызвать стадию хирургиче- Глава 10. Средства для наркоза (общие анестетики) 287 ского наркоза. Наркоз наступает быстро (через 3–5 мин), без выраженной стадии возбуждения, легко управляем. После прекращения ингаляции пациенты начинают приходить в сознание через 3–5 мин. Галотан обладает достаточной наркотической широтой. Пары галотана не раздражают дыхательные пути. Аналгезия и миорелаксация при примене- Рис. 10.1. Химическая нии галотана выражены меньше, чем при эфир- структура галотана ном наркозе, поэтому его комбинируют с азота закисью♠ и курареподобными средствами. Галотан используют для наркоза при оперативных вмешательствах, в том числе при полостных операциях. При применении галотана возникают некоторые побочные эффекты. Препарат снижает сократимость миокарда, вызывает брадикардию (результат стимуляции центра блуждающего нерва). АД снижается в связи с угнетением сосудодвигательного центра, симпатических ганглиев (ганглиоблокирующее действие), а также прямого миотропного воздействия на стенки сосудов. Галотан сенсибилизирует миокард к катехоламинам — адреналину и норадреналину: введение эпинефрина и норэпинефрина на фоне галотанового наркоза вызывает нарушения сердечного ритма (при необходимости повышения АД применяют фенилэфрин). Галотан потенцирует гипотензивное действие ганглиоблокаторов, β-адреноблокаторов, диазоксида и диуретиков. Существуют данные о гепатотоксическом действии галотана, связанном с образованием токсичных метаболитов (не рекомендуют применять при заболеваниях печени), возможно нефротоксическое действие. При сочетании галотана с суксаметония йодидом (дитилином ♠) существует опасность возникновения злокачественной гипертермии (повышение температуры тела до 42–43 °С в результате спазма скелетных мышц), что связано с выходом ионов кальция из саркоплазматического ретикулума в цитоплазму миоцитов. В этом случае применяют дантролен, снижающий выделение кальция из саркоплазматического ретикулума. Энфлуран сходен по свойствам с галотаном, но менее активен. Наркоз при применении энфлурана наступает быстрее и характеризуется более выраженной миорелаксацией. Важным свойством энфлурана считают то, что он в меньшей степени сенсибилизирует миокард к адреналину и норадреналину (меньше опасность возникновения аритмий), снижен риск гепатотоксического и нефротоксического эффектов. 288 Часть II. Частная фармакология Изофлуран — изомер энфлурана, менее токсичен — не провоцирует развитие аритмий, не обладает гепатотоксическими и нефротоксическими свойствами. Относительно новый препарат из группы фторсодержащих соединений — севофлуран. Препарат действует быстро, характеризуется легкой управляемостью и быстрым выходом больного из наркоза, практически не оказывает отрицательного действия на функцию внутренних органов, мало влияет на сердечно-сосудистую систему и дыхание. Севофлуран используют как в стационарах, так и в амбулаторной практике. Эфир диэтиловый (эфир для наркоза♠) обладает высокой активностью и большой наркотической широтой. Вызывает выраженную аналгезию и миорелаксацию, но при его применении возникает большое количество нежелательных эффектов. Наркоз при применении эфира♠ развивается медленно; выражена длительная стадия возбуждения, характерен медленный выход из наркоза (примерно в течение 30 мин). Для полного восстановления функций головного мозга после прекращения наркоза необходимо несколько часов. Эфир диэтиловый раздражает дыхательные пути, в связи с чем усиливается секреция слюнных и бронхиальных желез, возможны рефлекторное угнетение дыхания и уменьшение ЧСС, рвота. Пары эфира♠ легко воспламеняются и образуют с воздухом взрывоопасные смеси. В настоящее время эфир для наркоза♠ применяют крайне редко. К газообразным средствам для наркоза относят динитроген оксид (азота закись♠) (N2O) — бесцветный газ без запаха. Сама азота закись♠ не горит и не взрывается, однако поддерживает горение и с парами эфира♠ образует взрывоопасные смеси. Азота закись♠ обладает низкой наркотической активностью и может вызывать стадию хирургического наркоза только в гипербарических условиях. В концентрации 20% во вдыхаемой смеси азота закись♠ проявляет аналгетическое действие. При увеличении концентрации до 80% она способна вызвать поверхностный наркоз. Для предупреждения гипоксии в медицинской практике применяют газовые смеси, содержащие не более 80% азота закиси♠ и 20% кислорода (что соответствует его содержанию в воздухе). При использовании указанной смеси быстро возникает поверхностный наркоз без стадии возбуждения, характеризующийся хорошей управляемостью, но отсутствием миорелаксации. Пробуждение наступает практически в первые минуты после прекращения ингаляции. Азота закись♠ используют для обезболивания кратковременных операций в стоматологии, гинекологии, для обезболивания родов, купирования болей при инфаркте миокарда и острой коронарной недостаточности, Глава 10. Средства для наркоза (общие анестетики) 289 остром панкреатите. В связи с низкой наркотической активностью используют в комбинации с более активными средствами для наркоза. Азота закись♠ не метаболизируется в организме, выводится практически полностью через легкие. Побочные эффекты при кратковременном применении практически отсутствуют, но при длительных ингаляциях возможно развитие лейкопении, мегалобластической анемии, нейропатии. Эти эффекты связаны с окислением кобальта в молекуле витамина В12 под действием азота закиси♠, что приводит к недостаточности витамина. При комбинировании со средствами, используемыми в анестезиологической практике (наркотические анальгетики, нейролептики), возможно снижение АД и сердечного выброса. Ксенон — инертный газ, обеспечивающий вследствие очень низкого коэффициента распределения кровь/газ быстрое развитие наркоза с высокой степенью аналгезии. Не оказывает токсического действия и не влияет на сократимость миокарда. Существуют сведения о нейропротекторном действии ксенона. Механизм наркотического действия обусловлен неконкурентной блокадой NMDA-рецепторов, влиянием на ГАМКА-рецепторы и отличные от NMDA глутаматные рецепторы. К недостаткам можно отнести высокую стоимость препарата и необходимость использования специальной аппаратуры. 10.2. СРЕДСТВА ДЛЯ НЕИНГАЛЯЦИОННОГО НАРКОЗА Идея использования средств для неингаляционного наркоза впервые была высказана Н.И. Пироговым еще в 1847 г., когда он предложил и испытал в клинике наркоз при ректальном введении эфира. Идеи Н.И. Пирогова нашли практическое применение после получения активных нелетучих средств для наркоза. Первым таким средством стал гедонал — вещество, предложенное в 1909 г. Н.П. Кравковым для внутривенного наркоза и испытанное в хирургической клинике С.П. Федорова. К средствам для неингаляционного наркоза относят вещества разного химического строения, различной продолжительности действия. Как правило, эти ЛС вводят внутривенно, реже — ректально. Современные средства для неингаляционного наркоза имеют латентный период короче, чем ингаляционные общие анестетики. При этом для использования неингаляционных средств не нужна сложная и дорогостоящая аппаратура, нет необходимости в очистке воздуха от выдыхаемого ингаляционного анестетика. 290 Часть II. Частная фармакология В отличие от ингаляционного внутривенный наркоз протекает практически без стадии возбуждения. Высокая липофильность позволяет препаратам этой группы легко проникать в мозг. Необходимо учитывать, что в случае использования средств для внутривенного наркоза управляемость глубиной наркоза низкая. Современные средства для внутривенного наркоза по продолжительности действия могут быть разделены на следующие группы: ● препараты кратковременного действия (продолжительность наркоза до 15 мин): пропанидид, пропофол, этомидат, кетамин; ● препараты средней продолжительности действия (продолжительность наркоза 20–30 мин): тиопентал натрия, гексобарбитал (гексенал♠); наркоза ● препараты длительного действия (продолжительность 60 мин и более): натрия оксибутират (натрия оксибат♠). Пропанидид — маслянистая жидкость, выпускают в виде раствора в ампулах. После внутривенного введения наркоз наступает через 20–40 с без стадии возбуждения и продолжается 3–4 мин (препарат «ультракороткого» действия, поскольку быстро гидролизуется холинэстеразой плазмы крови). Пропанидид используют для вводного наркоза (введение в состояние наркоза без стадии возбуждения), а также для кратковременного наркоза при биопсии, вправлении вывихов, удалении зубов. Вследствие быстрого выхода из наркоза (сознание восстанавливается через 2–3 мин, а через 20–30 мин восстанавливаются психомоторные функции) пропанидид удобен для амбулаторной практики. При применении пропанидида сначала возникает кратковременная гипервентиляция с последующим угнетением дыхания (апноэ продолжительностью 10–30 с), возможны тахикардия и снижение АД. В начале наркоза у некоторых больных возникают мышечные подергивания. Пропанидид оказывает некоторое раздражающее действие, вследствие чего возникают болевые ощущения по ходу вены. Возможны аллергические реакции. Пропофол — 2,6-диизопропилфенол, в воде не растворим, вводят внутривенно в виде эмульсии. При внутривенном введении пропофол вызывает быстрое развитие наркоза (через 30–40 с) с минимальной стадией возбуждения. Возможно кратковременное угнетение дыхания. Пробуждение быстрое (сознание восстанавливается через 4 мин). Продолжительность наркоза после однократного введения — 3–10 мин. Препарат вводят дробно или капельно для введения в наркоз или его поддержания в комбинации со средствами для ингаляционного наркоза. Пропофол лишен Глава 10. Средства для наркоза (общие анестетики) 291 анальгетических свойств, поэтому его часто комбинируют с наркотическими анальгетиками. Препарат также применяют как седативное средство (в дозах, меньше наркотических в 2–5 раз) при кратковременных хирургических манипуляциях, искусственной вентиляции легких. Действие связывают с потенцированием эффектов, обусловленным связыванием пропофола с β2- или β3-субъединицами ГАМКА-рецепторов. Препарат метаболизируется в печени путем конъюгации с глюкуроновой кислотой и сульфатирования, метаболиты выделяются почками. Пропофол вызывает брадикардию, снижает АД, не исключено отрицательное инотропное действие. Пациенты отмечают лучшую переносимость препарата по сравнению с другими средствами для наркоза. Препарат не вызывает рвоту в посленаркозном периоде, не нарушает функции печени и почек. В месте введения возможна болезненность по ходу вены, реже возникают флебиты или тромбозы, возможны аллергические реакции. Этомидат относят к группе карбоксилированных имидазолов, используют для вводного или сбалансированного наркоза. Этомидат — весьма активное средство для наркоза ультракороткого действия (продолжительность 3–5 мин), не обладает анальгетической активностью, что часто приводит к необходимости его комбинирования с наркотическими анальгетиками. При внутривенном введении этомидат вызывает потерю сознания на 5 мин, сопровождающуюся понижением АД. Во время наркоза возможны спонтанные мышечные сокращения. Действие этомидата, как и пропофола, связывают с потенцированием эффектов ГАМК. В послеоперационном периоде часто возникает рвота, особенно при комбинированном применении с наркотическими анальгетиками. Этомидат угнетает стероидогенез в коре надпочечников, что приводит к снижению содержания гидрокортизона и альдостерона в плазме крови даже после однократного введения препарата. Длительное введение этомидата может привести к недостаточности коры надпочечников (гипотензии, нарушению электролитного баланса, олигурии). Кетамин — арилциклогексиламин, производное фенциклидина (рис. 10.2). Кетамин — уникальный препарат, вызывающий так называемую «диссоциативную анестезию», обусловленную тем, что кетамин угнетает одни структуры мозга и не влияет на другие. При введении кетамина возникают выраженная аналгезия, легкий снотворный эффект, амнезия (потеря памяти) с сохранением самостоятельного дыхания, мышечного тонуса, гортанного, глоточного и кашлевого рефлексов; сознание утрачивается лишь частично. Стадию хирургического наркоза кетамин не вызывает. Механизм действия кетамина связан с блокадой NMDA-рецепторов 292 Часть II. Частная фармакология нейронов головного мозга, вследствие чего устраняется возбуждающее действие глутамата на определенные структуры ЦНС. Кетамин применяют как для вводного наркоза, так и самостоятельно для обезболивания при кратковременных болезненных процедурах (в частности, при обработке ожоговой поверхности). Кетамин обладает высокой липофильностью, вследствие чего легко проникает в мозг, а его центральное действие развивается в течение 30–60 с после внутривенного введения и продолжается 5–10 мин. Препарат также вводят внутримышечно. При внутримышечном введении действие наступает через 2–6 мин и продолжается 15–30 мин. Среди средств для неингаляционного наркоза только кетамин увеличивает ЧСС, сердечный выброс и повышает АД. Максимальное влияние на сердечно-сосудистую систему отмечено на 2–4-й минуте и постепенно снижается через 10–20 мин. Механизм этого воздействия связан со стимуляцией симпатической иннервации и нарушением обратного нейронального захвата норадреналина. В послеоперационном периоде после применения кетамина возникают яркие, нередко кошмарные, сновидения, психомоторное возбуждение, галлюцинации, которые устраняются диазепамом. Возможность возникновения послеоперационных психозов ограничивает широкое применение препарата. Тиопентал натрия — производное барбитуровой кислоты (см. рис. 10.2). Механизм действия обусловлен взаимодействием тиопентала натрия с комплексом ГАМКА-рецептор–хлорный канал и усилением действия эндогенной ГАМК — основного тормозного медиатора в ЦНС. Взаимодействуя со специфическими местами связывания (барбитуратными рецепторами) на ГАМКА-рецепторном комплексе, тиопентал натрия вызывает изменение конформации ГАМКА-рецептора, при этом повышается чувствительность рецептора к ГАМК, что приводит к более длительной активации хлорных каналов (ионы хлора входят в клетку, и наступает гиперполяризация мембраны нейрона). Препарат обладает некоторым прямым ГАМК-миметическим действием, отличается высо- Рис. 10.2. Химические структуры кетамина и тиопентала натрия Глава 10. Средства для наркоза (общие анестетики) 293 кой наркотической активностью и быстрым развитием наркотического действия. Вследствие высокой липофильности тиопентал натрия быстро проникает в ткани мозга и уже через 1 мин после внутривенного введения вызывает наркоз без стадии возбуждения. Длительность наркоза после однократного введения составляет 15–25 мин. После выхода из наркоза развивается продолжительный посленаркозный сон. Этот феномен связан с особенностью фармакокинетики препарата: тиопентал натрия накапливается в жировой ткани, при этом его концентрация в тканях мозга снижается. Это определяет небольшую продолжительность действия препарата. Последующее медленное выделение вещества из жировой ткани в кровь определяет способность тиопентала натрия вызывать посленаркозный сон. Тиопентал натрия применяют для наркоза при кратковременных хирургических вмешательствах, для купирования психомоторного возбуждения и судорожных припадков. Наиболее часто тиопентал натрия применяют для вводного наркоза (введение в состояние наркоза без стадии возбуждения). Препарат выпускают во флаконах в виде порошка, который растворяют перед внутривенным введением (рН раствора около 10, при увеличении кислотности возможно образование осадка барбитуровой кислоты). Тиопентал натрия необходимо вводить медленно, так как при быстром введении возможно угнетение дыхательного и сосудодвигательного центров (вплоть до развития апноэ и коллапса). Метаболизм тиопентала натрия значительно длительнее, чем его перераспределение. В печени за 1 ч метаболизируется 12–16% вещества. Препарат противопоказан при нарушениях функции печени и почек (значительно увеличиваются продолжительность действия и токсичность). Натрия оксибутират (натрия оксибат♠) по химическому строению и свойствам близок ГАМК. Препарат в малых дозах оказывает седативное и мышечнорасслабляющее действие, а в больших — вызывает сон и наркоз. Наркотическая активность натрия оксибутирата ниже, чем у тиопентала натрия. Препарат медленно проникает в мозг, и, как следствие, наркотическое действие развивается медленно, после внутривенного введения хирургическая стадия наркоза наступает только через 30–40 мин. Подобно всем неингаляционным средствам для наркоза натрия оксибутират не вызывает стадию возбуждения. Длительность наркотического эффекта после однократного применения составляет 2–4 ч. Для наркотического действия натрия оксибутирата характерна выраженная миорелаксация. Препарат повышает устойчивость организма к гипоксии. Натрия оксибутират применяют внутривенно, ректально и внутрь для вводного и базисного наркоза, а также для купирования психическо- 294 Часть II. Частная фармакология го возбуждения, профилактики и лечения гипоксического отека мозга и эклампсии. Вопросы и задания для самоконтроля 1. Для галотана характерно: а) высокая наркотическая активность; б) брадикардия; в) огнеопасность; г) сенсибилизация адренорецепторов миокарда катехоламинам; д) разражение дыхательных путей. 2. Азота закись: а) обладает высокой наркотической активностью; б) используют в концентрации 90 об.%; в) обладает очень коротким последействием; г) вызывает хирургическую стадию наркоза; д) гепатотоксична. 3. Укажите средства для неингаляционного наркоза: а) лидокаин; б) морфин; в) пропанидид; г) севофлуран; д) кетамин. 4. Укажите побочные эффекты кетамина: а) апноэ; б) гипотензия; в) брадикардия; г) гипертензия; д) зрительные галлюцинации. 5. Для тиопентала натрия характерно: а) быстрое развитие наркоза; б) выраженная стадия возбуждения; в) прямое возбуждение ГАМКА-рецепторов; г) накопление в жировой ткани; д) взаимодействие с барбитуратными рецепторами. Глава 11 СНОТВОРНЫЕ СРЕДСТВА Снотворные средства — ЛВ, вызывающие у человека состояние, близкое к естественному сну. Применяют при бессоннице для облегчения засыпания и обеспечения нормальной продолжительности сна. Сон неоднороден по своей структуре. Выделяют две основные составляющие сна, различающиеся характером волновых колебаний электрической активности клеток мозга на электроэнцефалограмме (ЭЭГ): медленноволновый сон и быстроволновый сон. ● Медленноволновый сон (медленный, ортодоксальный, синхронизированный, non-REM-sleep) имеет продолжительность до 75–80% от общего времени сна и четыре последовательно развивающиеся фазы — от дремоты (первая фаза) до фазы δ-сна (четвертая фаза), характеризующейся возникновением на ЭЭГ медленных высокоамплитудных δ-волн. ● Быстроволновый сон (быстрый, парадоксальный, десинхронизированный) повторяется каждые 80–90 мин, сопровождается сновидениями и быстрыми движениями глаз (rapid eye movement sleep, REM-sleep). Длительность быстроволнового сна составляет 20–25% от общего времени сна. Соотношения фаз сна и их ритмическую смену регулируют серотонин (основной фактор, индуцирующий сон), мелатонин (фактор, обеспечивающий синхронизацию фаз сна), а также ГАМК, энкефалины и эндорфины, пептид 5-сна, ацетилхолин, дофамин, адреналин, гистамин. Чередования фаз медленноволнового и быстроволнового сна характерны для нормального сна, при этом человек чувствует себя бодрым и выспавшимся. Расстройства естественного сна могут быть связаны с нарушением засыпания, глубины сна (поверхностный сон, тревожные сновидения, частые пробуждения), продолжительности сна (недосыпание, длительное окончательное пробуждение), структуры сна (изменением соотношения медленного и быстрого сна). Основное действие снотворных средств направлено на облегчение процесса засыпания и/или на удлинение продолжительности сна. В за- 296 Часть II. Частная фармакология висимости от этого используют снотворные средства разной продолжительности действия. В небольших дозах снотворные средства оказывают седативный (успокаивающий) эффект. Снотворные средства действуют угнетающе на синаптическую передачу в ЦНС, причем одни из них относительно избирательно угнетают отдельные структуры и функции мозга (снотворные с ненаркотическим типом действия), а другие оказывают общее угнетающее действие на ЦНС, т.е. действуют неизбирательно (средства наркотического типа действия). В соответствии с такими различиями в действии, а также исходя из различий в химической структуре выделяют следующие основные группы снотворных средств. ● Снотворные средства с ненаркотическим типом действия. —Агонисты бензодиазепиновых рецепторов: ✧ производные бензодиазепина: нитразепам (радедорм♠, эуноктин♠), флунитразепам (рогипнол♠), триазолам (хальцион♠), мидазолам (дормикум♠); ✧ препараты другой химической структуры (небензодиазепины): зопиклон (имован♠, пиклодорм♠), эсзолпидем (ивадал♠, санвал♠), залеплон. —Блокаторы Н1-рецепторов: доксиламин (донормил♠). —Агонисты мелатониновых рецепторов: рамелтеон. ● Снотворные средства с наркотическим типом действия. —Производные барбитуровой кислоты (барбитураты): фенобарбитал (люминал♠). —Алифатические соединения: хлоралгидрат. Сон, возникающий при применении снотворных средств, несколько отличается от естественного (физиологического) сна. В первую очередь это касается изменения продолжительности быстроволнового сна: увеличивается латентный период в развитии этой фазы и уменьшается ее общая продолжительность. При отмене снотворных средств латентный период фазы быстрого сна временно укорачивается, а быстроволновый сон на какое-то время удлиняется. При этом возникает обилие сновидений, имеющих характер ночных кошмаров, что приводит к частым пробуждениям. Эти явления, связанные с прекращением применения снотворного препарата, называют феноменом «отдачи». Снотворные средства в неодинаковой мере нарушают соотношение между быстрой и медленной фазами сна (нарушают структуру сна). В большей степени это характерно для производных барбитуровой кислоты и в меньшей степени — для бензодиазепинов. Мало изменяют структуру сна золпидем и зопиклон, и практически не влияет на нее хлоралгидрат. Глава 11. Снотворные средства 297 К снотворным средствам предъявляют следующие основные требования: ● они должны быстро вызывать сон и поддерживать его оптимальную продолжительность; ● не должны нарушать естественного соотношения между фазами сна (не нарушать структуру сна); ● не должны вызывать угнетения дыхания, нарушения памяти, привыкания, физической и психической зависимости. В настоящее время нет снотворных средств, которые бы в полной мере удовлетворяли всем этим требованиям. 11.1. СНОТВОРНЫЕ СРЕДСТВА С НЕНАРКОТИЧЕСКИМ ТИПОМ ДЕЙСТВИЯ 11.1.1. Агонисты бензодиазепиновых рецепторов Производные бензодиазепина Производные бензодиазепина обладают анксиолитической активностью (устраняют чувство тревоги, беспокойства, напряженности (см. раздел «Анксиолитические средства») и оказывают снотворное, а в небольших дозах — успокаивающее (седативное) действие. Устранение психического напряжения способствует успокоению и развитию сна. Кроме того, бензодиазепины снижают тонус скелетных мышц (эффект связан с подавлением полисинаптических рефлексов на уровне спинного мозга) и проявляют противосудорожную активность, потенцируют действие веществ, угнетающих ЦНС, в том числе алкоголя и средств для наркоза, и оказывают амнестическое действие (вызывают антероградную амнезию). Анксиолитическое и снотворное действия бензодиазепинов обусловлены их угнетающим действием на лимбическую систему и активирующим ретикулярную формацию ствола мозга. Механизм этих эффектов связывают со стимуляцией бензодиазепиновых (ω) рецепторов, агонистами которых они являются. Выделяют 3 подтипа ω-рецепторов — ω1, ω2, ω3. Полагают, что снотворное действие бензодиазепинов обусловлено преимущественным связыванием с ω1-рецепторами. Бензодиазепиновые рецепторы образуют комплекс с ГАМКА-рецепторами, непосредственно образующими хлорный канал. ГАМКА-рецептор — гликопротеин, состоящий из 5 субъединиц (2α, 2β и γ), непосредственно образующих хлорный канал. ГАМК связывается с α- и β-субъединицами рецептора и вызывает открытие хлорного канала (рис. 11.1). Стимуляция бензодиазепиновых рецепторов, расположенных на γ-субъединице 298 Часть II. Частная фармакология ГАМКА-рецептора, сопровождается повышением чувствительности ГАМКА-рецепторов к ГАМК и увеличению эффективности этого медиатора. При этом активность ГАМК не возрастает, что обусловливает отсутствие наркотического действия у бензодиазепинов. При повышении чувствительности ГАМК-рецепторов к ГАМК под воздействием бензодиазепинов увеличивается частота открытия хлорных каналов, в результате большее число отрицательно заряженных ионов хлора поступает внутрь нейрона, что приводит к гиперполяризации нейрональной мембраны и развитию тормозных процессов. Бензодиазепины используют при бессоннице, связанной с тревогой, стрессовой ситуацией, резкой сменой часовых поясов и характеризующейся трудностью засыпания, частыми ночными и/или ранними утренними пробуждениями. Их также применяют в анестезиологии для премедикации перед хирургическими операциями. Бензодиазепины различают по продолжительности действия на: ● препараты длительного действия: флунитразепам; ● препараты средней продолжительности действия: нитразепам; ● препараты короткого действия: триазолам, мидазолам. Препараты длительного действия и препараты средней продолжительности действия вызывают сон, продолжающийся 6–8 ч. Продолжительность действия некоторых препаратов (флуразепам, диазепам) связана с образованием активных метаболитов. При применении бензодиазепинов, особенно длительно действующих препаратов, возможны явления последействия в течение дня, реализующиеся в виде сонливости, вялости, замедления реакций. В связи с этим бензодиазепины не следует назначать пациентам, профессиональная деятельность которых требует быстроты ГАМК-рецептор Cl– ГАМК-рецептор Бензодиазепиновый рецептор + – Рис. 11.1. Механизм действия бензодиазепинов (пояснения в тексте) Глава 11. Снотворные средства 299 реакции и повышенного внимания. При повторных применениях происходит кумуляция вещества. Явления последействия менее характерны для препаратов короткого действия. Однако при резкой отмене кратковременно действующих препаратов чаще возникает феномен «отдачи». Для уменьшения этого эффекта бензодиазепины следует отменять постепенно. При повторном применении бензодиазепинов развивается привыкание, при этом для получения такого же снотворного эффекта необходимо увеличить дозу препарата. Возможно развитие лекарственной зависимости (как психической, так и физической). В случае развития физической зависимости синдром отмены протекает менее тягостно, чем при зависимости от барбитуратов. По выраженности снотворного эффекта бензодиазепины уступают барбитуратам, однако они обладают рядом преимуществ: ● в меньшей степени нарушают структуру сна; ● обладают большей широтой терапевтического действия (меньше опасность острого отравления); ● вызывают меньше побочных эффектов; ● вызывают менее выраженную индукцию микросомальных ферментов печени; ● к ним медленнее развиваются толерантность и лекарственная зависимость. Нитразепам наиболее широко применяют при бессоннице (рис. 11.2). Препарат выпускают в виде таблеток, назначают на ночь за 30–40 мин до сна. Действие после введения внутрь наступает через 30–60 мин и продолжается 6–8 ч (t½ — 24–36 ч). Кроме того, нитразепам применяют для премедикации перед хирургическими операциями и в связи с его противосудорожным действием при некоторых формах судорожных припадков (особенно у детей). Для нитразепама в связи с его значительной продолжительностью действия характерны явления последействия: слабость, сонливость, наруше- Рис. 11.2. Химические структуры нитразепама и флунитразепама 300 Часть II. Частная фармакология ние концентрации внимания, замедление психических и двигательных реакций. Нитразепам потенцирует действие алкоголя и других средств, угнетающих ЦНС, вызывает снижение АД, возможно угнетение дыхания. Встречаются парадоксальные реакции (особенно на фоне приема алкоголя) — повышенная агрессивность, острые состояния возбуждения со страхом, расстройства засыпания и сна. Нитразепам обладает способностью к кумуляции, при длительном применении к нему развивается привыкание. Противопоказания: гиперчувствительность к бензодиазепинам, миастения, закрытоугольная форма глаукомы, лекарственная зависимость, острые отравления средствами, угнетающими ЦНС (в том числе алкоголем), беременность и лактация. Флунитразепам — препарат длительного действия (см. рис. 11.2). Снотворный эффект развивается через 20–45 мин и продолжается 6–8 ч (при этом увеличивается глубина сна). Препарат метаболизируется в печени, выводится почками (t½ — 24–36 ч). Побочные эффекты такие же, как у нитразепама. Противопоказания: поражения печени и почек, миастения, беременность, грудное вскармливание. Не рекомендуют совместное использование с ингибиторами МАО. Триазолам — препарат короткого действия (t½ — 1–5 ч), при повторном применении кумулирует незначительно, последействие выражено в меньшей степени, чем у длительно действующих бензодиазепинов. Мидазолам — препарат короткого действия (t½ — 1–5 ч). В качестве снотворного средства назначают внутрь для облегчения засыпания. Препарат не кумулирует при повторных введениях, явления последействия выражены незначительно. Мидазолам в основном используют в анестезиологии для премедикации перед хирургическими операциями (вводят внутрь и внутримышечно) и введения в наркоз (вводят внутривенно). При внутривенном введении мидазолама возможно угнетение дыхания вплоть до его остановки (в особенности при быстром введении). Антагонист бензодиазепинов — флумазенил. По химической структуре — это имидазобензодиазепин, конкурентно блокирующий бензодиазепиновые рецепторы и устраняющий эффекты бензодиазепинов, в том числе снотворное и седативное действия (например, при выведении из наркоза). Восстанавливает дыхание и сознание при передозировке бензодиазепинов. Препарат вводят внутривенно. Препараты другой химической структуры Высокой гипногенной эффективностью обладают препараты, по химической структуре отличающиеся от бензодиазепинов, однако их снот- Глава 11. Снотворные средства 301 ворный эффект также связан со стимуляцией бензодиазепиновых рецепторов. При стимуляции бензодиазепиновых рецепторов происходит повышение чувствительности ГАМКА-рецепторов к ГАМК, увеличивается частота открытия хлорных каналов, повышается поступление в нервную клетку ионов хлора и возникает гиперполяризация мембраны. Это приводит к развитию тормозных процессов, проявляющихся в виде снотворного и седативного (в меньших дозах) эффектов. К препаратам этой группы относят залеплон, зопиклон и золпидем. Отличительная особенность этих препаратов — они в меньшей степени, чем бензодиазепины нарушают структуру сна. Залеплон — производное пиразолопиримидина, взаимодействует с местами связывания бензодиазепинов ГАМКА-рецепторов. Используют для лечения транзиторной бессонницы в течение 7–10 дней. Действие связано с влиянием на латентный период сна; t½ составляет 2 ч, что достаточно для обеспечения 8-часового сна. Зопиклон — производное циклопирролона, снотворное средство средней продолжительности действия. Эффект развивается через 20–30 мин и продолжается 6–8 ч. Стимулирует ГАМК-ергические механизмы синаптической передачи в головном мозге вследствие возбуждения ω1- и ω2бензодиазепиновых рецепторов. Не влияет на общую продолжительность «быстрого» сна. В настоящее время используют также эсзопиклон — S-изомер зопиклона. Побочные действия: ощущение горького и металлического вкуса во рту, тошнота, раздражительность, подавленное настроение, аллергические реакции, при пробуждении возможны головокружение и нарушение координации движений. Феномен «отдачи» выражен в незначительной степени. При длительном применении возникают привыкание и лекарственная зависимость, в связи с чем курс применения зопиклона не должен превышать 4 нед. Противопоказания: гиперчувствительность, декомпенсированная дыхательная недостаточность, возраст до 15 лет. Не рекомендуют применение при беременности и кормлении грудью. Золпидем — производное имидазопиридина, снотворное средство средней продолжительности действия (t½ — 2–3 ч). Агонист ω1бензодиазепиновых рецепторов. Мало влияет на структуру сна. Золпидем не оказывает выраженных анксиолитического, противосудорожного и миорелаксирующего действий. Среди побочных эффектов отмечают головную боль, сонливость в дневное время, кошмарные сновидения, галлюцинации, атаксию. Феномен «отдачи» выражен в незначительной степени. При длительном применении препарата развиваются привыкание 302 Часть II. Частная фармакология и лекарственная зависимость, в связи с чем курс применения золпидема не должен превышать 4 нед. Антагонист золпидема, залеплона и зопиклона — флумазенил. 11.1.2. Блокаторы Н1-рецепторов Блокаторы Н1-рецепторов, проникающие в ЦНС, обладают снотворными свойствами. Так, противоаллергический препарат дифенгидрамин (димедрол♠), блокирующий Н1-рецепторы, оказывает выраженное снотворное действие. Из этой группы препаратов в качестве только снотворного средства используют доксиламин. К положительным качествам этого препарата относят отсутствие влияния на структуру сна, низкую токсичность. 11.1.3. Агонисты мелатониновых рецепторов Мелатонин имеет важное значение в регуляции цикла сон–бодрствование. Рамелтеон — агонист МТ1- и МТ2-мелатониновых рецепторов, расположенных в головном мозге (стимуляция мелатонином MT1- и МТ2рецепторов супрахиазматического ядра регулирует цикл сон–бодрствование). В результате у больных с хронической бессонницей укорачивается латентный период сна. Рамелтеон не вызывает синдрома «отдачи». Из числа побочных эффектов отмечают сонливость, снижение концентрации тестостерона и увеличение содержания пролактина. 11.2. СНОТВОРНЫЕ СРЕДСТВА С НАРКОТИЧЕСКИМ ТИПОМ ДЕЙСТВИЯ Эти средства оказывают неизбирательное угнетающее действие на ЦНС. В небольших дозах они вызывают седативный эффект, при увеличении дозы проявляют снотворное действие, а в больших дозах могут вызвать наркоз. Снотворные средства наркотического типа действия в основном представлены производными барбитуровой кислоты. 11.2.1. Производные барбитуровой кислоты (барбитураты) Барбитураты обладают седативным, снотворным и противосудорожным свойствами. В больших дозах они вызывают состояние наркоза, поэтому некоторые барбитураты короткого действия (тиопентал натрия) применяют для неингаляционного наркоза. В меньших дозах барбитура- Глава 11. Снотворные средства 303 ты оказывают выраженное снотворное действие, способствуют засыпанию и увеличивают общую продолжительность сна. Седативный эффект (без снотворного) барбитураты оказывают в меньших дозах. Угнетающее действие барбитуратов обусловлено их взаимодействием со специфическими участками связывания (барбитуратными рецепторами), располагающимися на комплексе ГАМКА–рецепторхлорный канал. Участки связывания барбитуратов этого комплекса отличаются от мест связывания бензодиазепинов. При связывании барбитуратов с этим рецепторным комплексом происходит повышение чувствительности ГАМКА-рецептора к ГАМК. При этом увеличивается время открытия хлорных каналов, в результате больше ионов хлора поступает через мембрану нейрона в клетку, развивается гиперполяризация мембраны и происходит усиление тормозного эффекта ГАМК. Полагают, что действие барбитуратов не ограничивается их потенцирующим воздействием на ГАМКА-рецепторы. Эти вещества способны непосредственно стимулировать ГАМКА-рецепторы. Выраженное ГАМК-миметическое действие в большей степени характерно для средств для наркоза (например, тиопентала натрия). Кроме того, барбитураты проявляют антагонизм в отношении глутамата и, возможно, других возбуждающих медиаторов. Барбитураты в значительной степени изменяют структуру сна — уменьшают продолжительность быстрого (парадоксального) сна. Резкая отмена препаратов приводит к удлинению фазы быстрого сна, однако сновидения при этом носят характер ночных кошмаров (феномен «отдачи»). Барбитураты обладают небольшой терапевтической широтой действия, поэтому при их применении высока опасность развития токсических эффектов (возможно угнетение дыхательного центра). Для барбитуратов характерно последействие, проявляющееся сонливостью в течение дня, вялостью, нарушением внимания, психических и двигательных реакций. Эти явления можно наблюдать даже после однократного приема препарата. При повторных применениях барбитураты кумулируют, и явления последействия усиливаются. Длительное применение барбитуратов может привести к нарушению высшей нервной деятельности. Барбитураты (особенно фенобарбитал) индуцируют микросомальные ферменты печени, вследствие чего ускоряют метаболизм многих ЛВ. Повышается также скорость метаболизма самих барбитуратов, с чем связывают развитие толерантности при их длительном применении (может возникнуть через 2 нед после начала приема). Длительное применение барбитуратов может также привести к развитию лекарственной зависимости (при применении достаточно высоких доз лекарственная зависимость может развиться в течение 1–3 мес). При применении барбитуратов воз- 304 Часть II. Частная фармакология никает как психическая, так и физическая лекарственная зависимость, при этом отмена препарата сопровождается беспокойством, страхом, рвотой, судорогами, нарушениями зрения, ортостатической гипотензией, в тяжелых случаях возможен смертельный исход. В связи с неблагоприятными эффектами барбитураты в настоящее время имеют ограниченное применение. Производные барбитуровой кислоты, широко применявшиеся в прошлом в качестве снотворных препаратов, в настоящее время в основном исключены из Государственного реестра ЛС. Иногда в качестве снотворного средства используют препарат длительного действия фенобарбитал. Фенобарбитал — длительно действующий барбитурат, оказывающий снотворное, седативное и противоэпилептическое действия (рис. 11.3). В основном фенобарбитал применяют при эпилепсии (см. главу «Противоэпилептические средства»). В качестве снотворного средства фенобарбитал имеет ограниченное применение. В небольших количествах фенобарбитал входит в состав комбинированного препарата валокордин♠ и оказывает седативное действие. Фенобарбитал выводится из организма медленно (способен Рис. 11.3. Химическая структура фенобарбитала к кумуляции). Продолжительность действия препарата — 8 ч. Побочные эффекты: гипотония, аллергические реакции (кожная сыпь). Как и все барбитураты, фенобарбитал вызывает нарушение структуры сна. При применении фенобарбитала можно наблюдать выраженное последействие — общее угнетение, ощущение разбитости, сонливость, двигательные расстройства. Фенобарбитал вызывает выраженную индукцию микросомальных ферментов печени и поэтому ускоряет метаболизм ЛС, в том числе ускоряет метаболизм самого фенобарбитала. При повторных применениях препарат вызывает развитие толерантности и лекарственной зависимости. Циклобарбитал (гексобарбитал, фанодорм♠) — барбитурат короткой/ средней продолжительности действия. До появления бензодиазепинов препарат широко использовали как снотворное средство. Препарат действует 5–6 ч, t½ составляет 20–25 ч. Последействие по сравнению с фенобарбиталом выражено незначительно. В настоящее время используют в составе комбинированного препарата реладорм♠ (циклобарбитал + диазепам). При передозировке барбитуратов (препараты с малой широтой терапевтического действия) возникают явления острого отравления, связан- Глава 11. Снотворные средства 305 ные с общим угнетением ЦНС. В тяжелых случаях развивается коматозное состояние, подавляется рефлекторная активность, выключается сознание. В связи с угнетением центров продолговатого мозга (дыхательного и сосудодвигательного) снижается объем дыхания и АД, кроме того, барбирутаты обладают угнетающим действием на ганглии и прямым миотропным влиянием на сосуды. Смерть наступает от остановки дыхания. При лечении острых отравлений основные действия направлены на ускорение выведения препарата из организма и поддержание адекватного дыхания и кровообращения. Для предупреждения всасывания вещества из ЖКТ промывают желудок, дают солевые слабительные, адсорбирующие средства. Для удаления всосавшегося препарата применяют форсированный диурез (внутривенно вводят 1–2 л 0,9% раствора натрия хлорида и сильнодействующий диуретик — фуросемид или маннитол, что приводит к быстрому увеличению диуреза). Также полезно назначение щелочных растворов (рН почечного фильтрата сдвигается в щелочную сторону, что препятствует реабсорбции барбитуратов). При высоких концентрациях барбитуратов в крови применяют гемосорбцию, гемодиализ. Для стимуляции дыхания при легких формах отравления назначают аналептики (бемегрид, см. главу «Аналептики»), однако в тяжелых случаях они противопоказаны, так как могут только ухудшить состояние пациента, в таких случаях проводят искусственное дыхание. При гипотензии, развитии коллапса вводят кровезаменители, сосудосуживающие средства (норадреналин♠). 11.2.2. Алифатические соединения Хлоралгидрат относят к снотворным наркотического типа действия. Механизм действия связывают с образованием в процессе метаболизма трихлорэтанола, вызывающего снотворный эффект. Препарат мало влияет на структуру сна. Поскольку хлоралгидрат обладает выраженным раздражающим действием, его применяют в основном в лекарственных клизмах вместе со слизями. В качестве снотворного средства назначают редко. В настоящее время используют преимущественно в геронтологии. Иногда назначают для купирования психомоторного возбуждения. В качестве снотворного средства применяют также клометиазол, по химической структуре составляющий фрагмент тиамина (витамин В1). Препарат не обладает витаминными свойствами, а оказывает седативное, снотворное, миорелаксирующее и противосудорожное действия. Механизм действия клометиазола связывают с его способностью повышать чувствительность ГАМК-рецепторов к ГАМК, что, возможно, обуслов- 306 Часть II. Частная фармакология лено его взаимодействием с местами связывания барбитуратов. Препарат выпускают в капсулах и в виде порошка для приготовления инфузионного раствора. В качестве снотворного средства применяют внутрь перед сном при всех видах расстройств сна, состояниях возбуждения и беспокойства (особенно у пожилых больных). При алкогольных психозах, эклампсии препарат вводят внутривенно в виде инфузий. Побочные эффекты: аллергические реакции, расстройства дыхания, тахикардия, тошнота, рвота. Противопоказания: гиперчувствительность, острая дыхательная недостаточность, детский возраст. Вопросы и задания для самоконтроля 1. Снотворное действие золпидема обусловлено: а) блокадой NMDA рецепторов; б) блокадой потенциалозависимых натриевых каналов; в) нарушением обратного нейронального захвата ГАМК; г) стимуляцией бензодиазепиновых рецепторов; д) блокадой кальциевых каналов N-типа. 2. Укажите снотворное средство из группы барбитуратов: а) нитразепам; б) зопиклон; в) рамелтеон; г) хлоралгидрат; д) фенобарбитал. 3. Хлоралгидрат: а) выводится почками в неизмененном виде; б) незначительно нарушает структуру сна; в) обладает раздражающим действием; г) относят к бензодиазепинам; д) относят к снотворным ненаркотического типа действия. 4. К снотворным наркотического типа действия относят: а) рамелтеон; б) нитразепам; в) фенобарбитал; г) золпидем; д) клометиазол. Глава 12 ПРОТИВОЭПИЛЕПТИЧЕСКИЕ СРЕДСТВА Противоэпилептические средства применяют при эпилепсии для предупреждения или купирования эпилептических припадков (судорог или соответствующих им эквивалентов). Эпилепсия — хроническое заболевание, характеризующееся повторными относительно стереотипными припадками. Эпилептические припадки обусловлены возникновением патологических электрических разрядов в головном мозге. Поскольку эти разряды могут возникать в различных отделах коры и подкорковых структур, эпизоды судорожной активности нейронов головного мозга могут сопровождаться различными психическими, двигательными и вегетативными проявлениями (припадками). Выделяют следующие основные формы эпилепсии. ● Генерализованные припадки (на ЭЭГ регистрируют судорожные волны в обоих полушариях): —большие судорожные припадки (grand mal) — генерализованные тонико-клонические судороги с потерей сознания, заканчивающиеся общим расслаблением мышц. Если у больного большие припадки следуют один за другим более 5 мин без восстановления сознания, то такое состояние считают эпилептическим статусом; —малые судорожные припадки (petit mal, абсансы) — кратковременные эпизоды потери сознания (длительностью 5–10 с) с характерными изменениями на ЭЭГ; —миоклонус-эпилепсия — одиночные или повторяющиеся непроизвольные подергивания групп мышц (обычно кратковременные, без потери сознания). ● Парциальные (фокальные) судорожные припадки (изменения на ЭЭГ наблюдают в ограниченной области головного мозга) включают различные формы эпилептических припадков, возникающих с потерей или без потери сознания. ● Эпилептический статус — следующие один за другим судорожные припадки, при этом в интервале между припадками больной не приходит в сознание. 308 Часть II. Частная фармакология Патогенез развития эпилепсии недостаточно изучен. Важное значение в развитии эпилептогенной активности головного мозга придают нарушению баланса между тормозными (ГАМК, глицин) и возбуждающими (глутамат, аспартат) нейромедиаторами. Медиаторы возбужающего характера (аминокислоты) взаимодействуют с нейрональными рецепторами (NMDA и AMPA — альфа-амино-3-гидрокси-5-метил4-изоксазол-пропионовая кислота), связанными с быстрыми натриевыми каналами. Показано, что содержание глутамата в тканях мозга в области эпилептогенного очага повышено, а содержание ГАМК в тканях мозга больных эпилепсией понижено. Выделяют наследственные формы эпилепсии, сязанные с полиморфизмом потенциалозависимого натриевого канала. По механизму действия выделяют следующие группы противоэпилептических средств (рис. 12.1). ● Блокаторы потенциалозависимых натриевых каналов: —фенитоин; —карбамазепин; —ламотриджин. ● Средства, усиливающие тормозные эффекты в ЦНС (повышающие эффективность ГАМК-ергической системы): —барбитураты: ✧ фенобарбитал; ✧ бензобарбитал; ✧ примидон; —бензодиазепины: ✧ диазепам; ✧ клоназепам; ✧ лоразепам; —средства, влияющие на метаболизм ГАМК в ЦНС: ✧ вигабатрин; ✧ тиагабин; ✧ габапентин. ● Средства, угнетающие действие возбуждающих аминокислот: фелбамат. ● Средства блокирующие нейрональные низкопороговые кальциевые каналы (Т-типа): этосуксимид. ● Средства, обладающие комбинированным действием: —вальпроевая кислота; —топирамат. Этосуксимид Кальциевый канал (Т-тип) ГАМКрецептор ГАМК-Т Бензодиазепины Фенобарбитал Рис. 12.1. Механизм действия противоэпилептических средств (пояснения в тексте) Габапентин 1 Фелбамат Тиагабин 1 — потенциалзависимый кальциевый канал Вальпроевая кислота Потенциалзависимый Глицин натриевый канал Ламотриджин Карбамазепин Вигабатрин AMPAрецептор NMDAрецептор Фенитоин Глава 12. Противоэпилептические средства 309 310 Часть II. Частная фармакология 12.1. БЛОКАТОРЫ ПОТЕНЦИАЛОЗАВИСИМЫХ НАТРИЕВЫХ КАНАЛОВ Фенитоин (дифенин♠) относят к производным гидантоина (рис. 12.2). Это первый противосудорожный препарат, используемый в клинической практике с 1938 г. Оказывает выраженное противосудорожное действие, не вызывая снотворного или седативного эффектов. Наряду с этим препарат обладает антиаритмической активностью, особенно при нарушениях сердечного ритма, вызванных передозировкой сердечных гликозидов (см. гл. «Антиаритмические средства»), оказывает анальгетическое действие (при невралгии тройничного нерва). Механизм противосудорожного действия фенитоина связывают с блокадой потенциалозависимых натриевых каналов и, как следствие, — снижением степени деполяризации нейронов, обусловленной уменьшением поступления ионов натрия. Фенитоин связывается с натриевыми каналами, находящимися в инактивированном состоянии, и замедляет переход этих каналов в активную (открытую) форму. При этом степень связывания препарата с рецепторами прямо пропорционально зависит от степени деполяризации мембраны нейрона и частоты открываний натриевых каналов (это увеличивает вероятность связывания препарата с каналом в неактивном состоянии). Таким образом, фенитоин связвается с натриевыми каналами наиболее активных нейронов (эпилептический фокус). Этим объясняется отсутствие у фенитоина седативного действия. Блокада натриевых каналов препятствует генерации и распространению высокочастотных импульсов, а также снижает возбудимость нейронов и препятствует их активации при поступлении к ним импульсов из эпилептогенного очага. Зависимое от частоты возбуждения взаимодейстие называют use-dependent. Такое взаимодействие блокатора и рецептора характерно для некоторых антиаритмических средств и местных анестетиков. Фенитоин применяют для лечения различных форм эпилепсии (за исключением малых судорожных припадков), в частности для профилактики парциальных и больших судорожных припадков. Для предупреждения судорожных припадков фенитоин назначают внутрь в виде таблеток, для куРис. 12.2. Химическая структура пирования эпилептического статуса фенитоина вводят внутривенно. При приеме внутрь Глава 12. Противоэпилептические средства 311 скорость всасывания препарата из ЖКТ в значительной степени зависит от лекарственной формы, состава таблеток (размера частиц, вспомогательных веществ), при этом время достижения максимальной концентрации вещества в крови может варьировать от 3 до 12 ч. 90% фенитоина устойчиво связывается с белками плазмы крови. Препарат метаболизируется в печени, основной неактивный метаболит [5-(п-гидроксифенил)-5-фенилгидантоин] образует конъюгаты с глюкуроновой кислотой. Для фенитоина характерна кинетика «нулевого порядка», что обусловливает высокий риск передозировки препарата и возникновения токсических реакций. Этот тип кинетики характерен для насыщения ферментной системы субстратом. Фенитоин выводится из организма почками в основном в виде метаболитов, t½ варьирует от 12 до 36 ч в зависимости от концентрации препарата в плазме крови (большие значения t½ наблюдают при высоких концентрациях вещества в крови, что связано с насыщением ферментов печени, метаболизирующих фенитоин). Фенитоин вызывает многочисленные побочные эффекты: головокружение, возбуждение, тошноту, рвоту, тремор, нистагм, атаксию, диплопию, гирсутизм, гиперплазию десен (особенно у молодых людей), снижение уровня фолатов и мегалобластную анемию, остеомаляцию (связано с нарушением метаболизма витамина D), аллергические реакции и др. Отмечено тератогенное действие. Фенитоин вызывает индукцию микросомальных ферментов в печени и, таким образом, ускоряет метаболизм ряда ЛВ (глюкокортикоидов, эстрогенов, теофиллина), повышает их концентрацию в крови. Карбамазепин (тегретол♠, финлепсин♠) — производное иминостильбена, наряду с выраженным противоэпилептическим эффектом оказывает нормотимическое (улучшение настроения) и антидепрессивное действия (рис. 12.3). Кроме того, карбамазепин обладает выраженной анальгетической активностью. Противосудорожное действие препарата связано с блокадой потенциалозависимых натриевых каналов мембран нервных клеток. Карбамазепин уменьшает способность нейронов поддерживать высокочастотную импульсацию, типичную для эпилептогенной активности. Не исключено, что препарат может оказывать эффект пресинаптически, препятствуя высвобождению медиатора за счет блокады преси- Рис. 12.3. Химическая структура карбамазепина наптических натриевых каналов. 312 Часть II. Частная фармакология Карбамазепин — препарат выбора для предупреждения парциальных судорог и больших судорожных припадков. Его применяют для ослабления нейропатической боли, в частности при невралгии тройничного нерва (препарат выбора), а также для профилактики маниакально-депрессивных состояний. Карбамазепин при приеме внутрь почти полностью всасывается из ЖКТ в кровь. Скорость всасывания препарата индивидуальна и подвержена колебаниям. Максимальная концентрация в плазме после приема внутрь достигается в течение 4–5 ч. В грудном молоке концентрация вещества достигает 60% концентрации в плазме крови матери. Препарат метаболизируется в печени, повышает скорость собственного метаболизма за счет индукции микросомальных ферментов печени, выводится в основном почками (более 70%). Один из метаболитов (карбамазепин10,11-эпоксид) обладает противосудорожной, антидепрессивной и антиневралгической активностью. Карбамазепин вызывает многочисленные побочные эффекты: потерю аппетита, тошноту, головную боль, сонливость, атаксию, нарушение аккомодации, диплопию (двоение в глазах), нарушения сердечного ритма, гипонатриемию, гипокальциемию, гепатит, аллергические реакции, лейкопению, тромбоцитопению, агранулоцитоз (требуется контроль картины крови). Отмечают риск тератогенного воздействия. Применение препарата при беременности возможно только по жизненным показаниям. Поскольку карбамазепин угнетает психомоторные реакции, его не следует назначать лицам, деятельность которых требует повышенного внимания (например, водителям автотранспорта). Карбамазепин повышает скорость метаболизма, вследствие чего снижает в крови концентрацию некоторых ЛВ, в том числе противоэпилептических препаратов (клоназепама, ламотриджина, этосуксимида и др.). Ламотриджин (ламиктал ♠) блокирует потенциалозависимые натриевые каналы мембран нейронов, а также уменьшает выделение глутамата из пресинаптических окончаний (что связывают с блокадой натриевых каналов пресинаптических мембран). Ламотриджин применяют практически при всех формах эпилепсии: для предупреждения парциальных судорог, больших судорожных припадков, малых приступов эпилепсии. Препарат назначают для лечения эпилепсии, устойчивой к другим противоэпилептическим средствам, или в качестве дополнения к терапии другими препаратами. Побочные эффекты ламотриджина: сонливость, диплопия, головная боль, атаксия, тремор, тошнота, кожные высыпания. Глава 12. Противоэпилептические средства 313 12.2. СРЕДСТВА, ПОВЫШАЮЩИЕ ЭФФЕКТИВНОСТЬ ГАМК-ЕРГИЧЕСКОЙ СИСТЕМЫ К этим средствам относят ЛВ, усиливающие тормозной эффект ГАМК (барбитураты и бензодиазепины), и средства, влияющие на метаболизм ГАМК (тиагабин, вигабатрин). 12.2.1. Барбитураты Из числа барбитуратов в качестве противоэпилептических средств используют фенобарбитал, бензобарбитал и примидон. Фенобарбитал (люминал♠) в качестве противоэпилептического средства применяют достаточно давно (его противосудорожные свойства были обнаружены в 1910 г.). Помимо противоэпилептической активности фенобарбитал обладает седативными и снотворными свойствами (см. главу «Снотворные средства»). Механизм действия фенобарбитала связывают с усилением тормозного действия ГАМК в ЦНС. Фенобарбитал взаимодействует со специфическими местами связывания (барбитуратными рецепторами), находящимися на ГАМКА-рецепторном комплексе, и, вызывая аллостерические изменения ГАМКА-рецептора, повышает его чувствительность к нейромедиатору. При этом увеличивается поступление ионов хлора через мембрану нейрона в клетку, что приводит к гиперполяризации мембраны и снижению возбудимости нейронов эпилептогенного очага. Кроме того, предполагают, что фенобарбитал вызывает изменения проницаемости мембраны нейрона и для других ионов (натрия, калия, кальция), а также проявляет антагонизм в отношении глутамата. Фенобарбитал применяют для предупреждения больших судорожных припадков (тонико-клонических судорог) и парциальных судорог. Препарат назначают внутрь в таблетках и растворах (для детей). Для купирования эпилептического статуса фенобарбитал в виде натриевой соли вводят внутривенно. В качестве противоэпилептического средства фенобарбитал назначают в дозах, не вызывающих снотворный эффект, однако возможно развитие седативного эффекта. При приеме внутрь фенобарбитал всасывается полностью (относительно медленно). Максимальную его концентрацию в крови определяют через 1–2 ч после приема, при этом 50% препарата связывается с белками плазмы крови. Фенобарбитал медленно и равномерно распределяется в различных органах и тканях, метаболизируется в печени, его неактивный метаболит (4-оксифенобарбитал) выводится почками в виде глюкуронида, 314 Часть II. Частная фармакология около 25% препарата — в неизмененном виде (t½ у взрослых составляет 2–4 сут, у новорожденных — до 7 сут). При приеме препарата отмечают кумулятивный эффект. Побочные эффекты применения фенобарбитала: гипотония, аллергические реакции (кожная сыпь), ощущение разбитости, сонливость, депрессия, атаксия, тошнота, рвота. Фенобарбитал вызывает выраженную индукцию микросомальных ферментов печени и поэтому ускоряет метаболизм ЛВ (в том числе собственный метаболизм). При повторном применении вызывает развитие толерантности и лекарственной зависимости. Противопоказания к применению фенобарбитала — беременность, грудное вскармливание, гиперчувствительность к препарату, заболевания печени и почек, миастения. Бензобарбитал (бензонал♠) применяют для предупреждения больших судорожных припадков. Побочные эффекты: сонливость, слабость, головная боль, атаксия, нарушения речи. Примидон (гексамидин♠) — производное пиримидина, сходен с фенобарбиталом по химической структуре. В меньшей степени, чем фенобарбитал, вызывает сонливость, менее токсичен. Применяют для предупреждения больших судорожных припадков. Выделяют несколько форм эпилептического статуса, наибольшую опасность представляет генерализованный тонико-клонический эпилептический статус. Наиболее эффективными препаратами для купирования эпилептического статуса считают диазепам и фенитоин, вводимые внутривенно. 12.2.2. Бензодиазепины Бензодиазепины обладают выраженной противосудорожной активностью, анксиолитическими, седативными, снотворными и мышечнорасслабляющими свойствами (см. раздел «Анксиолитические средства», главу «Снотворные средства»). Мышечнорасслабляющий эффект обусловлен угнетением полисинаптических рефлексов на уровне спинного мозга. Противосудорожное действие бензодиазепинов объясняют повышением тормозных эффектов ГАМК. Стимуляция аллостерических бензодиазепиновых рецепторов приводит к повышению чувствительности ГАМКА-рецепторов к ГАМК, увеличению частоты открытия хлорных каналов, в результате большее число отрицательно заряженных ионов хлора поступает внутрь нейрона. Это приводит к гиперполяризации нейрональной мембраны и усилению тормозных эффектов (см. рис. 11.1). Глава 12. Противоэпилептические средства 315 В качестве противоэпилептического средства, эффективного практически при всех формах эпилепсии, используют клоназепам. При эпилептическом статусе в основном применяют диазепам и лоразепам (вводят внутривенно). Клоназепам (антелепсин ♠) — производное бензодиазепина (рис. 12.4). Противосудорожное действие клоназепама выражено сильнее, чем у других бензодиазепинов. Препарат применяют для предупреждения малых припадков эпилепсии, а также при парциальных судорогах, миоклонус-эпилепсии, вводят внутрь и парентерально. Препарат применяют также для купирования эпилептического статуса, вводят внутривенно. Среди побочных эффектов клоназепама от- Рис. 12.4. Химическая мечают повышенную утомляемость, голово- структура клоназепама кружение, нарушение координации движений, мышечную слабость, депрессивные состояния, нарушения мышления и поведения, аллергические реакции (кожную сыпь). При длительном применении у детей клоназепам может вызвать нарушения умственного и физического развития. При парентеральном введении возможны угнетение дыхания (вплоть до апноэ), гипотензия, брадикардия. Диазепам — препарат выбора при эпилептическом статусе. Препарат вводят внутривенно, иногда ректально. К недостаткам относят непродолжительный эффект, высокий риск угнетения дыхания, возникновение артериальной гипотензии. Лоразепам по эффективности купирования эпилептического статуса превосходит диазепам. Препарат также применяют для лечения парциальной эпилепсии в сочетании с другими противоэпилептическими препаратами. При парентеральном введении может вызвать гипотензию, угнетение дыхания, апноэ, остановку сердца. 12.2.3. Средства, влияющие на метаболизм ГАМК в центральной нервной системе Вигабатрин — структурный аналог ГАМК, необратимо ингибирует ГАМК-трансаминазу, участвующую в метаболизме ГАМК, увеличивает содержание ГАМК в головном мозге. Вигабатрин обладает широким спектром противосудорожного действия, его используют для профилактики больших судорожных припадков. Препарат эффективен также при парциальных припадках. 316 Часть II. Частная фармакология Тиагабин создан в результате направленного синтеза. Препарат блокирует транспортную систему, обеспечивающую обратный нейрональный захват ГАМК из синаптической щели. Тиагабин используют как вспомогательный препарат для профилактики генерализованных и парциальных припадков. Потенцирует действие барбитуратов, бензодиазепинов и этанола. Среди побочных эффектов выделяют седативное действие, сонливость, депрессию и психозы. Габапентин (нейронтин♠) был синтезирован как липофильный структурный аналог ГАМК для стимуляции ГАМКА-рецепторов в головном мозге, однако его противосудорожный эффект не связан с ГАМК-миметической активностью. Габапентин взаимодействует с α2δ-субъединицей стенки потенциалозависимого кальциевого канала N-типа. Эти кальциевые каналы расположены на пресинаптических окончаниях и при деполяризации обеспечивают поступление ионизированного кальция, стимулирующего выделение медиатора. За счет блокады кальциевых каналов уменьшается выделение медиаторов (в основном возбуждающих). Предполагают, что габапентин способствует также высвобождению ГАМК, не исключена возможность блокады транспортных систем возбуждающих аминокислот. Габапентин применяют в качестве дополнительного средства при лечении парциальных припадков. Препарат эффективен при нейропатических болях. Побочные эффекты габапентина: сонливость, головокружение, атаксия, тремор, головная боль. 12.3. СРЕДСТВА, УГНЕТАЮЩИЕ ДЕЙСТВИЕ ВОЗБУЖДАЮЩИХ АМИНОКИСЛОТ Фелбамат блокирует глутаматные NMDA-рецепторы. Действие обусловлено связыванием с NR2B субъединицей рецептора. Поскольку эта субъединица не является универсальным компонентом всех NMDA-рецепторов, фелбамат обладает определенной избирательностью действия и отличается по свойствам от других блокаторов этих рецепторов. Потенцирование противосудорожного действия других препаратов не сопровождается седативным эффектом. Серьезные побочные эффекты препарата (апластическая анемия и печеночная недостаточность) ограничивают его применение. 12.4. БЛОКАТОРЫ КАЛЬЦИЕВЫХ КАНАЛОВ Т-ТИПА Этосуксимид (суксилеп♠) относят к химическому классу сукцинимидов (производных янтарной кислоты). Препарат блокирует кальциевые каналы Т-типа в нейронах таламокортикальной области, участвующих в развитии Глава 12. Противоэпилептические средства 317 эпилептической активности. Этосуксимид — препарат выбора при лечении малых припадков эпилепсии (рис. 12.5). Препарат практически полностью всасывается при приеме внутрь из ЖКТ в кровь, биодоступность составляет около 100%, метаболизируется в печени. В основном выводится из организма Рис. 12.5. Химическая почками в виде метаболитов и около 20% вве- структура этосуксимида денной дозы — в неизмененном виде. Побочные эффекты: тошнота, рвота, дискинезия; головная боль, сонливость нарушения сна, снижение психической активности, состояние тревоги, аллергические реакции; редко — лейкопения, агранулоцитоз. Механизм действия некоторых противоэпилептических средств включает несколько компонентов (при этом не установлено, какой из них преобладает). В связи с этим данные препараты трудно отнести к одной из вышеприведенных групп. К таким препаратам относят вальпроевую кислоту, топирамат. 12.5. СРЕДСТВА, ОБЛАДАЮЩИЕ КОМБИНИРОВАННЫМ ДЕЙСТВИЕМ Вальпроевую кислоту (ацедипрол♠, апилепсин♠, депакин♠) применяют также в виде вальпроата натрия (рис. 12.6). Вальпроевая кислота блокирует натриевые каналы нейронов. В результате снижается возбудимость нейронов в эпилептогенном очаге. Кроме того, препарат увеличивает содержание ГАМК в тканях головного мозга, что связывают как с ингибированием фермента, метаболизирующего ГАМК (ГАМК-трансаминазы), так и с повышением активности фермента, участвующего в синтезе ГАМК (глутаматдекарбоксилаза). Вальпроевая кислота блокирует кальциевые каналы Т-типа. Вальпроевую кислоту применяют практически при всех формах эпилепсии — при генерализованных (малых и больших судорожных припадках, миоклонус-эпилепсии), а также при парциальных судорогах. Препарат хорошо всасывается при приеме внутрь, максимальная концентрация в плазме крови отмечается в среднем через 2 ч (зависит от лекарственной формы). Выводится главным образом почками в виде конъюгатов Рис. 12.6. Химическая струкс глюкуроновой кислотой или в виде про- тура вальпроевой кислоты 318 Часть II. Частная фармакология дуктов окисления. Среди побочных эффектов отмечают тошноту, рвоту, диарею, боли в желудке, атаксию, тремор, кожные аллергические реакции, диплопию, нистагм, анемию, тромбоцитопению, ухудшение свертываемости крови; сонливость. Препарат противопоказан при нарушении функции печени и поджелудочной железы. Топирамат (топамакс♠) обладает сложным, до конца не выясненным механизмом действия. По некоторым данным, он блокирует потенциалозависимые натриевые каналы, а также активирует взаимодействие ГАМК с ГАМКА-рецепторами. Не исключено, что топирамат снижает активность рецепторов возбуждающих аминокислот (предположительно, глутаматных рецепторов каинатного подтипа). Препарат используют как дополнительный при терапии парциальных и генерализованных тонико-клонических судорог в сочетании с другими противоэпилептическими средствами. Характерные побочные эффекты применения топирамата — сонливость, заторможенность, снижение аппетита (анорексия), диплопия, атаксия, тремор, тошнота. Противоэпилептические средства можно классифицировать в соответствии с показаниями к их применению при различных формах эпилепсии и типах эпилептических припадков: ● средства для предупреждения больших судорожных припадков: карбамазепин, вальпроевая кислота, фенитоин, ламотриджин, фенобарбитал, топирамат, вигабатрин, примидон, бензобарбитал; ● средства для предупреждения малых припадков эпилепсии: этосуксимид, вальпроевая кислота, клоназепам, ламотриджин; ● средства для предупреждения миоклонус-эпилепсии: вальпроевая кислота, клоназепам, ламотриджин; ● средства для предупреждения парциальных судорог (фокальных припадков): карбамазепин, фенитоин, вальпроевая кислота, фенобарбитал, клоназепам, ламотриджин, топирамат, габапентин, вигабатрин; ● средства для купирования эпилептического статуса: диазепам, лоразепам, клоназепам, фенитоин, фенобарбитал. Вопросы и задания для самоконтроля 1. Эффективность ГАМК-ергической передачи повышают: а) фенитоин; б) фенобарбитал; в) клоназепам; г) карбамазепин; д) этосуксимид. Глава 12. Противоэпилептические средства 319 2. Укажите противоэпилептические средства — блокаторы натриевых каналов: а) топиромат; б) этосуксимид; в) фенобарбитал; г) карбамазепин; д) фенитоин. 3. Препарат выбора для лечения малых судорожных припадков: а) фенобарбитал; б) фенитоин; в) этосуксимид; г) карбамазепин; д) топирамат. 4. Для купирования эпилептического статуса применяют: а) диазепам; б) фенитоин; в) этосуксимид; г) вигабатрин; д) фенобарбитал. 5. Для фенитоина характерно: а) блокада NMDA рецепторов; б) отсутствие седативного эффекта; в) тератогенное действие; г) кинетика первого порядка; д) гиперпластический гингивит. Глава 13 ПРОТИВОПАРКИНСОНИЧЕСКИЕ СРЕДСТВА Противопаркинсонические средства — ЛС, применяемые для лечения болезни Паркинсона, синдрома паркинсонизма, включая лекарственный паркинсонизм. Болезнь Паркинсона (дрожательный паралич) — хроническое нейродегенеративное заболевание, обусловленное поражением пигментсодержащих дофаминергических нейронов черной субстанции головного мозга. Основные клинические проявления болезни: ● брадикинезия (скованность движений); ● ригидность (повышенный тонус скелетных мышц); ● тремор (дрожание) рук, головы. Симптоматика заболевания возникает при гибели 70–80% дофаминергических нейронов головного мозга. Болезнь Паркинсона (впервые описана английским врачом Джеймсом Паркинсоном в 1817 г.) диагностируют у 1% населения в возрасте до 60 лет и у 5% — старше 60 лет. Возможная причина заболевания — образование свободных кислородных радикалов при окислительном метаболизме дофамина в черной субстанции мозга, приводящих к разрушению дофаминергических нейронов. Некоторые данные свидетельствуют о генетической предрасположенности к болезни Паркинсона. Причинами синдрома паркинсонизма, также проявляющегося дрожательным параличом, могут быть инфекционные заболевания нервной системы (вирусный энцефалит), сосудистые заболевания головного мозга, травмы черепа, интоксикации (оксидом углерода, марганцем, этанолом), сифилитические поражения мозга. Лекарственный паркинсонизм — форма синдрома паркинсонизма, развивающаяся при длительном применении некоторых ЛС, например типичных нейролептиков (фенотиазины, бутирофеноны). Паркинсонизм относят к экстрапирамидным расстройствам. Экстрапирамидная система, в частности хвостатое ядро и скорлупа, объединяемые в неостриатум, осуществляет корректировку и уточнение движений. В регуляции моторных зон коры головного мозга участвует таламус. До- Глава 13. Противопаркинсонические средства 321 фаминергические нейроны черной субстанции подавляют активность холинергических нейронов неостриатума, контролирующих функции таламуса. Аксоны дофаминергических нейронов выделяют дофамин, который, стимулируя D2-рецепторы холинергических нейронов неостриатума, оказывает на них тормозное влияние (рис. 13.1). При болезни Паркинсона количество дофаминергических нейронов уменьшается, что приводит к усилению холинергических влияний в неостриатуме и возникновению характерных двигательных нарушений. По некоторым данным, активность холинергических нейронов контролируется глутаматергическими нейронами. Аксоны этих нейронов образуют синапсы в неостриатуме и выделяют глутамат. Глутамат стимулирует NMDA-рецепторы холинергических нейронов и таким образом повышает их активность, что приводит к увеличению секреции ацетилхолина в неостриатуме. Рис. 13.1. Локализация действия противопаркинсонических средств: A — дофаминергическое пресинаптическое окончание; Б — постсинаптические дофаминовые D2-рецепторы (D2-P); МАО-В — моноаминоксидаза В 322 Часть II. Частная фармакология При болезни Паркинсона в связи с уменьшением тормозных дофаминергических влияний в неостриатуме начинают преобладать стимулирующие глутаматергические влияния. Это приводит к большему повышению активности холинергических нейронов неостриатума и развитию характерных симптомов заболевания. Для снятия этих симптомов необходимо восстановить нарушенный баланс между указанными медиаторными системами в неостриатуме: обеспечить повышение дофаминергических влияний, снижение влияния глутаматергических нейронов, уменьшение влияния холинергических нейронов. В настоящее время при лечении болезни Паркинсона используют следующие противопаркинсонические средства. ● Средства, стимулирующие дофаминергическую систему головного мозга: —предшественник дофамина: леводопа; —ингибиторы МАО-B: селегилин (элдеприл♠, юмекс♠); —средства, повышающие выделение дофамина: амантадин (мидантан♠); —средства, стимулирующие дофаминовые рецепторы (агонисты дофаминовых рецепторов): бромокриптин (парлодел♠), перголид (пермакс♠), прамипексол (мирапекс♠). ● Средства, угнетающие холинергическую систему головного мозга: ♠ —центральные холиноблокаторы: тригексифенидил (циклодол ), бипериден (акинетон♠). 13.1. СРЕДСТВА, СТИМУЛИРУЮЩИЕ ДОФАМИНЕРГИЧЕСКУЮ ПЕРЕДАЧУ Леводопа — левовращающий изомер ДОФА (L-ДОФА), непосредственный предшественник дофамина, не проникающего через ГЭБ. Леводопа проникает через ГЭБ путем активного транспорта (с помощью транспортной системы, переносящей через эндотелий капилляров мозга ароматические аминокислоты). В неповрежденных дофаминергических нейронах леводопа под влиянием ДОФА-декарбоксилазы превращается в дофамин, который выделяется из окончаний дофаминергических волокон и стимулирует D 2-рецепторы на холинергических нейронах неостриатума (см. рис. 13.1). В результате снижение активности этих нейронов приводит к купированию симптомов паркинсонизма. Леводопа — наиболее эффективное противопаркинсоническое средство: оно уменьшает брадикинезию, мышечную ригидность и в меньшей степени тремор. Глава 13. Противопаркинсонические средства 323 Леводопа всасывается из кишечника путем активного транспорта, используя систему трансмембранного транспорта для ароматических аминокислот. Вследствие этого пища, богатая белками, снижает всасывание леводопы. В стенке кишечника и печени леводопа подвергается интенсивному метаболизму под действием ДОФА-декарбоксилазы: более 90% введенного препарата превращается в дофамин. Образовавшийся в периферических тканях дофамин не проникает в мозг и вызывает ряд побочных эффектов. Часть леводопы метаболизируется с участием МАО и катехол-орто-метилтрансферазы, в результате только 1–3% леводопы поступает в ЦНС. Для предотвращения декарбоксилирования леводопы в периферических тканях используют ингибиторы ДОФА-декарбоксилазы, не проникающие через ГЭБ, — карбидопу и бенсеразид. При этом большее количество леводопы (около 10%) поступает в ткани головного мозга для реализации своего действия. Это позволяет снизить назначаемую дозу препарата. Кроме того, снижение образования дофамина в периферических тканях уменьшает периферические побочные эффекты леводопы. В настоящее время леводопа выпускается в комбинации с блокаторами периферической ДОФА-декарбоксилазы и входит в состав комбинированных препаратов наком♠, синемет♠, дуэллин♠ (леводопа и карбидопа) и мадопар♠ (леводопа и бенсеразид). Показания к назначению препаратов с содержанием леводопы — болезнь Паркинсона и паркинсонизм (за исключением лекарственного паркинсонизма). Препараты принимают внутрь только после еды. Леводопа хорошо всасывается из ЖКТ. Действие развивается медленно (через 1 нед) и достигает максимума через 1 мес. Лечение леводопой проводят длительно. При применении леводопы возможно возникновение ортостатической гипотензии, тахикардии, сердечной аритмии, связанной с образованием дофамина в периферических тканях и действием дофамина на сосуды и сердце. Кроме того, частые побочные эффекты — тошнота и рвота, обусловленные стимуляцией D2-рецепторов рвотного центра дофамином. Эти эффекты уменьшаются при одновременном применении с леводопой ингибиторов периферической ДОФА-декарбоксилазы. Для профилактики тошноты и рвоты назначают блокатор дофаминовых рецепторов домперидон. Этот препарат не проникает через ГЭБ, однако воздействует на пусковую зону рвотного центра (пусковая зона рвотного центра не защищена ГЭБ). При этом домперидон не блокирует дофаминовые рецепторы в неостриатуме и поэтому не снижает эффективность леводопы. 324 Часть II. Частная фармакология Такие побочные эффекты, как психозы, галлюцинации, бессонница, тревожность, депрессия, нарушение координации движений, связаны с действием дофамина на ЦНС. Для устранения психозов применяют блокатор дофаминовых рецепторов центрального действия клозапин (атипичный нейролептик, в большей степени блокирующий D4-, чем D2-рецепторы, в ЦНС и поэтому в меньшей степени, чем типичные нейролептики, влияет на эффективность леводопы). У некоторых больных при длительном применении леводопы наблюдают дискинезии (непроизвольные хореиформные движения лица, шеи, конечностей). При длительном применении наблюдают синдром «включения/выключения» (внезапное резкое усиление симптомов болезни, а при снижении дозы препарата — резкое уменьшение эффективности). Последующее незначительное увеличение дозы снова приводит к появлению симптомов передозировки. Для уменьшения выраженности этого синдрома используют комбинированные препараты леводопы (леводопа + бенсеразид и леводопа + карбидопа) с замедленным высвобождением действующих веществ (мадопар♠, синемет♠). Противопоказания к применению леводопы: закрытоугольная форма глаукомы, сахарный диабет, психозы, нарушения сердечного ритма, функции печени и почек, беременность, лактация, возраст до 25 лет. При длительном (более 5 лет) приеме леводопы эффективность препарата снижается, что связано с прогрессирующей дегенерацией дофаминергических нейронов. Учитывая, что нейротоксическое действие на дофаминергические нейроны могут оказывать метаболиты дофамина, весьма актуальной задачей следует считать повышение эффективности имеющегося в мозге дофамина. Для повышения эффективности леводопы помимо ингибиторов ДОФА-декарбоксилазы одновременно с леводопой назначают ингибиторы катехол-О-метилтрансферазы — энтакапон (комтан♠). Этот препарат не проникает через ГЭБ и снижает скорость метаболизма леводопы в периферических тканях, особенно в условиях компенсаторного повышения активности периферической катехол-О-метилтрансферазы. В итоге поступление леводопы в ЦНС повышается. Кроме того, метаболиты леводопы, образующиеся при разрушении вещества, конкурируют с ней за транспортные системы ГЭБ. Блокада катехол-О-метилтрансферазы позволяет снизить дозу леводопы. Полагают, что применение ингибиторов катехол-О-метилтрансферазы уменьшает выраженность синдрома «включения/выключения». Толкапон, подобно энтакапону, блокирует катехол-О-метилтрансферазу, однако проникает в мозг, уменьшая метаболизм дофамина в ЦНС. Глава 13. Противопаркинсонические средства 325 Для повышения эффективности проводимого лечения леводопу обычно сочетают с другими противопаркинсоническими средствами, улучшающими дофаминергическую передачу и способствующими сохранению эндогенного дофамина. К ним относят селегилин (ингибитор МАО-В), амантадин (препарат, высвобождающий дофамин из пресинаптических окончаний) и агонисты дофаминовых рецепторов. Селегилин — селективный ингибитор МАО-В (фермента, преимущественно инактивирующего дофамин). Селегилин, угнетая МАО-В, уменьшает разрушение дофамина в нейронах черной субстанции и потенцирует действие леводопы. Это позволяет уменьшить дозу леводопы в среднем на 30%. Селегилин назначают в сочетании с леводопой. В качестве монотерапии препарат используют только на ранних стадиях болезни. Полагают, что селегилин обладает нейропротекторным действием, задерживая разрушение дофаминергических нейронов мозга. Этот эффект объясняют тем, что, угнетая окислительный метаболизм дофамина, селегелин уменьшает образование свободных кислородных радикалов, вызывающих гибель дофаминергических нейронов. Побочные эффекты применения селегилина: тошнота, рвота, артериальная гипотензия, нарушение концентрации внимания и спутанность сознания. Амантадин усиливает высвобождение дофамина из неповрежденных нейронов в синаптическую щель и нарушает обратный нейрональный захват дофамина (см. рис. 13.1). Повышение концентрации дофамина в ЦНС приводит к уменьшению выраженности симптомов болезни Паркинсона. Не исключено взаимодействие амантадина с NMDAрецепторами холинергических нейронов базальных ядер. Полагают, что амантадин, блокируя эти рецепторы, препятствует стимулирующему действию глутамата на холинергические нейроны. Это приводит к угнетению холинергической передачи в неостриатуме. Полагают также, что амантадин обладает некоторой антихолинергической активностью. Возможен и нейропротекторный эффект амантадина в отношении дофаминергических нейронов черной субстанции. Это связывают с блокадой NMDA-рецепторов, локализованных на этих нейронах, и уменьшением поступления ионов кальция в нервные клетки, препятствующим их разрушению. Амантадин оказывает умеренный противопаркинсонический эффект. Его применяют при болезни Паркинсона и паркинсонизме (за исключением лекарственного паркинсонизма), особенно при противопоказаниях к леводопе. Амантадин назначают также в сочетании с леводопой. К препарату быстро развивается привыкание. 326 Часть II. Частная фармакология К побочным эффектам амантадина относят возбуждение, раздражительность, бессонницу, головокружение, ортостатическую гипотензию, судороги. В отличие от леводопы агонисты дофаминовых рецепторов непосредственно возбуждают дофаминовые рецепторы в неостриатуме (см. рис. 13.1). Неселективные агонисты D1- и D2-рецепторов — перголид, бромокриптин, прамипексол. Препараты этой группы проявляют большую активность в отношении дофаминовых D2-рецепторов (прамипексол стимулирует D2- и D3-рецепторы). Перголид и бромокриптин относят к производным алкалоидов спорыньи. Бромокриптин — полусинтетическое производное алкалоида спорыньи эргокриптина. Противопаркинсоническая активность бромокриптина связана со стимуляцией D2-рецепторов неостриатума. Кроме того, бромокриптин, стимулируя D2-рецепторы, уменьшает высвобождение пролактина из передней доли гипофиза (см. раздел «Препараты гормонов гипоталамуса и гипофиза»). Бромокриптин и перголид в основном применяют в сочетании с леводопой, когда не удается получить удовлетворительных результатов, а также в случае возникновения синдрома «включения/выключения». Прамипексол по эффективности превосходит бромокриптин. Препараты назначают как в виде монотерапии, так и в сочетании с леводопой. По продолжительности действия агонисты дофаминовых рецепторов превосходят леводопу. В начале применения агонистов дофаминовых рецепторов возможны побочные эффекты: ортостатическая гипотензия, тошнота, рвота (вследствие стимуляции D2-рецепторов пусковой зоны рвотного центра). Эти проявления можно предупредить, назначая периферический блокатор дофаминовых рецепторов домперидон. При длительном применении возможно развитие галлюцинаций, психозов, дискинезии. При применении прамипексола характерны повышенная сонливость и внезапное засыпание. 13.2. СРЕДСТВА, УГНЕТАЮЩИЕ ХОЛИНЕРГИЧЕСКУЮ ПЕРЕДАЧУ Тригексифенидил — центральный холиноблокатор, блокирует М1холинорецепторы неостриатума, уменьшая симптомы паркинсонизма. Препарат оказывает умеренное противопаркинсоническое действие — преимущественно уменьшает тремор и мышечную ригидность, мало влияя на брадикинезию. Глава 13. Противопаркинсонические средства 327 Бипериден — центральный холиноблокатор, близкий по свойствам тригексифенидилу. Показания к назначению центральных холиноблокаторов: начальные стадии болезни Паркинсона и паркинсонизм. Их также применяют при лекарственном паркинсонизме, в частности назначают с целью профилактики и купирования экстрапирамидных расстройств, вызванных антипсихотическими средствами. Среди побочных эффектов, связанных с блокадой периферических М-холинорецепторов, отмечают нарушение аккомодации, сухость во рту, сердцебиение, запоры, задержку мочеиспускания. Побочные эффекты, связанные с блокадой центральных М-холинорецепторов, — нарушение памяти и концентрации внимания (особенно у пожилых пациентов). При передозировке биперидена возможны возбуждение, галлюцинации. Противопоказания к применению биперидена: глаукома, гипертрофия предстательной железы, гиперчувствительность к препарату, кормление грудью. Вопросы и задания для самоконтроля 1. К противопаркинсоническим средствам относят: а) карбамазепин; б) карбидопу; в) тригексифенидил; г) бромокриптин; д) амантадин. 2. Противопаркинсоническое действие леводопы обусловлено: а) холиноблокирующим действием; б) блокадой NMDA-рецепторов; в) блокадой МАО-В; г) стимуляцией дофаминергической передачи; д) правильных ответов нет. 3. Леводопу комбинируют с карбидопой с целью: а) получения аддитивного эффекта; б) блокады декарбоксилазы ароматических аминокислот в ЦНС; в) блокады декарбоксилазы ароматических аминокислот в периферических тканях; г) стимуляции декарбоксилазы ароматических аминокислот в ЦНС; д) предотвращения метаболизма леводопы МАО-В. 328 4. Побочные эффекты леводопы: а) сонливость; б) тахикардия; в) рвота; г) сердечные аритмии; д) психозы. 5. Агонист дофаминовых рецепторов: а) леводопа; б) карбидопа; в) бромокриптин; г) селегилин; д) тригексифенидил. Часть II. Частная фармакология Глава 14 АНАЛЬГЕЗИРУЮЩИЕ СРЕДСТВА (АНАЛЬГЕТИКИ) Анальгетики — ЛС, основной эффект которых представлен избирательным уменьшением или устранением болевой чувствительности (анальгезия) в результате резорбтивного действия ЛВ. В терапевтических дозах препараты не вызывают потерю сознания, не угнетают другие виды чувствительности (температурную, тактильную и др.) и не нарушают двигательных функций. Этим они отличаются от средств для наркоза, которые устраняют ощущение боли, но при этом выключают сознание и другие виды чувствительности, а также от местных анестетиков, которые неизбирательно угнетают все виды чувствительности. Боль — сложная защитная реакция. Болевые ощущения воспринимаются специальными рецепторами (ноцицепторами), которые расположены в коже, мышцах, капсулах суставов и внутренних органов, надкостнице и могут стимулироваться механическими, термическими и химическими раздражителями. В развитии болевой перцепции важное значение имеют ванилоидные рецепторы, возбуждаемые брадикинином, протонами, АТФ, ванилоидами (капсаицином острого перца). Эндогенные соединения (брадикинин, гистамин, серотонин, простагландины) могут сенсибилизировать эти рецепторы к внешним раздражителям, а также непосредственно вызывать боль (например, при воспалении). Ноцицептивные импульсы распространяются по С- и Аδ-волокнам афферентных нервов и поступают в ЦНС к нейронам задних рогов спинного мозга. Здесь через систему вставочных нейронов возбуждение направляется по 3 путям. ● В передние рога спинного мозга на двигательные мотонейроны. Их возбуждение проявляется быстрым защитным двигательным рефлексом со стороны скелетных мышц. ● В боковые рога спинного мозга на вегетативные нейроны симпатического отдела нервной системы, стимуляция которой приводит 330 Часть II. Частная фармакология к функциональной адаптации внутренних органов (например, повышение АД). ● В головной мозг по восходящим афферентным трактам к высшим структурам восприятия и оценки боли — стволу головного мозга, ретикулярной формации, таламусу, лимбической системе, коре головного мозга. Очевидно, что нейроны задних рогов спинного мозга имеют ключевое значение в восприятии и оценке болевой информации. Активность этих нейронов находится под контролем супраспинальной антиноцицептивной системы (так называемый контроль афферентного входа). В подкорковых структурах головного мозга (околоводопроводное серое вещество, большое ядро шва, голубое пятно) расположены нейроны, аксоны которых образуют нисходящие тормозные пути, заканчивающиеся на нейронах задних рогов спинного мозга. Активация нисходящей тормозной системы приводит к уменьшению выделения «ноцицептивных» медиаторов (субстанция Р, глутамат) и снижению активации вставочных нейронов, передающих информацию о боли. Таким образом, активация супраспинальной антиноцицептивной системы вызывает торможение проведения болевых импульсов по афферентным путям спинного мозга, что приводит к повышению порога болевой чувствительности. Иммунохимический анализ показал, что на нейронах околоводопроводного серого вещества, большого ядра шва и задних рогов спинного мозга находятся так называемые опиоидные рецепторы — специфические места связывания, с которыми взаимодействуют эндогенные анальгетические пептиды: энкефалины (содержат 5 аминокислот), динорфины (содержат 17 аминокислот) и эндорфины (содержат 31 аминокислоту). Выделяют несколько подтипов опиоидных рецепторов, различающиеся по чувствительности к вышеперечисленным эндогенным лигандам и эффектам, вызываемым активацией этих рецепторов. ● μ-Рецепторы активируются β-эндорфином, при возбуждении этих рецепторов развиваются анальгезия, седативный (успокаивающий) эффект, угнетение дыхательного центра, эйфория (положительные эмоции, повышенное настроение, ощущение душевного комфорта, не связанные с реальной действительностью) и лекарственная зависимость, брадикардия, миоз, снижение моторики ЖКТ. ● δ-Рецепторы активируются мет-энкефалином и лей-энкефалином, при стимуляции этих рецепторов развиваются анальгезия, угнетение дыхания, снижение моторики ЖКТ. Глава 14. Анальгезирующие средства (анальгетики) 331 ● κ-Рецепторы, эндогенными лигандами которых являются динорфины. Стимуляция этих рецепторов сопровождается угнетением проведения болевых импульсов на уровне спинного мозга (спинальная анальгезия), развиваются седативный эффект, миоз. Для агонистов κ-рецепторов характерна дисфория (отрицательные эмоции, ощущение дискомфорта), возможно развитие физической зависимости, возникает небольшое снижение моторики ЖКТ. Опиоидные рецепторы связаны с G-белками, стимуляция которых вызывает угнетение активности аденилатциклазы и снижение концентрации цАМФ в клетке. Кроме того, опиоиды открывают связанные с G-белками калиевые каналы, при этом повышается выход ионов калия из клетки, что приводит к гиперполяризации постсинаптической мембраны нейрона и, как следствие, снижает его возбудимость. В пресинаптических окончаниях нейронов опиоиды способны угнетать за счет блокады кальциевых каналов N-типа вход ионов кальция в клетку и уменьшать выделение медиаторов в синаптическую щель. Таким образом, из окончаний первичных афферентов в задних рогах спинного мозга уменьшается выделение «ноцицептивных» медиаторов (медиаторов боли) — глутамата, нейрокининов, вещества Р и снижается активирующее воздействие на вставочные нейроны, участвующие в передаче болевых импульсов в высшие центры. Кроме того, гиперполяризация мембран вставочных нейронов приводит к угнетению их активности. Нисходящие тормозные пути антиноцицептивной системы образованы аксонами норадренергических и серотонинергических и пуринергических нейронов. В развитии анальгетического действия определенную роль играют каннабиноидные рецепторы. По механизму и локализации действия выделяют несколько групп анальгезирующих средств. ● Анальгезирующие средства преимущественно центрального действия. —Опиоидные (наркотические) анальгетики: ✧ агонисты; ✧ частичные агонисты; ✧ агонисты-антагонисты. —Неопиоидные препараты с анальгетической активностью. —Анальгетики смешанного действия (опиоидный и неопиоидный компоненты). ● Анальгезирующие средства преимущественно периферического действия. 332 Часть II. Частная фармакология 14.1. СРЕДСТВА ПРЕИМУЩЕСТВЕННО ЦЕНТРАЛЬНОГО ДЕЙСТВИЯ 14.1.1. Опиоидные (наркотические) анальгетики Агонисты опиоидных рецепторов антиноцицептивной системы оказывают обезболивающее действие без утраты сознания или погружения в сон и угнетения других видов чувствительности. Основные механизмы анальгетического действия опиоидных анальгетиков реализуются на уровне спинного мозга, подкорковых структур и коры головного мозга: ● угнетение проведения болевых импульсов в афферентных путях ЦНС (нарушение передачи импульсов с окончаний первичных афферентов на вставочные нейроны спинного мозга); ● усиление тормозного влияния нисходящей антиноцицептивной системы на проведение болевых импульсов в афферентных путях ЦНС; ● изменение эмоциональной оценки боли. Действие наркотических анальгетиков опосредуется через опиоидные рецепторы. В результате блокирования кальциевых каналов N-типа в пресинаптических окончаниях аксонов первичных афферентов уменьшается выделение субстанции P и глутамата, при этом нарушается передача болевых импульсов на вставочные нейроны задних рогов спинного мозга. Вследствие стимуляции постсинаптических опиоидных рецепторов нарушается процесс деполяризации постсинаптической мембраны и угнетается активация вставочных нейронов под действием медиатора. Все это приводит к нарушению передачи болевых импульсов на уровне спинного мозга (спинальное действие) (рис. 14.1). При стимуляции опиоидных рецепторов в сером околоводопроводном веществе и некоторых других отделах ствола мозга за счет угнетения ингибирующих ГАМК-ергических нейронов происходит активация нисходящей антиноцицептивной системы, оказывающей тормозное влияние на передачу болевых импульсов по афферентным путям спинного мозга (супраспинальное действие). Нисходящие тормозные влияния на уровне спинного мозга осуществляются при участии серотонина и норадреналина (см. рис. 14.1). В результате действия наркотических анальгетиков на высшие отделы ЦНС изменяется эмоциональная оценка боли, снижается ее восприятие (даже если чувство боли сохраняется, оно меньше беспокоит больного). Рис. 14.1. Механизм действия наркотических анальгетиков: НА — норадреналин, 5-НТ — серотонин; ● — опиоидный рецептор; ЭНК — энкефалин Глава 14. Анальгезирующие средства (анальгетики) 333 334 Часть II. Частная фармакология Среди веществ, стимулирующих опиоидные рецепторы, выделяют: ● полные агонисты опиоидных рецепторов (вещества, способные вызвать максимальный для данной системы эффект); ● частичные агонисты опиоидных рецепторов (вещества, всегда вызывающие эффект, меньший максимального); ● агонисты-антагонисты опиоидных рецепторов (стимулирующие рецепторы одного подтипа и блокирующие рецепторы другого подтипа). К полным агонистам опиоидных рецепторов относят: анальгетики (опиаты): морфин (морфина ● природные наркотические гидрохлорид, долтард♠, морфилонг♠), омнопон♠ кодеин + морфин + носкапин + папаверин + тебаин, пантопон♠), кодеин; наркотические анальгетики: тримеперидин (проме● синтетические дол♠), фентанил, ремифентанил, метадон. К частичным агонистам и агонистам-антагонистам опиоидных рецепторов относят пентазоцин (фортрал♠), буторфанол (бефорал♠, стадол♠), налбуфин (нубаин♠), бупренорфин. Полные агонисты опиоидных рецепторов Природные наркотические анальгетики (опиаты) Источник получения морфина и других природных наркотических анальгетиков — опий (от греч. opos — млечный сок растения). Опий — высохший на воздухе млечный сок, который получают из надрезов на незрелых коробочках мака снотворного (Papaver somniferum). Опий использовали в качестве болеутоляющего средства более 6000 лет назад в Древнем Египте, Греции и Риме. Его способность вызывать физическую и психическую зависимость (пристрастие) стала являться предметом озабоченности начиная с XVIII в. Опий содержит более 20 алкалоидов. Алкалоиды фенантренового ряда (морфина гидрохлорид, кодеин) обладают анальгезирующей и противокашлевой активностью; алкалоиды изохинолинового ряда — не анальгетики, обладают спазмолитическим эффектом (папаверин). Морфина гидрохлорид — производное фенантрена, основной алкалоид опия (10% от общей массы). Морфина гидрохлорид был выделен из опия в 1806 г. немецким ученым Сертюрнером, который назвал его по имени бога сна Морфея (Morpheus). Химическая структура морфина гидрохлорида была установлена в 1925 г., а в 1952 г. был осуществлен его синтез, однако в промышленных масштабах более целесообразным оказалось его получение из растительного сырья. Глава 14. Анальгезирующие средства (анальгетики) 335 Действие морфина (рис. 14.2) на организм связано с возбуждением опиоидных рецепторов, расположенных как в ЦНС, так и в периферических тканях. Рис. 14.2. Химические структуры морфина и кодеина Эффекты, вызываемые возбуждением центральных опиоидных рецепторов ● Анальгезия. ● Эйфория — возникновение приятных ощущений и немотивированного состояния свободы от тревог и проблем. При этом возникает чувство комфорта и устраняются чувства голода, жажды и др. Это становится причиной развития лекарственной зависимости — непреодолимого желания повторного приема морфина (морфинизм). У некоторых больных и здоровых людей, не испытывающих боли, может развиться ощущение беспокойства и разбитости, плохое самочувствие (дисфория). ● Седативный эффект — состояние покоя, сонливость, нарушение способности к рассуждению (без утраты памяти) и поверхностный сон. ● Противокашлевое действие вследствие угнетения кашлевого центра (к эффекту быстро развивается привыкание). ● Угнетение дыхания связано в основном со снижением чувствительности дыхательного центра к двуокиси углерода и зависит от дозы. Дыхание становится редким и глубоким при введении даже терапевтических доз морфина. При приеме в токсических дозах развивается очень редкое поверхностное дыхание, вплоть до полной его остановки (при отравлении морфином смерть наступает от паралича дыхательного центра). Для восстановления дыхания используют антагонисты опиоидных рецепторов — налоксон и налтрексон. ● Миоз (сужение зрачков) — характерный диагностический признак приема морфина, возникает в результате возбуждения центра гла- 336 Часть II. Частная фармакология зодвигательного нерва. Привыкание в отношении миоза развивается медленно. ● Брадикардия вследствие повышения тонуса центра блуждающих нервов. ● Тошнота и рвота (усиливающиеся при движении) развиваются за счет стимуляции рецепторов пусковой (триггерной) зоны рвотного центра, расположенной на дне IV желудочка мозга. Непосредственно рвотный центр морфин угнетает. ● Влияние на продукцию гормонов. Повышение продукции пролактина, антидиуретического гормона (вазопрессина), гормона роста, что связано со стимуляцией центров в гипоталамусе. Усиление выделения вазопрессина приводит к уменьшению диуреза. Снижается секреция гонадотропных гормонов, адренокортикотропного гормона, а также тестостерона и гидрокортизона. ● Снижение температуры тела ниже нормы (не зависимо от исходного уровня). Эффект связан с угнетением центра теплорегуляции в гипоталамусе и снижением теплопродукции. Гипотермия отчетливо проявляется при применении больших доз морфина. ● Повышение тонуса скелетных мышц (преимущественно мышцсгибателей и дыхательных мышц). Эффект реализуется на уровне спинного мозга. ● Лекарственная зависимость (психическая и физическая) развивается при повторных приемах морфина. Желание повторного приема морфина сначала связано с вызываемой морфином эйфорией. Затем развивается физическая зависимость, проявляющаяся абстинентным синдромом. Явления абстиненции возникают при отмене морфина: сначала слезотечение, насморк, потливость, «гусиная кожа», затем беспокойство, тахикардия, тремор, тошнота, рвота, диарея, сильные боли в животе, спине и др. Эти явления исчезают при приеме морфина. Эффекты, вызываемые возбуждением периферических опиоидных рецепторов ● Стимуляция выделения гистамина приводит к расширению сосудов кожи и конъюнктивы глаз, крапивнице. У больных бронхиальной астмой морфин может вызвать бронхоспазм (повышение тонуса бронхов связано с действием на опиоидные рецепторы бронхиальных мышц). ● Снижение пропульсивной моторики желудка и кишечника, повышение тонуса сфинктеров кишечника, уменьшение секреции поджелудочной железы и выделения желчи (вследствие повышения тонуса сфинктера Глава 14. Анальгезирующие средства (анальгетики) 337 Одди и желчных протоков) нарушают продвижение содержимого по кишечнику и приводят к развитию обстипации (запора). Вследствие повышения тонуса гладких мышц желчевыводящих путей могут возникнуть спастические боли (колики). ● Повышение под действием морфина тонуса мочеточников может вызвать приступ почечной колики, а повышение тонуса сфинктеров уретры может вызвать задержку мочеиспускания. При повторном применении морфина к его действию развивается привыкание. Снижается его анальгетическое действие, и для получения прежнего эффекта необходимо увеличивать дозу. Привыкание развивается и к некоторым другим эффектам морфина (к возникновению эйфории, угнетению дыхания). Практически не развивается привыкание к действию на величину зрачка и ЖКТ. При постоянном приеме морфина развивается привыкание (толерантность) к его токсическому действию (угнетение дыхательного центра), поэтому у лиц с зависимостью к морфину высокие и даже смертельные дозы не вызывают токсических эффектов. При прекращении приема морфина (например, во время лечения в стационаре) толерантность быстро исчезает, и введение высокой, но прежде переносимой дозы может вызвать смертельный исход. Морфин применяют как болеутоляющее средство при выраженных болях, связанных с тяжелыми травмами, ожогами, такими заболеваниями, как злокачественные опухоли, инфаркт миокарда. В анестезиологии морфин используют для подготовки больных к операции (премедикации), а также при послеоперационных болях. Снимая сильные боли, морфин препятствует развитию болевого шока. Препарат можно применять при почечной, кишечной коликах, связанных со спазмом гладких мышц, однако поскольку морфин повышает тонус гладких мышц, в этих случаях его назначают вместе со спазмолитическими средствами (атропином, папаверином). Морфин также применяют при остром отеке легких. Морфин вводят парентерально (внутривенно, подкожно) и внутрь в виде таблеток. Поскольку морфин всасывается из ЖКТ недостаточно хорошо, а также в значительной степени метаболизируется при первом прохождении через печень, для достижения быстрого и выраженного действия его вводят парентерально. Действие морфина развивается через 10–15 мин после введения под кожу и через 20–30 мин после приема внутрь (максимальная концентрация в плазме крови достигается через 10–30 мин после подкожного введения и через 1–2 ч после приема внутрь). Действие однократной дозы продолжается 3–6 ч. 338 Часть II. Частная фармакология Морфин накапливается в наибольших концентрациях в хорошо кровоснабжаемых органах (печень, легкие, селезенка). В скелетных мышцах он накапливается в меньшей степени, но вследствие большой общей массы они служат основным его резервуаром. Морфин плохо преодолевает тканевые барьеры, однако при ацетилировании двух гидроксильных групп он превращается в активный метаболит — диацетилморфин (героин), который значительно быстрее проникает через ГЭБ в ткани мозга, где оказывает анальгетический эффект. В мозге диацетилморфин гидролизуется до моноацетилморфина и далее до морфина. У новорожденных, вследствие слабого развития ГЭБ, морфин в высоких концентрациях способен проникать в мозг и вызывать интоксикацию с угнетением дыхания. Наличие в молекуле морфина двух свободных гидроксильных групп позволяет ему легко конъюгировать с глюкуроновой кислотой. Морфин6-глюкуронид обладает большей анальгетической активностью и действует несколько продолжительнее, чем морфин. У больных с почечной недостаточностью может происходить накопление активных метаболитов, что ведет к более продолжительной и выраженной анальгезии. Значительная часть морфина превращается в полярные метаболиты, которые быстро экскретируются почками (85%), 9–12% морфина выводится с мочой в неизмененном виде. Небольшая часть глюкуронидов морфина (7–10%) экскретируется с желчью и поступает в просвет ЖКТ, откуда морфин может снова всасываться в кровь или (при лечении отравления морфином) может быть удален при промывании желудка слабым раствором калия перманганата. Побочные эффекты морфина: тошнота, рвота, спазм гладких мышц, обстипация, брадикардия, артериальная гипотония, урежение дыхания; при повторном применении — привыкание, лекарственная зависимость. Морфин противопоказан при артериальной гипотензии, дыхательной недостаточности, паралитических, спастических и обструктивных заболеваниях ЖКТ, гипертрофии простаты, повышенном внутричерепном давлении, беременности. Морфин не рекомендуют применять для обезболивания родов (возможно угнетение дыхательного центра у плода), у детей до 2 лет и лиц старше 60 лет, не назначают кормящим матерям. При остром отравлении морфином развиваются коматозное состояние, редкое поверхностное дыхание, брадикардия, резко суживаются зрачки (диагностический признак интоксикации опиоидами), однако при асфиксии зрачки расширяются. Тяжелое отравление приводит к смертельному исходу вследствие остановки дыхания. Глава 14. Анальгезирующие средства (анальгетики) 339 При отравлении морфином основные мероприятия направлены на удаление его из организма (промывают желудок 0,05% раствором калия перманганата, который вызывает окисление морфина, и теплой водой со взвесью активированного угля, адсорбирующего морфин, затем назначают солевое слабительное) и на восстановление дыхания (вводят внутримышечно или внутривенно антагонист опиоидных рецепторов — налоксон). При глубоком угнетении дыхания проводят искусственную вентиляцию легких. Омнопон♠ — препарат, содержащий смесь алкалоидов опия: морфин, кодеин, тебаин (алкалоиды фенантренового ряда), наркотин, папаверин (алкалоиды изохинолинового ряда). Омнопон♠ близок по фармакологическим свойствам к морфину (содержит 48–50% морфина). Применяют по тем же показаниям, что и морфин. Вводят парентерально (подкожно, внутривенно). За счет папаверина обладает спазмолитическими свойствами, поэтому имеет преимущества перед морфином при болях, связанных со спазмом гладкомышечных органов (почечная, печеночная, кишечная колики). Кодеин — производное фенантрена (см. рис. 14.2), алкалоид опия (0,5% концентрации). Для использования в качестве ЛС кодеин синтезируют из морфина (метилированное производное). Обладает всеми свойствами наркотических анальгетиков. По анальгетическому действию примерно в 10 раз слабее морфина. По сравнению с морфином кодеин в большей степени угнетает кашлевой центр, поэтому его применяют чаще как противокашлевое средство (см. раздел «Противокашлевые cредства»). Синтетические наркотические анальгетики Тримеперидин — синтетический наркотический анальгетик (рис. 14.3), производное N-метилпиперидина-(гидрохлорид-1,2,4-триметил-4пропионилокси-4-фенилпиперидин), оригинальный отечественный препарат (Машковский М.Д., Ищенко В.И.). Тримеперидин по анальгетической активности в 2–4 раза уступает морфину, но в меньшей степени угнетает дыхательный центр (может быть использован при беременности, для обезболивания родов и у детей), несколько повышает тонус и сократительную активность миометрия. В отличие от морфина тримеперидин оказывает спазмолитический эффект (на мочеточники, бронхи) и менее выраженное спазмогенное действие (на кишечник, желчевыводящие пути), в связи с чем может использоваться при почечных и печеночных коликах. Фармакокинетические параметры тримеперидина аналогичны морфину. Показания: выраженный болевой синдром (травмы, злокачественные новообразования, послеоперационный период и др.), подготовка к операции, обезболивание родов, почечная, кишечная и печеночная колики. 340 Часть II. Частная фармакология Рис. 14.3. Химические структуры некоторых синтетических наркотических анальгетиков Побочные действия: тошнота, рвота, слабость, головокружение. Возможно развитие лекарственной зависимости. Противопоказания: дыхательная недостаточность. Фентанил — производное фенилпиперидина, полный агонист опиоидных рецепторов (в большей степени стимулирует μ-рецепторы). В 100– 400 раз активнее морфина (для получения такого же анальгетического эффекта необходимы в 100–400 раз меньшие дозы фентанила), а также эффективнее морфина (устраняет боли, при которых морфин неэффективен). Вследствие высокой липофильности быстро проникает в ткани мозга, но затем перераспределяется и накапливается в жировой ткани, где подвергается медленному метаболизму. Фентанил оказывает очень быстрое (эффект наступает через 1–3 мин после внутривенного введения) и кратковременное (20–30 мин) обезболивающее действие. Препарат применяют парентерально (внутривенно, внутримышечно) главным образом для быстрого обезболивания перед и во время хирургических операций, при инфаркте миокарда, отдельно и в комбинации с нейролептиком дроперидолом (комбинированный препарат — таламонал♠). Таламонал♠ применяют для нейролептанальгезии (метод обезболивания с сохранением сознания при хирургических операциях), а также для снятия болей при инфаркте миокарда, травмах. Дроперидол потенцирует обезболива- Глава 14. Анальгезирующие средства (анальгетики) 341 ющее действие фентанила, а также устраняет у пациента чувство страха, тревоги, напряжения перед оперативным вмешательством. Противопоказания и побочные эффекты фентанила такие же, как и у морфина. Фентанил сильнее, чем морфин угнетает дыхательный центр, кроме того, после его применения возможна кратковременная ригидность (повышение тонуса) мышц грудной клетки. Метадон — производное фенилгептиламина, сходен по эффектам с морфином, оказывает более слабое, но продолжительное действие. Назначают внутрь. Привыкание и физическая зависимость развиваются значительно медленнее, чем при применении морфина. Абстиненция после прекращения приема метадона менее выражена (мягкая абстиненция), но продолжительнее, чем при отмене морфина. Эти свойства метадона позволяют использовать его в ряде стран для детоксикации и поддерживающего лечения лекарственной зависимости к опиоидам, в частности к героину. Родственное метадону соединение левометадил ацетат (1-а-ацетилметадон) оказывает еще более продолжительное действие, его применяют внутрь 1 раз в 2–3 дня (наиболее оптимальный препарат для проведения детоксикации). Частичные агонисты и агонисты-антагонисты опиоидных рецепторов Бупренорфин — частичный агонист μ-опиоидных рецепторов. По анальгетической активности в 20–30 раз превышает морфин и действует длительнее (6–8 ч). По эффективности сравним с морфином, вводят парентерально и сублингвально. При сублингвальном применении бупренорфина действие наступает через 25–35 мин и продолжается 8–12 ч (применяют для неотложной помощи при массовых травматических поражениях — оказывает противошоковое действие, облегчает транспортировку пострадавших). Пентазоцин — производное бензоморфана, синтетический наркотический анальгетик из группы агонистов-антагонистов опиоидных рецепторов (агонист κ- и δ-рецепторов, антагонист μ-рецепторов). Анальгетические свойства обусловлены в основном стимуляцией κ-рецепторов. По анальгетической активности уступает морфину, но в меньшей степени угнетает дыхание. Поскольку пентазоцин не вызывает эйфорию (связанную со стимуляцией μ-рецепторов), а может вызывать дисфорию, при его применении меньше риск возникновения лекарственной зависимости. Пентазоцин проявляет свойства антагониста — вытесняет морфин из связи с опиоидными рецепторами, обусловливая развитие абстинентного синдрома у лиц с физической зависимостью к наркотическим анальгетикам. Препарат вводят парентерально (внутривенно, внутримышечно, 342 Часть II. Частная фармакология подкожно) и внутрь. Хорошо всасывается при всех путях введения. Начало эффекта и его максимальная выраженность отмечаются соответственно через 2–3 мин и 15–30 мин после внутривенного введения, через 15– 30 мин и 30–60 мин после внутримышечного и энтерального введения. Продолжительность действия составляет до 3 ч. Пентазоцин вызывает повышение давления в легочной артерии, при этом увеличивается преднагрузка на сердце, усиливается работа сердца. В связи с этим препарат не рекомендуют применять при инфаркте миокарда. Для пентазоцина характерны дисфория, галлюцинации, тахикардия, повышение АД. Препарат противопоказан при бронхиальной астме, черепно-мозговой травме, эпилепсии, желчно- и мочекаменной болезни, недостаточности печени и почек. Ограничения к применению: беременность, грудное вскармливание, возраст до 1 года. Буторфанол — агонист-антагонист опиоидных рецепторов (стимулирует к-рецепторы и выступает в роли антагониста μ-рецепторов). Применяют для премедикации перед хирургическими операциями, во время операций и для послеоперационного обезболивания. В меньшей степени, чем морфин, угнетает дыхание, меньше вызывает риск лекарственной зависимости. Так же как пентазоцин, буторфанол повышает давление в легочной артерии и увеличивает работу сердца, в связи с чем его не рекомендуют применять при инфаркте миокарда. Вызывает такие же побочные эффекты, что и пентазоцин. Налбуфин также агонист κ-рецепторов и антагонист μ-рецепторов. По анальгетической активности близок к морфину, но меньше угнетает дыхание. В отличие от пентазоцина и буторфанола не влияет на гемодинамику, применяют для обезболивания при болевых синдромах, в том числе при инфаркте миокарда. Продолжительность действия до 6 ч. В качестве антагонистов наркотических анальгетиков используют налоксон (наркан♠) и налтрексон. Эти препараты блокируют μ-, δи κ-рецепторы, поэтому устраняют как анальгезирующее действие наркотических анальгетиков, так и вызываемые ими эйфорию, угнетение дыхания и другие эффекты. Налоксон — производное фенантрена, полный конкурентный антагонист μ-, δ- и κ-рецепторов (рис. 14.4). Препарат вводят внутривенно при интоксикации наркотическими анальгетиками для конкурентного вытеснения их из связи с дыхательным центром. Налоксон действует 2–4 ч, устраняет не только угнетение дыхания, но и многие другие эффекты опиоидных анальгетиков, в том числе агонистов-антагонистов. Менее эффективен при передозировке бупренорфина. Налоксон Глава 14. Анальгезирующие средства (анальгетики) вызывает синдром абстиненции у лиц с лекарственной зависимостью к опиоидам. Налтрексон эффективен при приеме внутрь, действует до 24 ч. Уменьшает или предотвращает эффекты, вызываемые опиоидами. Снижает потребление алкоголя. Используется при алкогольной зависимости, а также для предотвращения эйфории, вызываемой опиоидами, при лечении наркоманов. 343 Рис. 14.4. Химическая структура налоксона 14.1.2. Неопиоидные препараты с анальгетической активностью Препараты различных фармакологических групп с анальгетическим компонентом действия Парацетамол (ацетаминофен♠, панадол♠, тайленол♠, эффералган♠) — ненаркотический анальгетик, производное пара-аминофенола, активный метаболит фенацетина. Обладает болеутоляющим и жаропонижающим действиями. Противовоспалительная активность отсутствует. Анальгетическое действие связано с ингибированием циклооксигеназы в ЦНС. Не исключено влияние на серотонинергическую и каннабиноидную системы головного мозга. Используют при головной боли, для снижения температуры при лихорадке, миалгии, невралгии, суставных болях. По эффективности парацетамол сопоставим с ацетилсалициловой кислотой. В терапевтических дозах редко вызывает побочные эффекты. Однако токсическая доза парацетамола лишь в 3 раза превышает терапевтическую. При передозировке препарат оказывает гепатотоксическое действие, вызывая некроз клеток печени, что связано с образованием токсичного метаболита ацетаминофена — N-ацетил-п-бензохинонимина. С целью предупреждения развития токсических эффектов парацетамола в течение первых 12 ч после отравления вводят ацетилцистеин или метионин. Клонидин (клофелин♠) — α2-адреномиметик, используют в качестве антигипертензивного средства. Обладает выраженным анальгетическим действием, которое объясняют усилением нисходящих тормозных влияний (опосредуемых α2-адренорецепторами) на проведение болевых импульсов в афферентных путях спинного мозга. Препятствует развитию вегетативных нарушений, вызванных болью. Используют для уменьшения боли при оперативных вмешательствах, в послеоперационном периоде, при инфаркте миокарда, злокачественных опухолях. 344 Часть II. Частная фармакология Амитриптилин и имипрамин (имизин♠) — трициклические антидепрессанты. За счет угнетения обратного нейронального захвата норадреналина и серотонина активируют нисходящую антиноцицептивную систему, угнетающую передачу болевых импульсов на уровне спинного мозга. Эффективны при хронических болях. Используют при невралгии различной этиологии, фантомных болях. Карбамазепин (тегретол♠) и фенитоин (дифенин♠) — блокаторы натриевых каналов, применяют в качестве противоэпилептических средств. В качестве анальгетиков эффективны при невралгии тройничного нерва, сопровождающейся приступами сильных болей. Габапентин применяют в качестве противоэпилептического средства. Анальгетическое действие связано со стимуляцией ГАМК-ергической передачи в головном мозге. В качестве анальгетика применяют при мигрени, нейропатических болях. Баклофен — агонист ГАМК-рецепторов, применяют при болезненных мышечных спазмах, спастичности. Кетамин — производное фенциклидина, неконкурентный антагонист NMDA-рецепторов, обладает выраженным анальгетическим действием, применяют для общего обезболивания (диссоциативной анестезии). Азота закись♠ применяют ингаляционно (см. раздел «Средства для ингаляционного наркоза»), обладает выраженными анальгетическими свойствами, используют для уменьшения болей при инфаркте миокарда, для обезболивания родов, в послеоперационном периоде. Кроме того, некоторое анальгетическое действие оказывают антагонисты Н1-рецепторов, например дифенгидрамин, а также соматостатин, кальцитонин. 14.1.3. Анальгетики со смешанным механизмом действия (опиоидный и неопиоидный компоненты) Трамадол (трамал♠) — центральный неселективный агонист μ-, δи κ-рецепторов. Анальгетический эффект дополнительно опосредуется за счет влияния на адренергическую и серотонинергическую передачу (нарушается нейрональный захват норадреналина и серотонина) в нисходящих антиноцицептивных путях, в результате чего усиливаются нисходящие тормозные влияния на проведение болевых импульсов на уровне спинного мозга. Трамадол по активности уступает морфину. Анальгетическое действие практически не сопровождается угнетением дыхания, снижением моторики ЖКТ, повышением тонуса мочевыводящих путей. В сравнении с морфином препарат обладает незначительным наркоген- Глава 14. Анальгезирующие средства (анальгетики) 345 ным потенциалом (меньше риск лекарственной зависимости), не входит в перечень наркотиков. Препарат применяют при послеоперационных болях и других болевых синдромах (при инфаркте миокарда, злокачественных опухолях, травмах). Назначают парентерально, внутрь и ректально. 14.2. АНАЛЬГЕЗИРУЮЩИЕ СРЕДСТВА ПРЕИМУЩЕСТВЕННО ПЕРИФЕРИЧЕСКОГО ДЕЙСТВИЯ (НЕСТЕРОИДНЫЕ ПРОТИВОВОСПАЛИТЕЛЬНЫЕ СРЕДСТВА) Нестероидные противовоспалительные средства (НПВС) — большая группа соединений, обладающих противовоспалительным, анальгезирующим и жаропонижающим свойствами. Эти свойства НПВС связаны с их способностью нарушать образование простагландинов E2 и I2 — медиаторов воспаления, вызывающих следующие эффекты: ● расширяют артериолы, а также усиливают действие других медиаторов воспаления гистамина и брадикинина на проницаемость сосудов, что приводит к экстравазации плазмы, инфильтрации и отеку тканей; ● повышают чувствительность болевых рецепторов к брадикинину, гистамину и некоторым другим веществам, являющимся медиаторами боли; ● простагландин E2 оказывает стимулирующее действие на центр теплорегуляции в гипоталамусе и повышает температуру тела. Простагландины E2 и I2 образуются из арахидоновой кислоты. Происходит это следующим образом: вначале из арахидоновой кислоты под действием циклооксигеназы (ЦОГ) синтезируются циклические эндопероксиды, а из нестабильных циклических эндопероксидов далее образуются простагландины E2, I2, другие простагландины и тромбоксан. НПВС ингибируют ЦОГ и таким образом нарушают образование простагландинов E2 и I2, что и определяет их основные фармакологические эффекты — противовоспалительный, анальгезирующий и жаропонижающий. В качестве противовоспалительных и болеутоляющих средств при артритах, миозитах, невритах применяют различные НПВС, в том числе ацетилсалициловую кислоту (аспирин♠), ибупрофен (бруфен♠), диклофенак (вольтарен♠), при этом используют противовоспалительное и анальгетическое свойства этих препаратов. В качестве болеутоляющих средств НПВС (ацетилсалициловую кислоту) применяют при головных болях. НПВС эффективны также при болях, связанных с менструацией (воз- 346 Часть II. Частная фармакология никновение болей обусловлено простагландинами). Подробнее об этих препаратах см. раздел «Нестероидные противовоспалительные средства». Только в качестве болеутоляющих средств используют метамизол натрия и кеторолак. Метамизол натрия (анальгин♠) применяют при болях, связанных с воспалительными процессами: миалгиях, невралгиях, артралгиях, а также при головной боли, болях, вызванных менструациями. Вводят внутрь и парентерально. Входит в состав комбинированного препарата баралгин♠ (спазмалгин♠), который помимо метамизола натрия содержит вещества спазмолитического действия — питофенон и фенпивериния бромид. Препарат применяют при болях, связанных со спазмом гладких мышц (почечная, печеночная, кишечная колики), вводят внутривенно или внутримышечно. При систематическом применении метамизол натрия может вызвать лейкопению, возможен агранулоцитоз, в связи с чем его применение ограничено. Препарат не рекомендуется принимать длительно. Кеторолак (кетанов♠) обладает выраженной анальгетической активностью, но оказывает меньшее по сравнению с другими НПВС противовоспалительное действие. Кеторолак хорошо всасывается из ЖКТ, при введении внутрь биодоступность составляет 80–100%. Препарат применяют внутрь и парентерально для купирования послеоперационных болей (в качестве альтернативы опиоидным анальгетикам), а также при болях, вызванных травмами, при опухолевых заболеваниях и др. Препарат показан при невралгии тройничного нерва. Кеторолак при длительном применении вызывает побочные эффекты, характерные для других НПВС: изъязвление слизистой оболочки желудка и двенадцатиперстной кишки (в связи с угнетением синтеза простагландинов E2 и I2, обладающих гастропротекторным действием), нарушение функции почек (изменение фильтрации в почечных клубочках) и др. (подробнее о побочных эффектах НПВС см. раздел «Нестероидные противовоспалительные средства»). Вопросы и задания для самоконтроля 1. Укажите полные агонисты опиоидных рецепторов: а) бупренорфин; б) ремифентанил; в) пентазоцин; г) морфин; д) трамадол. Глава 14. Анальгезирующие средства (анальгетики) 347 2. Морфин вызывает: а) тошноту и рвоту; б) миоз; в) тахикардию; г) тремор; д) эйфорию. 3. Антагонисты опиоидных рецепторов: а) налтрексон; б) налбуфин; в) налоксон; г) омнопон; д) кетамин. 4. Неопиоидные анальгетики: а) фентанил; б) карбамазепин; в) бупренорфин; г) габапентин; д) амитриптилин. 5. Морфин вызывает анальгезию за счет: а) нарушения передачи возбуждения в передних рогах спинного мозга; б) нарушения передачи возбуждения в задних рогах спинного мозга; в) угнетения полисинаптических рефлексов спинного мозга; г) активации нисходящей тормозной системы; д) изменения эмоциональной оценки боли. Глава 15 ПСИХОТРОПНЫЕ СРЕДСТВА Психотропные средства — ЛВ с направленным действием на психическую деятельность человека. Введение в клиническую практику хлорпромазина и резерпина в середине ХХ в. открыло эру психофармакологии, сегодня это динамично развивающаяся область фармакологии. В зависимости от характера изменений психических функций, возникающих в результате применения психотропных средств, их можно разделить на следующие основные группы: ● антипсихотические средства (нейролептики); ● антидепрессанты; ● анксиолитические средства (транквилизаторы); ● седативные средства; ● психостимуляторы; ● ноотропные средства. 15.1. АНТИПСИХОТИЧЕСКИЕ СРЕДСТВА Антипсихотические средства (нейролептики) — ЛС, обладающие антипсихотическими свойствами, т.е. способностью устранять продуктивную и негативную симптоматику психозов в виде расстройства мышления (бред), восприятия (слуховые, зрительные, обонятельные галлюцинации), двигательной активности (психомоторное возбуждение). Психоз — признак ряда заболеваний головного мозга с наиболее тяжелым течением у больных шизофренией. Шизофрения (от греч. «расщепление души»: шизо — расщепляю, френ — душа) — хроническое заболевание, с психотическими симптомами и характерными признаками: потерей связи с реальностью, галлюцинациями, бредом, нарушениями мышления и жизнедеятельности человека в профессиональной и социальной сферах (регистрируют примерно у 1% населения). Выделяют следующие симптомы шизофрении: Глава 15. Психотропные средства 349 ● «позитивные» (дополнительно к норме) или «психотическую сферу» — бред, галлюцинации, причудливое поведение; ● «негативные» (дефицитарные по отношению к норме) — снижение мотивации, эмоциональная индифферентность, бедность речи, ангедония (снижение способности испытывать удовольствие), асоциальность; ● нарушения мышления (снижение памяти, способности логически мыслить, решать проблемы). Существуют различные теории возникновения психозов и сопутствующих им заболеваний. По-видимому, генетические нарушения играют ключевую роль в этиологии шизофрении, однако тип наследования заболевания относится не к разряду моногенных («менделевских»), а к комбинированной патологии многих генов, зависящей и от окружающей среды. Патогенез шизофрении связан с нарушениями развития отдельных областей головного мозга, вызванных неправильной миграцией нейронов у плода, в результате возникают нарушения цитоархитектоники коры головного мозга, а происходящие при этом в головном мозге процессы иллюстрирует так называемая биохимическая, «дофаминовая» теория. Согласно этой теории, гиперактивность мезолимбической части дофаминергической систмы головного мозга приводит к перевозбуждению D2-рецепторов этой области, вызывая «позитивные» симптомы. При этом активность мезокортикальной дофаминергической системы снижена, что приводит к гипостимуляции соответствующих D1-рецепторов и развитию «негативных» симптомов (в последнее время активно изучаются роль центральных серотонинергических структур в развитии психотических реакций и реципрокные отношения этих структур с дофаминергической системой головного мозга). Основной эффект нейролептиков — антипсихотическое действие — связывают с блокадой дофаминовых D2-рецепторов мезолимбической системы, этим действием определяется их способность устранять позитивную симптоматику психозов (бред, галлюцинации). Некоторые нейролептики представляют собой антагонисты 5-НТ2-рецепторов. Блокада данных рецепторов уменьшает выраженность негативной симптоматики и когнитивных нарушений у больных шизофренией. Седативное действие связывают с блокадой центральных гистаминовых Н1-рецепторов и α-адренорецепторов. Побочные эффекты, вызываемые нейролептиками, определяются блокадой дофаминергической системы головного мозга, в их число входят экстрапирамидные нарушения в виде лекарственного паркинсонизма, острой (лицо и шея) и поздней дискинезии мышц. 350 Часть II. Частная фармакология По способности вызывать лекарственный паркинсонизм выделяют «типичные» нейролептики, блокирующие дофаминовые рецепторы, и, как следствие, его вызывающие, и «атипичные» нейролептики — препараты второго поколения, механизм действия которых в меньшей степени связан с блокадой дофаминовых рецепторов, а лекарственный паркинсонизм для них не характерен. Типичные нейролептики практически не влияют на негативную симптоматику психоза и когнитивные нарушения, способствуют развитию экстрапирамидных расстройств за счет блокады D2-рецепторов нигростриатной системы. Эта группа препаратов устраняет в основном позитивные симптомы психоза за счет блокады дофаминовых D2-рецепторов в мезолимбической области головного мозга. ● Блокируя D2-рецепторы тубероинфундибулярной системы, эти препараты вызывают в гипоталамусе и гипофизе гормональные сдвиги, увеличивая секрецию пролактина (вызывают галакторею у женщин и гинекомастию у мужчин), снижая секрецию гормона роста и другие гормональные нарушения. ● Противорвотное (антиэметическое) действие типичных антипсихотических средств обусловлено в основном блокадой D2-рецепторов триггерной зоны рвотного центра. ● При однократном приеме нейролептики снижают тонус скелетной мускулатуры и двигательную активность, ослабляя активирующее влияние ретикулярной формации на спинной мозг. ● Понижают температуру тела, угнетая центр терморегуляции, увеличивая теплоотдачу за счет расширения кровеносных сосудов кожи и других механизмов. ● Потенцируют действие средств для наркоза, снотворных, наркотических анальгетиков, что используется для нейролептаналгезии. ● Периферические нарушения, обусловлены влиянием на вегетативную иннервацию воздействиями, обусловленными м-холиноблокирующим («атропиноподобным») и адреноблокирующим влияниями в виде гипотензии. ● Блокада периферических гистаминовых рецепторов может сопровождаться угнетением моторики гладкой мускулатуры, снижением АД и антигистаминными эффектами. Атипичные нейролептики эффективны в отношении продуктивной симптоматики психозов и активны в отношении негативной симптоматики и когнитивных расстройств. Препараты этой группы не вызывают экстрапирамидных расстройств. Глава 15. Психотропные средства 351 Механизм антипсихотического действия атипичных нейролептиков до конца не ясен. Было предложено несколько гипотез, включая блокаду 5-НТ-рецепторов подтипа 5НТ2А, реципрокное взаимодействие серотонинергических 5НТ2А-рецепторов и дофаминергических D2-рецепторов, что позволяет достигнуть оптимального сбалансирования воздействия на D2-рецепторы атипичным нейролептиком, быструю неустойчивую связь с D2-рецепторами и другие. 15.1.1. Типичные антипсихотические средства ● Производные фенотиазина: —алифатические производные: хлорпромазин (аминазин♠, ларгактил♠, плегомазин♠); левомепромазин (тизерцин♠, нозинан); —пиперазиновые производные: перфеназин (этаперазин♠); трифлуоперазин (трифтазин♠, стелазин♠); флуфеназин (фторфеназин♠, модитен♠, флуфеназин-деканоат♠, модитен-депо♠); —пиперидиновые производные: тиоридазин (сонапакс♠); пипотиазин (пипортил♠). бутирофенона: галоперидол (галдол♠, галофен♠, тран● Производные ♠ кодол ); дроперидол. ● Производные тиоксантена: хлорпротиксен (труксал♠). Производные фенотиазина Алифатические производные Хлорпромазин — один из основных представителей нейролептиков группы фенотиазинов. Препарат оказывает антипсихотическое, выраженное седативное, анксиолитическое действия, потенцирует действие наркозных, снотворных и ряда других средств, угнетающих ЦНС (рис. 15.1). Антипсихотическое действие препарата определяется его способностью устранять позитивную симптоматику психоза и реализуется путем блокады постсинаптических D2-рецепторов в мезолимбической системе. Седативное действие связано с угнетающим влиянием на восходящую Рис. 15.1. Химическая структура ретикулярную формацию ствола мозга хлорпромазина 352 Часть II. Частная фармакология вследствие блокады α-адренорецепторов и проявляется в виде общего успокоения, устранения аффективных реакций, понижения двигательной активности при эмоциональном, психическом и двигательном возбуждении. Способность хлорпромазина блокировать центральные гистаминергические и холинергические структуры усиливает седативное действие, в больших дозах вызывает снотворный эффект (поверхностный сон). Результат анксиолитического действия наблюдают в снижении страха, тревоги, беспокойства, психической напряженности. Центральное мышечнорасслабляющее действие обусловлено угнетением супраспинальной регуляции мышечного тонуса. Препарат оказывает противорвотное действие, вызванное блокадой дофаминовых D2-рецепторов в пусковой (триггерной) зоне рвотного центра; этот эффект иногда используют для купирования тяжелой рвоты. Гипотермическое действие связано с угнетением центра терморегуляции в гипоталамусе, препарат увеличивает теплоотдачу и способствует развитию гипотермии при снижении температуры окружающей среды. Этот эффект может быть использован при искусственной гипотермии (охлаждении организма на фоне выключения центра терморегуляции хлорпромазином). Усилению гипотермического действия способствует вызываемая им блокада α-адренорецепторов сосудов кожи, что увеличивает теплоотдачу с кожных покровов. Хлорпромазин увеличивает секрецию пролактина в передней доле гипофиза, что связано с блокадой дофаминовых D2-рецепторов и устранением действия дофамина на продукцию этого гормона (дофамин — гипоталамический фактор, угнетающий высвобождение пролактина). Повышение уровня пролактина в крови приводит к усилению лактации, снижению продукции гонадотропных гормонов и расстройству менструального цикла, развитию галактореи, гинекомастии, импотенции. Для хлорпромазина характерны экстрапирамидные нарушения (лекарственный паркинсонизм), связанные с блокадой дофаминовых D2рецепторов в неостриатуме. Блокада периферических α-адренорецепторов сосудов приводит к понижению АД, ортостатической гипотензии. Гипотензия может привести к рефлекторной тахикардии (в механизме гипотензивного эффекта играет роль угнетение препаратом активирующего влияния сосудодвигательного центра на периферические сосуды). Для периферического м-холиноблокирующего эффекта характерно снижение секреции слюнных, бронхиальных и пищеварительных желез, снижение моторики ЖКТ (возможно развитие других атропиноподобных эффектов). Глава 15. Психотропные средства 353 Препарат обладает антигистаминным действием, что связано с его способностью блокировать гистаминовые Н1-рецепторы (противоаллергическое действие). При приеме внурь препарат плохо всасывается из ЖКТ, связывается с белками плазмы крови примерно на 90%; метаболизируется в печени, образуя свыше 150 метаболитов, половина из которых — фармакологически активны. Выводится в основном почками в виде метаболитов и в неизмененном виде, а также через ЖКТ. Продолжительность терапевтического действия хлорпромазина составляет 6 ч, при длительном применении развивается привыкание. Показания: шизофрения и другие психозы, психомоторное возбуждение, маниакальное состояние у больных маниакально-депрессивным психозом, острые галлюцинаторно-бредовые состояния, психозы с агрессивностью, состояния тревоги, страха, эмоционального напряжения, при подготовке к анестезии (премедикации), для потенцирования наркоза, купирования тяжелой рвоты, икоты. Побочные эффекты: сонливость, дезориентация, снижение АД, ортостатическая гипотензия, нейроэндокринные нарушения (гипотермия, галакторея, аменорея, импотенция), атропиноподобный эффект (нарушение аккомодации, сухость во рту, задержка мочеиспускания, констипация). Возможны аллергические реакции на коже и слизистых оболочках, агранулоцитоз, гемолитическая анемия, фотосенсибилизация и пигментация кожи, контактный дерматит. Наиболее частый и тяжелый побочный эффект — экстрапирамидные расстройства, включающие симптомы паркинсонизма (тремор, мышечную ригидность, двигательную заторможенность) с постепенным нарастанием. Эти симптомы исчезают после отмены препарата или могут быть уменьшены при назначении центральных холиноблокаторов (см. главу «Противопаркинсонические средства»). К другим признакам таких расстройств относят острую дистонию (спастические сокращения лица, шеи, спины), которую регистрируют после приема первых доз препарата, и акатизию (неусидчивость, двигательное беспокойство). При длительном применении (в течение нескольких лет) возможна поздняя (тардивная) дискинезия (непроизвольные чрезмерные движения лица, губ, шеи). Отсроченная дискинезия не исчезает после отмены препарата и не поддается лечению. Опасное осложнение терапии — злокачественный нейролептический синдром (повышение тонуса скелетных мышц, гипертермия, нестабильность АД, тахикардия и др.). 354 Часть II. Частная фармакология Противопоказания: коматозные состояния, депрессии, тяжелые заболевания печени и почек; нарушение функции кроветворных органов; микседема; беременность. Левомепромазин по механизму действия и фармакологическим свойствам Рис. 15.2. Химическая структура близок к хлорпромазину, но по способлевомепромазина ности потенцировать эффекты наркотических веществ и анальгетиков, гипотермическому, адреноблокирующему, противогистаминному действиям превосходит хлорпромазин, а по холиноблокирующей активности и противорвотному действию ему уступает. Способен вызывать обезболивающий эффект (рис. 15.2). Пиперазиновые производные Флуфеназин-деканоат♠ (рис. 15.3) — препарат пролонгированного действия, его получают этерификацией флуфеназина остатком каприновой кислоты, что увеличивает относительную молекулярную массу препарата и придает высокую липофильность (после однократной внутримышечной инъекции масляного раствора препарат постепенно высвобождается и обеспечивает лечебный эффект в течение 1–2 нед и более). Рис. 15.3. Химические структуры флуфеназина гидрохлорида и трифлуоперазина гидрохлорида Глава 15. Психотропные средства 355 Пиперидиновые производные Для препаратов этой группы характерны умеренная антипсихотическая активность и слабо выраженная по сравнению с хлорпромазином способность вызывать экстрапирамидные расстройства и нейроэндокринные побочные эффекты, умеренные седативный эффект и холиноблокирующая активность. В связи с меньшей частотой развития побочных эффектов по сравнению с другими производными фенотиазина пиперидиновые производные интересны для применения у пациентов пожилого возраста. Представители данной группы препаратов — тиоридазин и пипотиазин. Тиоридазин по сравнению с хлорпромазином обладает менее выраженными антипсихотическими и седативными свойствами, не вызывает сонливости, подавленности, оказывает антидепрессивное действие при эндогенных депрессиях, имеет выраженную холиноблокирующую активность. По сравнению с хлорпромазином в меньшей степени вызывает экстрапирамидные расстройства. Двигательные нарушения при его применении наступают реже, чем при использовании других нейролептиков. В связи с меньшей частотой развития побочных эффектов по сравнению с другими производными фенотиазина препарат показан для пациентов пожилого возраста. При употреблении препарата в высоких дозах возможны кардиотоксический эффект и дегенерация сетчатки. Пипотиазин (рис. 15.4) в низких дозах блокирует пресинаптические дофаминовые D2-рецепторы, что вызывает облегчение дофаминергической передачи и приводит к активирующему эффекту. В больших дозах приводит к блокаде постсинаптических D2-рецепторов, что снижает активность дофаминергических влияний (антибредовый и антигаллюцинаторный эффекты). Длительность антипсихотического эффекта составляет 3–4 нед, что делает его удобным для назначения больным шизофренией в амбулаторных условиях. Рис. 15.4. Химическая структура пипотиазина 356 Часть II. Частная фармакология Производные бутирофенона Производные бутирофенона обладают выраженной антипсихотической активностью, превосходящей активность фенотиазиновых производных в 50–100 раз, оказывают седативное и противорвотное действия, не обладают холиноблокирующими свойствами, в меньшей степени блокируют периферические α-адренорецепторы, практически не вызывают снижения АД. Особенность фармакокинетики — хорошая всасываемость из ЖКТ. Производные бутирофенона — антагонисты по отношению к психостимуляторам, потенцируют действие наркозных, снотворных средств, наркотических анальгетиков, алкоголя и других препаратов, угнетающих ЦНС, вызывают выраженные экстрапирамидные нарушения. Представители данной группы препаратов — галоперидол и дроперидол. Галоперидол (рис. 15.5) применяют по тем же показаниям, что и хлорпромазин, чаще используют в качестве активного антипсихотического препарата с седативным эффектом для купирования психомоторного возбуждения. Эффективен у больных, резистентных к другим нейролептическим средствам, часто вызывает экстрапирамидные расстройства, поэтому противопоказанием для его применения считают заболевания ЦНС с экстрапирамидной симптоматикой. Дроперидол (см. рис. 15.5) оказывает сильное, но кратковременное антипсихотическое действие, его 0,25% раствор применяют для купирования острых приступов психомоторного возбуждения. В сочетании с наркотическим анальгетиком фентанилом дроперидол в составе комбинированного препарата таламонал♠ используют для нейролептаналгезии (см. раздел «Опиоидные анальгетики»). Рис. 15.5. Химические структуры галоперидола и дроперидола Глава 15. Психотропные средства 357 Производные тиоксантена Хлорпротиксен (рис. 15.6) по спектру фармакологических свойств близок к хлорпромазину (оказывает седативное действие, потенцирует действие наркозных, снотворных средств, наркотических анальгетиков, обладает значительной противорвотной активностью, адреноблокирующими и холиноблокирующими свойствами), но уступает ему по выраженности Рис. 15.6. Химическая структуантипсихотического действия и обладает ра хлорпротиксена слабой антидепрессивной активностью. Побочные действия у препарата аналогичны тем, что возникают при приеме хлорпромазина, однако менее выражены, в частности препарат в обычных дозах практически не вызывает экстрапирамидных расстройств. Показания к применению: психозы, включая алкогольный. Препарат можно использовать как противорвотное средство. Наличие седативного компонента у препарата считается показанием для назначения его в малых дозах при невротических расстройствах, нарушениях сна и кожном зуде. Среди антипсихотических средств есть производные других химических соединений. Производное дифенилбутилпиперидина пимозид по спектру действия близок к галоперидолу, при приеме внутрь оказывает продолжительное действие. При острых психозах препарат не применяют, так как он не оказывает седативного действия. Производное индола дикарбин (карбидин♠) — нейролептик с мягким антипсихотическим действием и антидепрессивной активностью. Применяют при лечении различных форм шизофрении с выраженным депрессивным компонентом, алкогольных психозов. Препарат обладает центральной адреноблокирующей активностью. 15.2.2. Атипичные антипсихотические средства Данные препараты в отличие от «типичных» нейролептиков реже и в меньшей степени вызывают экстрапирамидные расстройства и нейроэндокринные нарушения, связанные с блокадой дофаминовых D2-рецепторов, эффективны при терапии негативной симптоматики и ряда когнитивных нарушений. 358 Часть II. Частная фармакология Отсутствие у атипичных нейролептиков существенных экстрапирамидных расстройств обусловлено высокими значениями соотношения вызываемой ими блокады 5-НТ2А-/D2-рецепторов. Известно, что центральные серотонинергические и дофаминергические структуры находятся в реципрокных отношениях. Блокада 5-НТ2А-рецепторов в нигростриатной и тубероинфундибулярной системах реципрокно повышает дофаминовую активность в этих структурах, что уменьшает выраженность побочных эффектов (экстрапирамидных расстройств, гиперпролактинемии и др.), свойственных типичным нейролептикам. Производные дибензодиазепина Клозапин (азалептин♠, лепонекс♠) — высокоэффективное антипсихотическое средство с седативным эффектом, не вызывает заторможенность, редко (1% случаев) вызывает экстрапирамидные нарушения. Препарат устраняет негативную симптоматику у больных шизофренией (интравертность, бедность речи), преимущественно блокируя D4-рецепторы (в сравнении с D2-рецепторами) и 5-НТ 2А-рецепторы, а также м-холинорецепторы и α1-адренорецепторы головного мозга (рис. 15.7). Клозапин быстро и почти полностью всасывается из ЖКТ, после чего быстро распределяется, легко проникает через ГЭБ; Рис. 15.7. Химическая струк- с белками плазмы крови связывается на 95%; тура клозапина в печени инактивируется до неактивных метаболитов; t½ составляет около 12 ч. Показания к применению: шизофрения (все формы), маниакальная фаза маниакально-депрессивного психоза. Побочные эффекты: мышечная слабость, сонливость, гипотензия, в том числе ортостатическая, атропиноподобные эффекты (тахикардия, нарушение аккомодации, обстипация), повышение аппетита и массы тела, возможна гиперсаливация. Наиболее серьезный побочный эффект — гранулоцитопения (вплоть до агранулоцитоза), поэтому лечение препаратом следует проводить под контролем состава периферической крови. Оланзапин (зипрекса♠) блокирует 5-HT2А-рецепторы, D1-, D2-, D3-, D4-рецепторы, α1-адренорецепторы, М-холинорецепторы, гистаминовые H1-рецепторы. Глава 15. Психотропные средства 359 В сравнении с галоперидолом оланзапин оказывает сходное по величине действие по устранению продуктивной симптоматики шизофрении, но более эффективен в лечении негативной симптоматики. По сравнению с рисперидоном оланзапин более эффективен в устранении негативных симптомов шизофрении. В дозе 17,5 мг/кг препарат вызывает минимальные экстрапирамидные нарушения, однако при увеличении дозы отмечается дозозависимое усиление этих побочных эффектов. В сравнении с клозапином в меньшей степени влияет на картину крови. Производные бензизоксазола Рисперидон (рисполепт♠) селективно блокирует центральные 5-НТ2Арецепторы, дофаминовые D2-рецепторы, α-адренорецепторы, а также гистаминовые Н 1-рецепторы. Препарат практически не влияет на м-холинорецепторы, поэтому, возможно, он чаще, чем другие атипичные нейролептики, вызывает экстрапирамидные расстройства (рис. 15.8). В экспериментах на животных по коэффициенту блокирования серотонин/дофамин и по способности вызывать экстрапирамидные расстройства в малых дозах препарат относится к атипичным нейролептикам, в высоких дозах проявляет себя как типичный нейролептик. Рисперидон в процессе метаболизма образует активный метаболит 9-гидроксирисперидон, отличающийся от исходного препарата бо́льшим t½ (23 ч), что позволяет применять его 1 раз в сутки. Эффективен в лечении резистентных форм шизофрении, в том числе резистентных к терапии галоперидолом. Рис. 15.8. Химическая структура рисперидона 360 Часть II. Частная фармакология Производные бензамида Амисульприд — атипичный нейролептик, отличающийся от других антипсихотических препаратов тем, что он блокирует только D2-рецепторы и D3-рецепторы и не обладает сродством к каким-либо другим типам рецепторов, блокирует пре- и постсинаптические дофаминовые рецепторы. В малых дозах препарат блокирует прежде всего пресинаптические рецепторы, что повышает выделение дофамина и облегчает дофаминергическую передачу. В лобной коре это приводит к ослаблению негативной симптоматики, поскольку препарат не влияет на D1-рецепторы. С другой стороны, в лимбической системе препарат блокирует постсинаптические D2- и D3-рецепторы, что нейтрализует дофаминергическое влияние блокады пресинаптических рецепторов и уменьшает позитивные признаки психоза (рис. 15.9). Амисульприд в качестве препарата выбора может применяться для купирования острых эпизодов шизофрении, а в низких дозах — это один из лучших препаратов для лечения негативной симптоматики и средство с доказательно установленной лечебной эффективностью для коррекции нейрокогнитивных нарушений. D3 Усиление воздействия на D1-рецепторы Корковая активация Ослабление негативной Усиление D1 D2 высвобожсимптоматики Лобная кора дения дофамина Вентральная часть покрышки D3 D2 Лимбическая система D3 D2 Ослабление позитивной (продуктивной) симптоматики Блокада дофаминовой нейропередачи Рис. 15.9. Механизм действия амисульприда Глава 15. Психотропные средства 361 Доклинические и клинические исследования показали, что арипипразол может служить прототипом атипичных антипсихотиков нового поколения со стабилизирующим эффектом дофаминергических систем. Доклинические исследования классифицировали арипипразол как частичный агонист с сильным сродством к D2-рецепторам, а его стабилизирующий эффект на дофаминовую систему объясняется одновременным воздействием на пресинаптические (ауторецептор) и постсинаптические D2-рецепторы. Последующее нейрохимическое изучение профиля рецепторного связывания арипипразола позволило расширить представление о фармакологическом действии препарата, включая частичное агонистическое влияние на 5-HT1A- и 5-HT2C-рецепторы и антагонизм к 5-HT2A-рецепторам. Краткосрочные рандомизированные контролируемые исследования арипипразола показали его эффективность и безопасность при лечении обострений у больных шизофренией и шизоаффективным психозом. Препарат практически не вызывает экстрапирамидных расстройств. При шизофрении отмечается расстройство глутаматергических функций. В настоящее время на клинических испытаниях находятся препараты d-серина и d-циклосерина — частичные агонисты аллостерического глицинового рецептора, входящего в структуру N-метил-d-аспартамового глутаминового рецептора. Применение полных агонистов способно запускать процесс токсического возбуждения и поэтому не рассматривается как перспективное направление поиска новых препаратов с влиянием на глутаматергические функции. В РФ для клинических испытаний разрешен препарат блонансерин, в случае успешных испытаний поступит новая оригинальная группа нейролептиков, не влияющая на дофаминергические структуры. На стадии клинических испытаний находится препарат бифепрунокс, классифицированный как частичный агонист D2- и 5-НТ1А-рецепторов и во многом сходный по фармакологическим свойствам с арипипразолом. В исследованиях на животных обнаружилась способность бифепрунокса редуцировать шизофреноподобные и экстрапирамидные симптомы. 15.2. АНТИДЕПРЕССАНТЫ Антидепрессанты — ЛС, применяемые для лечения депрессий. Депрессия (от лат. depressio — подавление, угнетение) — психическое расстройство, оказывающее существенное отрицательное влияние на социальную адаптацию, качество жизни, характеризующееся патологически 362 Часть II. Частная фармакология пониженным настроением (гипотимией) с пессимистической оценкой себя и своего положения в окружающей действительности (может сопровождаться суицидальными попытками), торможением интеллектуальной и моторной деятельности, снижением побуждений и соматовегетативными нарушениями. Механизмы депрессии окончательно не выяснены, наиболее разработана биохимическая теория возникновения этого заболевания, согласно которой при депрессивных состояниях снижается уровень моноаминергической активности (в частности, понижение содержания моноаминов норадреналина и серотонина в головном мозге), что приводит к измению чувствительности и увеличению числа рецепторов, воспринимающих воздействие этих нейромедиаторов и количество постсинаптических адренорецепторов (up-регуляция). В последнее время уделяется большое внимание изучению биохимических процессов в постсинаптической мембране (роли G-белков), связанных с развитием депрессий. Перспективно генетическое направление исследования причин возникновения депрессии, в пользу которого свидетельствует тот факт, что эта психическая патология может передаваться по наследству из поколения в поколение. Кроме того, в возникновении реактивных депрессий могут играть важную роль отрицательные эмоциональные и социальные факторы. Антидепрессанты, обладающие стимулирующим компонентом в спектре фармакологического действия, применяемые для лечения депрессии с признаками стойкого угнетения, называют тимеретиками. Препараты с седативным компонентом, используемые для лечения депрессий с признаками возбудительных процессов (ажитации и др.), называют тимолептиками. Антидепрессанты различают по механизму действия и подразделяю на следующие группы (рис. 15.10): ● ингибиторы нейронального захвата моноаминов: —средства неизбирательного действия (преимущественно угнетающие нейрональный захват серотонина и норадреналина); ✧ трициклические антидепрессанты (TCA): амитриптилин (триптизол♠), имипрамин, кломипрамин, пипофезин; —средства избирательного действия: ✧ ингибиторы нейронального захвата серотонина (SSRI): пароксетин, флуоксетин, флувоксамин, сертралин; ✧ ингибиторы нейронального захвата норадреналина (NaRI): мапротилин; ✧ ингибиторы нейронального захвата серотонина и норадреналина (SNRI): венлафаксин; (миансерин) (миртазапин) блокаторы – NASSA (амитриптилин) Рис. 15.10. Механизмы действия антидепрессантов (пояснения в тексте) (мапротилин) (ребоксетин) (венлафаксин) (моклобемид) (ниаламид) (тразодон) (пароксетин) 5-HT – SSRI (тианептин) (кломипрамин) Глава 15. Психотропные средства 363 364 Часть II. Частная фармакология ● ингибиторы моноаминоксидазы (МАО): —ингибиторы МАО неизбирательного действия (МАО-А и МАО-В): ниаламид; —ингибиторы избирательного действия (МАО-А): моклобемид; ● антидепрессанты из других групп: тианептин, миртазапин. 15.2.1. Ингибиторы нейронального захвата моноаминов Средства неизбирательного действия (неизбирательные ингибиторы нейронального захвата моноаминов) Препараты этой группы неизбирательно ингибируют обратный нейрональный захват моноаминов, преимущественно норадреналина и серотонина. Основные представители этой группы препаратов — трициклические антидепрессанты амитриптилин и имипрамин, обладающие антидепрессивными, седативными или психостимулирующими свойствами: ● седативные свойства наиболее выражены у амитриптилина, в то время как имипрамин на фоне угнетенного настроения может оказывать психостимулирующее действие; ● антидепрессивный эффект препаратов развивается через 2–3 нед постоянного приема, седативный эффект проявляется раньше антидепрессивного. Трициклические антидепрессанты конкурентно блокируют транспортную систему пресинаптических окончаний, обеспечивающую обратный нейрональный захват норадреналина, серотонина и дофамина, при этом медиаторы более продолжительное время находятся в синаптической щели, стимулируя постсинаптические рецепторы; через 2–3 нед плотность центральных адренорецепторов понижается (down-регуляция). Препараты этой группы обладают м-холиноблокирующей и α-адреноблокирующей активностью, поэтому для них характерны такие побочные эффекты, как нарушение аккомодации, сухость во рту, тахикардия, констипация, задержка мочеиспускания, снижение АД. Трициклические антидепрессанты нельзя назначать вместе с неизбирательными ингибиторами МАО, интервал между приемом этих препаратов должен составлять не менее 2 нед. Амитриптилин — один из наиболее эффективных препаратов, его антидепрессивное действие сочетается с выраженным седативным эффектом, значительной холиноблокирующей активностью и антигистаминными свойствами (рис. 15.11). Ингибируя обратный нейрональный захват моноаминов, преимущественно норадреналина и серотонина, препарат ос- Глава 15. Психотропные средства 365 лабляет или устраняет тревогу, ажитацию и собственно депрессивные проявления. Блокада пресинаптических М-холинорецепторов адренергических синапсов способна усиливать высвобождение норадреналина из адренергических нервных окончаний, что теоретически может усиливать антидепрессивное действие препарата. Амитриптилин назначают при эндоген- Рис. 15.11. Химическая струкных депрессиях, тревожно-депрессивных тура амитриптилина и невротических состояниях, кроме того, он обладает анальгетической активностью и применяется при хронических болях. Антидепрессивное действие развивается при систематическом приеме препарата в среднем через 2–3 нед. При приеме внутрь препарат хорошо всасывается, в значительной степени (более 90%) связывается с белками плазмы крови и тканей, метаболизируется в печени, выводится главным образом почками в виде метаболитов (период полувыведения — 10–26 ч). Побочные эффекты амитриптилина и противопоказания к его применению в ряде случаев обусловлены блокадой периферических М-холинорецепторов: сухость во рту, расширение зрачка, нарушение аккомодации, задержка мочеиспускания; нарушение сердечного ритма вследствие блокады α-адренорецепторов, возможна ортостатическая гипотензия. Иногда препарат вызывает сонливость, головокружение, аллергические реакции. Имипрамин (рис. 15.12) относят к группе трициклических антидепрессантов 1-го поколения, в отличие от амитриптилина обладает еще и стимулирующими свойствами. Седативный компонент, а также холиноблокирующее действие выражены в меньшей степени, чем у амитриптилина. Кломипрамин обладает антидепрессивным, седативным и психостимулирующим действиями, преимущественно блокирует обратный нейрональный захват серотонина, периферические α1-рецепторы и М-холинорецепторы. Пипофезин — отечественный препарат трициклических антидепрессантов, выгод- Рис. 15.12. Химическая струкно отличается от указанных выше препа- тура имипрамина 366 Часть II. Частная фармакология ратов отсутствием М-холиноблокирующей активности, оказывает умеренное антидепрессивное действие. Применяют при депрессиях легкой и средней тяжести. Средства избирательного действия Избирательные ингибиторы нейронального захвата серотонина Механизм антидепрессивного действия препаратов этой группы преимущественно связан с усилением серотонинергической активности в ЦНС в результате ингибирования обратного захвата серотонина нейронами мозга. Препараты этой группы отличаются от неизбирательных ингибиторов обратного захвата моноаминов прежде всего меньшим (вплоть до полного отсутствия) холиноблокирующим действием, а также незначительным влиянием на α-адренорецепторы и гистаминовые рецепторы. Это выгодно отличает их от препаратов неизбирательного действия меньшим количеством побочных эффектов, связанных с блокадой этих структур. Пароксетин (рис. 15.13) преимущественно блокирует обратный нейрональный захват серотонина, что приводит к усилению серотонинергических влияний в ЦНС. Препарат считается наиболее сильным ингибитором обратного захвата серотонина из всех известных препаратов с подобным механизмом действия. Антидепрессивное действие препарата при его регулярном приеме развивается в среднем через 10–14 дней, в сравнении с трициклическими антидепрессантами оказывает менее выраженное м-холиноблокирующее действие и незначительный антигистаминный эффект. На фоне лечения пароксетином наблюдают уменьшение состояния тревоги, депрессии и расстройства сна. Показаниями для применения препарата считают тяжелые депрессивные расстройства. При приеме внутрь препарат полностью всасывается из ЖКТ, но подвергается интенсивному метаболизму при первом прохождении через печень, что приводит к снижению его биодоступности (пища не влияет на биодоступность препарата). Назначают 1 раз в сутки; пик плазменной концентрации достигает через 2–8 ч, Рис. 15.13. Химическая струк- равновесная концентрация в плазме крови тура пароксетина устанавливается через 7–14 дней. Препарат Глава 15. Психотропные средства 367 распределяется в основном в тканях, и только 1% от введенной дозы находится в плазме крови. Побочные эффекты: тошнота, головная боль, кожный зуд, крайне редко — нарушение аккомодации, расширение зрачка; тахикардия, нарушение сердечного ритма, ортостатическая гипотензия. Флуоксетин по механизму действия близок к пароксетину: блокирует обратный нейрональный захват серотонина, мало влияет на нейрональный захват норадреналина (избирательность действия по отношению к нейрональному захвату серотонина ниже, чем у пароксетина). Антидепрессивный эффект при постоянном приеме препарата развивается через 1–3 нед. В отличие от трициклических антидепрессантов у флуоксетина практически отсутствует седативный эффект (оказывает некоторое психостимулирующее действие). Обладает незначительной м-холиноблокирующей активностью (меньше, чем у пароксетина), практически отсутствуют α-адреноблокирующие и антигистаминные свойства, в отличие от трициклических антидепрессантов характеризуется низкой токсичностью. Побочные эффекты: нарушение аппетита (анорексигенное действие), тошнота, акатизия (неусидчивость, беспокойство), нервозность, бессонница, головные боли, кожные высыпания. Флувоксамин практически не обладает седативным действием, не влияет на периферические холинергические и адренергические рецепторы. Сертралин наряду с антидепрессивным обладает седативным действием, не влияет на периферическую иннервацию. Препараты этой группы нельзя применять вместе с неизбирательными ингибиторами МАО, так как это может вызвать избыточное повышение концентрации серотонина («серотониновый синдром»), что приводит к спутанности сознания, психомоторным возбуждениям, мышечной ригидности, гипертермии, сердечно-сосудистым коллапсам. Интервал между приемом препаратов этой группы и приемом неизбирательных ингибиторов МАО должен составлять не менее 2 нед. Избирательные ингибиторы обратного нейронального захвата норадреналина Мапротилин (рис. 15.14) по фармакологическим свойствам близок к трициклическим антидепрессантам, блокирует обратный захват моноаминов. Однако в отличие от других антидепрессантов этой группы он сильнее тормозит обратный захват норадреналина пресинаптическими нервными окончаниями по сравнению с обратным захватом серотонина, что позволяет говорить об избирательном ингибировании обратного нейронального захвата норадреналина. 368 Часть II. Частная фармакология Холиноблокирующая и α-адреноблокирующая активность выражены умеренно, антидепрессивное действие мапротилина сопровождается анксиолитическим и умеренным седативным эффектами. Мапротилин медленно всасывается из ЖКТ (9–16 ч), биодоступность составляет 75%. На 88% препарат связывается с белРис. 15.14. Химическая струк- ками плазмы крови, метаболизируется в печени, выводится в основном почками (57% тура мапротилина в виде метаболитов, 24% — в неизмененном виде). Период полувыведения составляет в среднем 43–51 ч. Показания к применению: различные формы депрессий, в том числе сопровождающихся страхом, раздражительностью. Побочные эффекты обусловлены как периферическим действием препарата, в том числе связанным с м-холиноблокирующим действием (сухость во рту, запор, задержка мочеиспускания), так и центральным (головная боль, головокружение, парестезии, общая слабость, вялость, сонливость); наиболее частые признаки — кожная сыпь, зуд, крапивница. Игибиторы нейронального захвата серотонина и норадреналина Венлафаксин одновременно блокирует захват из синаптической щели норадреналина и серотонина, действие вызвано как самим препаратом, так и его метаболитом. Препарат вызывает сонливость, головокружение, сухость во рту, нарушения эйякуляции. 15.2.2. Ингибиторы моноаминоксидазы При участии МАО происходит окислительное дезаминирование моноаминов — норадреналина, серотонина, дофамина, что приводит к их инактивации. Известны две разновидности МАО — МАО-А и МАО-В. Значение ингибирования их неоднозначно для лечения депрессий: МАО-А преимущественно инактивирует норадреналин и серотонин, а МАО-В — дофамин. Селективные ингибиторы МАО-В (селегилин) не влияют на течение депрессии и используются для лечения паркинсонизма. Антидепрессивное действие ингибиторов МАО зависит от ингибирования МАО-А, в результате чего в тканях мозга повышается содержание норадреналина и серотонина. Глава 15. Психотропные средства 369 Антидепрессанты из группы ингибиторов МАО подразделяют на препараты неизбирательного (ингибируют МАО-А и МАО-В) и избирательного (в основном ингибируют МАО-А) действия. Ингибиторы моноаминооксидазы неизбирательного действия Ниаламид (рис. 15.15) необратимо и неизбирательно блокирует как МАО-А, так и МАО-В, оказывает антидепрессивное и психостимулирующее действия (может вызывать возбуждение, бессонницу). Рис. 15.15. Химическая структура ниаламида Антидепрессивное действие препарата развивается через 7–14 дней при его систематическом приеме вследствие необратимого ингибирования МАО. Активность этого фермента восстанавливается через 2 нед (время, необходимое для синтеза новых молекул фермента). Показания к применению: депрессивные состояния, сочетающиеся с вялостью, заторможенностью. Ниаламид уменьшает ощущение боли при стенокардии. Препарат применяется редко из-за большого количества серьезных побочных эффектов, связанных с неселективным ингибированием МАО в организме (как в ЦНС, так и в периферических тканях). Вследствие возбуждающего действия на ЦНС возникают беспокойство, бессонница, возможны тремор, судороги, снижение АД, ортостатическая гипотензия, гепатотоксическое действие. Ниаламид нельзя назначать вместе с трициклическими антидепрессантами (между приемом препаратов должен быть перерыв в 2–3 нед). Ниаламид усиливает прессорный эффект симпатомиметиков (эфедрина), амфетамина, тирамина, так как при ингибировании МАО в окончаниях симпатических нервов накапливается норадреналин, который высвобождается из них под действием вышеназванных веществ. В сыре и некоторых других пищевых продуктах (бананы, бобы сои, пиво, дрожжевые экстракты и др.) в большом количестве содержится тирамин. Употребление этих продуктов при приеме ниаламида приводит к гипертензивному кризу («сырный эффект»). 370 Часть II. Частная фармакология Ингибиторы моноаминооксидазы избирательного действия Моклобемид (рис. 15.16) — препарат избирательного действия в отношении ингибирования МАО-А с обратимым ингибированием этого фермента, в результате временно ингибируется метаболизм биогенных аминов, преимущественно серотонина Рис. 15.16. Химическая струк- и норадреналина. Препарат действует более тура моклобемида кратковременно, чем ниаламид. При сохранении достаточной активности препарат оказывает менее выраженные побочные эффекты, характерные для ингибиторов МАО неизбирательного и необратимого действия, не обладает ярко выраженными токсическими свойствами, в частности гепатотоксичностью. В сочетании с симпатомиметиками (в том числе при употреблении продуктов, содержащих тирамин) не повышает АД. 15.2.3. Антидепрессанты из других групп Тианептин (коаксил♠) (рис. 15.17) — антидепрессант с анксиолитическими свойствами, повышает обратный нейрональный захват серотонина нейронами коры головного мозга и гиппокампа. На обмен дофамина и норадреналина в ЦНС влияет относительно слабо. Тианептин нормализует поведение, повышает настроение и уменьшает соматические симптомы, способствует восстановлению нормального физиологического сна, улучшает концентрацию внимания, не оказывает отрицательного влияния на память, не подавляет либидо, к препарату не развивается привыкания. Тианептин быстро и полностью всасывается из ЖКТ (биодоступность 99%), связывается с белками плазмы крови приблизительно на 94%, метаболизируется в печени путем β-окисления и N-деметилирования. Период полувыведения составляет 2,5 ч, у пациентов пожилого возраста — 3,5 ч. Рис. 15.17. Химическая структура тианептина Глава 15. Психотропные средства 371 Выводится из организма почками, в основном в виде метаболитов (8% в неизмененном виде). Показания к применению: депрессивные состояния невротического и реактивного происхождения; тревожно-депрессивные состояния с соматическими жалобами; тревожно-депрессивные состояния у больных хроническим алкоголизмом. Побочные эффекты (возникают сравнительно редко и не носят угрожающего жизни характера): боли в эпигастральной области, сухость во рту, анорексия, тошнота, рвота, запоры, метеоризм; нарушение сна, сонливость, астения, головокружение, головная боль, тремор; тахикардия, экстрасистолия, загрудинные боли; затруднение дыхания; мышечные боли, боли в пояснице. Миртазапин (ремерон♠) блокирует пресинаптические α2-адренорецепторы в адренергических и серотонинергических синапсах, в результате чего увеличивается высвобождение норадреналина и серотонина из окончаний нервных волокон. Препарат оказывает седативное действие, что связывают с блокадой гистаминовых Н1-рецепторов, слабо блокирует холинорецепторы и периферические α1-адренорецепторы. Препарат применяется при эндогенных депрессиях, психомоторной заторможенности. Антидепрессивный эффект развивается через 1–2 нед. По эффективности миртазапин превосходит трициклические антидепрессанты. Побочные эффекты: повышение аппетита, повышение массы тела, чрезмерный седативный эффект, апатия, подергивание мышц, редко возникают ортостатическая гипотензия (вследствие блокады α2-адренорецепторов), агранулоцитоз, лейкопения, возможен синдром отмены. 15.3. НОРМОТИМИЧЕСКИЕ СРЕДСТВА (СОЛИ ЛИТИЯ) Препараты лития нормализуют настроение при мании (антиманиакальное действие) и применяют для предупреждения развития маниакальной и депрессивной симптоматики при маниакально-депрессивном психозе. Маниакально-депрессивный психоз — биполярное аффективное расстройство, характеризующееся чередованием фаз депрессии и мании со светлыми промежутками. Мания характеризуется болезненно повышенным возбуждением, чрезмерной веселостью (патологической), чрезмерной физической активностью, ускорением движений и речи. 372 Часть II. Частная фармакология Циклотимия — психическое расстройство, неглубокое и не резко выраженное, как при маниакально-депрессивном психозе, характеризуется хронической нестабильностью настроения с частой сменой сниженного и повышенного настроения. Наиболее распространенные нормотимические средства — соли лития и противоэпилептические средства — карбамазепин и вальпроевая кислота. Механизм действия солей лития до конца не выяснен. Считают, что ионы лития через быстрые натриевые каналы проникают в нейроны, накапливаются и блокируют трансмембранный транспорт ионов натрия, в итоге нарушаются процессы деполяризации мембран нейронов. По некоторым данным, литий влияет на обмен моноаминов в ЦНС, нарушает продукцию вторичных мессенджеров — диацилглицерола, инозитол1,4,5-трифосфата, цАМФ. Из солей лития наиболее часто используется лития карбонат, препарат хорошо всасывается из ЖКТ, лечебный эффект развивается медленно, через 2–3 нед. Лития карбонат применяется с профилактической целью (для предупреждения маниакальной и депрессивной фаз) при маниакально-депрессивном психозе, назначается длительное время (годами). Характеризуется избирательным действием в отношении маниакальной симптоматики, отсутствием выраженного седативного эффекта. Побочные эффекты: тошнота, жажда, полиурия, тремор, мышечная слабость. Препарат обладает малой широтой терапевтического действия; токсическое действие развивается при превышении терапевтической концентрации в 2–3 раза, в этом случае возникают рвота, атаксия, судороги, возможна коматозная реакция. Лития карбонат в основном выводится почками, при нарушении их выделительной функции резко повышается риск развития токсических эффектов, вследствие чего препарат противопоказан при патологии почек. Контемнол♠ — пролонгированная форма лития карбоната, выпускается в виде таблеток с замедленным высвобождением лития, отличается от лития карбоната низкой гигроскопичностью и слабым раздражающим действием на кишечник. Микалит♠ — отечественный препарат лития карбоната пролонгированного действия. Помимо лития карбоната, применяют также лития оксибутират♠ (лития оксибат). Карбамазепин и вальпроевую кислоту применяют для профилактики маниакально-депрессивных состояний в сочетании с солями лития при их недостаточной эффективности. Глава 15. Психотропные средства 373 15.4. АНКСИОЛИТИЧЕСКИЕ СРЕДСТВА (ТРАНКВИЛИЗАТОРЫ) Анксиолитические средства (транквилизаторы, от лат. tranquillare — спокойствие, покой) — ЛС, устраняющие чувство страха, тревогу, внутреннюю эмоциональную напряженность. Изначально отчетливые транквилизирующие свойства были открыты у бензодиазепинов, однако впоследствии оказалось, что подобными свойствами обладают препараты, относящиеся и к другим фармакологическим группам, например нейролептикам, р-блокаторам и другим. Это обстоятельство послужило причиной того, что всем препаратам, обладающим способностью устранять страх, тревогу, напряжение, было дано название анксиолитики, или анксиолитические средства (от лат. аnxius — тревожный, находящийся в страхе и Lysis — растворение, устранение). Классификация анксиолитических средств ● Производные бензодиазепина: —короткого действия (1–12 ч): триазолам, мидазолам; —средней продолжительности действия (12–40 ч): темазепам, нитразепам, алпразолам, феназепам♠, оксазепам; —длительного действия (40–250 ч): диазепам, флуразепам, хлордиазепоксид. ● Анксиолитические средства небензодиазепиновой структуры: —производные азаспиродекандиона: буспирон; —производные других химических групп: бензоклидин, мебикар♠. 15.4.1. Производные бензодиазепина Бензодиазепины оказывают анксиолитическое (транквилизирующее), седативное, снотворное, противосудорожное и миорелаксирующее действия. Механизм анксиолитического действия связан с усилением ГАМКергического торможения в ЦНС. Комплекс ГАМК-рецептор–Cl–-канал содержит аллостерический бензодиазепиновый рецепторный участок («бензодиазепиновый рецептор»). Его стимуляция бензодиазепинами вызывает конформационные изменения ГАМК-рецептора, способствуя повышению чувствительности к ГАМК, с усилением влияния ГАМК на проницаемость мембран нейронов для ионов хлора (увеличивается частота открывания хлорных каналов, в клетку поступает больше ионов хлора), возникает гиперполяризация мембраны с торможением нейрональной активности. 374 Часть II. Частная фармакология Место связывания бензодиазепинов определяется α- и γ-субъединицами рецепторного комплекса. Выделено несколько изоформ α-субъединицы с их расположением в различных отделах головного мозга. Рецепторы, содержащие α1-субъединицу, условно называют ω1-бензодиазепиновыми рецепторами (обусловливают седативный и амнестический эффект); ω2-рецепторы (но не ω3), включающие α2-, α3- или α5-субъединицу, обусловливают развитие анксиолитического действия. Бензодиазепины неизбирательно взаимодействуют со всеми типами бензодиазепиновых рецепторов, в то время как золпидем (см. главу «Снотворные средства») связывается только с ω1-рецепторами. Противосудорожное действие бензодиазепинов связано с подавлением эпилептогенной активности, что объясняют усилением тормозных ГАМК-ергических процессов в ЦНС. Некоторые препараты этой группы применяются в качестве противоэпилептических средств (см. главу «Противоэпилептические средства»). Механизм миорелаксирующего действия бензодиазепинов точно не установлен, однако считают, что в развитии этого эффекта важную роль играет угнетение спинальных полисинаптических рефлексов и нарушение их супраспинальной регуляции (центральное миорелаксирующее действие). Агонисты бензодиазепиновых рецепторов в небольших дозах оказывают седативное, а при увеличении доз — снотворное действие, потенцируют угнетающее действие на ЦНС средств для наркоза, наркотических анальгетиков, алкоголя (см. главы «Снотворные средства», «Средства для наркоза»), в больших дозах могут вызвать антероградную амнезию. Бензодиазепины применяют при невротических и неврозоподобных состояниях, сопровождаемых страхом, тревогой, и в качестве седативных (успокаивающие) и снотворных средств при бессоннице; широко используются для премедикации перед хирургическими операциями и для введения в наркоз, в качестве противоэпилептических средств и при заболеваниях с повышенным мышечным тонусом. Бензодиазепины вызывают эйфорию (приятное состояние, ощущение душевного комфорта) — такое изменение эмоционального статуса некоторые пациенты стремятся получить вновь, злоупотребляя препаратами этой группы. При систематическом применении формируется лекарственная зависимость (см. главу «Снотворные средства»), поэтому при неврозах и бессоннице бензодиазепины следует принимать не более 3–4 нед, а их применение вызывает привыкание, вследствие чего для достижения того же терапевтического эффекта дозу препарата приходится постоянно увеличивать. Глава 15. Психотропные средства 375 После прекращения приема препарата может возникнуть синдром отмены, этот феномен отмечают даже у пациентов с коротким периодом приема препарата. Тревога, депрессия, бессонница, тошнота, расстройство восприятия могут отмечаться на протяжении нескольких недель и даже месяцев после отмены бензодиазепинов. Для предупреждения синдрома отмены дозы препарата следует снижать постепенно. Бензодиазепины хорошо всасываются из ЖКТ (липофильные соединения), основной процесс происходит в двенадцатиперстной кишке, быстрее всасывается диазепам (рис. 15.18), его биотрансформация преимущественно происходит в печени (подвергается окислению, а затем конъюгирует с глюкуроновой кислотой). Метаболизация бензодиазепинов происходит в печени до активных метаболитов с более продолжительным анксиолитическим действием, чем исходное вещество. Так, Рис. 15.18. Химическая например, диазепам превращается в активный структура диазепама N-дезметилдиазепам, его t½ составляет 40–200 ч. Бензодиазепины и их метаболиты в основном хорошо связываются с белками плазмы крови (70–90%) и, будучи липофильными соединениями, в значительных количествах депонируются в жировой ткани. Выводятся главным образом почками, в меньшей степени — кишечником в виде метаболитов и конъюгатов. Различия в продолжительности действия бензодиазепинов обусловлены превращением некоторых из них в активные метаболиты. Так, например, диазепам (как и хлордиазепоксид) в процессе метаболизма превращается в активный метаболит дезметилдиазепам, который, в свою очередь, превращается в активный метаболит оксазепам. В настоящее время оксазепам используется в качестве лекарственного препарата. Бензодиазепины усиливают угнетащее действие на ЦНС других ЛС (например, средств для наркоза, что используется в анестезиологии). Диазепам и феназепам♠ оказывают наиболее сильное анксиолитическое и снотворное действия, причем феназепам♠ превосходит диазепам и хлордиазепоксид, по сравнению с этими препаратами обладает менее выраженным анксиолитическим действием. Медазепам — «дневной» транквилизатор, его седативное, снотворное и миорелаксирующее действия выражены в минимальной степени. Побочные эффекты: сонливость, головная боль, нарушение памяти, внимания, координации движений, особенно у пожилых людей. При употреблении бензодиазепинов не рекомендуют заниматься деятельностью, 376 Часть II. Частная фармакология требующей повышенного внимания (вождение автомобиля). Бензодиазепины угнетают ЦНС у новорожденных, проникают в грудное молоко и вызывают седативный эффект, что приводит к трудностям с кормлением и потере массы у ребенка. Передозировка бензодиазенинов приводит к развитию отравления, проявляющегося сонливостью, вялостью, атаксией. В тяжелых случаях возможно развитие комы. В этих случаях применяют специфический конкурентный антагонист бензодиазепинов — флумазенил (см. главу «Снотворные средства»). 15.4.2. Анксиолитические средства небензодиазепиновой структуры Производные азаспиродекандиона Буспирон отличается от других анксиолитиков по механизму действия, точка его приложения — серотонинергическая система мозга. Буспирон — частичный агонист 5-НТ1А-рецепторов, на бензодиазепиновые рецепторы не влияет и вследствие этого не оказывает (в отличие от бензодиазепинов) стимулирующего эффекта на ГАМК-ергическую систему. Буспирон — анксиолитик без седативной активности, не обладающий противосудорожной и миорелаксирующей активностью. Недостаток препарата — «отсроченный» анксиолитический эффект, который определяется через 2 нед от начала приема. Препарат практически не вызывает лекарственную зависимость, развитие привыкания выражено слабо. Производные других химических групп Бензоклидин (оксилидин♠) (рис. 15.19) в отличие от других транквилизаторов не обладает центральными миорелаксирующими свойствами, умеренно блокирует ганглии и адренорецепторы и обладает гипотензивным действием, поэтому может рассматриваться в качестве препарата выбора у больных гипертонической болезнью и с расстройствами мозгового кровообращения. Особенность мебикара♠ — умеренная транквилизирующая активность. Препатат не вызывает центрального миорелакРис. 15.19. Химическая структу- сирующего действия, применяется в качера бензоклидина гидрохлорида стве «дневного» транквилизатора. Глава 15. Психотропные средства 377 15.5. СЕДАТИВНЫЕ СРЕДСТВА Седативные средства усиливают процессы торможения в ЦНС, их применяют при вегетососудистых дистониях и неврозах. Согласно теории нервизма И.П. Павлова, нормальное течение психических процессов обусловлено равновесием между возбуждающими и тормозящими процессами в ЦНС и их достаточной для адекватного реагирования на внешние раздражители лабильностью. При наличии чрезмерных внешних раздражений возможен срыв нервной деятельности, связанный с преобладанием процессов возбуждения в ЦНС. В таких ситуациях показано назначение седативных препаратов. Механизм действия заключается в усилении процессов торможения в ЦНС, что приводит их в соответствие с патологически увеличенными процессами возбуждения. Среди седативных средств выделяют препараты брома и препараты растительного происхождения. Препараты брома представлены солями брома — калия бромидом и натрия бромидом. Они обладают умеренным седативным действием, хорошо всасываются в ЖКТ и медленно выводятся из организма (период полувыведения — 10–12 дней), в основном почками, но также кишечником, потовыми и молочными железами. Применяют при неврастении и других неврозах, повышенной раздражительности. Бромиды при длительном применении кумулируют в организме, для побочных эффектов, особенно при длительном применении, характерен комплекс патологических реакций, называемых бромизмом: общая заторможенность, сонливость, нарушение памяти, кожные высыпания. Препарат может оказывать раздражающее действие на слизистые оболочки, вызывая понос, кашель, конъюнктивит. Для ускорения выведения бромидов из организма назначают большие количества натрия хлорида (10–20 г/сут) и обильное питье. Более безопасны в применении седативные средства растительного происхождения — препараты валерианы лекарственной, пиона, пустырника и др. Они обладают большим терапевтическим действием, практически не вызывая серьезных побочных эффектов. Наиболее широко применяют препараты валерианы лекарственной: настой корневища с корнями валерианы, настойку валерианы, экстракт валерианы густой. Эти препараты оказывают выраженное седативное действие, усиливают действие снотворных средств, оказывают спазмолитическое действие. Применяют при неврозах, спазмах гладких мышц внутренних органов. 378 Часть II. Частная фармакология В качестве седативных средств используют комбинированные препараты, содержащие фенобарбитал. В состав валокордина♠ входят фенобарбитал, этилбромизовалерианат, масло мяты перечной, хмеля шишек масло, этанол. Валокордин♠оказывает седативное, умеренное сосудорасширяющее и спазмолитическое действия. Корвалол♠ близок по составу и аналогичен валокордину♠ по действию. Помимо этих препаратов применяют близкие по составу и действию комплексные препараты — валосердин♠, новопассит♠ и др. Сравнивать клиническую эффективность седативных средств довольно сложно в связи с тем, что на оценку препарата влияет не только состав входящих в него компонентов, но и субъективная оценка пациентом ценности употребляемого препарата. Приверженность к валокордину♠ может способствовать отрицанию больным терапевтической ценности аналогичного по составу препарата корвалол♠ и, наоборот, у лиц с более слабым типом нервной системы эффект седативных средств более ярок. 15.6. ПСИХОСТИМУЛЯТОРЫ Психостимуляторы оказывают стимулирующее влияние на функции головного мозга (преимущественно кора головного мозга), облегчают межнейронную передачу в виде повышения психической и двигательной активности. При применении психостимуляторов повышаются умственная работоспособность, концентрация внимания, увеличивается скорость рефлексов, физическая выносливость, снижаются усталость, потребность во сне и пище. В высоких дозах психостимуляторы обладают аналептическим (пробуждающим) действием. Выделяют следующие группы психостимуляторов, различающиеся по химической структуре и механизму действия: ♠ ); ● производные фенилалкиламина: амфетамин (фенамин ♠ ● производные сиднонимина: мезокарб (сиднокарб ); ● производные метилксантина: кофеин. 15.6.1. Производные фенилалкиламина Амфетамин (рис. 15.20) представляет собой рацемическую смесь правои левовращающего изомеров (левовращающий изомер относительно неактивен). Механизм психостимулирующего действия амфетамина связывают с его способностью вызывать высвобождение норадреналина и дофамина из пресинаптических окончаний. Кроме того, амфетамин несколько Глава 15. Психотропные средства 379 уменьшает нейрональный захват норадреналина и дофамина и обладает некоторой ингибирующей активностью в отношении МАО. Препарат вызывает эйфорию, повышает физическую и умственную работоспособность (однако точность выполнения работы, Рис. 15.20. Химическая как правило, снижается), устраняет усталость, структура амфетамина уменьшает аппетит (снижение чувства голода связано с влиянием на пищевой центр в гипоталамусе). В ряде случаев, особенно под действием высоких доз, могут наступать чувство тревоги, нервозность, физическое напряжение и ряд других эффектов. Результат регулярного употребления амфетамина — сравнительно быстрое истощение энергетических ресурсов организма и развитие связанных с этим серьезных негативных процессов. Существуют отличия в реакции на амфетамин у детей и взрослых: у детей развивается седативное действие, а у взрослых — психостимулирующее. Вегетативное действие препарата — повышение АД, тахикардия, повышение потребности миокарда в кислороде, что связано с повышением высвобождения норадреналина из окончаний симпатических нервных волокон в периферических синапсах и усилением в связи с этим стимулирующего действия на адренорецепторы сердца и сосудов. Амфетамин легко всасывается при любом способе введения, частично метаболизируется в печени и элиминируется почками главным образом в неизмененном виде (период полувыведения — 12 ч). Подкисление мочи значительно ускоряет элиминацию препарата, эту особенность кинетики используют при проведении мероприятий по устранению передозировки или предупреждения опасных побочных эффектов. При передозировке препарата возникают возбуждение, бессонница, тахикардия, повышение АД, возможны аритмии. Препарат кумулирует в организме, вызывает привыкание и лекарственную зависимость (психическую и физическую). При отмене препарата после его длительного применения возникает абстинентный синдром. Возможные показания к применению амфетамина: нарколепсия и другие состояния, сопровождающиеся сонливостью, вялостью, повышенной утомляемостью. В настоящее время применение амфетамина ограничено. 15.6.2. Производные сиднонимина Препараты этой группы обладают достаточно выраженной, но по сравнению с амфетамином меньшей психостимулирующей активностью, од- 380 Часть II. Частная фармакология новременно обладают менее резким отрицательным влиянием на вегетативные функции организма, в меньшей степени повышают АД. Основной представитель этой группы препаратов в нашей стране — мезокарб. Мезокарб (рис. 15.21) вытесняет норадреналин из везикул в синаптическую щель, вызывая активацию норадренергических влияний в ЦНС. Периферическое симпатомиметическое действие препарата по сравнению с амфетамином выражено незначительно, поэтому он мало влияет на гемодинамику. ± Рис. 15.21. Химическая структура мезокарба Действие мезокарба развивается постепенно, препарат оказывает мягкое психостимулирующее действие без начальной стадии эйфории и последующего истощения энергетических ресурсов организма. Лекарственная зависимость к препарату развивается медленнее, чем к производным фенилалкиламина. Препарат применяют при общей слабости, астении, нарколепсии (патологической сонливости), некоторых субдепрессивных состояниях. Побочные эффекты: беспокойство, повышенная раздражительность (не следует назначать на ночь); возможно повышение АД; гипертермия, потеря аппетита, запоры, аллергические реакции. 15.6.3. Производные метилксантина Рис. 15.22. Химическая структура кофеина К метилксантинам относят алкалоиды — кофеин, теобромин, теофиллин. Наибольшее влияние на ЦНС оказывает кофеин. Кофеин (рис. 15.22) — алкалоид, содержащийся в листьях чая, семенах кофе, какао, орехах кола и других растениях. Оказывает сложное влияние на содержание внутриклеточного кальция, аденозиновые рецепторы и фосфодиэстеразу, разрушающую цАМФ. Кофеин ингибирует фосфоди- Глава 15. Психотропные средства 381 эстеразу цАМФ, повышая концентрацию цАМФ в тканях мозга, сердца и других органов. В последнее время выделяют механизм действия препаратов этой группы, связанный с блокадой аденозиновых рецепторов. Аденозин, стимулируя соответствующие α-рецепторы, вызывает гиперполяризацию клеток миокарда и ЦНС. Этот эффект имеет защитный характер, поскольку аденозин накапливается при гипоксии, а кофеин блокирует аденозиновые рецепторы и устраняет процесс торможения, связанный с действием аденозина. Кофеин стимулирует кору головного мозга, с чем связывают его психостимулирующее действие. Для него характерно повышение умственной и физической работоспособности, устранение чувства усталости, уменьшение потребности во сне, однако действие кофеина зависит от типа нервной деятельности (у некоторых людей кофеин усиливает процессы торможения, вызывая сонливость). Кофеин оказывает не только психостимулирующее, но и аналептическое действие, связанное со стимулирующим влиянием на центры продолговатого мозга — дыхательный и сосудодвигательный (см. главу «Аналептики»). Кофеин оказывает сложное влияние на сердечно-сосудистую систему, которое зависит от соотношения его центральных и периферических эффектов, а именно — прямое стимулирующее влияние на миокард (периферическое действие). Однако, возбуждая центры блуждающего нерва, кофеин вызывает брадикардию (центральное действие). Результат применения зависит от преобладающего влияния (центрального или периферического). В больших дозах кофеин, как правило, вызывает тахикардию, увеличивает потребность миокарда в кислороде, может вызвать аритмии. На тонус сосудов кофеин оказывает неоднозначное действие: стимуляция сосудодвигательного центра приводит к повышению сосудистого тонуса (центральное действие), а непосредственное действие на гладкие мышцы сосудов вызывает их расслабление. При этом сосудорасширяющее действие происходит не на все сосудистые области: кофеин расширяет коронарные сосуды и сосуды почек, но суживает сосуды других внутренних органов. Сосуды мозга под влиянием кофеина суживаются (особенно если они были расширены) — с этим связывают благоприятное действие препарата при мигрени. Кофеин расслабляет гладкие мышцы других органов, в том числе бронхов. Препарат несколько повышает диурез вследствие угнетения реабсорбции электролитов в почечных канальцах. 382 Часть II. Частная фармакология При гипотензии кофеин повышает АД, способствуя его нормализации. Это действие связано со стимуляцией сосудодвигательного центра; нормальное давление кофеин практически не изменяет или незначительно его повышает. Препарат применяют в качестве легкого психостимулятора для повышения работоспособности, устранения усталости, сонливости, при гипотензии. При мигрени назначают в составе комплексных препаратов в комбинации с алкалоидами спорыньи («Кофетамин»♠). Кофеин вместе с анальгетиками входит в состав таблеток «Пиркофен»♠, «Цитрамон»♠», «Седальгин»♠, «Колдрекс»♠, применяемых в качестве болеутоляющих и жаропонижающих средств. Кофеин выпускается в виде кофеин-бензоата натрия♠ в таблетках и растворах для парентерального введения; в комбинации с анальгетиками входит в состав комплексных таблеток «Пенталгин»♠. Побочные эффекты: бессонница, беспокойство, возбуждение, тошнота, рвота, осложнения со стороны сердечно-сосудистой системы (тахикардия, аритмии); при длительном применении возможно медленное развитие привыкания и психической зависимости (теизм). Противопоказания: выраженная гипертензия, бессонница, атеросклероз, глаукома. 15.7. НООТРОПНЫЕ СРЕДСТВА Термин «ноотропные средства» происходит от двух греческих слов: noos — ум, разум, мышление и tropos — стремление. В названии отражена направленность действия препаратов этой группы на высшие интегративные функции головного мозга — интеллектуальные и мнестические. Ноотропные средства (рис. 15.23) составляют особую группу нейропсихотропных препаратов. Их специфический эффект определяется способностью улучшать процессы памяти и обучения, когнитивные функции, нарушенные при различных заболеваниях и агрессивных воздействиях. Рис. 15.23. Химические структуры гамма-аминомасляной кислоты и пирацетама Глава 15. Психотропные средства 383 Целый ряд ноотропных средств по химической структуре имеет сходство с ГАМК. Пирацетам (ноотропил♠) — производное пирролидина, циклическое производное ГАМК. Препарат обладает антигипоксическими свойствами и умеренным противосудорожным действием. Ноотропное действие препарата связано с улучшением метаболических процессов в нервной клетке: повышением синтеза фосфолипидов (ФЛ) и белка, активацией аденилатциклазы, повышением уровня АТФ, усилением утилизации глюкозы в мозге, усилением микроциркуляции без сосудорасширяющего действия. С пирролидиновым кольцом связывают способность препарата стимулировать рецепторы для взаимодействия с нейропептидами (вазопрессин, субстанция Р, адренокортикотропный гормон). Улучшение мнестических функций ноотропила♠ связано с активацией AMPA-рецепторов. Препарат хорошо всасывается в кишечнике (биодоступность 95%), не связывается с белками плазмы крови. В отличие от ГАМК пирацетам хорошо проходит через ГЭБ, а также другие гистогематические барьеры, в том числе через плацентарный. Препарат избирательно накапливается в тканях коры головного мозга; выводится преимущественно в неизмененном виде почками. При почечной недостаточности дозу препарата следует корректировать в сторону уменьшения. Действие препарата развивается постепенно, поэтому он вводится внутрь или парентерально длительно (1–6 мес). Пирацетам применяют при деменции (слабоумии), развившейся вследствие нарушения мозгового кровообращения и дегенеративных поражений головного мозга (атеросклероз сосудов мозга, постинсультный период, черепно-мозговые травмы), и других заболеваниях со снижением памяти, концентрации внимания, а также при хроническом алкоголизме для снятия абстинентного синдрома и в комплексной терапии эпилепсии; в педиатрии — при умственной отсталости у детей. Побочные эффекты: головная боль, тошнота, нервозность, раздражительность, расстройства сна, функции ЖКТ (диарея, рвота, тошнота). Однако в большинстве случаев препарат хорошо переносится. Никотиноил гамма-аминомасляная кислота (пикамилон♠) (рис. 15.24) — комбинированный препарат ноотропных средств с витаминами, представляет собой молекулу ГАМК, соединенную с никотиновой кислотой. Введение никотиновой кислоты в комплекс позволяет получить вазодилатирующий эффект, улучшить мозговое кровообращение и доставку ГАМК в мозг. Кроме того, никотиновая кислота обладает гиполипидемическим действием, что может оказывать положительное действие при атеросклерозе сосудов мозга. Препарат следует назначать с осторожностью 384 Часть II. Частная фармакология Рис. 15.24. Химическая структура никотиноил гамма-аминомасляной кислоты больным с повышенной чувствительностью к никотиновой кислоте. Среди побочных эффектов отмечают аллергические реакции, тошноту, головную боль, возбуждение, раздражительность. Гопантеновая кислота (пантогам♠) и пиритинол (пиридитол♠, энцефабол♠) обладают антигипоксическим действием, имеют широкое терапевтическое действие, оказывают благоприятное влияние на метаболические процессы в тканях мозга. Пиритинол представляет собой остатки двух молекул пиридоксола, соединенные между собой дисульфидным мостиком, поэтому его следует назначать с осторожностью пациентам с гиперчувствительностью к пиридоксину. Мемантин (Акатинол Мемантин♠) — ЛВ для лечения болезни Альцгеймера. По механизму действия является неконкурентным низкоаффинным антагонистом NMDA-рецепторов. Обладая большим сродством к этим рецепторам, чем ионы магния, мемантин укорачивает входящий деполяризующий ток ионов кальция, что снижает нейротоксическое действие глутамата. Наряду с этим показано его взаимодействие с никотиновыми рецепторами головного мозга (тип alpha-7nAChR) и 5-НТ3рецепторами, возможно выполняющими аналогичную с NMDAрецепторами функцию. Препарат обладает умеренной активностью в отношении восстановления мнестических возможностей, поведения и возможности выполнять действия по самообслуживанию. Препарат рекомендован при умеренных и тяжелых формах болезни Альцгеймера. Среди побочных эффектов выделяют заторможенность, ажитацию, бессонницу, галюцинации, гипертензию, цистит. В настоящее время исследуется возможная эффективность мемантина при других заболеваниях ЦНС. Вопросы и задания для самоконтроля 1. Укажите «типичные» антипсихотические средства: а) хлорпромазин; б) арипипразол; Глава 15. Психотропные средства 385 в) галоперидол; г) оланзапин; д) хлорпротиксен. 2. Укажите «атипичные» антипсихотические средства: а) клозапин; б) флуфеназин; в) рисперидон; г) дроперидол; д) тиоридазин. 3. Экстрапирамидные расстройства не типичны для: а) галоперидола; б) арипипразола; в) хлорпромазина; г) рисперидона; д) оланзапина. 4. «Типичные» антипсихотические средства: а) вызывают антипсихотическое действие, сопровождающееся экстрапирамидными расстройствами; б) эффективны в отношении «негативных» признаков психоза; в) вызывают гиперпролактинемию; г) обладают противорвотным действием; д) потенцируют действие наркотических аналгетиков. 5. Хлорпромазин: а) обладает более выраженным антипсихотическим дейстием, чем галоперидол; б) не влияет на вегетативную нервную систему; в) вызывает галакторею*; г) обладает м-холиноблокирующей активностью; д) не вызывает экстрапирамидные расстройства. 6. К трициклическим антидепрессантам относят: а) миртазапин; б) амитриптилин; в) моклобемид; г) флуоксетин; д) имипрамин. 386 Часть II. Частная фармакология 7. Избирательно блокирует МАО-А: а) кломипрамин; б) моклобемид; в) ниаламид; г) амитриптилин; д) имипрамин. 8. Механизм действия ниаламида: а) избирательная блокада МАО-А; б) блокада обратного нейронального захвата норадреналина; в) блокада обратного нейронального захвата серотонина; г) неизбирательная блокада МАО-А и МАО-В; д) блокада пресинаптических α2-адренорецепторов. 9. Флуоксетин: а) ингибитор МАО-А; б) нарушает обратный нейрональный захват серотонина; в) повышает аппетит; г) вызывает акатизию; д) оказывает психостимулирующий эффект. 10. Амитриптилин: а) избирательно нарушает обратный нейрональный захват норадреналина; б) неизбирательно блокирует МАО; в) вызывает сухость во рту; г) снижает артериальное давление; д) способен вызвать аритмию. 11. Бензодиазепины: а) блокируют потенциалозависимые натриевые каналы; б) активируют калиевые каналы; в) связываются с ГАМКА-рецепторами; г) связываются с аллостерическими бензодиазепиновыми рецепторами; д) увеличивают частоту открывания каналов для хлора. 12. Бензодиазепиновые рецепторы: а) являются местами связывания ГАМК; б) относят к аллостерическим рецепторам; Глава 15. Психотропные средства в) увеличивают сродство ГАМК-рецептора к ГАМК; г) блокируются налоксоном; д) блокируются флумазенилом. 13. Для бензодиазепинов характерно: а) психостимулирующее действие; б) анксиогенное действие; в) периферическое мышечнорасслабляющее действие; г) снотворное действие; д) лекарственная зависимость. 14. К бензодиазепинам длительного действия относят: а) диазепам; б) бензоклидин; в) темазепам; г) зопиклон; д) хлордиазепоксид. 15. Укажите анксиолитики небензодиазепиновой структуры: а) мидазолам; б) триазолам; в) буспирон; г) флумазенил; д) мебикар. 387 Глава 16 АНАЛЕПТИКИ Аналептические средства (аналептики) — группа ЛС, оказывающая оживляющее (аналептическое) действие путем стимуляции жизненно важных центров продолговатого мозга (дыхательного и сосудодвигательного). Препараты с аналептическим действием имеют различную тропность к отдельным структурам ЦНС. К аналептикам относят такие препараты, как никетамид (кордиамин♠), бемегрид, камфора, кофеин, стрихнин. Кофеин имеет преобладающее влияние на кору головного мозга, в связи с чем его используют и как аналептик, и как психостимулирующее средство. Стрихнин (алкалоид из семян чилибухи Strychnos nux vomica) действует на центры продолговатого мозга и на спинной мозг. В настоящее время стрихнин имеет ограниченное применение. Аналептики понижают порог возбудимости дыхательного и сосудодвигательного центров, в результате чего возрастает чувствительность к гуморальным и нервным раздражителям. Этот эффект особенно ярко заметен на фоне угнетения этих центров, например при отравлении снотворными наркотического типа действия, этиловым спиртом и другими средствами, угнетающими ЦНС, при асфиксии новорожденных и др. Кроме того, аналептики проявляют функциональный антагонизм к веществам наркотического типа действия и могут способствовать выведению больных из состояния наркоза в послеоперационном периоде. Однако этот эффект возникает при применении достаточно больших доз, которые могут вызвать судороги (токсическое действие). Аналептики в основном имеют малую широту терапевтического действия. Увеличение дозы приводит к генерализации процессов возбуждения в ЦНС, повышению рефлекторной возбудимости. При передозировке препаратов наиболее опасное осложнение — судороги. При этом препараты, действующие преимущественно на головной мозг (бемегрид, камфора), вызывают клонические судороги, а действующие на спинной мозг (стрихнин) — тонические судороги. По этой причине аналептики иногда называют судорожными ядами. В высоких дозах вещества, дей- Глава 16. Аналептики 389 ствующие преимущественно на головной мозг, могут вызывать тоникоклонические судороги. По механизму действия различают: ● препараты прямого действия (бемегрид, камфора) — непосредственно стимулируют дыхательный и сосудодвигательный центры; ● препараты рефлекторного (непрямого) действия (цитизин, лобелин) — возбуждают H-холинорецепторы синокаротидной зоны, импульсы от которых по афферентным путям поступают в продолговатый мозг и стимулируют его центры. Эти препараты неэффективны при угнетении рефлекторной возбудимости дыхательного центра средствами для наркоза, снотворными наркотического типа действия (например, барбитуратами). Лобелин и цитизин могут стимулировать дыхание при асфиксии новорожденных, отравлении угарным газом. Препараты вводят внутривенно; действия — никетамид. ● препараты смешанного Бемегрид (этимидин♠) — препарат прямого действия, применяемый при легкой степени отравления барбитуратами, снотворными наркотического типа действия, некоторыми средствами для наркоза. При тяжелых отравлениях барбитуратами бемегрид противопоказан. Побочные эффекты: тошнота, рвота, мышечные подергивания, судороги. Препарат противопоказан при психомоторном возбуждении. Выпускают в виде 0,5% раствора в ампулах по 10 мл. Сульфокамфокаин♠ — комплексное соединение, состоящее из сульфокамфорной кислоты и новокаина♠. Механизм действия сульфокамфокаина♠ аналогичен камфоре, но в отличие от нее он растворяется в воде и быстро всасывается при подкожном и внутримышечном введении (при этом не вызывает образования инфильтратов). Препарат оказывает положительное влияние на вентиляцию легких, улучшает легочный кровоток и функцию миокарда. Сульфокамфокаин♠ применяют при угнетении дыхательного и сосудодвигательного центров (при инфекционных заболеваниях, кардиогенном шоке). Кофеин непосредственно возбуждает дыхательный и сосудодвигательный центры. Может использоваться в качестве аналептического средства (как правило, в высоких дозах) или психостимулятора (см. раздел «Психостимуляторы»). Кофеин-бензоат натрия♠ по фармакологическим свойствам и показаниям аналогичен кофеину, но лучше растворим в воде и быстрее выводится из организма. 390 Часть II. Частная фармакология Никетамид (кордиамин ♠) — 25% раствор диэтиламида никотиновой кислоты, относится к аналептикам смешанного типа действия (прямого и рефлекторного одновременно). С одной стороны, никетамид Рис. 16.1. Химическая струк- (рис. 16.1) оказывает аналептическое действие, непосредственно возбуждая дыхательтура никетамида ный и сосудодвигательный центры, особенно при их сниженном тонусе. С другой стороны, его аналептическое действие дополняется рефлекторным — с хеморецепторов каротидных клубочков. Показания к применению: нарушения кровообращения, понижение сосудистого тонуса и ослабление дыхания у больных с инфекционными заболеваниями, коллапс и асфиксия (в том числе асфиксия новорожденных), шоковые состояния. Никетамид вводят парентерально или по 15–40 капель на прием 2–3 раза в день за 30–40 мин до еды, запивая достаточным количеством жидкости. Препарат хорошо всасывается при любом пути введения, но вызывает болезненность в месте инъекции. Побочные эффекты: мышечные подергивания, тревожность, рвота, аритмии. При передозировке препарата возникают тонико-клонические судороги. Препарат противопоказан при предрасположенности к судорожным реакциям, эпилепсии. Стрихнина нитрат♠ в первую очередь действует на спинной мозг. Механизм его действия связан с ослаблением тормозного влияния нейромедиатора глицина на передачу возбуждения в спинном мозге, по-видимому, в результате блокады глициновых рецепторов. Устраняя постсинаптическое торможение, стрихнин оказывает таким образом стимулирующий эффект на спинной мозг. Повышение рефлекторной возбудимости приводит к тому, что рефлекторные реакции становятся более генерализованными, и при введении препарата в больших дозах различные раздражители вызывают появление болезненных тетанических судорог. Применение препарата ограничено в связи с небольшой широтой терапевтического действия. Препарат оказывает тонизирующее действие при понижении процессов обмена, быстрой утомляемости, гипотонии, парезах, параличах, в терапевтических дозах стрихнин оказывает стимулирующее действие на органы чувств, улучшает зрение, слух, тактильную чувствительность. Препарат способен к кумуляции. Глава 16. Аналептики 391 Побочные эффекты: тошнота, рвота, мышечные подергивания, судороги. Ослабление действия нейромедиатора ГАМК — механизм действия пикротоксина, который блокирует хлорные каналы, связанные с ГАМКрецепторами, препятствуя гиперполяризации мембраны и устраняя тормозные эффекты ГАМК в ЦНС. Избирательность действия аналептиков на различных уровнях ЦНС была использована при разработке аналептической смеси (Кудрин А.Н.). В состав этой смеси входят: кофеин, преимущественно стимулирующий кору головного мозга; пикротоксин, стимулирующий промежуточный мозг; коразол, обладающий аналептическим действием на продолговатый мозг, а также стрихнин, облегчающий передачу импульсов в спинном мозге. Смесь более эффективна, чем любой из входящих в нее компонентов, за счет эффекта суммирования действия на разных уровнях организации ЦНС. Вопросы и задания для самоконтроля 1. Аналептиками являются: а) никетамид; б) налоксон; в) кофеин; г) флумазенил; д) кетамин. 2. Бемегрид противопоказан при тяжелых отравлениях снотворными, потому что: а) вызывает судороги; б) снижает АД; в) увеличивает потребление кислорода нейронами головного мозга до устранения гипоксии; г) вызывает рвоту; д) неизбирательно одновременно активирует все нейроны ЦНС. СРЕДСТВА, ВЛИЯЮЩИЕ НА ФУНКЦИИ ИСПОЛНИТЕЛЬНЫХ ОРГАНОВ И СИСТЕМ Глава 17 СРЕДСТВА, ВЛИЯЮЩИЕ НА ФУНКЦИИ ОРГАНОВ ДЫХАНИЯ Выделяют следующие группы ЛС, влияющих на функции органов дыхания: ● стимуляторы дыхания; ● противокашлевые средства; ● отхаркивающие средства; ● бронхолитические средства; ● препараты сурфактантов. Система органов дыхания представлена воздухоносными путями — носовой полостью, носоглоткой, гортаноглоткой, гортанью, трахеей, бронхами, а также легочными альвеолами, в которых происходит газообмен. Центры, регулирующие функции системы органов дыхания, — дыхательный центр, центр кашлевого рефлекса и ядро блуждающего нерва. Эфферентная иннервация дыхательных мышц осуществляется соматической нервной системой по двигательным нервам через Nм-холинорецепторы, расположенные на мышечных волокнах. Дыхательный акт осуществляется при сокращении поперечнополосатых дыхательных мышц (диафрагма и межреберные мышцы). Гладкие мышцы бронхов и бронхиальных желез получают парасимпатическую эфферентную иннервацию из центра блуждающего нерва через М3-холинорецепторы. Кроме того, на гладких мышцах бронхов расположены β2-адренорецепторы, которые не иннервируются, а имеют внесинаптическую локализацию и стимулируются циркулирующим в крови адреналином. Секреторные клетки слизистой оболочки дыхательных путей имеют симпатическую и парасимпатическую иннервацию. Регуляция тонуса сосудов бронхов осуществляется симпа- Глава 17. Средства, влияющие на функции органов дыхания 393 тическими волокнами через α1- и β2-рецепторы гладкомышечных клеток сосудов. Афферентные импульсы от органов дыхания поступают в ЦНС по чувствительным волокнам блуждающего и языкоглоточного нервов. Основные показания к применению ЛС, влияющих на функции органов дыхания: ● угнетение дыхания (применяют стимуляторы дыхания и антагонисты средств, угнетающих дыхание); ● кашель (применяют отхаркивающие и противокашлевые средства); ● бронхиальная астма (применяют бронхолитики, средства с противовоспалительным и противоаллергическим действиями); ● дыхательная недостаточность и дистресс-синдром (применяют препараты сурфактантов). 17.1. СТИМУЛЯТОРЫ ДЫХАНИЯ Стимуляторы дыхания — группа ЛС, применяемых при угнетении дыхания. По механизму действия выделяют 3 группы стимуляторов дыхания: ● центрального действия — бемегрид, кофеин (см. главу «Аналептические средства»); ● рефлекторного действия — лобелин, цитизин (см. раздел «Холиномиметики»); ● смешанного типа действия — никетамид (кордиамин♠) (см. главу «Аналептические средства»). Стимуляторы дыхания центрального типа действия непосредственно стимулируют дыхательный центр. Эти соединения (никетамид, бемегрид, кофеин) называют аналептиками, они уменьшают угнетающее действие на дыхательный центр снотворных средств, средств для наркоза. Их применяют при легких степенях отравления снотворными средствами наркотического действия, а также для ускорения выведения из наркоза в послеоперационном периоде. Вводят внутривенно или внутримышечно. При тяжелых отравлениях веществами, угнетающими дыхательный центр, аналептики противопоказаны, так как в этом случае дыхание не восстанавливается и повышается потребность тканей мозга в кислороде, что усиливает гипоксию. Стимуляторы дыхания рефлекторного действия (лобелин, цитизин) активируют H-холинорецепторы каротидных клубочков, усиливают афферентную импульсацию, поступающую в продолговатый мозг к дыхательному центру, и повышают его активность. Эти препараты неэффективны 394 Часть II. Частная фармакология при нарушении возбудимости дыхательного центра, т.е. при угнетении дыхания снотворными средствами, средствами для наркоза. Их применяют при асфиксии новорожденных, отравлении угарным газом (вводят внутривенно). В качестве стимулятора дыхания смешанного типа действия, который кроме непосредственного влияния на дыхательный центр оказывает стимулирующее воздействие на хеморецепторы каротидных клубочков, ингаляционно применяют карбоген♠ (смесь 5–7% углекислого газа и 93–95% кислорода). Стимулирующее действие карбогена♠ на дыхание развивается в течение 5–6 мин. Эффект карбогена♠ обусловлен содержащимся в нем углекислым газом. Стимуляторы дыхания применяют нечасто. При гипоксических состояниях обычно используют вспомогательную или искусственную вентиляцию легких. Нарушение дыхания может быть обусловлено передозировкой препаратов, угнетающих функции ЦНС (опиоидных анальгетиков и агонистов бензодиазепиновых рецепторов). При отравлении опиоидными (наркотическими) анальгетиками угнетение дыхания — это результат угнетения дыхательного центра за счет стимуляции μ-опиоидных рецепторов нейронов этого центра. В таком случае для восстановления дыхания применяют специфические антагонисты μ-опиоидных рецепторов: налоксон (вводят внутривенно, действует до 1 ч) и налтрексон (можно принимать внутрь, действует до 36 ч). В случае отравления бензодиазепинами для восстановления дыхания применяют антагонист бензодиазепиновых рецепторов — флумазенил (анексат♠). Он эффективен также при передозировке золпидема (небензодиазепиновый агонист бензодиазепиновых рецепторов). Препарат вводят внутривенно капельно. 17.2. ПРОТИВОКАШЛЕВЫЕ СРЕДСТВА Кашель — защитный рефлекс, возникающий в ответ на раздражение слизистой оболочки дыхательных путей. При кашле из дыхательных путей удаляется раздражающий агент — мокрота (избыточный секрет бронхиальных желез) или инородное тело. Противокашлевые средства, действуя на разные звенья кашлевого рефлекса, уменьшают частоту и интенсивность кашля. Кашлевой рефлекс инициируется с чувствительных рецепторов слизистой оболочки бронхов и верхних дыхательных путей. Афферентная им- Глава 17. Средства, влияющие на функции органов дыхания 395 пульсация поступает в продолговатый мозг (от бронхов — по афферентным волокнам блуждающего нерва, от гортани — по афферентным волокнам языкоглоточного нерва). Увеличение афферентации приводит к стимуляции центра кашлевого рефлекса. Эфферентная импульсация от центра кашлевого рефлекса достигает дыхательных мышц (межреберных и диафрагмы) по соматическим двигательным волокнам и вызывает их сокращения, проявляющиеся форсированными выдохами. При воспалительных заболеваниях дыхательных путей, сопровождающихся повышенной секрецией бронхиальных желез (бронхит, трахеит), кашель способствует дренированию бронхов и ускоряет процесс выздоровления (продуктивный кашель). В подобных случаях целесообразным представляется не подавление кашля противокашлевыми средствами, а назначение средств, облегчающих отделение мокроты (отхаркивающие средства). Однако при некоторых заболеваниях (хронические воспалительные заболевания, плеврит, злокачественные новообразования) кашель не выполняет защитных функций (непродуктивный кашель) и изнуряет больного, возникая в ночное время. В таких случаях целесообразно назначение противокашлевых средств. Противокашлевые средства различают по локализации и механизму действия. ● Противокашлевые средства центрального действия: —средства с наркотическим действием — кодеин, этилморфин; —ненаркотические средства — глауцин, окселадин. ● Противокашлевые средства периферического действия: преноксдиазин. Механизм противокашлевого действия кодеина и этилморфина обусловлен снижением возбудимости центра кашлевого рефлекса и дыхательного центра за счет стимуляции опиоидных рецепторов в продолговатом мозге. При этом прямой корреляции между способностью препаратов угнетать дыхание и кашлевой центр не отмечают. Стимуляция опиоидных рецепторов в мезолимбической и мезокортикальной системах мозга приводит к развитию эйфории и, как следствие, развитию лекарственной зависимости (см. главу «Анальгезирующие средства»). В связи с последним свойством (наркогенный потенциал) отпуск кодеина и этилморфина регламентирован. Кодеин — алкалоид опия, по структуре метилморфин, обладает выраженным противокашлевым, а также анальгезирующим действиями. Выпускают в виде основания и в виде кодеина фосфата. Применяют в составе комбинированных препаратов: таблетки «Коделак»♠, таблетки «Терпинкод»♠ (кодеин и отхаркивающие средства: натрия гидрокарбо- 396 Часть II. Частная фармакология нат и терпингидрат), входит в состав микстуры Бехтерева (настой травы горицвета, натрия бромид и кодеин) и др. В терапевтических дозах кодеин практически не угнетает дыхательный центр либо это действие мало выражено. При систематическом применении препарат может вызвать констипацию. При длительном использовании кодеина развиваются привыкание и лекарственная зависимость. Этилморфин (дионин♠) получают полусинтетическим путем из морфина. Этилморфин действует подобно кодеину, оказывает выраженное угнетающее влияние на кашлевой центр. Препарат применяют внутрь в случае сухого изнуряющего непродуктивного кашля при плеврите, бронхите, трахеите. Препараты ненаркотического действия (глауцин, окселадин) непосредственно угнетают центр кашлевого рефлекса. При этом они не активируют опиоидергическую систему мозга и не вызывают лекарственную зависимость, в меньшей степени угнетают дыхание. Глауцин (глаувент♠) — препарат растительного происхождения (алкалоид мачка желтого), блокирует центральные звенья кашлевого рефлекса. Хорошо всасывается при приеме внутрь, эффект возникает через 30 мин и длится около 8 ч. Среди побочных эффектов возможны гипотензия, головокружение и тошнота. Окселадин (тусупрекс♠) — синтетический препарат (рис. 17.1). Блокирует центральные звенья кашлевого рефлекса. Быстро и полно всасывается при приеме внутрь, максимальная концентрация в крови достигается через 4–6 ч после приема. По свойствам близок к глауцину. Преноксдиазин (либексин♠) относят к противокашлевым средствам периферического действия (см. рис. 17.1). Оказывает местноанестезирующее действие на слизистую оболочку бронхов, препятствуя возникновению кашлевого рефлекса. Препарат практически не влияет на ЦНС, обладает бронхолитическим и противовоспалительным действиями. Применяют внутрь, противокашлевой эффект продолжается 3–4 ч. В качестве побочных эффектов может вызывать онемение языка, сухость ротовой полости, диарею. 17.3. ОТХАРКИВАЮЩИЕ СРЕДСТВА Данная группа веществ облегчает отделение секрета бронхиальных желез и назначается при кашле с трудноотделяемой мокротой. Интенсивность отделения мокроты зависит от ее реологических свойств — вязкости и адгезивности, от объема секреции бронхиальных желез, от функции мерцательного эпителия. Среди отхаркивающих средств выделяют: Глава 17. Средства, влияющие на функции органов дыхания 397 Рис. 17.1. Химические структуры окселадина и преноксдиазина ● муколитические средства — препараты, уменьшающие вязкость и адгезивность мокроты за счет деполимеризации ее молекул; ● секретомоторные средства — препараты, увеличивающие секрецию мокроты, что делает ее менее вязкой, и стимулирующие подвижность мерцательного эпителия. 17.3.1. Муколитические средства К препаратам этой группы относят ацетилцистеин, карбоцистеин, амброксол, бромгексин и ряд ферментных препаратов — трипсин, химотрипсин, рибонуклеаза, дезоксирибонуклеаза и др. Ацетилцистеин (АЦЦ♠, мукосольвин♠, мукобене♠) — эффективный муколитический препарат, производное аминокислоты цистеина, от которой отличается тем, что один водород аминогруппы замещен остатком уксусной кислоты (N-ацетил-L-цистеин). Муколитическое действие препарата обусловлено несколькими механизмами. Ацетилцистеин (рис. 17.2) содержит в структуре сульфгидрильные группы, которые разрывают дисульфидные связи протеогликанов мокроты, вызывая их деполимеризацию, что приводит к снижению вязкости и адгезивности мокроты. Препарат стимулирует секрецию мукозных клеток, секрет которых лизирует фибрин, что также 398 Часть II. Частная фармакология способствует разжижению мокроты. Ацетилцистеин увеличивает объем секреции мокроты, что приводит к уменьшению вязкости и облегчает ее отделение. Кроме того, препарат подавляет образование свободных радикалов, уменьшая воспалительную реакцию в бронхах. Ацетилцистеин стимулирует образование глутатиона, в связи с чем обладает детоксицирующим действием. Препарат вводят внутрь (шипучие таблетки, гранулы для приготовления раствора), парентерально (внутримышечно и внутривенно), интратрахеально (в виде медленной инстилляции) и ингаляционно. При применении внутрь быстро и полно абсорбируется, однако биодоступность не превышает 10%, поскольку при первом же прохождении через печень дезацетилируется, превращаясь в цистеин. Латентный период составляет 30–90 мин, продолжительность действия — 2–4 ч. Ацетилцистеин применяют в качестве муколитика при воспалительных заболеваниях органов дыхания (хронические бронхиты и трахеобронхиты, пневмония и др.), а также при бронхиальной астме и хронических обструктивных болезнях легких (ХОБЛ, см. ниже). Кроме того, ацетилцистеин как поставщик глутатиона применяют при передозировке парацетамола (ацетаминофена) с целью профилактики гепатотоксического действия последнего (см. гл. «Анальгезирующие средства»). Препарат обычно хорошо переносится. В отдельных случаях возможны тошнота, рвота, шум в ушах, крапивница. Осторожность следует соблюдать при применении препарата у больных бронхиальной астмой (при внутривенном введении возможен бронхоспазм). Ацетилцистеин противопоказан при язвенной болезни желудка и двенадцатиперстной кишки, склонности к легочным кровотечениям, заболеваниях печени, почек, дисфункции надпочечников, при беременности, лактации. Смешивание растворов ацетилцистеина с растворами антибиотиков и протеолитических ферментов нежелательно во избежание инактивации препарата. Препарат несовместим с некоторыми материалами (железо, медь, резина), при контакте с которыми образует сульфиды с характерным запахом. Препарат уменьшает всасывание пенициллинов, цефалоспоринов, тетрациклина, усиливает эффект нитроглицерина (интервал между приемами должен быть не менее 2 ч). Карбоцистеин (мукодин♠, мукосол♠) по структуре и действию сходен с ацетилцистеином (представляет собой S-карбоксиметилцистеин). Карбоцистеин применяют по тем же показаниям, что и ацетилцистеин, назначают внутрь. Амброксол (амбробене♠, амброгексал♠, лазолван♠, халиксол♠) оказывает муколитическое действие (см. рис. 17.2) за счет изменения структуры мукополисахаридов мокроты и увеличения секреции гликопротеидов (мукокинетическое действие). Кроме того, амброксол стимулирует двигательную активность мерцательного эпителия. Одна из особенностей действия пре- Глава 17. Средства, влияющие на функции органов дыхания 399 Рис. 17.2. Химические структуры ацетилцистеина, амброксола и бромгексина парата — его способность стимулировать образование и уменьшать распад эндогенных сурфактантов, что, в свою очередь, изменяет реологические свойства мокроты и облегчает ее отделение. При введении внутрь эффект развивается через 30 мин и продолжается 10–12 ч. Препарат применяют при остром и хроническом бронхите, пневмонии, бронхиальной астме, бронхоэктатической болезни. Имеются указания, что амброксол может быть использован для стимуляции образования сурфактанта при респираторном дистресс-синдроме у новорожденных и недоношенных детей. В качестве побочных эффектов может вызывать тошноту, рвоту, кишечные расстройства. Бромгексин (солвин♠, бизолвон♠) по химической структуре и фармакологическому действию сходен с амброксолом (см. рис. 17.2). При метаболических процессах в организме из бромгексина образуется амброксол, оказывающий муколитическое и отхаркивающее действия. Кроме того, у бромгексина присутствует собственное противокашлевое действие. Применяют бромгексин при заболеваниях дыхательных путей, сопровождающихся затруднением отделения вязкой мокроты: бронхитах и трахеобронхитах, в том числе осложненных бронхоэктазами, пневмонии, бронхиальной астме. Назначают внутрь в таблетках или растворах, в тяжелых случаях внутривенно. Препарат хорошо переносится. В отдельных случаях возможны аллергические реакции (кожная сыпь, ринит и др.). При длительном приеме возможны диспептические расстройства. В качестве муколитических средств иногда используют ферментные препараты (трипсин, химотрипсин, рибонуклеазу, дезоксирибонуклеазу 400 Часть II. Частная фармакология и др.). Протеолитические ферменты разрывают пептидные связи в белковых молекулах. Рибонуклеаза и дезоксирибонуклеаза вызывают деполимеризацию молекул РНК и ДНК. Выпускают рекомбинантный препарат α-дезоксирибонуклеаза (α-ДНКазы) — пульмозим♠. Ферментные препараты применяют ингаляционно. 17.3.2. Средства, стимулирующие секрецию бронхиальных желез Выделяют секретомоторные средства рефлекторного и прямого действия. К отхаркивающим средствам рефлекторного действия относят: ● средства растительного происхождения (препараты термопсиса, ипекакуаны, солодки, алтея, истода); ● синтетические средства (терпингидрат). Отхаркивающие средства рефлекторного действия при приеме внутрь оказывают раздражающее действие на рецепторы слизистой оболочки желудка, рефлекторно повышают секрецию бронхиальных желез и подвижность мерцательного эпителия. В результате увеличения объема секреции мокрота становится более жидкой, менее вязкой и адгезивной. Увеличение активности мерцательного эпителия и перистальтических движений бронхиол способствует продвижению мокроты из нижних в верхние отделы дыхательных путей и ее выведению. Большинство отхаркивающих средств рефлекторного действия относят к препаратам из растительного лекарственного сырья, не имеющим МНН. Трава термопсиса ланцетного♠ (hеrba Thermopsidis Lanceolata) содержит алкалоиды (цитизин, метилцитизин, пахикарпин, анагирин, термопсин, термопсидин), сапонины, эфирное масло и другие вещества. Содержащиеся в растении вещества оказывают отхаркивающее (в концентрациях 1:300–1:400), а в больших дозах (1:10–1:20) — рвотное действие. Препараты термопсиса применяют в виде настоев, сухого экстракта, в составе порошков, таблеток и микстуры от кашля. Корни солодки♠ (radices Glycyrrhizae), или лакричный корень (radix Liquiritiae), содержат ликуразид, глицирризиновую кислоту (тритерпеноидный гликозид, обладающий противовоспалительными свойствами), флавоноиды, слизистые вещества и др. Ликвиритозид (флавоновый гликозид) и 2,4,4-триоксихалкон оказывают спазмолитическое действие. Экстракт солодкового корня густой (extractum Glycyrrhizae spissum) входит в состав грудного эликсира. Препарат глицирам♠ (монозамещенная аммониевая соль глицирризиновой кислоты) оказывает противовоспалительное и некоторое отхаркивающее действия. Глава 17. Средства, влияющие на функции органов дыхания 401 Корни алтея♠ (radices Althаeae) применяют в виде порошка, настоя, экстракта и сиропа как отхаркивающее и противовоспалительное средство при заболеваниях дыхательных путей. Входит в состав грудных сборов (species pectoralis), из которых готовят настои, и в состав сухой микстуры от кашля для детей (mixtura sicca contra tussim pro infantibus). Мукалтин♠ — таблетки, содержащие смесь полисахаридов из травы алтея лекарственного. Корни истода♠ (radices Polygalae) содержат сапонины, применяют в виде отвара в качестве отхаркивающего средства. Некоторые отхаркивающие средства растительного происхождения оказывают не только рефлекторное, но и прямое действие — содержащиеся в них эфирные масла и другие вещества выделяются через дыхательные пути и вызывают усиление секреции и разжижение мокроты. Такие средства входят в состав комбинированных лекарственных препаратов. Пертуссин♠ (Pertussinum) состоит из 12 частей экстракта чабреца или экстракта тмина, 1 части калия бромида, 82 частей сиропа сахарного, 5 частей 80% спирта. Таблетки от кашля♠ (tabulettae contra tussim) содержат 0,01 г травы термопсиса в мелком порошке и 0,25 г натрия гидрокарбоната. Сухая микстура от кашля для взрослых♠ (mixtura sicca contra tussim pro adultis) состоит из смеси сухих экстрактов травы термопсиса и корней солодки, натрия гидрокарбоната, натрия бензоата и аммония хлорида, с добавлением масла анисового и сахара. Применяют в виде водного раствора. К синтетическим отхаркивающим средствам рефлекторного действия относят терпингидрат, представляющий собой пара-ментандиол-1,8-гидрат. Назначают внутрь как отхаркивающее средство при хроническом бронхите. Терпингидрат не следует назначать при гиперацидных состояниях. К отхаркивающим средствам прямого действия относят калия йодид и натрия гидрокарбонат. Эти препараты принимают внутрь, они всасываются и затем выделяются слизистой оболочкой дыхательных путей, за счет этого стимулируя секрецию бронхиальных желез и повышая двигательную активность мерцательного эпителия. Калия йодид и натрия гидрокарбонат могут назначаться ингаляционно. 17.4. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ БРОНХИАЛЬНОЙ АСТМЕ Бронхиальная астма — инфекционно-аллергическое заболевание, характеризующееся периодически возникающими приступами бронхоспазма и хроническим воспалительным процессом в стенке бронхов. Рис. 17.3. Механизмы действия средств, применяемых при бронхиальной астме (пояснения в тексте) 402 Часть II. Частная фармакология Глава 17. Средства, влияющие на функции органов дыхания 403 Хроническое воспаление приводит к повреждению эпителия дыхательных путей и развитию гиперреактивности бронхов. В результате увеличивается чувствительность бронхов к стимулирующим факторам (вдыхание холодного воздуха, воздействие аллергенов). К наиболее распространенным в окружающей среде аллергенам относят пыльцу растений, домашнюю пыль, химические вещества (сернистый газ), инфекционные агенты, пищевые аллергены и т.д. Их воздействие приводит к возникновению бронхоспазма, проявляющегося в виде характерных приступов удушья (экспираторная одышка). В развитии бронхиальной астмы значительную роль играют аллергический и аутоиммунный процессы. Аллергический компонент заболевания развивается по механизму реакции гиперчувствительности немедленного типа. Антигены при попадании в организм поглощаются макрофагами, и это вызывает ряд последовательных реакций, которые приводят к активации пролиферации В-лимфоцитов и их дифференцировке в плазматические клетки, продуцирующие антитела, в том числе IgE (см. рис. 17.1). Антитела циркулируют в системном кровотоке, и при повторном попадании в организм того же антигена связывают его и выводят из организма. Пролиферация и дифференцировка В-лимфоцитов регулируются интерлейкинами (ИЛ), которые вырабатываются сенсибилизированными макрофагами и регуляторными Т-лимфоцитами, так называемыми Т-хелперами. Т-хелперы выделяют различные ИЛ, в том числе ИЛ-3, увеличивающий клон тучных клеток, ИЛ-5, увеличивающий клон эозинофилов и др. ИЛ-4 стимулирует пролиферацию и дифференцировку В-лимфоцитов (и, следовательно, продукцию IgE). Кроме того, ИЛ-4 вызывает сенсибилизацию тучных клеток и базофилов, т.е. экспрессию в их мембранах рецепторов к IgE (рис. 17.3). Эти рецепторы называют Fсε-рецепторами и подразделяют на высокоаффинные FcεRI (находятся в тучных клетках) и низкоаффинные FcεRII. С высокоаффинными рецепторами FcεRI связываются IgE. При взаимодействии антигена с IgE, фиксированными на поверхности тучных клеток, происходит дегрануляция тучных клеток, из которых выделяются биологически активные вещества: ● с бронхоконстрикторными свойствами (вызывающие бронхоспазм), к которым относят цистеиниловые лейкотриены LtС 4, LtD 4, LtE4 (медленно реагирующая субстанция анафилаксии), фактор активации тромбоцитов, гистамин и др.; ● с хемотаксическими свойствами, вызывающие эозинофильную инфильтрацию бронхов (лейкотриен B4, фактор активации тромбоцитов); 404 Часть II. Частная фармакология ● с проаллергическими и провоспалительными свойствами (простагландины Е2, I2, D2, гистамин, брадикинин, лейкотриены, фактор активации тромбоцитов). Эти вещества расширяют кровеносные сосуды и повышают их проницаемость, вызывая отек слизистой оболочки, способствуют инфильтрации слизистой оболочки бронхов лейкоцитами (в том числе эозинофилами). Из активированных эозинофилов выделяются вещества, обладающие цитотоксическими свойствами (эозинофильные белки), повреждающие эпителиальные клетки. Таким образом, эти вещества поддерживают в бронхах воспалительный процесс, на фоне которого развивается гиперреактивность бронхов к факторам, вызывающим бронхоспазм. Выделяют несколько групп ЛС, применяемых при бронхиальной астме. ● Бронхолитические средства: —средства, стимулирующие β2-адренорецепторы; —средства, блокирующие М-холинорецепторы; —спазмолитики миотропного действия. ● Средства с противовоспалительным и противоаллергическим действиями: —препараты глюкокортикоидов; —стабилизаторы мембран тучных клеток; —средства с антилейкотриеновым действием: ✧ блокаторы лейкотриеновых рецепторов; ✧ ингибиторы синтеза лейкотриенов (ингибиторы 5-липоксигеназы); —препараты моноклональных антител к IgE. 17.4.1. Бронхолитические средства Средства, стимулирующие β2-адренорецепторы В качестве бронхолитиков можно использовать селективные агонисты β2-адренорецепторов — фенотерол, сальбутамол, тербуталин, гексопреналин, салметерол, формотерол и кленбутерол, а также неселективные агонисты — орципреналин и изопреналин (стимулируют β1и β2-адренорецепторы). Среди бронхолитических препаратов группа веществ селективного действия используется наиболее часто. Данная группа препаратов имеет ряд положительных качеств: β2-адреномиметики удобны в применении (вводят ингаляционно), имеют короткий латентный период (несколько минут), высокую эффективность, препятствуют дегрануляции тучных клеток, а также способствуют отделению мокроты (увеличивают му- Глава 17. Средства, влияющие на функции органов дыхания 405 коцилиарный клиренс). Высокая эффективность β2-адреномиметиков при экспираторной одышке связана с тем, что они способны расширять мелкие бронхи. Это обусловлено неравномерностью распределения β2адренореактивных структур в бронхах (плотность β2-адренорецепторов тем выше, чем дистальнее бронх, таким образом, максимальную плотность β2-адренорецепторов наблюдают в мелких бронхах и бронхиолах). Помимо бронхолитического действия β2-адреномиметики препятствуют дегрануляции тучных клеток. Это связано со снижением концентрации ионов Са2+ в тучных клетках (за счет повышения концентрации цАМФ в результате активации аденилатциклазы). Приступ бронхиальной астмы обычно заканчивается отхождением вязкой мокроты. β2-Адреномиметики облегчают отделение мокроты, что связано с устранением антигензависимого подавления мукоцилиарного транспорта и увеличением объема секреции вследствие расширения сосудов слизистой оболочки. Сальбутамол (вентодиск♠, вентолин♠), фенотерол (беротек♠), тербуталин (бриканил♠), гексопреналин (ипрадол♠) действуют 4–6 ч (рис. 17.4). Бронхолитическое действие начинается быстро (латентный период 2–5 мин) и достигает максимума через 40–60 мин. Эти препараты можно использовать для купирования и профилактики бронхоспазма. Кленбутерол (спиропент♠), формотерол (форадил♠), салметерол (серевент♠, сальметер♠) действуют продолжительно (около 12 ч), основное показание к их применению — профилактика бронхоспазма. Формотерол, кроме того, обладает коротким латентным периодом (1–2 мин). Однако использовать эти препараты для купирования бронхоспазма нерационально, поскольку в виду большой продолжительности действия существует риск передозировки. Наряду с бронхолитическим все перечисленные препараты оказывают также токолитическое действие (см. главу «Средства, влияющие на миометрий»). Побочные эффекты: снижение АД, тахикардия, мышечный тремор, отек слизистой оболочки бронхов, потливость, тошнота, рвота. Орципреналин (алупент♠, астмопент♠) отличается от вышеперечисленных бронхолитиков отсутствием селективности. Он стимулирует β1и β2-адренорецепторы. В связи с β1-адреномиметическим действием он обладает положительным дромотропным (поэтому может использоваться при атриовентрикулярном блоке и брадиаритмиях) и положительным хронотропным действиями, вызывая более выраженную тахикардию, чем селективные β2-адреномиметики. В ряде ситуаций для купирования бронхоспазма в качестве средства скорой помощи применяют адреналин♠ (стимулирует β1-, β2-, α1-, α2- 406 Часть II. Частная фармакология Рис. 17.4. Химические структуры некоторых бронхолитических средств Глава 17. Средства, влияющие на функции органов дыхания 407 адренорецепторы). Для того чтобы бронхолитический эффект адреналина♠ не сопровождался выраженным прессорным действием, препарат следует вводить подкожно. Характерный набор свойств (прессорное действие в комбинации с бронхолитическим) делает адреналин♠ средством выбора при анафилактическом шоке (при этом для достижения выраженного прессорного эффекта препарат вводят внутривенно). Бронхолитическое действие присуще симпатомиметику эфедрину. Однако из-за способности вызывать лекарственную зависимость его используют не самостоятельно, а в составе комбинированных препаратов с бронхолитическим действием. 17.4.2. Средства, блокирующие М-холинорецепторы М-холиноблокаторы в качестве бронхолитиков уступают по эффективности β2-адреномиметикам. Это обусловлено несколькими причинами. Во-первых, распределение М-холинорецепторов в бронхиальном дереве таково, что чем дистальнее расположен бронх, тем меньше в нем М-холинорецепторов (таким образом, М-холиноблокаторы устраняют спазм не столько мелких, сколько крупных бронхов). Вовторых, снижение тонуса бронхов представляет собой результат блокады М3-холинорецепторов гладкомышечных клеток бронхов, в то же время на пресинаптической мембране холинергических синапсов находятся М2-холинорецепторы (ауторецепторы), блокада которых (по принципу обратной отрицательной связи) приводит к усилению выделения ацетилхолина в синаптическую щель. При повышении концентрации ацетилхолина в синаптической щели он конкурентно вытесняет М-холиноблокаторы из связи с М3-холинорецепторами на мембране гладкомышечных клеток, препятствуя его бронхолитическому действию. Кроме того, М-холиноблокаторы уменьшают секрецию бронхиальных желез, что при бронхиальной астме нежелательно (снижение объема секреции делает мокроту более вязкой и трудноотделяемой). В связи с вышеизложенным блокаторы М-холинорецепторов рассматривают как вспомогательные средства. Ипратропия бромид (атровент♠, итроп♠) имеет в структуре четвертичный атом азота и обладает низкой липофильностью, поэтому при ингаляционном применении практически не всасывается в системный кровоток. Бронхолитический эффект развивается через 30 мин после ингаляции, достигает максимума через 1,5–2 ч и продолжается 5–6 ч. Побочные эффекты: сухость во рту. Системных побочных (атропиноподобных) эффектов практически не вызывает. 408 Часть II. Частная фармакология Тиотропия бромид (спирива♠) отличается от ипратропия бромида тем, что блокирует постсинаптические М3-холинорецепторы в большей степени, чем пресинаптические М2-холинорецепторы, поэтому более эффективно снижает тонус бронхов. Тиотропия бромид оказывает более быстрое (максимальный эффект развивается через 1,5–2 ч) и более продолжительное действие (около 12 ч), чем ипратропия бромид. Назначают ингаляционно 1 раз в сутки. Бронхолитическим действием обладают все атропиноподобные средства, однако их применение в качестве бронхолитиков ограничено в связи с большим количеством побочных эффектов. Спазмолитики миотропного действия К бронхолитикам миотропного действия относят метилксантины: теофиллин и аминофиллин. Теофиллин (рис. 17.5) малорастворим в воде (1:180). Аминофиллин (эуфиллин♠) представляет собой смесь 80% теофиллина и 20% этилендиамина, что обусловливает более легкую растворимость этого вещества в воде (см. рис. 17.5). Метилксантины в качестве бронхолитических средств не уступают по эффективности β2-адреномиметикам, но в отличие от β2-адреномиметиков их не вводят ингаляционно. Механизм бронхолитического действия метилксантинов связывают с блокадой аденозиновых А1-рецепторов гладкомышечных клеток, а также с неизбирательным ингибированием фосфодиэстеразы (типов III, IV). Угнетение фосфодиэстеразы в гладкомышечных клетках бронхов (фосфодиэстераза IV) приводит к накоплению в клетках цАМФ и снижению внутриклеточной концентрации Са2+, в результате чего в клетках уменьшается активность киназы легких цепей миозина и нарушается взаимодействие актина и миозина. Это приводит к расслаблению гладких мышц бронхов (спазмолитическое действие). Аналогичным образом теофиллин действует на гладкие мышцы кровеносных Рис. 17.5. Химические структуры теофиллина и аминофиллина Глава 17. Средства, влияющие на функции органов дыхания 409 сосудов, вызывая расширение сосудов. Под действием теофиллина в тучных клетках также увеличивается концентрация цАМФ (за счет угнетения фосфодиэстеразы IV) и снижается концентрация Са2+. Это препятствует дегрануляции тучных клеток и высвобождению медиаторов воспаления и аллергии. Ингибирование фосфодиэстеразы в кардиомиоцитах (фосфодиэстераза III) приводит к накоплению в них цАМФ и повышению концентрации Са2+ (повышение силы сердечных сокращений, тахикардия). При действии на систему органов дыхания помимо бронхолитического действия отмечается усиление мукоцилиарного клиренса, уменьшение легочного сосудистого сопротивления, стимуляция дыхательного центра и улучшение сокращений дыхательных мышц (межреберных и диафрагмы). Кроме того, теофиллин оказывает слабое антиагрегантное и диуретическое действия. При приеме внутрь быстро и полно всасывается из кишечника (биодоступность выше 90%). Максимальная концентрация в крови достигается через 2 ч. Метаболизируется в печени с образованием неактивных метаболитов. Скорость метаболизма и продолжительность действия неодинаковы у разных пациентов (в среднем около 6 ч). Побочные эффекты: беспокойство, нарушение сна, тремор, головная боль (связаны с блокадой аденозиновых рецепторов в ЦНС), тахикардия, аритмии (связаны с блокадой аденозиновых рецепторов сердца и ингибированием фосфодиэстеразы III), тошнота, рвота, понос. Разработаны таблетированные лекарственные формы теофиллина пролонгированного действия: эуфиллин ретард Н♠, эуфилонг♠, уни-дур♠, вентакс♠, спофиллин ретард♠, теопэк♠, теодур♠ и др. Ретард-форма отличается более медленным высвобождением действующего начала в системный кровоток. При применении пролонгированных форм теофиллина максимальная концентрация достигается через 6 ч, а общая продолжительность действия увеличивается до 12 ч. К пролонгированным формам аминофиллина можно отнести ректальные суппозитории (применяют по 360 мг 2 раза в сутки). Фармацевтическая промышленность выпускает комбинированные препараты с бронхолитическим действием. Для ингаляций применяют: ● дитэк♠ (дозированный аэрозоль, содержащий в 1 дозе 50 мкг фенотерола и 1 мг кромоглициевой кислоты); ● инталплюс♠ (дозированный аэрозоль, содержащий в 1 дозе сальбутамола 100 мкг и динатриевой соли кромоглициевой кислоты 1 мг); ● беродуал♠ (раствор для ингаляций и дозированный аэрозоль, содержащий в 1 дозе фенотерола гидробромида 50 мкг и ипратропия бромида 20 мкг); 410 Часть II. Частная фармакология ● комбивент♠ (дозированный аэрозоль, содержащий в 1 дозе сальбутамола сульфата 120 мкг и ипратропия бромида 20 мкг); ● серетидмультидиск♠, содержащий салметерол с флутиказоном. Для применения внутрь используют: ● таблетки теофедрин Н♠ (одна таблетка содержит: теофиллина 100 мг, эфедрина гидрохлорида 20 мг, экстракта красавки сухого 3 мг, парацетамола 200 мг, фенобарбитала 20 мг, цитизина 100 мкг); ● капсулы и сироп трисолвин♠ (1 капсула содержит: теофиллина безводного 60 мг, гвайфенезина 100 мг, амброксола 30 мг; 5 мл сиропа содержат: теофиллина безводного 50 мг, гвайфенезина 30 мг, амброксола 15 мг); ● капли солутан♠ (1 мл соответствует 34 каплям и содержит: алкалоида корня красавки радобелина 100 мкг, эфедрина гидрохлорида 17,5 мг, прокаина гидрохлорида 4 мг, экстракта толуанского бальзама 25 мг, натрия йодида 100 мг, сапонина 1 мг, масла укропного 400 мкг, воды горькоминеральной 30 мг). Течению бронхиальной астмы часто сопутствуют такие проявления гиперчувствительности немедленного типа, как крапивница, аллергический ринит, аллергический конъюнктивит и ангионевротический отек (отек Квинке). Их вызывает гистамин, выделяющийся из сенсибилизированных тучных клеток в ходе дегрануляции. Для устранения этих симптомов применяют антигистаминные препараты, блокирующие гистаминовые Н1-рецепторы (см. раздел «Противоаллергические средства»). 17.4.3. Средства с противовоспалительным и противоаллергическим действием Препараты глюкокортикоидов Глюкокортикоиды имеют сложный механизм антиастматического действия, в котором можно выделить несколько компонентов: противовоспалительный, противоаллергический и иммуносупрессивный. Противовоспалительное действие глюкокортикоидов имеет несколько механизмов. За счет экспрессии соответствующего гена они стимулируют продукцию липокортинов, естественных ингибиторов фосфолипазы А2, что приводит к уменьшению продукции в тучных клетках фактора активации тромбоцитов, лейкотриенов и простагландинов. Кроме того, глюкокортикоиды подавляют синтез ЦОГ-2 (за счет репрессии соответствующего гена), что также приводит к снижению синтеза простагландинов в очаге воспаления (см. рис. 17.3). Глюкокортикоиды угнетают синтез молекул межклеточной адгезии, что затрудняет проникновение Глава 17. Средства, влияющие на функции органов дыхания 411 моноцитов и лейкоцитов в очаг воспаления. Все это приводит к уменьшению воспалительной реакции, препятствует развитию гиперреактивности бронхов и возникновению бронхоспазма. Глюкокортикоиды оказывают иммуносупрессивное действие, угнетая продукцию ИЛ (за счет репрессии соответствующих генов), в том числе ИЛ-1, ИЛ-2 и ИЛ-4 и др. В связи с этим они подавляют пролиферацию и дифференцировку В-лимфоцитов и препятствуют образованию антител, в том числе IgE. Глюкокортикоиды уменьшают количество и сенсибилизацию тучных клеток (за счет уменьшения продукции ИЛ-3 и ИЛ-4), препятствуют биосинтезу в тучных клетках цистеиниловых лейкотриенов (за счет активации липокортина-1 и угнетения фосфолипазы А2), а также стабилизируют мембраны тучных клеток, препятствуя их дегрануляции (см. рис. 17.3). Это приводит к подавлению аллергической реакции немедленного типа. Глюкокортикоиды также сенсибилизируют β2-адренорецепторы бронхов к циркулирующему в крови адреналину, в результате чего они усиливают бронхолитическое действие адреналина. Глюкокортикоиды для резорбтивного действия (преднизолон, дексаметазон, бетаметазон и др.) высокоэффективны при бронхиальной астме. Однако большое количество возникающих при этом побочных эффектов делает целесообразным использование препаратов глюкокортикоидов для ингаляционного введения. К препаратам этой группы относят беклометазон, флутиказон, флунизолид и будесонид. Эти препараты мало всасываются в системный кровоток и имеют высокие показатели пресистемной элиминации, вследствие чего удается избежать побочных эффектов, связанных с их резорбтивным действием. Антиастматическое действие глюкокортикоидов нарастает постепенно при их регулярном применении. Их обычно используют для систематического лечения. В последние годы эти препараты стали выпускаться в порошковых (не содержащих фреона) дозированных аэрозолях, активируемых вдохом. Беклометазон (см. рис. 17.4) выпускают в ингаляторах различных модификаций: ● бекотид♠♠(дозированный аэрозоль, 200 доз); 200 доз во флаконе); ● беклазон (дозированный аэрозоль, ● беклазон — легкое дыхание♠ (дозированный аэрозоль, 200 доз во флаконе с оптимизатором дозы); ● бекломет-изихалер♠ (порошок для ингаляций, 200 доз в дозирующем устройстве изихалер); ● бекодиск♠ (порошок для ингаляций, 120 доз в комплекте с дискхалером). 412 Часть II. Частная фармакология Беклометазон применяют главным образом для профилактики приступов бронхоспазма, он эффективен только при регулярном применении. Эффект развивается постепенно и достигает максимума на 5–7-е сут от начала использования. Обладает выраженным противоаллергическим, противовоспалительным и противоотечным действиями. Уменьшает эозинофильную инфильтрацию легочной ткани, снижает гиперреактивность бронхов, улучшает показатели функции внешнего дыхания, восстанавливает чувствительность бронхов к бронхолитическим средствам. Применяют 2–4 раза в сутки. Поддерживающая доза — 100–200 мкг. Побочные действия: дисфония (изменение или охриплость голоса), чувство жжения в зеве и гортани, крайне редко — парадоксальный бронхоспазм. При длительном применении может развиться кандидамикоз ротовой полости и глотки. Кроме того, препараты беклометазона (беконазе♠) могут применяться для лечения аллергического ринита. Флунизолид (ингакорт♠) по фармакологическим свойствам и применению сходен с беклометазоном. Отличается от него более интенсивным всасыванием в системный кровоток, однако вследствие выраженного пресистемного метаболизма биодоступность флунизолида не превышает 40%, t½ составляет 1–8 ч. Так же, как и беклометазон, можно применять при аллергическом рините. Будесонид (будесонид мите♠, будесонид форте♠, пульмикорт турбухалер♠) по фармакологическим свойствам и применению сходен с беклометазоном, но имеет ряд отличий. Будесонид имеет более продолжительное действие, в связи с этим его применяют 1–2 раза в сутки. Нарастание эффекта до максимума происходит более продолжительное время (в течение 1–2 нед). При ингаляционном введении около 28% препарата попадает в системный кровоток. Будесонид применяют не только при бронхиальной астме, но и в дерматологии в составе мазей и кремов апулеин♠. Местные побочные эффекты такие же, как у беклометазона. Кроме того, могут наблюдаться побочные эффекты со стороны ЦНС в виде депрессии, нервозности, возбудимости. Флутиказон применяют при бронхиальной астме (дозированный аэрозоль фликсотид♠), при аллергическом рините (спрей для носа фликсоназе♠), при заболеваниях кожи (мазь и крем кутивейт♠). При бронхиальной астме препарат применяют 2 раза в сутки ингаляционно (20% от введенной дозы всасывается в системный кровоток). По свойствам и фармакокинетике сходен с будесонидом. При ингаляционном применении глюкокортикоидов нельзя исключить их системную абсорбцию и риск угнетения инкреции эндогенных глюкокортикоидов (по механизму обратной отрицательной связи). Не- Глава 17. Средства, влияющие на функции органов дыхания 413 прерывно ведется изыскание более совершенных препаратов глюкокортикоидов. Одна из новых групп — «мягкие» глюкокортикоиды. К ним относят лотепреднола этабонат♠ (применяющийся в офтальмологии) и циклесонид, рекомендующийся к применению при бронхиальной астме. Циклесонид — эстерифицированный стероид, не содержащий галогенов, пролекарство. При введении внутрь подвергается интенсивному пресистемному метаболизму, в связи с чем его биодоступность составляет 0,5–1%. При ингаляционном введении контактирует с эстеразами легочной ткани и преобразуется в дезциклесонид (С21дезметилпропионилциклесонид), который обладает выраженной противовоспалительной активностью. Применяют 1 раз в сутки, хорошо переносится, в меньшей степени, чем флутиказон, подавляет образование естественных глюкокортикоидов. Стабилизаторы мембран тучных клеток К препаратам этой группы относят кромоглициевую кислоту, недокромил, кетотифен. Кромоглициевая кислота стабилизирует мембраны тучных клеток, препятствуя входу в них ионов кальция. В связи с этим уменьшается дегрануляция сенсибилизированных тучных клеток (прекращается высвобождение лейкотриенов, фактора активации тромбоцитов, гистамина и других медиаторов воспаления и аллергии). Очевидно, что препараты кромоглициевой кислоты эффективны как средство профилактики, но не купирования бронхоспазма. При ингаляционном применении кромоглициевой кислоты в системный кровоток всасывается 5–15% от введенной дозы, t½ составляет 1–1,5 ч. Действие после однократного ингаляционного применения продолжается около 5 ч. При систематическом применении эффект нарастает постепенно, достигая максимума через 2–4 нед. При бронхиальной астме используют следующие препараты кромоглициевой кислоты: кромолин♠, интал♠, кропоз♠, талеум♠ и др. Все указанные препараты применяют ингаляционно, обычно 4 раза в сутки. В связи с тем, что кромоглициевая кислота практически не всасывается в системный кровоток, препараты практически не оказывают системных побочных эффектов. Местные побочные эффекты проявляются в виде раздражения слизистой оболочки дыхательных путей: жжения и першения в горле, кашля, возможен кратковременный бронхоспазм. Препараты кромоглициевой кислоты также применяют при аллергическом рините в виде капель в нос или интраназального спрея (вивидрин♠, кромоглин♠, кромосол♠) и аллергическом конъюнктивите в виде глазных капель (вивидрин♠, кромогексал♠, хай-кром♠, лекролин♠). 414 Часть II. Частная фармакология Недокромил (тайлед♠, тайлед минт♠) выпускают в виде кальциевой и динатриевой соли (недокромил натрий). По свойствам препарат сходен с кромоглициевой кислотой, но имеет отличную химическую структуру. Применяют ингаляционно, в системный кровоток всасывается 8–17% вещества. Используют как средство профилактики, но не купирования бронхоспазма. Эффект нарастает постепенно, достигая максимума к концу 1-й нед регулярного применения. Назначают по 4 мг 4 раза в сутки. Кетотифен (задитен♠, зетифен♠) имеет свойства стабилизатора мембран тучных клеток и блокатора Н1-рецепторов. Практически полностью всасывается из кишечника. Не очень высокая биодоступность препарата (около 50%) объясняется эффектом первого прохождения через печень; t½ составляет 3–5 ч. Применяют внутрь по 1 мг 2 раза в сутки (во время еды). Побочные эффекты: седативное действие, замедление психомоторных реакций, сонливость, сухость во рту, увеличение массы тела, тромбоцитопения. Средства с антилейкотриеновым действием Блокаторы лейкотриеновых рецепторов Бронхоспазм, вызываемый цистеинилсодержащими лейкотриенами LTC4, LTD4 и LTE4 (ранее известными как медленно реагирующая субстанция анафилаксии), — это результат стимуляции специфических лейкотриеновых рецепторов бронхиол (CysLT-рецепторы). Бронхоконстрикторное действие лейкотриенов устраняют конкурентные блокаторы лейкотриеновых рецепторов (см. рис. 17.3). К ним относят: зафирлукаст, монтелукаст, пранлукаст. Зафирлукаст (аколат♠) не только устраняет бронхоспазм, вызываемый цистеиниловыми лейкотриенами (LTC4LTD4LTE4), но и оказывает противовоспалительное действие, уменьшая проницаемость сосудов, экссудацию и отек слизистой оболочки бронхов (рис. 17.6). Из кишечника всасывается медленно и неполно, t½ составляет около 10 ч. Применяют внутрь натощак (за 1 ч до еды) или через 2 ч после последнего приема пищи, 2 раза в сутки. Действие препарата развивается медленно, около суток, поэтому зафирлукаст применяют для профилактики приступов бронхиальной астмы, при длительном лечении бронхиальной астмы. Препарат применяют также при аллергическом рините. Побочные эффекты: диспепсия, фарингит, гастрит, головная боль. Зафирлукаст ингибирует микросомальные ферменты печени, поэтому удлиняет действие некоторых ЛС. Монтелукаст (сингулер ♠) — избирательный антагонист CysLTрецепторов (см. рис. 17.6). В отличие от зафирлукаста не ингибирует Глава 17. Средства, влияющие на функции органов дыхания O OCH3 415 COOH Cl HN S N S O N O CH3 H3C H N O O H3C H 3C C OH Рис. 17.6. Химические структуры зафирлукаста и монтелукаста микросомальные ферменты печени (не изменяет продолжительность действия других ЛС). Ингибиторы синтеза лейкотриенов Зилеутон избирательно угнетает 5-липоксигеназу, препятствуя биосинтезу лейкотриенов (см. рис. 17.3). Применяют внутрь, зилеутон быстро всасывается из кишечника, t½ составляет 1–2,3 ч. Механизм действия препарата определяет основную сферу его применения: профилактика приступов бронхоспазма при бронхиальной астме и профилактика бронхоспазма, вызванного применением нестероидных противовоспалительных средств. Неизбирательные ингибиторы ЦОГ (особенно ацетилсалициловая кислота) могут спровоцировать бронхоспазм вследствие «субстратного шунтирования» арахидоновой кислоты (накапливающаяся при угнетении ЦОГ арахидоновая кислота расходуется на биосинтез лейкотриенов, которые и вызывают бронхоспазм). Побочные эффекты: лихорадка, миалгия, диспепсия, головокружение. Препараты моноклональных антител к IgE Омализумаб (ксолар♠) — препарат рекомбинантных человеческих моноклональных антител к IgE. Омализумаб связывается с циркулирующими в плазме крови IgE (см. рис. 17.3) и уменьшает их количество, тем самым препятствуя связыванию IgE с высокоаффинными рецепторами FcεRI на мембранах тучных клеток. Кроме того, при регулярном применении омализумаба количество FcεRI в мембранах тучных клеток уменьшается. Вероятно, это вторичная реакция на уменьшение коли- 416 Часть II. Частная фармакология чества IgE в плазме крови. Омализумаб не связывается с уже фиксированными к тучным клеткам антителами и не вызывает агглютинацию тучных клеток. При применении препарата урежаются приступы бронхиальной астмы и восстанавливается чувствительность к ингаляционным глюкокортикоидам (что особенно ценно при развитии резистентности к глюкокортикоидам). Препарат вводят подкожно в дозе 150–375 мг 1 раз в 2–4 нед. В качестве побочных эффектов наблюдают инфекции верхних дыхательных путей (в том числе вирусные) и осложнения в местах инъекций (покраснение, боль и зуд). Возможны также головная боль и аллергические реакции. 17.5. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ ХРОНИЧЕСКИХ ОБСТРУКТИВНЫХ БОЛЕЗНЯХ ЛЕГКИХ Хронические обструктивные болезни легких (ХОБЛ) — это группа заболеваний, ведущим клиническим синдромом которых является обструкция дыхательных путей. ХОБЛ (наряду с сахарным диабетом, онкологическими заболеваниями, гипертензией и др.) включен в список пандемии неинфекционных заболеваний ВОЗ. Одна из главных причин их возникновения — курение. ХОБЛ характеризуются развитием хронического воспалительного процесса дыхательных путей, легочной паренхимы и сосудов, при котором отмечается повышенное количество макрофагов, Т-лимфоцитов и нейтрофилов в легочной ткани. Основные клетки воспаления при ХОБЛ — нейтрофилы (в отличие от бронхиальной астмы, где основные участники патологического процесса — эозинофилы и тучные клетки). Нейтрофилы вызывают катаральное и катарально-гнойное воспаление бронхиального эпителия. Оно проявляется бокаловидноклеточной гиперплазией (что приводит к увеличению секреции слизи) и сквамозной метаплазией (что приводит к нарушению мукоцилиарного клиренса и способствует застою слизи в бронхах). Таким образом, первый клинический симптом ХОБЛ — кашель с отделением мокроты (секреторную активность бокаловидных клеток стимулируют лейкотриены, интерлейкины, в том числе ИЛ1β, ИЛ6, ФНО-α протеиназы и нейропептиды, выделяющиеся в ходе нейтрофильного воспаления). Позже присоединяется разрушение соединительнотканной стромы легких (экстрацеллюлярного матрикса). В основе этого процесса лежит дисбаланс между ферментами, вызывающими деградацию экстрацеллюлярного матрикса (матриксных металлопротеаз, например коллагеназы, Глава 17. Средства, влияющие на функции органов дыхания 417 желатиназы и пр.), и их тканевыми ингибиторами. При ХОБЛ ферменты семейства матриксных металлопротеаз продуцируются нейтрофилами и другими клетками, участвующими в воспалении. Кроме того, нейтрофилы вырабатывают активные формы кислорода, которые прямо разрушают соединительнотканную строму легких. Нарушение эластических свойств легочной ткани приводит к эмфиземе, при которой альвеолы увеличиваются в размерах и сдавливают соседние бронхиолы и легочные капилляры, что приводит к усилению бронхиальной обструкции, нарушению легочного кровотока и гипертензии в малом круге кровообращения. На этом этапе развития заболевания к кашлю добавляются признаки дыхательной недостаточности: одышка, снижение жизненной емкости легких, снижение объема форсированного выдоха. Наиболее мощный фактор в развитии и прогрессировании ХОБЛ — курение. Никотин является хемоаттрактантом, привлекающим в бронхи нейтрофилы. Кроме того, продукты пиролиза содержат активные формы кислорода и усиливают оксидативный стресс. Задачи фармакотерапии при ХОБЛ — предупреждение прогрессирования заболевания, уменьшение выраженности клинических симптомов, предупреждение обострений и осложнений. Объем лечения увеличивается по мере нарастания тяжести болезни, а его уменьшение при ХОБЛ, в отличие от бронхиальной астмы, как правило, невозможно. Кроме того, в развитии ХОБЛ (в отличие от бронхиальной астмы) отсутствует аллергический компонент, поэтому среди фармакотерапевтических средств отсутствуют стабилизаторы мембран тучных клеток и антагонисты лейкотриенов. Непременное условие лечения ХОБЛ — отказ от курения. Основная группа фармакотерапевтических препаратов — бронхолитики. У пожилых пациентов, имеющих сопутствующие заболевания сердечно-сосудистой системы (ИБС, нарушения сердечного ритма, артериальная гипертензия и др.), в качестве препаратов первого ряда предпочтительны холиноблокаторы. При назначении бронхолитиков из группы β2-адреномиметиков предпочтение отдают препаратам с длительным действием (формотерол). Среди миотропных бронхолитиков — ингибиторов фосфодиэстеразы — также предпочтительны пролонгированные формы. Кроме того, в настоящее время появилась группа избирательных ингибиторов фосфодиэстеразы IV — циломиласт (арифло♠) и рофлумиласт (даксас♠). Эти препараты обладают не только бронхолитическим действием. При их применении уменьшаются количество и активность нейтрофилов, Т-лимфоцитов CD8+, уменьшается пролиферация Т-хелперов CD4+ и синтез ими цитокинов (ИЛ-2, ИЛ-4, ИЛ-5), подавляется продукция фактора некроза опухолей моноцитами, а также синтез лейкотриенов. 418 Часть II. Частная фармакология В результате уменьшается воспалительный процесс в стенке бронхов. Общая проблема всех разрабатываемых препаратов — высокая частота тошноты и рвоты, что способно значительно ограничить их клиническое применение. Рофлумиласт (даксас♠) почти в 100 раз более активен, чем циломиласт (рис. 17.7). Избирательно ингибирует D-изоформу фосфодиэстеразы 4, что позволяет снизить количество побочных эффектов. При приеме в стандартной дозе 500 мг его максиCl мальная концентрация в системном N O кровотоке (Сmax) устанавливается чеO рез час. Метаболизируется цитохроN F H мом р450 с образованием активного Cl O F метаболита рофлумиласт-N-оксид, который оказывает такое же действие, как рофлумиласт, но более Рис. 17.7. Химическая структура длительно циркулирует в системном кровотоке (рофлумиласт — 3–4 дня, рофлумиласта рофлумиласт-N-оксид — 6 дней). Длительный период полуэлиминации рофлумиласт-N-оксида позволяет назначать препарат не чаще 1 раза в сутки. Препараты глюкокортикоидов при ХОБЛ менее эффективны, чем при бронхиальной астме. Их применение ограничено только периодами обострения, и курс не должен превышать 10–14 дней. Ацетилцистеин, обладающий как муколитической, так и антиоксидантной активностью, способен снижать продолжительность и частоту обострений ХОБЛ. Этот препарат может использоваться у больных в течение длительного времени (3–6 мес) в дозе 600 мг/сут. Эффективность прочих муколитиков неясна. В настоящее время разрабатывается ряд новых групп ЛВ для лечения ХОБЛ. Среди них — антиоксиданты, блокаторы провоспалительных интерлейкинов (ИЛ1β, ИЛ6, ФНОα). 17.6. ПРЕПАРАТЫ СУРФАКТАНТОВ В эту группу входят ЛС, временно замещающие природные сурфактанты при нарушении их образования. Эндогенные сурфактанты — поверхностно-активные вещества (фосфатидилхолины, сфингомиелины), синтезирующиеся в альвеолярных Глава 17. Средства, влияющие на функции органов дыхания 419 клетках и в виде тонкого слоя выстилающие внутреннюю поверхность легких. Легочные сурфактанты не позволяют альвеолам спадаться, обладают защитными свойствами в отношении альвеолярных клеток, а также регулируют реологические свойства бронхолегочного секрета и облегчают отделение мокроты. Нарушение биосинтеза сурфактантов у новорожденных проявляется респираторным дистресс-синдромом (множественные ателектазы и интерстициальный отек легочной паренхимы), а также может наблюдаться у взрослых при различных бронхолегочных заболеваниях. Основное показание к применению препаратов сурфактантов — респираторный дистресс-синдром у недоношенных детей. Куросурф♠ — препарат сурфактанта, содержащий фосфолипидные фракции (фосфатидилхолин) и низкомолекулярные гидрофобные протеины (1%), выделенные из легочной ткани свиней. Применяют при респираторном дистресс-синдроме, связанном с дефицитом сурфактантов у новорожденных (недоношенных) детей (с массой тела не менее 700 г). Применение препарата рассчитано на восстановление адекватного дыхания и допускается только в клинических условиях (учитывая необходимость искусственной вентиляции легких и мониторирования). Экзосурф♠ — препарат, действующее вещество которого представлено кольфосцерил пальмитатом. Экзосурф обладает свойствами сурфактанта и облегчает растяжимость легких. Применяют, подобно куросурфу♠, при респираторном дистресс-синдроме у новорожденных. Вводят в виде раствора в дозе 5 мл/кг через эндотрахеальную трубку. При необходимости повторяют введение в той же дозе через 12 ч. Вопросы и задания для самоконтроля 1. К стимуляторам дыхания относят: а) никетамид; б) кодеин; в) цитизин; г) ацетилцистеин; д) кофеин. 2. К противокашлевым средствам относят: а) ацетилцистеин; б) глауцин; в) окселадин; г) преноксдиазин; л) кодеин. 420 Часть II. Частная фармакология 3. К отхаркивающим средствам относят: а) глауцин; б) амброксол; в) преноксдиазин; г) ацетилцистеин; д) бромгексин. 4. Обладает болеутоляющим и противокашлевым действиями. Подавляет активность кашлевого и дыхательного центров за счет стимуляции опиоидных рецепторов. Обладает наркогенным потенциалом, применяется, главным образом, в составе комбинированных препаратов: а) ацетилцистеин; б) преноксдиазин; в) окселадин; г) глауцин; д) кодеин. 5. Облегчает отделение мокроты за счет разрушения дисульфидных мостиков протеогликанов. Оказывает противовоспалительное действие за счет подавления образования свободных радикалов. Оказывает дезинтоксицирующее действие за счет стимуляции образования глутатиона. При приеме внутрь обладает невысокой биодоступностью. Применяется при продуктивном кашле и передозировке парацетамола: а) амброксол; б) глауцин; в) преноксдиазин; г) бромгексин; д) ацетилцистеин. 6. При бронхиальной астме применяют: а) стимуляторы дыхания; б) стабилизаторы мембран тучных клеток; в) препараты глюкокортикоидов; г) бронхолитики; д) блокаторы лейкотриеновых рецепторов. 7. Препараты глюкокортикоидов эффективны при бронхиальной астме, так как они: Глава 17. Средства, влияющие на функции органов дыхания 421 а) препятствуют синтезу лейкотриенов; б) стабилизируют мембраны тучных клеток; в) оказывают противовоспалительное действие; г) уменьшают дифференцировку В-лимфоцитов и продукцию ими антител; д) обладают иммуностимулирующим действием. 8. Применяется ингаляционно для купирования бронхоспазма. Действует около 6 ч. В качестве побочных эффектов вызывает мышечный тремор и тахикардию: а) ипратропий; б) фенотерол; в) кромоглициевая кислота; г) беклометазон. 9. Применяется ингаляционно для купирования бронхоспазма. Действует около 6 ч. В качестве побочных эффектов может вызвать сухость во рту, тахикардию и нарушение зрения: а) фенотерол; б) будесонид; в) недокромил; г) ипратропий. 10. Применяется ингаляционно при бронхиальной астме. Эффект нарастает постепенно и достигает максимума через неделю. Применяется 2 раза в сутки. Мало эффективен как средство купирования бронхоспазма. В качестве побочных эффектов вызывает дисфонию и кандидоз ротовой полости: а) кромоглициевая кислота; б) фенотерол; в) будесонид; г) ипратропий. СРЕДСТВА, ВЛИЯЮЩИЕ НА СЕРДЕЧНО-СОСУДИСТУЮ СИСТЕМУ Группа ЛС, влияющих на сердечно-сосудистую систему, фармакологически весьма неоднородна. В нее входят вещества: ● воздействующие непосредственно на сердце (хинидиноподобные и кардиотонические средства); ● воздействующие непосредственно на сосудистую стенку (вазодилататоры миотропного действия); ● воздействующие на иннервацию сердца и сосудов (холиномиметики, адреноблокаторы). В связи с этим целесообразно использовать классификацию по клинико-фармакологическому принципу (с учетом патологии, при которой показаны данные препараты): ● средства, применяемые при сердечных аритмиях; ● средства, применяемые при недостаточности коронарного кровообращения; ● средства, применяемые при артериальной гипертензии; ● средства, применяемые при артериальной гипотензии; ● средства, применяемые при сердечной недостаточности. Глава 18 АНТИАРИТМИЧЕСКИЕ СРЕДСТВА Противоаритмические (антиаритмические) средства — клинико-фармакологическая группа препаратов, применяемых при нарушениях сердечного ритма и воздействующих непосредственно на сердце или на его иннервацию. Кардиомиоциты как основной структурный элемент сердца подразделяют на типичные (рабочие, сократительные) и атипичные. Типичные кардиомиоциты содержат сократительные белки (актин и миозин), благодаря которым осуществляется насосная функция органа. Атипичные кардиомиоциты не содержат сократительных белков. Их основные функции — проводимость и автоматизм (способность к самопроизвольной генерации импульса). Они образуют проводящую систему сердца (рис. 18.1). Проводящая система состоит из: ● синусового (синоатриального) узла; ● межузловых (интернодальных) проводящих пучков; ● атриовентрикулярного соединения (атриовентрикулярного узла); ● атриовентрикулярного пучка (пучка Гиса); ● волокон Пуркинье. Основная функция проводящей системы сердца — поддержание нормального ритма сердечных сокращений. Синусовый (синоатриальный) узел расположен в верхнелатеральной стенке правого предсердия, около устья верхней полой вены. Он состоит из пучка волокон длиной 15 мм, шириной 3 мм и толщиной 1 мм. Потенциал покоя кардиомиоцитов синусового узла составляет 55 мВ. Синусовый узел генерирует импульсы с частотой 70–80 в минуту. Это наиболее высокие значения во всей проводящей системе. Меньшая частота генерации импульсов нижерасположенных элементов проводящей системы подавляется («обнуляется») более высоким уровнем автоматизма синусового узла. Поэтому именно он в норме определяет ЧСС и выполняет функцию водителя ритма (pacemaker), а генерируемые им импульсы проводятся по проводящей системе к рабочим кардиомиоцитам желудочков. Рис. 18.1. Проводящая система сердца 424 Часть II. Частная фармакология Глава 18. Антиаритмические средства 425 Атриовентрикулярное соединение состоит из атриовентрикулярного узла, тонких прободающих и толстых дистальных волокон предсердножелудочкового пучка. Атриовентрикулярный узел находится в задней части стенки правого предсердия, непосредственно позади трехстворчатого клапана. Уровень его автоматизма достигает 40–60 импульсов в минуту и в норме подавляется синусовым узлом. Значения проводимости неодинаковы в разных отделах проводящей системы. Так, межузловые пучки проводят импульс со скоростью 0,3 м/с, а в прободающих волокнах атриовентрикулярного соединения отмечают наиболее низкие значения проводимости — 0,02–0,05 м/с. Оба узла проводящей системы находятся в стенках предсердий, отделенных от желудочков непроводящей фиброзной тканью, препятствующей ретроградному проведению возбуждения во время систолы. Через эту фиброзную преграду из предсердий в желудочки проходят медленно проводящие прободающие волокна атриовентрикулярного пучка. Таким образом, все атриовентрикулярное соединение функционирует как фильтр, передающий возбуждение только в одну сторону и ограничивающий проведение лишних импульсов от тканей предсердий, имеющих высокий автоматизм, на желудочки. Атриовентрикулярный пучок (пучок Гиса) расположен в межжелудочковой перегородке. Он обладает наиболее высокой проводимостью (1,5–5 м/с). Пучок Гиса делится на две ножки (правую и левую), проходящие субэндокардиально. От них отходят волокна Пуркинье, располагающиеся в толще стенок желудочков и контактирующие там с рабочими кардиомиоцитами. Волокна содержат некоторое количество филаментов миозина, что позволяет им сокращаться вместе с типичными кардиомиоцитами желудочков. Проводимость рабочих клеток желудочков составляет 0,6–1,0 м/с. Ее высокие значения в системе Гис–Пуркинье и сократительных кардиомиоцитах желудочков позволяют импульсам от водителей ритма с большой скоростью распространяться по миокарду. Это делает систолическое сокращение быстрым и скоординированным. Автоматизм и проводимость находятся под контролем эфферентной иннервации сердца. Парасимпатическая нервная система оказывает угнетающее влияние за счет стимуляции М2-холинорецепторов. Холинергическая иннервация сердца не распространяется на желудочки: правая ветвь блуждающего нерва иннервирует синусовый узел, левая — атриовентрикулярный. Симпатическая нервная система повышает автоматизм и проводимость за счет стимуляции β1-адренорецепторов. Адренергические во- 426 Часть II. Частная фармакология локна иннервируют как саму проводящую систему, так и сократительные кардиомиоциты, а следовательно, регулируют и проводимость, и автоматизм, и сократимость. Нарушения автоматизма и проводимости приводят к сердечным аритмиям. Среди их причин выделяют: ● гипоксические явления (в результате ухудшения коронарного кровотока); ● нарушения метаболических процессов в сердце; ● изменения нейроэндокринной регуляции вследствие различных заболеваний (тиреотоксикоз, пороки сердца, тромбоэмболия легочной артерии и др.). Кроме того, некоторые группы ЛВ способны повышать автоматизм и изменять проводимость, способствуя тем самым развитию аритмий (оказывая проаритмическое или аритмогенное действие). К таким препаратам относят адренергические, кардиотонические и даже антиаритмические средства. Аритмии классифицируют по: ● локализации: —наджелудочковые (суправентрикулярные); —желудочковые (вентрикулярные); ● влиянию на частоту сердечных сокращений: —брадисистолические (снижающие ЧСС); —тахисистолические (повышающие ЧСС); ● механизму развития: —блокады (развиваются в результате снижения проводимости); —экстрасистолии (развиваются в результате повышения автоматизма); —аритмии повторного входа («reentry-аритмии» — развиваются в результате нарушения проводимости). Блокады проводящей системы обычно протекают в брадиаритмической форме; экстрасистолии и аритмии повторного входа — в тахиаритмической форме (их часто объединяют в группу «экстрасистолии и тахиаритмии»). Для выяснения причин возникновения аритмий и понимания механизмов действия антиаритмических средств необходимо рассмотреть некоторые вопросы электрофизиологии миокарда. Моделью для изучения служит модель потенциала действия волокна Пуркинье. Механизм представлен на рис. 18.2. Потенциал покоя мембраны волокна Пуркинье составляет –90 мВ (см. рис. 18.2а). Однако он самопроизвольно увеличивается во время спонтанной медленной диастолической деполяризации (фаза 4). При дости- б д в Рис. 18.2. Механизм возникновения потенциала действия волокна Пуркинье (пояснения в тексте) а г Глава 18. Антиаритмические средства 427 428 Часть II. Частная фармакология жении порогового уровня возникает потенциал действия, вызывающий сокращение сердца. Спонтанная деполяризация обусловлена медленным входом ионов Na+ и выходом ионов К+. Продолжительность этой фазы прямо влияет на время генерации очередного импульса и в связи с этим отражает автоматизм волокна Пуркинье (чем она длительнее, тем ниже уровень автоматизма). В синоатриальном узле спонтанный диастолический входящий ток ионов Na+ и в меньшей степени К+ определяет ЧСС. Этот ток обозначается If и обеспечивается неизбирательными катионными f-(funny) каналами. В результате спонтанной диастолической деполяризации заряд на мембране достигает –60 мВ. В этот момент открываются потенциалзависимые натриевые каналы (см. рис. 18.2б) и возникает лавинообразный вход ионов Na+ в клетку. Заряд на мембране при этом мгновенно увеличивается до +35мВ, возникает быстрая деполяризация мембраны (фаза 0). Продолжительность этой фазы отражает проводимость волокна (чем она короче, тем выше проводимость). Во время фазы 0 (при уровне заряда на мембране –40мВ) открываются потенциалзависимые кальциевые каналы (см. рис. 18.2в), обеспечивающие ток ионов Са2+. Эти каналы функционируют примерно в 3 раза медленнее натриевых. Благодаря этому ток ионов Са2+ образует фазу «плато» (фаза 2) потенциала действия. Потенциалзависимые калиевые каналы открываются при достижении электроположительных значений заряда на мембране (см. рис. 18.2г). Через них ионы K+ выходят из кардиомиоцитов, что обусловливает реполяризацию мембраны: инициальную (фаза 1) и окончательную (фаза 3). Фаза 1 непродолжительна, так как повышение заряда, вызываемое выходом ионов K+, нивелируется противотоком ионов Са2+. Во время окончательной реполяризации кальциевые и натриевые каналы уже закрыты, поэтому изменение потенциала мембраны достигается только за счет тока ионов К+. В ходе фазы 3 заряд на мембране возвращается к исходному уровню потенциала покоя (–90 мВ). Смена фаз потенциала действия волокна Пуркинье представлена на рис. 18.2д. ● Фаза 0 — быстрая деполяризация (вход ионов Na+), отражает функцию проводимости. (выход ионов K+ из клетки). ● Фаза 1 — ранняя реполяризация 2+ ● Фаза 2 — «плато» (вход ионов Са ). ● Фаза 3 — окончательная реполяризация (выход ионов K+). ● Фаза 4 — спонтанная медленная диастолическая деполяризация (вход ионов Na+ и К+ через специфические f-(funny) каналы), отражает функцию автоматизма. Глава 18. Антиаритмические средства 429 Общая продолжительность потенциала действия составляет около 1 с. При этом половина времени приходится на фазу 4. В фазы 0, 1 и 2 волокно Пуркинье пребывает в состоянии возбуждения и абсолютной рефрактерности (невозбудимости). В фазу 3 абсолютная рефрактерность сменяется относительной (способностью генерировать внеочередной импульс в ответ на пришедшее извне раздражение). Этот период времени обозначают как эффективный рефрактерный период (ЭРП). Обычно ЭРП занимает ⅔ окончательной реполяризации, поэтому, чем она продолжительнее, тем длительнее ЭРП и, соответственно меньше риск возникновения потенциала действия в ответ на внеочередную стимуляцию. Электрофизиологические механизмы генерации потенциала действия в узлах проводящей системы существенно отличаются от таковых в волокнах Пуркинье (рис. 18.3). Синусовый и атриовентрикулярный узлы генерируют кальциевые потенциалы действия. Потенциал покоя пейсмейкерных клеток первого составляет –55 мВ. При этом большинство натриевых каналов находятся в инактивированном состоянии, а фаза 4 и фаза 0 обусловлены током ионов Са2+. Однако их проникновение в клетки инициируется входящим током ионов Na+ через специфические If-каналы. Это дает основание считать, что If-каналы имеют основополагающее значение для нормального автоматизма синусового узла. Основные способы терапии при экстрасистолиях и тахиаритмиях: ● угнетение автоматизма; ● угнетение проводимости; ● пролонгирование ЭРП. а б Рис. 18.3. Потенциалы действия водителей ритма (пояснения в тексте): а — синусового узла; б — атриовентриокулярного узла 430 Часть II. Частная фармакология Первый способ целесообразен при экстрасистолиях (внеочередных сокращениях желудочков), возникающих в результате несвоевременной и эктопической генерации импульса. Повышенное содержание калия в тканях, гипоксия и другие факторы приводят к увеличению уровня автоматизма волокон Пуркинье и сократительных кардиомиоцитов. Формируется эктопический очаг, генерирующий внеочередные импульсы, которые могут распространяться на миокард желудочков и вызывать экстрасистолию. Применение ЛС, пролонгирующих фазу 4, приводит к уменьшению автоматизма и подавлению активности таких очагов. Угнетение проводимости целесообразно при аритмиях повторного входа (reentry-аритмиях). Они возникают в результате неполных блокад в системе Гис–Пуркинье (рис. 18.4). Рис. 18.4. Механизм возникновения reentry-аритмий (пояснения в тексте) Глава 18. Антиаритмические средства 431 В норме импульс проходит по волокнам с одинаковой скоростью (см. рис. 18.4а). Однако если из-за вышеперечисленных патологических факторов одно из них находится в рефрактерном состоянии, то оно не способно к проведению ортодромного импульса, направленного от узла на периферию. В то же время потенциал действия, прошедший по неповрежденному волокну, может возвратиться антидромно в поврежденное, которое к этому моменту теряет рефрактерность и способно к проведению. Возникает так называемый односторонний блок. В этом случае антидромный импульс повторно входит в неповрежденное волокно, становясь внеочередным (см. рис. 18.4б). При применении средств, угнетающих проводимость, блок в пораженном волокне становится полным (двусторонне направленным), и волокно теряет способность проводить не только ортодромные, но и ретроградные импульсы. В результате повторный вход внеочередных потенциалов действия прекращается (см. рис. 18.4в). По механизму reentry возникают не только желудочковые аритмии (рис. 18.5). Так, синдром преждевременного возбуждения желудочков представляет собой упорядоченную циркуляцию возбуждения между предсердиями и желудочками, если между ними существует дополнительный проводящий пучок (см. рис. 18.5а). В этом случае ортодромный импульс, пройдя к желудочкам по пучку Гиса, может ретроградно возвратиться по дополнительному пучку к предсердиям и стать внеочередным. а б в Рис. 18.5. Различные варианты reentry-аритмий (а, б, в): С — синусовый узел; АВ — атриовентрикулярный узел; ПГ — пучок Гиса; ДП — дополнительный проводящий пучок 432 Часть II. Частная фармакология Кроме того, по принципу упорядоченной циркуляции возбуждения возникает трепетание предсердий (см. рис. 18.5б). В этом случае они сокращаются по очереди. Фибрилляция предсердий возникает по принципу беспорядочной циркуляции возбуждения (см. рис. 18.5в). Пролонгирование ЭРП целесообразно при экстрасистолиях и тахиаритмиях любой локализации. Применение средств, увеличивающих фазу 3, снижает риск генерации желудочками внеочередного импульса в ответ на несвоевременную стимуляцию. В соответствии с вышеописанным электрофизиологическим механизмом возникновения потенциала действия волокна Пуркинье можно заключить, что: ● пролонгирование фазы 0 достигают путем блокады натриевых каналов; ● пролонгирование фазы 3 достигают путем блокады калиевых каналов; ● пролонгирование фазы 4 достигают путем блокады как натриевых, так и калиевых каналов. Кроме того, снижение уровня автоматизма, проводимости и увеличение ЭРП возникает при блокаде β1-адренорецепторов. Необходимо также особо подчеркнуть, что угнетение сократимости не приводит к противоаритмическому эффекту и считается побочным действием антиаритмических средств. Противоаритмические средства классифицируют по применению и механизмам действия. ● Средства, применяемые при тахиаритмиях и экстрасистолии: —I класс — блокаторы натриевых каналов; —II класс — β-адреноблокаторы; —III класс — блокаторы калиевых каналов; —IV класс — блокаторы кальциевых каналов; —аденозин; —сердечные гликозиды; —препараты калия и магния. ● Средства, применяемые при брадиаритмиях и блокадах: —м-холиноблокаторы; —β-адреномиметики. Классификацию антиаритмических средств на 4 класса по механизму действия впервые предложил Vaughan Williams в 1970 г. В несколько модифицированном виде ей пользуются до сих пор. Помимо этих четырех групп при некоторых видах наджелудочковых тахиаритмий эффективны сердечные гликозиды и аденозин, а при экстрасистолиях, связанных с гипокалиемией, — препараты калия и магния. Глава 18. Антиаритмические средства 433 18.1. КЛАСС I — БЛОКАТОРЫ НАТРИЕВЫХ КАНАЛОВ Прежнее название этой группы препаратов — мембраностабилизирующие средства. В настоящее время блокаторы натриевых каналов принято разделять на 3 подкласса: IА, IВ и IС. Между ними существует ряд различий в механизме действия, влиянии на параметры сердца и применении. 18.1.1. Класс IА К этому классу противоаритмических препаратов относят хинидиноподобные средства (хинидин, прокаинамид и дизопирамид). Эти ЛВ блокируют натриевые и калиевые каналы и тем самым пролонгируют фазы 0, 3 и 4 потенциала действия (рис. 18.6). Это приводит к снижению автоматизма, проводимости и увеличению ЭРП. Хинидиноподобные средства действуют на все отделы сердца, поэтому они эффективны как при желудочковых, так и при суправентрикулярных экстрасистолии и тахиаритмиях. Общие побочные эффекты этой группы препаратов: ● снижение сократимости миокарда; ● снижение атриовентрикулярной проводимости; ● снижение АД; ● ваголитическое действие; ● аритмогенное действие. Последнее обусловлено несколькими причинами. Пролонгирование потенциала действия повышает риск появления ранней постдеполяризации, которая в свою очередь приводит к возник- Рис. 18.6. Влияние хинидина на потенциал действия волокна Пуркинье 434 Часть II. Частная фармакология новению аритмии torsades de pointes (русские названия: мультиформная желудочковая тахикардия, «пируэт»). ● Ваголитическое действие хинидиноподобных средств повышает уровень автоматизма синусового узла, тем самым облегчая проведение импульсов через атриовентрикулярное соединение и усиливая аритмию желудочков. ● Угнетение проводимости при reentry-аритмиях может привести к циркуляции возбуждения по более коротким траекториям, что, в свою очередь, способствует учащению повторного входа и усилению аритмии. Хинидин (Хинипэк♠, Кинидин Дурулес♠) — алкалоид коры хинного дерева (правовращающий изомер хинина). В медицинской практике препарат используют в виде хинидина сульфата. Он хорошо всасывается из ЖКТ, что обусловливает его применение внутрь. Биодоступность в среднем составляет 70–80%. Максимальная концентрация препарата в крови достигается через 2–3 ч, а связывание с белками плазмы происходит на 60–80%. Хинидин метаболизируется в печени, выводится почками (около 20% в неизмененном виде); период полуэлиминации составляет 6–7 ч. Почечная экскреция препарата увеличивается при изменении реакции мочи в кислую сторону. Хинидин оказывает следующие действия: ● снижает автоматизм, проводимость и увеличивает ЭРП кардиомиоцитов желудочков; ● подавляет автоматизм водителей ритма (в большей степени атриовентрикулярного и в меньшей — синусового узла); ● умеренно снижает скорость проведения возбуждения по атриовентрикулярному узлу и сократимость миокарда (в терапевтических дозах); ● оказывает ваголитическое (антихолинергическое) действие, угнетая передачу возбуждения с блуждающего нерва на сердце и таким образом вызывая повышение уровня автоматизма синоатриального узла и умеренную тахикардию; ● расширяет периферические сосуды (блокирует β-адренорецепторы), тем самым вызывая умеренное снижение АД. Хинидин эффективен при желудочковых и наджелудочковых нарушениях сердечного ритма. Его назначают при постоянной и пароксизмальной формах мерцательной аритмии предсердий, пароксизмальной тахикардии, экстрасистолии. Побочные эффекты хинидина: ● снижение сократимости миокарда, артериальная гипотензия; ● атриовентрикулярный блок; Глава 18. Антиаритмические средства 435 ● тошнота, рвота, диарея; ● головокружение, звон в ушах, нарушения слуха, зрения; ● тромбоцитопения. В 5% случаев хинидин оказывает аритмогенное действие, приводя к возникновению желудочковых нарушений ритма типа «пируэт». Прокаинамид (новокаинамид♠) воздействует на электрофизиологические механизмы работы сердца аналогично хинидину (рис. 18.7). В отличие от последнего он меньше снижает сократимость миокарда, обладает менее выраженными антихолинергическими свойствами и не блокирует β-адренорецепторы, однако обладает умеренным ганглиоблокирующим действием. Так же, как хинидин, прокаинамид применяют при желудочковых и наджелудочковых тахиаритмиях и экстрасистолии. Рис. 18.7. Химические структуры прокаинамида и дизопирамида Препарат назначают не только внутрь, но и внутривенно (при необходимости быстрого достижения эффекта). При применении внутрь прокаинамид всасывается быстрее, чем хинидин. Биодоступность составляет 75–95%. Препарат незначительно связывается с белками плазмы крови (15–20%). Метаболизируется в печени с образованием активного метаболита — N-ацетилпрокаинамида. По скорости метаболизма среди пациентов выделяют медленных и быстрых «ацетиляторов», существенно различающихся по продолжительности действия препарата. Прокаинамид выводится почками в неизмененном виде и в виде метаболитов. Активный метаболит экскретируется медленнее, чем сам препарат, и при нарушении функции почек может кумулировать в организме. Побочные эффекты: ● снижение АД (вследствие ганглиоблокирующего действия); ● нарушение проводимости; ● тошнота, рвота, диарея; ● судороги; 436 Часть II. Частная фармакология ● аллергические реакции в виде лихорадки, болей в суставах и мышцах, кожной сыпи, реже в виде агранулоцитоза или синдрома, подобного системной красной волчанке; (редко). ● нервно-психические расстройства Дизопирамид (ритмодан♠, ритмилен♠) — аналог хинидина, обладает выраженными антиаритмическими свойствами (см. рис. 18.7). Показания к применению аналогичны хинидину. Препарат снижает сократимость миокарда, а также обладает сильным М-холиноблокирующим действием. Хорошая всасываемость из кишечника (биодоступность 80%) позволяет применять препарат внутрь; связывается с белками плазмы крови, метаболизируется в печени. Выводится в основном почками (период полуэлиминации — 6–8 ч). Побочные эффекты: ● значительное снижение сократимости миокарда, атриовентрикулярный блок, аритмогенное действие; ● сухость во рту, паралич аккомодации, обстипация (запор), задержка мочеиспускания (в связи с выраженной М-холиноблокирующей активностью). 18.1.2. Класс IВ Препараты этого класса имеют ряд существенных отличий. ● Это селективные блокаторы натриевых каналов, пролонгирующие только фазы 0 и 4. Они угнетают проводимость и автоматизм волокон Пуркинье и сократительных кардиомиоцитов. Препараты класса IВ блокируют натриевые каналы в инактивированном состоянии и имеют скорость ассоциации/диссоциации менее 1 с, что не превышает продолжительности нормального потенциала действия. Таким образом, не урежая сердечного ритма, они препятствуют генерации и проведению внеочередных импульсов. ● Препараты класса IВ не блокируют калиевые каналы и не пролонгируют реполяризацию. Напротив, при их применении отмечено некоторое укорочение фазы 3 и ЭРП (возможно, за счет активации калиевых каналов). ● Препараты класса IВ не изменяют ток кальция (ни прямо, ни опосредованно). В связи с этим они воздействуют только на желудочки, не влияя на водители ритма и не снижая сократимость миокарда. Это обусловливает применение данных препаратов только при желудочковых экстрасистолиях. Лидокаин (ксикаин ♠, ксилокаин ♠), блокируя натриевые каналы клеточных мембран, оказывает противоаритмическое и местноане- Глава 18. Антиаритмические средства 437 стезирующее действия (рис. 18.8). Препарат уменьшает проводимость и автоматизм волокон Пуркинье и рабочих кардиомиоцитов, подавляя эктопические очаги в желудочках. Его считают препаратом выбора при желудочковых аритмиях, возникающих после инфаркта миокарда. Это обусловлено тем, что, оказывая противоаритмическое действие, лидокаин не влияет на сократимость, которая в этом случае обычно снижена. Лидокаин отличается высоким пресистемным метаболизмом. При первом прохождении через печень практически полностью выводится из портального кровотока. В связи с этой фармакокинетической особенностью препарат неэффективен при приеме внутрь, его вводят внутривенно капельно (связь с белками плазмы крови — 50–80%), t½ составляет около 2 ч. При патологии печени скорость метаболизма лидокаина снижается. Препарат легко переносится, однако возможны следующие побочные эффекты: ● нарушения со стороны ЦНС (сонливость, спутанность сознания, судорожные реакции); ● артериальная гипотензия, снижение атриовентрикулярной проводимости. Мексилетин (см. рис. 18.8) — структурный аналог лидокаина, эффективный при приеме внутрь (биодоступность — 90–100%). Действует длительно (период полуэлиминации — 12–16 ч). Возможно и внутривенное введение препарата. Применяют при желудочковых аритмиях. Побочные эффекты: ● брадикардия, артериальная гипотензия, нарушение атриовентрикулярной проводимости, аритмии; ● тошнота, рвота; ● неврологические нарушения. Фенитоин (дифенин♠) первоначально применяли в качестве противоэпилептического средства. Позже у него было обнаружено антиаритмическое действие. Фенитоин считают препаратом выбора при желудочковых аритмиях, вызванных сердечными гликозидами. Это связано с тем, Рис. 18.8. Химические структуры лидокаина гидрохлорида и мексилетина 438 Часть II. Частная фармакология что, оказывая антиаритмическое действие, он не уменьшает положительный инотропный эффект данных кардиотонических средств. Фенитоин медленно всасывается из ЖКТ, в крови связывается с белками плазмы на 80–96%. Метаболизируется в печени, для препарата характерна кинетика нулевого порядка, время полуэлиминации составляет 24–30 ч. Возможна кумуляция препарата. Метаболиты выводятся в основном почками. Из побочных эффектов отмечают атаксию, нистагм, диплопию, гиперпластический гингивит (гиперплазию десен). 18.1.3. Класс IС Как и препараты класса IА, эти вещества действуют и на желудочки, и на водители ритма. Однако применяют их только при вентрикулярных аритмиях, угрожающих жизни и резистентных (устойчивых) к другим противоаритмическим средствам. Такая специфическая ниша препаратов класса IС обусловлена их выраженной аритмогенной активностью. Кроме того, отдельные представители этой группы могут выраженно угнетать сократимость и атриовентрикулярную проводимость. Пропафенон — типичный представитель класса IС. Блокирует натриевые и в некоторой степени кальциевые каналы, а также обладает β-адреноблокирующей активностью. При введении внутрь хорошо всасывается, однако интенсивно метаболизируется при первом прохождении через печень (биодоступность варьирует от 4 до 40%), применяется внутривенно. Препарат используют при желудочковых аритмиях в случае неэффективности других антиаритмических средств. Побочные эффекты: ● тошнота, рвота, запор, сухость во рту; ● нарушение сна; ● брадикардия; ● бронхоспазм; аритмогенное действие. ● выраженное Флекаинид♠ выраженно угнетает проводимость в атриовентрикулярном узле, пучке Гиса и волокнах Пуркинье, незначительно снижает уровень автоматизма синусного узла, практически не увеличивает ЭРП в желудочках и мало влияет на сократимость миокарда. Препарат применяют в виде флекаинида ацетата при желудочковых аритмиях в случае неэффективности других антиаритмических средств. Вводят внутрь, иногда внутривенно. При применении внутрь максимальную концентрацию в плазме отмечают через 3–4 ч после приема. Флекаинид♠ практически Глава 18. Антиаритмические средства 439 не изменяется при первом прохождении через печень. 75–85% препарата связываются с белками плазмы. Побочные эффекты: ● аритмогенное действие (отмечают в 5–15% случаев); ● тошнота, головная боль, головокружение; ● нарушение зрения.♠ Морацизин (этмозин ) (рис. 18.9) — производное фенотиазина, препарат смешанного типа действия, обладает свойствами блокаторов натриевых каналов всех трех классов — IС, IВ, IА. Препарат угнетает проводимость в атриовентрикулярном узле, пучке Гиса и волокнах Пуркинье, мало влияет на автоматизм синусового узла и сократимость миокарда. Морацизин эффективен при желудочковых и наджелудочковых нарушениях ритма. Однако из-за выраженного аритмогенного действия его применяют только при вентрикулярных аритмиях, рефрактерных к другим ЛС. Вводят внутрь (биодоступность составляет около 40%) и внутривенно. На 95% связывается с белками плазмы, период полуэлиминации составляет 2 ч. Побочные эффекты: ● аритмогенное действие; ● головная боль; ● повышенная утомляемость и слабость; ● диарея ♠и др. Этацизин (см. рис. 18.9) близок по химическому строению к морацизину. Препарат блокирует не только натриевые, но и кальциевые каналы. Применяют аналогично морацизину, назначают внутрь и внутривенно. Побочные эффекты более выражены и проявляются чаще (в особенности при внутривенном введении). Лаппаконитина гидробромид (аллапинин♠) — гидробромид алкалоида борца белоустого (Aconitum Leucostomum). При приеме внутрь легко всасывается, но подвергается интенсивной пресистемной биотрансформации, в связи с чем биодоступность не превышает 39%. Эффект развивается че- Рис. 18.9. Химические структуры морацизина и этацизина 440 Часть II. Частная фармакология рез 40–60 мин, достигает максимума через 4–5 ч и продолжается 8 ч и более; t½ составляет 1–1,2 ч. При внутривенном введении антиаритмическое действие проявляется через 15–20 мин и длится 6–8 ч (с максимумом через 2 ч). Основные преимущества лаппаконитина гидробромида следующие: ● мало влияет на функции синусового узла; ● практически не угнетает атриовентрикулярную проводимость (хотя снижает проведение по предсердиям и желудочкам); ● практически не угнетает сократимость (в связи с чем может назнаРис. 18.10. Химическая структура лаппаконитина гидробромида чаться при инфаркте миокарда); ● практически не снижает АД. Препарат применяется при наджелудочковой и желудочковой экстрасистолии, в том числе на фоне инфаркта миокарда. Переносимость препарата в целом хорошая. Побочные эффекты: проаритмическое действие, головная боль, головокружение, диплопия, обусловленные проникновением препарата через ГЭБ. Эти реакции проходят при уменьшении дозы препарата. 18.2. КЛАСС II — β-АДРЕНОБЛОКАТОРЫ β1-Адренорецепторы локализованы как в водителях ритма, так и в рабочих кардиомиоцитах желудочков. При их блокаде снижаются проводимость, автоматизм, возбудимость и увеличивается ЭРП во всех отделах сердца. В связи с этим β-адреноблокаторы эффективны как при желудочковых, так и при суправентрикулярных тахиаритмиях. В качестве противоаритмических средств применяют кардиоселективные (атенолол, талинолол, метопролол) и неселективные β-адреноблокаторы (пропранолол). К побочным эффектам этих средств относят: ● атриовентрикулярный блок; ● снижение сократимости миокарда; ● брадикардию. Для пропранолола характерен также спазм бронхов и периферических сосудов. Глава 18. Антиаритмические средства 441 18.3. КЛАСС III — БЛОКАТОРЫ КАЛИЕВЫХ КАНАЛОВ Данный класс включает средства, пролонгирующие реполяризацию и, следовательно, увеличивающие продолжительность потенциала действия. Эта группа веществ очень неоднородна, традиционно к ней относят: ● амиодарон — препарат с многокомпонентным механизмом антиаритмического действия, включающим в том числе блокаду калиевых каналов; ● соталол — блокатор β-адренорецепторов и калиевых каналов. Кроме того, существуют средства, избирательно блокирующие калиевые каналы, — ибутилид и нибентан♠. Амиодарон (кордарон♠) обладает смешанным механизмом противоаритмического действия (сочетает свойства I, II, III и IV классов антиаритмических средств) (рис. 18.11). Препарат оказывает следующие действия: ● блокирует калиевые каналы волокон Пуркинье, тем самым увеличивая ЭРП и пролонгируя фазу 3 потенциала действия; ● блокирует натриевые каналы волокон Пуркинье, уменьшая таким образом проводимость и автоматизм желудочков (из-за увеличения фаз 0 и 4); ● блокирует кальциевые каналы, снижая уровень автоматизма, проводимости и увеличивая ЭРП в узлах проводящей системы; ● обладает неконкурентным β-адреноблокирующим действием; ● угнетает аденилатциклазу; ● выполняет функции антагониста глюкагоновых рецепторов миокарда. Амиодарон воздействует на все отделы сердца, поэтому он эффективен как при желудочковых, так и при суправентрикулярных тахиаритмиях Рис. 18.11. Химические структуры амиодарона и соталола 442 Часть II. Частная фармакология и экстрасистолии. При приеме внутрь препарат всасывается медленно и не полностью (степень всасывания варьирует от 20 до 50%). Связывается с белками плазмы на 96%. Терапевтический эффект при приеме внутрь развивается медленно, для достижения максимальной эффективности требуется несколько недель. Метаболизируется в печени с образованием активного метаболита N-дезэтиламиодарона. Выводится главным образом через кишечник, очень медленно. Элиминация носит двуфазный характер: ● начальная фаза — около 10 дней; ● терминальная фаза — до 100 дней. Период полуэлиминации составляет 10–50 сут. В связи с продолжительной циркуляцией в системном кровотоке амиодарон кумулирует (накапливается в тканях) и может откладываться в виде кристаллов в роговице. Препарат применяют в качестве противоаритмического, а также антиангинального средства, поскольку он оказывает следующие действия: ● уменьшает потребность миокарда в кислороде за счет блокады β1-адренорецепторов сердца; ● повышает доставку кислорода к миокарду за счет расширения коронарных сосудов в результате блокады α-адренорецепторов и кальциевых каналов. К побочным эффектам относят: ● снижение силы сердечных сокращений (в результате блокады кальциевых каналов сократительных кардиомиоцитов), уменьшение атриовентрикулярной проводимости, брадикардию, гипотензию; ● аритмогенное действие (однако он реже, чем хинидиноподобные средства, вызывает аритмию типа «пируэт»); ● нарушения функции щитовидной железы (гипо- или гипертиреоз), а также струмогенное (зобогенное) действие. Это обусловлено тем, что амиодарон содержит в структуре йод и активирует продукцию тиреотропного гормона аденогипофизом; ● фиброзный пневмонит, часто приводящий к летальному исходу. При длительном применении возможно: ● возникновение обменных нарушений (отложение липофусцина в роговице и коже, что придает ей серо-голубой оттенок); ● появление фотосенсибилизации; ● угнетение микросомальных ферментов печени; ● появление неврологических нарушений (тремор, атаксия, парестезии). Соталол (Соталекс♠, Лоритмик♠, Дароб♠) (см. рис. 18.11) сочетает в себе свойства II и III классов противоаритмических средств: Глава 18. Антиаритмические средства 443 ● блокирует калиевые каналы, что ведет к пролонгированию реполяризации и увеличению ЭРП; ● блокирует β-адренорецепторы, тем самым угнетая автоматизм синоатриального узла, снижает автоматизм и проводимость в атриовентрикулярном соединении и в других отделах проводящей системы. Поскольку соталол оказывает влияние на все отделы сердца, он эффективен как при желудочковых, так и при наджелудочковых тахиаритмиях и экстрасистолии. Вводят внутрь и внутривенно. Хорошо всасывается из ЖКТ — биодоступность составляет 90–100%; однако она снижается при приеме препарата вместе с молочными продуктами. Действие препарата начинается через 1 ч после приема внутрь и продолжается в течение 24 ч. Соталол не связывается белками плазмы крови, экскретируется почками в неизмененном виде. Период полуэлиминации составляет 7–12 ч. Побочные эффекты соталола связаны с неселективной β-адреноблокирующей активностью (см. раздел «Адреноблокаторы»). Бретилия тозилат (Орнид♠) — четвертичное аммониевое соединение, обладающее симпатолитическими свойствами. Увеличивает продолжительность потенциала действия и удлиняет ЭРП. Применяют при желудочковых аритмиях, рефрактерных к другим антиаритмическим средствам. Поскольку препарат плохо всасывается из ЖКТ, его вводят внутривенно или внутримышечно. Экскретируется преимущественно почками в неизмененном виде. Период полуэлиминации составляет 6–10 ч. Из побочных эффектов отмечают связанную с симпатолитическим действием препарата артериальную гипотензию (возможна ортостатическая гипотензия). Блокаторы калиевых каналов более избирательного действия — ибутилид (корверт♠) и нитрофенилдиэтиламинопентилбензамид (нибентан♠). Однако оба препарата оказались эффективными только при наджелудочковых тахиаритмиях. Это объясняют морфофункциональной особенностью волокон атриовентрикулярного пучка: их мембраны содержат большое количество калиевых каналов, поэтому фаза 3 потенциала действия значительно короче, чем в других отделах. Короткий период рефрактерности кардиомиоцитов пучка Гиса определяет высокий уровень проводимости. Избирательная блокада калиевых каналов ибутилидом или нибентаном♠ в первую очередь увеличивает ЭРП атриовентрикулярного пучка, и внеочередные импульсы с предсердий перестают проходить на желудочки. Необходимо отметить, что, по некоторым данным, ибутилид обладает также способностью блокировать натриевые каналы, что также ведет к снижению проводимости за счет увеличения фазы 0. 444 Часть II. Частная фармакология Оба препарата вводят внутривенно с целью купирования пароксизмов суправентрикулярных тахиаритмий (мерцание и трепетание предсердий). Дозы подбирают индивидуально с учетом массы тела пациента. Существенным недостатком обоих средств считают их высокую аритмогенную активность. В связи с этим данные препараты применяют только в условиях стационара при обязательном постоянном контроле ЭКГ. 18.4. КЛАСС IV — БЛОКАТОРЫ КАЛЬЦИЕВЫХ КАНАЛОВ Эта группа препаратов очень неоднородна, поскольку существует несколько типов кальциевых каналов (L, T и др.). В сердце и кровеносных сосудах локализованы преимущественно кальциевые каналы L-типа. Их чувствительность к веществам разного химического строения неодинакова. Поэтому блокаторы кальциевых каналов классифицируют на следующие группы: ● селективные блокаторы кальциевых каналов I класса (фенилалкиламины — верапамил). Эти препараты в большей степени блокируют кальциевые каналы L-типа кардиомиоцитов; ● селективные блокаторы кальциевых каналов II класса (дигидропиридины — нифедипин, амлодипин, лацидипин). Эти препараты в основном блокируют кальциевые каналы L-типа гладкомышечных клеток сосудов; ● селективные блокаторы кальциевых каналов III класса (бензотиазепин — дилтиазем). Они в равной степени блокируют кальциевые каналы L-типа в сердце и сосудах; ● блокаторы кальциевых каналов IV класса с преимущественным влиянием на сосуды головного мозга (циннаризин); ● блокаторы кальциевых каналов V класса (фенилалкиламин — прениламин); ● блокаторы кальциевых каналов VI класса (мибефрадил). Эти препараты блокируют преимущественно кальциевые каналы Т-типа. В качестве противоаритмических средств применяют верапамил, реже — дилтиазем. Препараты этой группы эффективны только при наджелудочковых аритмиях. Это связано с тем, что водители ритма генерируют кальциевые потенциалы действия, поэтому блокада этих каналов приводит к снижению уровня автоматизма и проводимости преимущественно в узлах проводящей системы. Блокада же кальциевых каналов кардиомиоцитов желудочков вызывает уменьшение сократимости, что считают побочным эффектом данных препаратов. Кроме того, представители этой группы Глава 18. Антиаритмические средства 445 воздействуют на ангиомиоциты, в результате чего расширяются кровеносные сосуды и возникает гипотензия. Верапамил (изоптин♠, финоптин♠) относят к группе фенилалкиламинов (рис. 18.12). При приеме внутрь хорошо всасывается (степень абсорбции — 90–92%). Максимальный эффект отмечают через 1,5–2 ч. До 90% препарата связываются с белками плазмы крови. Экскретируется с мочой и желчью в неизмененном виде и в виде конъюгатов, t½ составляет 3–7 ч. Побочные эффекты верапамила, связанные с блокадой кальциевых каналов: ● ослабление и урежение сокращений сердца; ● снижение атриовентрикулярной проводимости (вплоть до полного атриовентрикулярного блока); ● артериальная гипотензия. Кроме того, возможны констипация, тошнота, рвота, аллергические реакции. Дилтиазем (кардил♠, алтиазем PP♠) относят к группе бензодиазепинов (см. рис. 18.12). При приеме внутрь препарат хорошо всасывается (степень абсорбции до 90%), биодоступность составляет 40%. Пик концентрации в плазме наступает через 3 ч. Действие препарата развивается быстро — через 30 мин после приема. Дилтиазем ацетилируется и в неизмененном виде и в виде метаболитов выводится через кишечник, t½ 3–4 ч. Побочные эффекты дилтиазема, как правило, обусловлены его вазодилатирующим действием: ● головная боль; ● головокружение; ● тахикардия; ● отеки; ● мышечная слабость. Рис. 18.12. Химические структуры верапамила и дилтиазема 446 Часть II. Частная фармакология 18.5. ДРУГИЕ СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ ТАХИАРИТМИЯХ И ЭКСТРАСИСТОЛИИ 18.5.1. Аденозин Аденозин — естественный нуклеозид, агонист аденозиновых А1-рецепторов (рис. 18.13). Быстро захватывается эритроцитами (специфический нуклеозидный транспортер) и быстро разрушается энзимами люминальных мембран эндотелиоцитов, поэтому обладает чрезвычайно коротким периодом полуэлиминации (t½ 10 с) и невысокой продолжительностью действия (около 1 мин). Вводят внутривенно болюсно. В качестве противоаритмического средства применяется только при наджелудочковых тахиаритмиях (например, при синдроме преждевременного возбуждения желудочков). Антиаритмическое действие обусловлено способностью аденозина вызывать кратковременную атриовентрикулярную блокаду, приводящую к прекращению циркуляции возбуждения между желудочками и предсердиями по механизму reentry. Аденозиновые А1-рецепторы сопряжены с калиевыми каналами, и при их стимуляции возникает гиперполяризация, что приводит к угнетению проводимости атриовентрикулярного соединения, а также к подавлению автоматизма синусного узла и снижению сократимости миокарда. Последнее в виде кратковременной асистолии относят к побочным эффектам препарата. Рис. 18.13. Химическая струк- Кроме того, аденозин может вызывать натура аденозина рушения дыхания (диспноэ). 18.5.2. Сердечные гликозиды Противоаритмическим действием обладает препарат наперстянки дигоксин. Его назначают в основном при тахисистолической форме мерцательной аритмии. Мерцание предсердий обусловлено повышением их автоматизма и беспорядочной циркуляцией возбуждения (см. рис. 18.5в). Частота их сокращений достигает 300 в минуту. При этом естественная проводимость атриовентрикулярного соединения позволяет пропустить только каждый 2-й или каждый 4-й импульс. Частота сокращения желудочков в такой ситуации составляет 150 (тахисистолическая форма Глава 18. Антиаритмические средства 447 мерцательной аритмии) или 75 (нормосистолическая форма) ударов в минуту соответственно. Дигоксин снижает атриовентрикулярную проводимость — обладает отрицательным дромотропным эффектом. При этом даже неполная блокада (переход от тахисистолической формы мерцательной аритмии к нормосистолической) способна нормализовать частоту сокращения желудочков. В случае если дигоксин вызывает полную поперечную блокаду, мерцание предсердий переходит в трепетание, а желудочки начинают сокращаться в автономном (идиовентрикулярном) ритме. Роль водителя такого ритма берет на себя группа кардиомиоцитов пучка Гиса, генерирующая импульсы с частотой 40–60 в мин. Сокращения желудочков урежаются, однако при этом становятся более скоординированными, что положительно сказывается на гемодинамике. 18.5.3. Препараты калия и магния Калия хлорид, наиболее эффективный в виде поляризующих смесей, а также калия и магния аспарагинат (панангин♠ и аспаркам♠) используют только при тахиаритмиях и экстрасистолии, обусловленных сниженной концентрацией этих ионов в крови. Препарат вводят внутрь или внутривенно. Побочные эффекты: ● парестезии; ● диспепсия; ● атриовентрикулярный блок. Магния сульфат применяют для купирования мультиформной желудочковой тахикардии (аритмии типа «пируэт»). Вводят внутривенно. Вопросы и задания для самоконтроля 1. При экстрасистолии и тахиаритмиях применяют: а) блокаторы натриевых каналов; б) β-адреноблокаторы; в) блокаторы кальциевых каналов; г) М-холиноблокаторы; д) блокаторы калиевых каналов. 2. Только при наджелудочковых тахиаритмиях применяют: а) пропранолол; б) верапамил; 448 Часть II. Частная фармакология в) лаппаконитин; г) нибентан; д) аденозин. 3. Только при желудочковых экстрасистолиях применяют: а) амиодарон; б) мексилетин; в) лидокаин; г) лаппаконитин; д) фенитоин. 4. Блокирует калиевые каналы, увеличивает эффективный рефрактерный период. В некоторой степени блокирует натриевые и кальциевые каналы, а также β-адренорецепторы. Применяется при желудочковых и наджелудочковых нарушениях ритма, стенокардии. В качестве побочных эффектов вызывает брадикардию, нарушение атриовентрикулярной проводимости, нарушение функций щитовидной железы, отложение кристаллов в роговице: а) верапамил; б) амиодарон; в) пропранолол; г) лаппаконитин; д) дизопирамид. 5. Обладает противоаритмической и противосудорожной активностью. Существенно не влияет на сократимость миокарда и уровень артериального давления. Является препаратом выбора при аритмиях, вызванных передозировкой сердечных гликозидов: а) лидокаин; б) лаппаконитин; в) фенитоин; г) прокаинамид; д) амиодарон. Глава 19 СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ НЕДОСТАТОЧНОСТИ КОРОНАРНОГО КРОВООБРАЩЕНИЯ Недостаточность коронарного кровообращения (коронарная недостаточность) возникает в результате сужения просвета коронарных сосудов (окклюзия атеросклеротическими бляшками, тромбообразование в области бляшек в коронарных сосудах) или в результате коронароспазма (что выявляют гораздо реже). Патологическое состояние, обусловленное коронарной недостаточностью, обозначают термином «ишемическая болезнь сердца». Ее основные клинические формы: ● хроническая (проявляется стенокардией); ● острая (проявляется инфарктом миокарда). Кровоснабжение миокарда происходит через коронарные (венечные) артерии. Их устья расположены в аорте непосредственно над створками аортального клапана. Каждая из коронарных артерий делится на две ветви — субэндокардиальную (проходит между эндокардом и миокардом) и субэпикардиальную (проходит между эпикардом и миокардом) (рис. 19.1). Обе системы (субэпикардиальная и субэндокардиальная) сообщаются друг с другом через коллатеральные сосуды. Такое коллатеральное сообщение служит для локального перераспределения коронарного кровотока и способно (до некоторой степени) компенсировать возникающую ишемию. Коронарный кровоток возможен в период диастолы. Во время систолы он невозможен, поскольку устья коронарных артерий закрыты створками аортального клапана, а сами коронарные сосуды сдавлены сокращающимся миокардом. При «достаточном» коронарном кровотоке доставка кислорода к сердцу всегда адекватна потребности миокарда в кислороде. Потребность сердца в кислороде — вариабельный параметр (так, при физической или психоэмоциональной нагрузке она возрастает, в состоянии покоя и положении лежа она снижается). Адекватность доставки кислорода потребностям миокарда обусловлена взаимосвязанной регуляцией ра- 450 Часть II. Частная фармакология Рис. 19.1. Схема коронарного кровообращения (пояснения в тексте) боты сердца и тонуса коронарных сосудов. При увеличении работы сердца интенсифицируется метаболизм АТФ. Если оксигенация кардиомиоцитов достаточна, происходит ресинтез АТФ. Если кровоток недостаточен, вместо ресинтеза АТФ происходит дальнейший метаболизм циклических аденозиновых нуклеотидов до аденозиндифосфата (АДФ), аденозинмонофосфата и аденозина. Продукты распада АТФ накапливаются в миокарде (особенно в зоне ишемии) и за счет стимуляции пуриновых рецепторов расширяют коронарные сосуды, что приводит к восстановлению адекватной доставки кислорода (кроме того, за счет стимуляции А1-рецепторов аденозин снижает сократимость миокарда и проводимость). Таким образом, в норме доставка кислорода всегда адекватна потребности, поскольку вещества, регулирующие как потребность, так и доставку, — последовательные звенья одной и той же метаболической цепи и вырабатываются в эквивалентных количествах. При коронарной недостаточности (например, при стенозирующем атеросклерозе) просвет коронарного сосуда сужен атеросклеротической бляшкой (рис. 19.2). Доставка кислорода к тканям ниже места окклюзии затруднена. В результате локальной ишемии ниже атеросклеротической бляшки накапливаются метаболиты АТФ (аденозин и АДФ), расширяющие коронарные Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 451 Рис. 19.2. Нарушение коронарного кровотока при стенозирующем атеросклерозе сосуды. Таким образом, ниже места окклюзии в условиях гипоксии коронарные сосуды расширены максимально, а доставка кислорода к миокарду недостаточна (ввиду механического препятствия). В результате при физической нагрузке за счет дисбаланса между потребностью миокарда в кислороде и доставкой кислорода к миокарду может возникнуть приступ стенокардии напряжения. Ишемическая боль возникает в результате локальной стимуляции ванилоидных рецепторов протонами, образующимися при гипоксии. 19.1. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ СТЕНОКАРДИИ (АНТИАНГИНАЛЬНЫЕ СРЕДСТВА) Стенокардия (или angina pectoris — грудная жаба) проявляется приступами загрудинных болей сжимающего характера, локализованных за грудиной и иррадиирующих в левую лопатку и руку. Болевой синдром представляет собой приступ ишемической боли и возникает как следствие дисбаланса между потребностью миокарда в кислороде и доставкой кислорода к миокарду. В соответствии с причинами дисбаланса выделяют следующие формы стенокардии. 452 Часть II. Частная фармакология ● Стенокардия напряжения (стабильная стенокардия, классическая стенокардия). Недостаточное поступление кислорода к тканям сердца обусловлено стенозирующим атеросклерозом коронарных сосудов (см. рис. 19.2). Приступы стенокардии возникают во время физического или эмоционального напряжения, когда повышается потребность миокарда в кислороде, а пораженные атеросклерозом сосуды не могут адекватно расшириться и обеспечить необходимую доставку кислорода. При этой форме стенокардии целесообразно применение веществ, уменьшающих потребность миокарда в кислороде. ● Вазоспастическая стенокардия (вариантная стенокардия, стенокардия Принцметала). Дисбаланс между потребностью и доставкой обусловлен спазмом коронарных артерий. Коронароспазм (и, соответственно, приступ стенокардии) возникает спонтанно, без очевидных провоцирующих факторов (при обычной физической нагрузке, в покое и даже во сне). Возможной причиной коронароспазма считают снижение синтеза эндотелиального релаксирующего фактора. Вазоспастическая стенокардия встречается реже, чем стенокардия напряжения (не более 1% случаев стенокардии). При этой форме стенокардии целесообразно применение средств, расширяющих коронарные сосуды. ● Нестабильная стенокардия. Нарушения трофики обусловлены массированным тромбообразованием в коронарных сосудах. Приступ стенокардитической боли возникает в покое и плохо купируется антиангинальными препаратами. Нестабильную стенокардию расценивают как прединфарктное состояние, которое требует назначения средств, препятствующих тромбообразованию (антиагреганты, антикоагулянты, фибринолитики — см. гл. «Средства, влияющие на гемостаз и тромбообразование»). ЛС, применяемые при стенокардии, обозначают термином антиангинальные средства (от angina pectoris) и классифицируют в соответствии с механизмом их влияния на дисбаланс потребности и доставки. ● Средства, понижающие потребность миокарда в кислороде и повышающие доставку кислорода к миокарду: —органические нитраты; —блокаторы кальциевых каналов; —активаторы калиевых каналов; —разные средства. ● Средства, понижающие потребность миокарда в кислороде: —β-адреноблокаторы; —брадикардитические средства. Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 453 ● Средства, повышающие доставку кислорода к миокарду: —коронарорасширяющие средства миотропного действия; —коронарорасширяющие средства рефлекторного действия. Антиангинальные средства применяют как для купирования приступов (при возникшем приступе с целью его прекращения), так и для систематического лечения (препарат применяют в период между приступами с целью профилактики). Кроме антиангинальных средств, в лечении стенокардии применяют другие группы препаратов (антиагреганты, гиполипидемические средства, ангиопротекторы). Одна из новых, широко применяемых при стенокардии групп — средства, повышающие устойчивость кардиомиоцитов к ишемии (кардиопротекторные средства). 19.1.1. Средства, понижающие потребность миокарда в кислороде и повышающие доставку кислорода Органические нитраты Антиангинальное действие органических нитратов обнаружено в 1867 г. В настоящее время эта группа представлена препаратами нитроглицерина и органическими нитратами другой структуры. Нитроглицерин рассматривают как эталонное средство в группе органических нитратов. Его препараты широко применяют как для купирования, так и для профилактики приступов стенокардии. Препараты нитроглицерина Нитроглицерин (рис. 19.3) (нитроглицерол, глицерилтринитрат) — бесцветная маслянистая жидкость, плохо растворимая в воде и хорошо растворимая в спирте, эфире, хлороформе. При сублингвальном применении нитроглицерин быстро всасывается через слизистую оболочку ротовой полости, его действие развивается через 1–2 мин и продолжается около 30 мин (t½ составляет 1–4 мин). При таком пути введения Рис. 19.3. Химические структуры некоторых органических нитратов 454 Часть II. Частная фармакология нитроглицерин попадает в системный кровоток, минуя печень, и поэтому не подвергается пресистемному метаболизму. В случае приема внутрь при первом прохождении через печень инактивируется 40% нитроглицерина. В виду указанных фармакокинетических особенностей, препараты нитроглицерина для сублингвального применения используют для купирования приступов стенокардии. С этой целью применяют: капсулы с 1% масляным раствором нитроглицерина в дозе 0,000 5 и 0,001 г; таблетки нитроглицерина по 0,000 5 г; аэрозоль для сублингвального применения: (нитроспрей-ICN♠, нитролингвал-аэрозоль♠ и др.), (1 доза — 0,000 4 г нитроглицерина); 1% спиртовой раствор нитроглицерина (применяют по 1–2 капли на сахар). С целью увеличения продолжительности действия созданы пролонгированные формы нитроглицерина. Их применяют для систематического лечения стенокардии. В этом случае препарат используют в межприступный период с целью предупреждения (профилактики) приступов. К ним относят микрокапсулированные таблетки, мазь и трансдермальные системы. К таблетированным формам нитроглицерина относят нитрогранулонг♠, нитронг♠, сустак форте♠ и др. При приеме микроинкапсулированных таблеток действие нитроглицерина начинается через 10–20 мин и продолжается порядка 6 ч. Поскольку при приеме внутрь нитроглицерин подвергается интенсивному пресистемному метаболизму, микроинкапсулированные таблетки содержат более высокие дозы нитроглицерина, чем препараты для сублингвального введения (не менее 0,005 г). При применении дозированной 2% мази нитроглицерина (нитро-мазь и др.) нитроглицерин всасывается через кожу и начинает действовать через 30–40 мин. Продолжительность действия — до 5 ч. Трансдермальная терапевтическая система представляет собой специальный пластырь (нитроперкутен ТТС♠, депонит♠ и др.), который наклеивают на здоровый участок кожи на 10–12 ч (1 см2 пластыря выделяет в сутки 0,000 5 г нитроглицерина). При применении пластыря эффект развивается в среднем через 30 мин. Длительность эффекта зависит от срока пребывания пластыря на коже. Для купирования и профилактики приступов стенокардии можно применять пленки для наклеивания на десну, содержащие 0,001 и 0,002 г нитроглицерина (тринитролонг♠). При трансбуккальном (как и при сублингвальном) применении действие нитроглицерина начинается через 2 мин. При этом продолжительность действия тринитролонга♠ составляет около 4 ч. Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 455 Механизм антиангинального действия нитроглицерина заключается в одновременном уменьшении потребности миокарда в кислороде и увеличении доставки кислорода к миокарду. Уменьшение потребности миокарда в кислороде связано с уменьшением работы сердца за счет снижения нагрузки на миокард, т. е. обусловлено экстракардиальными влияниями нитроглицерина (прямо на сердце он не действует). Снижение нагрузки на сердце возникает вследствие того, что нитроглицерин уменьшает тонус сосудов. Известно, что нитроглицерин — донатор NO. NO (эндотелиальный релаксирующий фактор) выделяется в системный кровоток эндотелиоцитами капилляров в ответ на гипоксию. Из крови он поступает в ангиомиоциты (гладкомышечные волокна как емкостных, так и резистивных сосудов), где, частично преобразуясь в S-нитрозотиолы, активирует цитозольную гуанилатциклазу, способствуя накоплению циклического гуанозинмонофосфата. Это приводит к снижению содержания кальция в цитозоле гладкомышечных клеток и расслаблению ангиомиоцитов. Высвобождаемый из нитроглицерина в системном кровотоке NO оказывает сосудорасширяющее (вазодилатирующее) действие. В результате этого: ● снижается тонус емкостных сосудов (вен), уменьшаются венозный возврат крови к сердцу и конечное диастолическое давление, изза чего снижается преднагрузка на сердце; ● снижается тонус резистивных сосудов (артерий), уменьшается ОПСС и АД, и, как следствие, снижается постнагрузка на сердце. Снижение пред- и постнагрузки на сердце приводит к уменьшению работы сердца и, следовательно, его потребности в кислороде. Повышение доставки кислорода к миокарду происходит за счет нескольких механизмов. ● При применении нитроглицерина расширяются крупные коронарные сосуды и коллатерали, но не расширяются мелкие коронарные сосуды. Это приводит к перераспределению коронарного кровотока в пользу ишемизированного участка (рис. 19.4). ● Снижение венозного возврата приводит к уменьшению диастолического напряжения стенки желудочков и уменьшению экстравазальной компрессии субэндокардиальных сосудов. ● Применение нитроглицерина приводит к угнетению центральных звеньев коронаросуживающих рефлексов. Помимо антиангинального действия нитроглицерин обладает антиагрегантной активностью, оказывает спазмолитическое действие на глад- 456 Часть II. Частная фармакология Рис. 19.4. Перераспределение коронарного кровотока при действии нитроглицерина кие мышцы внутренних органов (снижает тонус бронхов, желчевыводящих путей, кишечника, мочеточников). Наиболее типичные побочные эффекты нитроглицерина обусловлены его сосудорасширяющим действием — гипотензия (вплоть до коллапса), как следствие — рефлекторная тахикардия, сильная головная боль, головокружение, возможны покраснение лица, ощущение жара. Подобные явления особенно выражены при первых приемах нитроглицерина. Впоследствии их интенсивность снижается. Так, головная боль со временем прекращается, при этом антиангинальное действие нитроглицерина сохраняется. Передозировка нитроглицерина может вызвать метгемоглобинемию. При резком прекращении приема нитроглицерина возможно развитие синдрома отмены, одно из проявлений которого — увеличение частоты и тяжести приступов стенокардии. Высвобождение NO из нитроглицерина (и прочих органических нитратов) происходит не спонтанно, а ферментативно, при участии тиоловых ферментов и тканевых сульфгидрильных групп. При истощении резерва тиоловых групп выделение NO из нитроглицерина уменьшается, и антиангинальное действие препарата снижается. Таким образом раз- Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 457 вивается толерантность к препаратам нитроглицерина. Она, как правило, не возникает при применении короткодействующих форм нитроглицерина, однако существенна при применении пролонгированных форм нитроглицерина или нитросорбида♠. Для восстановления чувствительности необходим 10–12-часовой интервал на протяжении суток, свободный от приема нитроглицерина. Нитроглицерин применяют не только при стенокардии. Внутривенное капельное введение нитроглицерина показано при инфаркте миокарда. Кроме того, в качестве вазодилататора его применяют при острой сердечной недостаточности. Для подобного применения существует раствор нитроглицерина в ампулах. Органические нитраты длительного действия К этой группе препаратов относят изосорбида динитрат и изосорбида мононитрат. Изосорбида динитрат (нитросорбид♠, изокет♠, изокард♠, пролонгированные формы — изосорб ретард♠, кардикет♠) по действию сходен с нитроглицерином. Эффективен как при сублингвальном применении, так и при приеме внутрь. При сублингвальном введении действие развивается через 2–5 мин, биодоступность составляет 60%. Продолжительность действия 1–2 ч (продолжительность действия таблеток для разжевывания от 15 мин до 2 ч). При приеме внутрь действие развивается через 15–40 мин, биодоступность (ввиду пресистемного метаболизма) ниже, чем при сублингвальном введении, и составляет 20%. Метаболизируется в печени с образованием фармакологически активного метаболита изосорбида-5-мононитрата (t½ составляет в среднем 2–4 ч). Продолжительность действия изосорбида динитрата при приеме обычных таблеток 4–6 ч, при приеме пролонгированных таблеток — 8–12 ч. Механизм антиангинального действия сходен с таковым нитроглицерина. Применяют изосорбида динитрат как для купирования, так и для профилактики приступов стенокардии. Возможно применение при хронической сердечной недостаточности (в качестве вазодилататора при невозможности применения ингибиторов ангиотензинпревращающего фермента). Побочные эффекты изосорбида динитрата сходны с побочными эффектами нитроглицерина, однако выражены в меньшей степени. Помимо таблетированных форм выпускают также препараты изосорбида динитрата для внутривенного введения и накожного применения (трансдермальная терапевтическая система нисоперкутен♠). Изосорбида мононитрат (изомонат♠, моночинкве♠, оликард♠, пролонгированные формы — эфокс лонг♠, моночинкве ретард♠, оликард 458 Часть II. Частная фармакология ретард♠) представляет собой активный метаболит изосорбида динитрата (изосорбид-5-мононитрат). В основном схож с изосорбида динитратом, однако обладает более высокой биодоступностью и имеет более продолжительный период полужизни. Антиангинальный эффект после приема препарата внутрь продолжается до 12 ч и более. Изосорбида динитрат и изосорбида мононитрат способны вызывать толерантность, а также перекрестную толерантность с препаратами нитроглицерина. К препаратам с нитратоподобным действием относят производное сиднонимина молсидомин (рис. 19.5) (корватон♠, сиднофарм♠) — пролекарство. Препарат применяют внутрь, биодоступность 60–70%. Выводится в основном почками, t½ составляет 0,85–2,35 ч. В процессе метаболических изменений в печени он превращается в фармакологически активное соединение SIN-1, которое затем неэнзиматическим путем преобразуется в нестойкий метаболит SIN-1А, непосредственно выделяющий NO. Механизм антиангинального действия аналогичен таковому у нитратов. Молсидомин применяют для профилактики приступов стенокардии. Побочные эффекты такие же, как у нитраРис. 19.5. Химическая струк- тов, но менее выражены, толерантность развивается в меньшей степени. тура молсидомина Блокаторы кальциевых каналов Общая классификация блокаторов кальциевых каналов представлена в предыдущей главе. При стенокардии применяют препараты первых трех классов (фенилалкиламины, дигидропиридины и бензотиазепины). Все три класса соединений избирательно блокируют потенциалзависимые кальциевые каналы L-типа. Потенциалзависимые кальциевые каналы открываются при деполяризации клеточной мембраны. Они состоят из α1-, α2-, β- и γ-субъединиц; α1-субъединица — потенциалочувствительный сенсор, его могут блокировать ЛВ. Потенциалзависимые кальциевые каналы L-типа расположены в кардиомиоцитах и ангиомиоцитах (гладкомышечных клетках) резистивных сосудов. Кальциевые каналы сердца и сосудов проявляют неодинаковую чувствительность к фенилалкиламинам, дигидропиридинам и бензотиазепинам. Так, фенилалкиламины действуют главным образом на сердце, дигидропиридины — на сосуды, бензотиазепины — и на сердце, и на сосуды. Все три группы блокаторов кальциевых каналов обладают Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 459 смешанным механизмом антиангинального действия (уменьшают потребность миокарда в кислороде и увеличивают доставку кислорода к миокарду). Однако, поскольку разные препараты неодинаково действуют на сердце и сосуды, существуют некоторые различия в механизмах антиангинального действия трех указанных групп блокаторов кальциевых каналов. Верапамил (изоптин♠) относят к группе фенилалкиламинов. При приеме внутрь легко и полно (90% от принятой дозы) всасывается в системный кровоток. Однако биодоступность верапамила составляет всего 20–35% (за счет пресистемной элиминации). Действие начинается через 1 ч, достигает максимума через 2 ч и продолжается 8–10 ч. Выделяется с мочой и желчью (3–4% в неизмененном виде), t½ составляет 3–7 ч. Механизм антиангинального действия верапамила включает следующие компоненты. ● Блокирует кальциевые каналы кардиомиоцитов, в результате чего уменьшаются сила и частота сердечных сокращений, уменьшается работа сердца и снижается потребность миокарда в кислороде. ● Блокирует кальциевые каналы ангиомиоцитов (гладких мышц) резистивных сосудов, они расширяются, снижаются ОПСС и постнагрузка, в результате чего уменьшается потребность миокарда в кислороде. ● Блокирует кальциевые каналы ангиомиоцитов коронарных сосудов, в результате они расширяются, и увеличивается доставка кислорода к миокарду. В качестве антиангинального средства верапамил эффективен при стенокардии напряжения и вазоспастической стенокардии. Кроме того, его применяют в качестве противоаритмического препарата при суправентрикулярных аритмиях (антиаритмическое средство IV класса). Побочные эффекты: атриовентрикулярная блокада, чрезмерное ослабление сердечных сокращений, гипотензия, головокружение, тошнота, рвота. Продолжительность действия препаратов (капсул и таблеток) с замедленным высвобождением верапамила —24 ч. Дилтиазем (алдизем♠, кардил♠) — производное бензотиазепина. Хорошо всасывается из ЖКТ в кровь, но биодоступность его при введении внутрь составляет 20–35% вследствие пресистемного метаболизма. Действие развивается через 30 мин. Выводится преимущественно кишечником, t½ составляет 3–4 ч. Механизм антиангинального действия и показания к применению аналогичны таковым верапамила. В качестве побочных эффектов вызывает гипотензию, тахикардию, головную боль, отеки, мышечные спазмы. 460 Часть II. Частная фармакология Нифедипин (адалат♠, коринфар♠, кордафлекс♠) — производное дигидропиридина (рис. 19.6). При приеме внутрь легко всасывается из кишечника (абсорбция 90%). Биодоступность составляет 50%. Максимальная концентрация в плазме крови устанавливается через 1–3 ч. Действие начинается через 15–20 мин, достигает максимума через 1–2 ч и продолжается 6–8 ч. Метаболизируется в печени, выводится в основном через почки (t½ состаляет 2–4 ч). Механизм антиангинального действия нифедипина (и прочих дигидропиридинов) отличается тем, что препарат не оказывает прямого влияния на сердце, и его антиангинальное действие связано с влиянием на резистивные сосуды большого круга кровообращения и коронарные сосуды. В качестве антиангинального препарата нифедипин эффективен при вазоспастической стенокардии и стенокардии напряжения. Кроме того, нифедипин применяют в качестве антигипертензивного средства. Побочные эффекты: гипотензия, рефлекторная тахикардия, отеки лодыжек. В настоящее время при терапии стенокардии предпочтение отдают дигидропиридиновым блокаторам кальциевых каналов длительного действия. При их применении менее выражены нежелательные гемодинамические и рефлекторные реакции. Кроме того, более редкое назначение позволяет увеличить комплаэнтность пациентов (четкое выполнение врачебных назначений). Амлодипин (норваск♠) — производное дигидропиридина (рис. 19.7). При приеме внутрь хорошо всасывается, биодоступность составляет 64–80%. Максимальная концентрация в крови достигается через 6–12 ч. С белками плазмы связывается на 97,5%. Характерная особенность амлодипина — большой период полуэлиминации (t½ —35–50 ч). Это позволяет назначать препарат 1 раз в сутки. Механизм действия, применение и побочные эффекты сходны с нифедипином. Рис. 19.6. Химическая структура нифедипина Рис. 19.7. Химическая структура амлодипина Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 461 Помимо органических нитратов и блокаторов кальциевых каналов в группу антиангинальных средств, одновременно снижающих потребность миокарда в кислороде и повышающих доставку кислорода к миокарду, относят амиодарон и новый активатор калиевых каналов никорандил*. Фармакокинетика и фармакодинамика амиодарона подробно описаны в предыдущей главе. Препарат эффективен при стабильной и вазоспастической стенокардии. Его применяют в качестве антиангинального средства, а также в качестве противоаритмического средства (III класса) при тахиаритмиях и экстрасистолии любой локализации. Никорандил (коронель) активирует (открывает) калиевые каналы гладкомышечных клеток сосудов (рис. 19.8). При этом происходит гиперполяризация мембран ангиомиоцитов. Это приводит к закрытию потенциалзависимых кальциевых каналов и снижению входа Са2+ в ангиомиоциты резистивных сосудов. Кроме того, никорандил обладает нитратоподобным действием (увеличивает высвобождение NO). В результате снижается тонус артерий и вен, расширяются коронарные сосуды. Препарат применяют при стенокардии напряжения, вазоспастической стенокардии, Рис. 19.8. Химическая структура никорандила артериальной гипертензии. 19.1.2. Средства, понижающие потребность миокарда в кислороде В эту группу входят β-адреноблокаторы и брадикардитические (брадикардические) средства. β-Адреноблокаторы β-Адреноблокаторы — препараты выбора при стенокардии напряжения. С них начинают лечение больных стабильной стенокардией. Механизм антиангинального действия связан с тем, что они блокируют β1-адренорецепторы сердца, что приводит к снижению силы и частоты сердечных сокращений. При этом работа сердца уменьшается и снижается его потребность в кислороде. Описанный механизм характерен как для кардиоселективных адреноблокаторов (атенолол, метопролол, бисопролол, талинолол), так и для неселективных β1-, β2-адреноблокаторов (пропранолол) (см. главу «β-Адреноблокаторы»). Для препаратов неселективного действия (пропранолол ) характерна также способность перераспределять коронарный кровоток 462 Часть II. Частная фармакология в пользу ишемизированного участка. Это связано с тем, что при блокаде β 2-адренорецепторов повышается тонус не пораженных атеросклерозом коронарных сосудов (просвет сосудов в области бляшки не изменяется). Кроме того, при длительном (не менее недели) применении β-адреноблокаторы снижают АД и уменьшают нагрузку на сердце, что также способствует уменьшению потребности миокарда в кислороде. В качестве антиангинальных средств β-адреноблокаторы применяют при стенокардии напряжения с целью профилактики приступов. При вазоспастической стенокардии они противопоказаны, поскольку могут повышать тонус коронарных сосудов. Кроме того, β-адреноблокаторы применяют в качестве противоаритмических средств (класс II), в качестве антигипертензивных средств, а также при хронической застойной сердечной недостаточности (II–III функциональные классы). Из побочных эффектов β-адреноблокаторов при их применении в качестве антиангинальных средств следует отметить снижение атриовентрикулярной проводимости, чрезмерное снижение силы и частоты сердечных сокращений, артериальную гипотензию. Для неселективных препаратов характерны бронхоспазм и спазм периферических сосудов (похолодание конечностей, головная боль). При резком прекращении приема β-адреноблокаторов может возникнуть синдром отмены в виде учащения и усиления приступов стенокардии. Небиволол (небилет ♠) не только селективно блокирует β 1-адренорецепторы, но и увеличивает высвобождение NО клетками эндотелия с последующей вазодилатацией. Индекс блокирования β-адренорецепторов (β1/β2) у небиволола составляет 293 (в то время как у атенолола — 15, а пропранолола — 1,9). Препарат обладает длительным действием, назначается 1 раз в сутки. Сходный режим дозирования (1 раз в сутки) имеет бисопролол (конкор♠). β-Адреноблокаторы часто комбинируют с нитратами. Такую комбинацию считают целесообразной, так как β-адреноблокаторы устраняют вызываемую нитратами рефлекторную тахикардию, а нитраты препятствуют вазоспастическому действию β-адреноблокаторов (в результате которого могут увеличиваться конечное диастолическое давление и объем левого желудочка). Брадикардитические средства К группе брадикардитических (брадикардических) средств относят ивабрадин, алинидин♠ и фалипамил♠. Они угнетают автоматизм синусового узла, в результате чего снижают частоту сердечных сокращений, не оказы- Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 463 вая существенного влияния на сократимость миокарда (что ценно при лечении стенокардии у лиц с сердечной недостаточностью). В результате брадикардии уменьшается потребность миокарда в кислороде. Ивабрадин (кораксан♠) (рис. 19.9) после приема внутрь быстро всасывается, биодоступность составляет 40%, максимальная концентрация в плазме крови устанавливается через 1,5 ч. С белками плазмы связывается на 70%. Метаболизируется в печени (цитохром р-4503А4), в ходе метаболических превращений образует активный метаболит (N-десметилированный дериват). Выводится с мочой и калом, t½ составляет 2 ч. Механизм брадикардитического действия ивабрадина связан с избирательной блокадой If-каналов клеток синусового узла, что приводит к пролонгированию спонтанной диастолической деполяризации. Брадикардия, вызываемая препаратом, не сопровождается снижением проводимости (внутрипредсердной, предсердно-желудочковой, внутрижелудочковой) и сократимости желудочков. АД также не снижается. Увеличение продолжительности диастолы приводит к увеличению магистрального и коллатерального кровотока. Типичный побочный эффект — фотопсии (изменение световосприятия, проявляющееся вспышками света в поле видения), связанные с блокадой If-каналов в сосудах сетчатки. Фотопсии не приводят к морфологическим изменениям сосудов и, как правило, самопроизвольно прекращаются (в течение 2 мес) на фоне приема препарата. Рис. 19.9. Химическая структура ивабрадина 464 Часть II. Частная фармакология 19.1.3. Средства, повышающие доставку кислорода к миокарду Эта группа препаратов представлена коронарорасширяющими средствами миотропного и рефлекторного действия. Дипиридамол (рис. 19.10) (курантил♠, персантин♠) — производное пиримидина, относят к коронарорасширяющим средствам миотропного действия. При приеме внутрь всасывается в желудке и кишечнике. Максимальная концентрация в крови устанавливается через 1 ч. Почти полностью связывается с белками плазмы. Основной путь метаболизма — глюкуронирование в печени. В виде глюкуронидов выводится через почки; t½ составляет 20–30 мин. Коронарорасширяющий эффект дипиридамола связан с его способностью повышать содержание аденозина в миокарде. Это обусловлено угнетением обратного захвата аденозина (кардиомиоцитами, эндотелиоцитами, эритроцитами), а также ингибированием фермента аденозиндезаминазы, разрушающей аденозин. Накапливающийся в результате действия дипиридамола аденозин за счет стимуляции аденозиновых А2-рецепторов расширяет коронарные сосуды и увеличивает доставку кислорода к миокарду. Следует особо подчеркнуть, что в очагах ишемии концентрация аденозина и так достаточно высока. Коронарные сосуды ниже мест атеросклеротической окклюзии уже расширены максимально. Таким образом, при применении дипиридамола накопление аденозина и расширение им коронарных сосудов происходит, главным образом, в неишемизированных участках миокарда. При этом возникают перераспределение коронарного кровотока в пользу неишемизированных участков и ухудшение кровоснабжения ишемизированных участков — феномен «обкрадывания» (рис. 19.11). Рис. 19.10. Химическая структура дипиридамола Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 465 Рис. 19.11. Перераспределение коронарного кровотока при действии дипиридамола В виду указанных выше особенностей дипиридамол показан к применению только при вазоспастической стенокардии. Кроме того, способность дипиридамола вызывать феномен «обкрадывания» используется для диагностики скрытых форм коронарной недостаточности. С этой целью пациенту после приема дипиридамола проводят велоэргометрическую пробу с регистрацией ЭКГ. При наличии скрытого атеросклероза коронарных сосудов на ЭКГ, при физической нагрузке выявляют признаки гипоксии миокарда (элевация сегмента S–T). Помимо антиангинального действия дипиридамол оказывает умеренное антиагрегантное действие (см. главу «Средства, снижающие агрегацию тромбоцитов»), что способствует улучшению микроциркуляции в миокарде и расценивается как положительное качество препарата. Валидол♠ представляет собой 25–30% раствор ментола в ментиловом эфире изовалериановой кислоты. Ментол раздражает рецепторы слизистой оболочки в подъязычной области, что может вызвать рефлекторное расширение коронарных сосудов. Препарат при стенокардии оказывает слабое и непостоянное действие. Применяют сублингвально для купирования приступов при легких и начальных формах стенокардии. Выпускают в растворе, капсулах или таблетках. 466 Часть II. Частная фармакология 19.1.4. Кардиопротекторные средства Кардиопротекторные средства повышают устойчивость кардиомиоцитов к ишемии, нормализуя их энергетический баланс. Ишемия вызывает в миокарде ряд метаболических изменений. Уменьшение количества АТФ приводит к нарушению функционирования АТФзависимых калиевых каналов и таким образом способствует нарушению трансмембранного транспорта калия, натрия и кальция. Усиление анаэробного гликолиза и активация окисления жирных кислот приводят к ацидозу и накоплению свободных радикалов, оказывающих цитотоксическое действие. Триметазидин (предуктал♠) при приеме внутрь хорошо всасывается из ЖКТ (рис. 19.12). Легко проникает через гистогематические барьеры, мало (1%) связывается с белками плазмы, выводится в основном через почки, t½ составляет 6 ч. При приеме по 20 мг 3 раза в сутки стационарная концентрация в плазме крови устанавливается через 24 ч. Триметазидин оптимизирует нарушенный энергетический метаболизм кардиомиоцитов. Препарат подавляет β-окисление жирных кислот в митохондриях за счет ингибирования 3-кетоацил-КоА-тиолазы и таким образом сдвигает окислительные реакции в митохондриях в сторону окисления глюкозы. Нормализация окисления глюкозы обеспечивает более полноценный синтез АТФ и восстанавливает ее запасы. Это приводит к нормализации трансмембранных ионных токов (нормализуется концентрация ионов К+ и уменьшается перегрузка кардиомиоцитов ионами Na+ и Са2+), что положительно сказывается на сократимости миокарда. Активация аэробного гликолиза уменьшает явления ацидоза, а угнетение окисления жирных кислот уменьшает накопление свободных радикалов, что приводит к цитопротекторному действию. Кровоснабжение миокарда и общая гемодинамика не изменяются. Применение триметазидина при стенокардии уменьшает частоту приступов, увеличивает толерантность к физической нагрузке и уменьшает потребление нитратов. Препарат хорошо переносится и редко вызывает побочные эффекты. Возможны расстройства функций ЖКТ. В комплексной терапии стенокардии с целью профилактики ишемических осложнений (инфаркт миокарда) широко применяют антиагреганты (ацетилсалициловая кислота, клопидогрел) и гиРис. 19.12. Химическая структура полипидемические средства из группы триметазидина статинов (симвастатин, правастатин). Глава 19. Средства, применяемые при недостаточности коронарного кровообращения 467 19.2. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ ОСТРОМ КОРОНАРНОМ СИНДРОМЕ Под термином «острый коронарный синдром» (ОКС) подразумевается наличие симптоматики, позволяющей заподозрить развитие у пациента либо нестабильной стенокардии, либо инфаркта миокарда. Выделяют следующие формы ОКС: ● острый коронарный синдром с подъемом сегмента ST, что характерно для развития трансмурального инфаркта миокарда; ● острый коронарный синдром без подъема сегмента ST, что предполагает наличие нестабильной стенокардии или мелкоочагового субэндокардиального инфаркта (при увеличении в крови уровня тропонина Т и I, а также креатинкиназы). Введение термина ОКС было обусловлено тем, что начальные проявления и некоторые принципы лечения вышеуказанных заболеваний часто схожи. Использование данного термина в клинической практике позволяет облегчить диагностику и, как следствие, ускорить оказание медицинской помощи на догоспитальном этапе и сразу после поступления больного в стационар. Для борьбы с болевым синдромом применяют: нейролептанальгезию (фентанил + дроперидол), опиоидные анальгетики (морфин и др.), неопиоидный анальгетик из группы средств для ингаляционного наркоза — азота закись♠. Частое осложнение острого периода инфаркта миокарда — желудочковые аритмии (экстрасистолы, способные привести к фибрилляции с последующей асистолией). С целью их купирования применяют противоаритмическое средство класса IВ — лидокаин, устраняющий экстрасистолы не снижая сократительную активность желудочков. Для гемодинамической разгрузки сердца и профилактики острой сердечной недостаточности внутривенно вводят нитроглицерин. С первых часов острого периода инфаркта миокарда проводят профилактику тромбообразования. С этой целью применяют антиагреганты (ацетилсалициловую кислоту, клопидогрел, прасугрел, тикагрелор), антикоагулянты прямого действия — гепарин (при ОКС с подъемом сегмента ST), надропарин кальция (фраксипарин♠), ривароксабан, дабигатрана этексилат, которые впоследствии могут быть заменены антикоагулянтами непрямого действия (аценокумарол, варфарин). При ОКС с подъемом сегмента ST для растворения свежих тромбов применяют фибринолитические средства (стрептокиназа, алтеплаза). Кроме того, могут применяться различные средства симптоматической терапии. Их подбор индивидуален в каждом конкретном случае. 468 Часть II. Частная фармакология Вопросы и задания для самоконтроля 1. Средства, одновременно уменьшающие потребность миокарда в кислороде и увеличивающие доставку кислорода к миокарду: а) дипиридамол; б) нитроглицерин; в) амлодипин; г) пропранолол; д) верапамил. 2. Антиангинальное действие нитроглицерина обусловлено: а) уменьшением преднагрузки на сердце; б) уменьшением постнагрузки на сердце; в) перераспределением коронарного кровотока в пользу ишемизированных участков миокарда; г) уменьшением экстравазальной компрессии субэндокардиальных сосудов; д) уменьшением потребности миокарда в кислороде за счет брадикардии. 3. Побочные эффекты нитроглицерина: а) ортостатический коллапс; б) головная боль; в) головокружение; г) рефлекторная тахикардия; д) рефлекторная брадикардия. 4. Антиангинальное действие пропранолола обусловлено: а) снижением силы сердечных сокращений; б) снижением ЧСС; в) расширением коронарных сосудов. 5. Уменьшает потребность миокарда в кислороде за счет снижения ЧСС. Не оказывает существенного влияния на силу сердечных сокращений и коронарный кровоток. В качестве побочного действия вызывает фотопсии: а) нитроглицерин; б) атенолол; в) никорандил; г) ивабрадин; д) амлодипин. Глава 20 СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ АРТЕРИАЛЬНОЙ ГИПЕРТЕНЗИИ (АНТИГИПЕРТЕНЗИВНЫЕ СРЕДСТВА) Уровень АД — одна из важнейших констант в организме. Его стабильный уровень обеспечивают: ● сердечный выброс; ● ОПСС; ● объем циркулирующей крови; ● в меньшей степени на АД влияют вязкость и электролитный состав крови. При изменении одного из указанных факторов другие стремятся к компенсаторному изменению в противоположном направлении. Ключевое значение в координации этих изменений имеют симпатоадреналовая и ренин-ангиотензиновая системы. В норме АД у человека составляет 120 мм рт.ст. (систолическое) и 80 мм рт.ст. (диастолическое). Под артериальной гипертензией понимают устойчивое повышение АД выше 140 мм рт.ст. (систолическое) и 90 мм рт.ст. (диастолическое). Артериальная гипертензия может быть первичной (эссенциальной) и вторичной (симптоматической). Эссенциальную гипертензию (гипертоническую болезнь) регистрируют у 15–30% населения индустриально развитых регионов. Повышение АД при этом — основной симптом заболевания. Вторичная гипертензия встречается при заболеваниях почек, феохромоцитоме, эндокринных расстройствах. Гипертензия приводит к гипертрофии левого желудочка, концентрическому отложению холестерина в сосудах (коронарных, почечных, мозговых, сетчатки) и, таким образом, создает риск развития инфаркта миокарда и инсультов. Для снижения и поддержания АД на нормальном уровне применяют антигипертензивные средства. Их классифицируют в соответствии с основными принципами антигипертензивного действия. 470 Часть II. Частная фармакология ● Антигипертензивные средства нейротропного действия: —средства, понижающие тонус вазомоторных центров; —ганглиоблокаторы; —симпатолитики; —адреноблокаторы. ● Средства, снижающие активность ренин-ангиотензиновой системы: —средства, угнетающие секрецию ренина; —ингибиторы ангиотензин-превращающего фермента (АПФ); —блокаторы ангиотензиновых рецепторов 1-го типа. ● Антигипертензивные средства миотропного действия: —блокаторы кальциевых каналов; —активаторы калиевых каналов; —донаторы оксида азота (NO); —препараты различных фармакологических групп. ● Диуретики. 20.1. АНТИГИПЕРТЕНЗИВНЫЕ СРЕДСТВА НЕЙРОТРОПНОГО ДЕЙСТВИЯ Нейротропные антигипертензивные средства уменьшают активирующее влияние адренергической иннервации на сердечно-сосудистую систему. Симпатическая нервная система оказывает стимулирующее действие на работу сердца и тонус кровеносных сосудов. За счет активации адренергической иннервации увеличивается работа сердца (стимуляция β1-адренорецепторов сократительных и атипичных кардиомиоцитов) и повышается ОПСС (стимуляция α1-адренорецепторов гладких мышц резистивных сосудов). Симпатическая нервная система оказывает автономное тоническое влияние, а кроме того, выступает как одно из эфферентных звеньев барорецепторного прессорно-депрессорного рефлекса. Барорефлекс возникает при внезапных изменениях АД и быстро (в течение нескольких секунд) их компенсирует. При внезапном повышении АД усиливается стимуляция барорецепторов дуги аорты и каротидных клубочков. Это приводит к увеличению афферентной импульсации в ядро солитарного тракта продолговатого мозга (импульс от дуги аорты идет по афферентным волокнам блуждающего нерва, а от синокаротидной зоны — по нерву Геринга, затем — по афферентным волокнам языкоглоточного нерва). Стимуляция ядра солитарного тракта активирует ядро блуждающего нерва и угнетает активность сосудодвигательного центра Глава 20. Средства, применяемые при артериальной гипертензии… 471 (с ним ядро солитарного тракта связано через тормозный интернейрон, рис. 20.1). Угнетение сосудодвигательного центра приводит к снижению нисходящих адренергических влияний на сердце (через β1 адренорецепторы) и резистивные сосуды (через α1-адренорецепторы). Работа сердца снижается, тонус резистивных сосудов уменьшается, и повышенное АД возвращается к норме (барорецепторный депрессорный рефлекс). При внезапном снижении АД (например, при резком переходе из положения лежа в положение стоя) барорефлекс действует в обратном направлении (как барорецепторый прессорный рефлекс). Барорефлекс — один из важнейших компенсаторных факторов, поддерживающих АД в диапазоне нормальных значений. Однако при неконтролируемом повышении АД чувствительность барорецепторов быстро (в течение 1–2 сут) понижается. В этом случае повышенное давление начинает восприниматься барорецепторами как норма, а медикаментозное понижение АД (до нормальных значений) — как гипотензия. Это приводит к включению компенсаторных систем повышения АД (рефлекторная тахикардия, задержка воды в организме и др.), снижающих эффективность антигипертензивной терапии. Поскольку симпатическая нервная система иннервирует как сердце, так и сосуды, большинство нейротропных средств снижают АД за счет одновременного уменьшения сердечного выброса и снижения ОПСС. В результате, как правило, в качестве побочного действия эти препараты вызывают задержку жидкости в организме (попытка компенсации снижения давления). По локализации действия нейротропные антигипертензивные средства подразделяют на средства центрального действия и средства периферического действия. Нейротропные антигипертензивные средства центрального действия понижают тонус вазомоторных центров. 20.1.1. Средства, понижающие тонус вазомоторных центров К антигипертензивным средствам центрального действия относят метилдопу, клонидин, гуанфацин, моксонидин. Принципиальный механизм их действия сходен. Они снижают тонус вазомоторных центров за счет активации ядра солитарного тракта (см. рис. 20.1). Вазомоторный (сосудодвигательный) центр — сложное образование, находящееся в ростральных и вентролатеральных отделах продолговатого мозга. Он состоит из нескольких функциональных зон, среди которых следует отметить вазоконстрикторную (С-1), кардиоакселерирующую и сенсорную. Сенсорная зона представлена ядром солитарного тракта, собирающим афферентную импульсацию от волокон блуждающего Рис. 20.1. Механизмы действия средств, снижающих тонус вазомоторных центров Метилнорадреналин 472 Часть II. Частная фармакология Глава 20. Средства, применяемые при артериальной гипертензии… 473 и языкоглоточного нервов. На телах нейронов ядра солитарного тракта обнаружены имидазолиновые I1-рецепторы (их эндогенный лиганд — агмантин — декарбоксилированный аргинин) и α2-адренорецепторы. При стимуляции I1-рецепторов и α2-адренорецепторов ядро солитарного тракта возбуждается и стимулирует тормозные интернейроны, подавляющие активность вазоконстрикторной и кардиоакселерирующей зон вазомоторного центра (они осуществляют симпатическую иннервацию сердца и сосудов). Таким образом, стимуляция ядра солитарного тракта приводит к подавлению тонуса симпатической иннервации сосудов и сердца. Активация ядра солитарного тракта происходит с помощью различных механизмов (см. рис. 20.1). Метилдопа в организме превращается в α-метилнорадреналин и стимулирует α2-адренорецепторы; клонидин и гуанфацин прямо стимулируют как α2-адренорецепторы, так и имидазолиновые I1-рецепторы, а моксонидин избирательно стимулирует имидазолиновые I1-рецепторы ядра солитарного тракта. Это приводит к угнетению вазомоторных центров, снижению импульсации адренергических волокон, иннервирующих кровеносные сосуды и сердце. Уменьшение тонуса сосудов приводит к снижению ОПСС, а уменьшение работы сердца приводит к снижению сердечного выброса. Кроме того, уменьшение импульсации симпатических волокон приводит к снижению секреции ренина. Клонидин (клофелин♠, гемитон♠) при приеме внутрь хорошо всасывается, выраженный гипотензивный эффект развивается в течение 2–4 ч и сохраняется около 5 ч. Общая продолжительность действия составляет 6–12 ч. Легко проникает через ГЭБ. Выводится почками в неизмененном виде, t½ составляет 12 ч. Клонидин применяют как для систематического лечения артериальной гипертензии, так и для купирования гипертензивных кризов. Для систематического лечения артериальной гипертензии препарат назначают в таблетках внутрь 2 раза в сутки. Кроме того, применяют трансдермальную терапевтическую систему (полимерный пластырь с дозировкой на 7 дней, наклеивают 1 раз в неделю). Для купирования гипертензивного криза клонидин назначают в таблетках под язык. Кроме того, возможно применение 0,01% раствора клонидина в вену, под кожу или в мышцу. Однако при внутривенном введении клонидин может вызвать кратковременное повышение АД (за счет стимуляции α2 адренорецепторов кровеносных сосудов). Из побочных эффектов при систематическом применении клонидина отмечают сухость во рту, седативный эффект, депрессию, сонливость, головокружение, задержку натрия, жидкости, появление отеков, запоры. Клонидин потенцирует действие снотворных средств наркотического типа и этилового спирта (это может быть использовано в криминальных целях). При резком прекращении 474 Часть II. Частная фармакология приема клонидина возникает синдром отмены, который выражается в развитии гипертензивного криза. Поэтому отмену препарата производят с постепенным снижением дозы в течение 7–10 дней. Из других свойств препарата следует отметить способность снижать внутриглазное давление за счет уменьшения секреции внутриглазной жидкости (применяют в виде глазных капель при открытоугольной форме глаукомы), а также болеутоляющее действие (применяют в качестве неопиоидного анальгетика центрального действия). Гуанфацин (эстулик♠) действует подобно клонидину, но более продолжительно (длительность гипотензивного эффекта — 24 ч). Назначают 1 раз в сутки. Побочные эффекты аналогичны таковым для клонидина. Седативный эффект, вероятность развития синдрома отмены при приеме гуанфацина менее выражены, чем у клонидина. Метилдопу применяют внутрь, биодоступность составляет не более 50%. После прохождения через ГЭБ метилдопа превращается в α-метилдофамин, а затем в α-метилнорадреналин, стимулирующий α2-адренорецепторы нейронов ядра солитарного тракта. Это приводит к снижению тонуса сосудодвигательного центра и уменьшению ОПСС и сердечного выброса, в связи с чем снижается АД. Антигипертензивный эффект наступает через 3–5 ч и продолжается в течение суток. В качестве побочных эффектов отмечают седативное действие, сонливость, сухость во рту, ортостатическую гипотензию, задержку натрия и воды в организме, отеки, запоры, возможны нарушение функции печени, паркинсонизм, лейкопения, гемолитическая анемия, нарушения половых функций и др. Наряду с клонидином и гуанфацином метилдопу относят к I поколению гипотензивных препаратов центрального действия. Присущие им побочные эффекты, трудности в дозировании и развитие синдрома отмены ограничивают их применение. Метилдопа — препарат для лечения гипертензии при беременности. Моксонидин (цинт♠, физиотенз♠) — производное имидазолина (рис. 20.2). При приеме внутрь препарат быстро всасывается из кишечника, биодоступность составляет 88%. Максимальную концентрацию в плазме крови регистрируют через 1 ч после приема, интервал между достижением максимальной концентрации в плазме и выраженным снижением АД составляет 2–4 ч. В ходе метаболических преобразований образуются 2 активных метаболита с низкой специфической активностью. 60% введенной дозы выводятся почками в неизмененном виде. Продолжительность действия моксонидина составляет более 12 ч. Моксонидин применяют для систематического лечения артериальной гипертензии, при этом суточную дозу (0,4 мг) делят на 2 приема. Из побочных эффектов отмечают седативное действие, сла- Глава 20. Средства, применяемые при артериальной гипертензии… бость, утомляемость, сухость во рту, ангионевротический отек, синдром отмены. Однако эти эффекты выражены в значительно меньшей степени, чем при применении клонидина или гуанфацина. Это обусловлено большей избирательностью моксонидина к имидазолиновым I1-рецепторам и относительно меньшим сродством к центральным α2 адренорецепторам. 475 Рис. 20.2. Химическая структура моксонидина 20.1.2. Ганглиоблокаторы Антигипертензивное действие ганглиоблокаторов связано с блокадой NN-холинорецепторов симпатических ганглиев (рис. 20.3). Это приводит к уменьшению тонуса артериальных сосудов и снижению АД, расширению вен и снижению венозного давления. Последнее может привести к ортостатическому коллапсу и рефлекторной тахикардии. Вместе с тем, не обладая избирательностью, ганглиоблокаторы блокируют также NN-холинорецепторы парасимпатических ганглиев. Это проявляется побочными эффектами, такими как паралич аккомодации, сухость во рту, атония кишечника и мочевого пузыря. В настоящее время ганглиоблокаторы (несмотря на высокую эффективность) в значительной мере утеряли самостоятельное значение как группа антигипертензивных средств. Азаметония бромид (пентамин♠) и гексаметоний♠ применяют для купирования гипертензивных кризов (продолжительность действия 2,5–3 ч), триметафан♠ и трепирия йодид♠ (гигроний♠) применяют для управляемой гипотензии (продолжительность действия 10–20 мин). 20.1.3. Симпатолитики Антигипертензивное действие симпатолитиков (резерпин, гуанетидин) связано с истощением запасов норадреналина в окончаниях адренергических волокон. В результате возникает симпатическая «денервация» сердца и сосудов. Снижение сердечного выброса и ОПСС приводит к понижению АД (см. рис. 20.3). Антигипертензивное действие симпатолитиков развивается постепенно и достигает максимума через 1–2 нед. Симпатическая денервация ЖКТ приводит к функциональному преобладанию парасимпатических влияний и, как результат, к побочным эффектам: увеличение секреции пищеварительных желез и моторики 476 Часть II. Частная фармакология Рис. 20.3. Механизмы действия средств, регулирующих тонус сосудов и объем циркулирующей крови Глава 20. Средства, применяемые при артериальной гипертензии… 477 (диарея, обострение язвенной болезни). Кроме того, симпатолитики могут вызывать заложенность носа и ортостатический коллапс (наиболее характерен для гуанетидина). Резерпин, истощая запасы норадреналина, дофамина и серотонина в ЦНС, может вызывать сонливость, паркинсонизм и психическую депрессию. В настоящее время симпатолитик резерпин применяют, главным образом, в составе комбинированных антигипертензивных средств: адельфан-эзидрекс♠, кристепин♠, трирезид К♠ (см. ниже). 20.1.4. Средства, блокирующие адренорецепторы В качестве антигипертензивных средств применяют α-адреноблокаторы, β-адреноблокаторы и α-, β-адреноблокаторы. β-Адреноблокаторы β-Адреноблокаторы представлены: блокаторами β1-адренорецепторов: атенолол (те● избирательными нормин♠), бетаксолол (локрен♠), бисопролол (конкор♠), небиволол (небилет♠), метопролол, талинолол; блокаторами β1-, β2-адренорецепторов: пропрано● неизбирательными лол (анаприлин♠), надолол (коргард♠). Общие для всех β-адреноблокаторов механизмы антигипертензивного действия следующие: ● блокада β1-адренорецепторов сердца приводит к снижению силы и частоты сердечных сокращений, уменьшению сердечного выброса и снижению систолического давления; ● блокада β1-адренорецепторов юкстагломерулярного аппарата почек приводит к уменьшению выделения ренина и снижению активности ренин-ангиотензин-альдостероновой системы; уменьшение вазоконстрикторного действия ангиотензина II обусловливает расширение кровеносных сосудов, снижение ОПСС и диастолического давления; ● применение β-адреноблокаторов восстанавливает чувствительность барорецепторного депрессорного рефлекса (см. рис. 20.3). Кроме того, блокада пресинаптических β2-адренорецепторов приводит к уменьшению высвобождения норадреналина из окончаний симпатических волокон и уменьшению адренергической иннервации кровеносных сосудов. В дополнение к этому пропранолол оказывает угнетающее действие на ЦНС, а небиволол (рис. 20.4) стимулирует выделение NO эндотелием сосудов. 478 Часть II. Частная фармакология α-, β-Адреноблокаторы Карведилол (дилатренд♠) (рис. 20.4) и лабеталол неселективно блокируют β-адренорецепторы и селективно α1-адренорецепторы. Блокада α1 адренорецепторов приводит к снижению тонуса кровеносных сосудов, а блокада β-адренорецепторов — к снижению секреции ренина и работы сердца. В результате АД снижается без выраженной рефлекторной тахикардии. Лабеталол применяют внутрь для систематического лечения артериальной гипертензии и внутривенно при гипертензивных кризах. Карведилол помимо антигипертензивного действия обладает также антиоксидантной и гиполипидемической активностью. Его применяют внутрь при гипертонической болезни и хронической застойной сердечной недостаточности (см. гл. «Средства, применяемые при сердечной недостаточности»). Рис. 20.4. Химические структуры небиволола и карведилола α-Адреноблокаторы В качестве антигипертензивных средств применяют селективные блокаторы α1-адренорецепторов празозин (минипресс♠), доксазозин (кардура♠), теразозин. Механизм антигипертензивного действия препаратов связан с блокадой постсинаптических α1-адренорецепторов кровеносных сосудов и устранением стимулирующего влияния симпатической нервной системы на их тонус (см. рис. 20.3). Расширение резистивных сосудов приводит к снижению ОПСС и АД, расширение емкостных сосудов — к снижению венозного давления. Антигипертензивное действие препаратов сопровождается побочными эффектами в виде рефлекторной тахикардии (рефлекс инициируется с барорецепторов), увеличения секреции ренина (в результате снижения перфузии почечной паренхимы) и ортостатической гипотензии (в результате расширения вен). Ортостатическая гипотензия наиболее выражена при первых приемах препарата Глава 20. Средства, применяемые при артериальной гипертензии… 479 («феномен первой дозы») и наблюдается у 25% пациентов. В связи с этим празозин назначают в вечернее время, начиная с пороговых доз (½ таблетки на ночь) с последующим их увеличением. Доксазозин (рис. 20.5) (кардура♠, артезин♠, камирен♠, тонокардин♠) — селективный конкурентный блокатор постсинаптических α1 адренорецепторов (сродство к α1-адренорецепторам в 600 раз выше, чем к α2 адренорецепторам). При приеме внутрь хорошо всасывается (системная абсорбция 80–90%) и подвергается пресистемному метаболизму, снижающему биодоступность препарата до 60–70%. Максимальная концентрация в плазме крови устанавливается через 3 ч в дневное время и через 5 ч при вечернем приеме (одновременный прием пищи замедляет всасывание на 1 ч). Интенсивно метаболизируется печенью с экскрецией метаболитов через кишечник, t½ составляет 19–22 ч. После однократного приема снижение АД развивается постепенно, максимальный эффект развивается через 2–6 ч и сохраняется в течение 24 ч. В качестве антигипертензивного средства применяют 1 раз в сутки как в форме монопрепарата, так и в комбинации с другими антигипертензивными средствами: тиазидными диуретиками, β-адреноблокаторами, блокаторами кальциевых каналов или ингибиторами АПФ. Наряду с антигипертензивным действием, доксазозин также снижает агрегацию тромбоцитов, уменьшает в плазме крови концентрации триглицеридов, общего холестерина и ЛПВП и увеличивает содержание активного плазминогена в тканях. В связи с тем, что доксазозин блокирует α1А-адренорецепторы, расположенные в строме и капсуле предстательной железы и в шейке мочевого пузыря, он облегчает мочеотделение при доброкачественной гиперплазии предстательной железы. Препарат хорошо переносим, среди побочных эффектов наиболее часто отмечается постуральная гипотензия в начале лечения. Рис. 20.5. Химическая структура доксазозина 480 Часть II. Частная фармакология Неселективные α1- и α2-адреноблокаторы фентоламин (регитин♠) и феноксибензамин для лечения эссенциальной гипертензии в настоящее время не применяют. Их антигипертензивное действие непродолжительно (лимитируется способностью блокировать пресинаптические α2-адренорецепторы и увеличивать выделение норадреналина из адренергических окончаний) и сопровождается выраженной тахикардией (как рефлекторной, так и развивающейся за счет блокады пресинаптических α2 адренорецепторов). Основная сфера применения фентоламина — диагностика и лечение вторичной артериальной гипертензии при феохромоцитоме (опухоль надпочечников, продуцирующая адреналин и норадреналин). Высокая эффективность фентоламина при феохромоцитоме обусловлена его способностью извращать прессорное действие адреналина (в условиях блокады постсинаптических α1-адренорецепторов и внесинаптических α2 адренорецепторов сосудов адреналин стимулирует β2-адренорецепторы в сосудах, что приводит к их расширению и снижению АД). 20.2. СРЕДСТВА, СНИЖАЮЩИЕ АКТИВНОСТЬ РЕНИН-АНГИОТЕНЗИНОВОЙ СИСТЕМЫ Эта группа представлена ЛС: ● угнетающими секрецию ренина (β-адреноблокаторы); ● нарушающими образование ангиотензина II (ингибиторы АПФ, ингибиторы вазопептидаз); ● препятствующими действию ангиотензина II (блокаторы АТ1-рецепторов). Ренин-ангиотензиновая система (более полное название — ренинангиотензин-альдостероновая система) регулирует тонус резистивных сосудов и объем циркулирующей крови (а также ее электролитный состав). Ренин — фермент, вырабатываемый юкстагломерулярными клетками (видоизмененными миоцитами вносящего сосуда), выделяется в системный кровоток, где переводит неактивный ангиотензиноген в ангиотензин I (предшественник ангиотензина II). Секреция ренина увеличивается при недостаточном кровотоке в почках или при стимуляции β1-адренорецепторов юкстагломерулярного аппарата. Ангиотензин I в системном кровотоке контактирует с АПФ (кининаза-II, пептидилдипептидгидролаза). Этот фермент отщепляет от ангиотензина I два аминокислотных остатка, превращая его в октапептид ангиотензин II. Ангиотензин II стимулирует АТ1-рецепторы гладких мышц резистивных сосудов, повышая ОПСС и АД. Кроме того, он стимулирует АТ1-рецепторы коры Глава 20. Средства, применяемые при артериальной гипертензии… 481 надпочечников, что приводит к увеличению выделения минералокортикоида альдостерона. Альдостерон стимулирует альдостероновые рецепторы клеток эпителия дистальных извитых канальцев и собирательных трубок, увеличивает в них количество натриевых каналов и таким образом задерживает реабсорбцию ионов натрия и соответствующих количеств воды. Указанные факторы приводят к увеличению объема внеклеточной жидкости, циркулирующей крови и повышению АД. 20.2.1. Ингибиторы ангиотензинпревращающего фермента К ингибиторам АПФ относят: ♠ ); ● действующий непродолжительно (4–8 ч) каптоприл (капотен ♠ длительно (24 ч и более): эналаприл (ренитек , энап♠, ● действующие ♠ ♠ ♠ ♠ эднит ), лизиноприл (синоприл , лизорил , скоприл ), фозиноприл (моноприл♠), периндоприл (престариум♠), рамиприл (тритаце♠), трандолаприл. Антигипертензивное действие этих препаратов обусловлено тем, что, ингибируя АПФ, они препятствуют образованию ангиотензина II из ангиотензина I. При этом количество ангиотензина II в крови уменьшается и снижается его вазоконстрикторное действие: кровеносные сосуды расширяются, снижаются ОПСС и АД. Кроме того, уменьшается опосредованная ангиотензином II секреция альдостерона. Это приводит к уменьшению задержки в организме натрия и воды. Не исключено, что определенный вклад в антигипертензивное действие ингибиторов АПФ может вносить уменьшение высвобождения норадреналина из окончаний симпатических волокон (его стимулирует ангиотензин II) и увеличение содержания брадикинина (АПФ его инактивирует). Накапливающийся брадикинин стимулирует β2-кининовые рецепторы эндотелия сосудов. Это приводит к быстрому высвобождению из эндотелия простациклина и других веществ, снижающих тонус гладких мышц кровеносных сосудов. Показания к применению ингибиторов АПФ — артериальная гипертензия (в особенности они эффективны при повышенной активности ренин-ангиотензиновой системы) и хроническая застойная сердечная недостаточность. К побочным эффектам ингибиторов АПФ относят артериальную и ортостатическую гипотензию, тахикардию, головокружение. Они связаны с влиянием препаратов на гемодинамику, и при продолжении лечения их интенсивность снижается. Наиболее типичные побочные эффекты ингибиторов АПФ — сухой кашель и ангионевротический отек. Они связаны с накоплением кининов 482 Часть II. Частная фармакология (в том числе брадикинина), не прекращаются при продолжении лечения и служат основанием для отмены препарата. Кашель, вызываемый ингибиторами АПФ, не купируется противокашлевыми средствами. Каптоприл (капотен♠) — первый введенный в клиническую практику ингибитор АПФ (рис. 20.6). При приеме внутрь хорошо всасывается из ЖКТ (всасывание препарата из ЖКТ в присутствии пищи уменьшается до 30–55%). Гипотензивный эффект развивается через 30–60 мин и сохраняется 4–8 ч. При применении каптоприла происходит расширение периферических артерий и некоторое расширение вен, снижение ОПСС и АД, уменьшение пред- и постнагрузки на сердце, улучшается кровообращение в малом круге и в почках. Каптоприл применяют при артериальной гипертензии (в особенности он эффективен при повышенном содержании Рис. 20.6. Химическая струк- ренина) и хронической сердечной недостаточности. Из побочных эффектов наблюдатура каптоприла ют сухой кашель, ангионевротический отек (связаны с повышением уровня брадикинина), гиперкалиемию (связана со снижением уровня альдостерона), протеинурию, головную боль, головокружение, кожную сыпь. Эналаприл (ренитек♠, энап♠) отличается от каптоприла большей продолжительностью действия (рис. 20.7). Хорошо всасывается при приеме внутрь, прием пищи не влияет на всасывание препарата. В ходе метаболических превращений эналаприл гидролизуется с образованием эналаприлата, который ингибирует АПФ и оказывает продолжительное антигипертензивное действие (24 ч). Побочные эффекты такие же, как у каптоприла, Рис. 20.7. Химические структуры эналаприла и фозиноприла Глава 20. Средства, применяемые при артериальной гипертензии… 483 но их отмечают реже. Это связывают с отсутствием в структуре эналаприла сульфгидрильных групп. Фозиноприл (моноприл♠) — пролекарство, содержащее в своей структуре атом фосфора (см. рис. 20.7). Медленно и не полностью всасывается из ЖКТ, биодоступность препарата составляет 30%. Метаболизируется в печени с образованием активного метаболита — фозиноприлата, оказывающего пролонгированное действие (до 24 ч). Трандолаприл (гоптен♠) при приеме внутрь всасывается не полностью, абсолютная биодоступность составляет 40–60%. При одновременном приеме с пищей биодоступность снижается. В ходе метаболических превращений образуется активный метаболит трандолаприлат. Отличительные особенности препарата — высокая продолжительность действия (эффект сохраняется до 48 ч) и высокая липофильность. В связи с этим трандолаприл и трандолаприлат легко преодолевают гистогематические барьеры и ингибируют АПФ не только в сосудистом русле, но и в тканях. Омапатрилат♠ — первый представитель нового класса — ингибиторов вазопептидаз. Препарат блокирует АПФ и нейтральную эндопептидазу, которая инактивирует эндогенные пептиды, обладающие сосудорасширяющими свойствами. Таким образом, омапатрилат♠ снижает АД, ингибируя активность ренин-ангиотензиновой системы (оказывающей вазопрессорное действие) и повышая действие вазодилатирующих систем. Препарат хорошо всасывается из ЖКТ, биодоступность составляет 30%, пик плазменной концентрации достигается через 2 ч, выводится с мочой в виде метаболитов; t½ составляет 14–19 ч, продолжительность действия — более 24 ч. Несмотря на положительные качества, в клинической практике омапатрилат♠ не нашел применения в виду того, что часто вызывает ангионевротический отек. 20.2.2. Блокаторы ангиотензиновых рецепторов 1-го типа В эту группу входят лозартан (козаар♠), ирбесартан (апровель♠), валсартан (диован♠), эпросартан (теветен♠). Препараты этой группы в большей степени, чем ингибиторы АПФ, предотвращают действие ангиотензина II на сердечно-сосудистую систему. Это связано с тем, что ангиотензин II может образовываться не только в сосудах под влиянием АПФ, но и в тканях под влиянием других ферментов. При этом в системный кровоток он выделяется в готовом виде. Блокаторы АТ1-рецепторов устраняют эффекты ангиотензина II независимо от того, где он образовался. Блокада АТ1-рецепторов кровеносных сосудов препятствует вазоспастическому действию ангиотензина II, что приводит 484 Часть II. Частная фармакология к снижению ОПСС и АД. Блокада АТ1-рецепторов коры надпочечников приводит к уменьшению выделения альдостерона. Кроме того, блокаторы АТ1-рецепторов устраняют другие эффекты ангиотензина II. Показания к применению блокаторов АТ1-рецепторов — артериальная гипертензия и хроническая застойная сердечная недостаточность. Препараты отличаются хорошей переносимостью. Среди побочных эффектов отмечают головную боль, головокружение, слабость, ангионевротический отек. Лозартан (козаар♠) — производное имидазола, при приеме внутрь быстро всасывается; биодоступность составляет 30–35% (рис. 20.8). Метаболизируется в печени с образованием активного метаболита, который в 10– 40 раз активнее лозартана; t½ лозартана 1,5–2 ч, а его метаболита — 6–9 ч. Лозартан — конкурентный антагонист, а его основной метаболит — неконкурентный антагонист АТ1-рецепторов. Применение препарата уменьшает тонус сосудов, снижает ОПСС, АД и нагрузку на сердце, уменьшает секрецию альдостерона, увеличивает диурез (натрийуретический эффект), отмечают урикозурическое действие. Применяют внутрь 1 раз в сутки. Среди побочных эффектов отмечают головокружение, головную боль, утомляемость, ангионевротический отек. Ирбесартан (апровель♠) — производное лозартана, в 2,5 раза прочнее связывается с АТ1-рецептором, чем лозартан. ЭффективРис. 20.8. Химическая струк- нее лозартана снижает АД, гипотензивный тура лозартана эффект сохраняется 24 ч и более. 20.3. АНТИГИПЕРТЕНЗИВНЫЕ СРЕДСТВА МИОТРОПНОГО ДЕЙСТВИЯ Тонус периферических кровеносных сосудов — один из трех основных факторов, определяющих уровень АД. Антигипертензивный эффект сосудорасширяющих средств миотропного действия связан с их спазмолитическим влиянием на гладкие мышцы сосудов. Кровеносные сосуды расширяются, снижается ОПСС и АД. При этом как типичный побочный эффект отмечают рефлекторную тахикардию различной степени (рефлекс инициируется с барорецепторов дуги аорты и каротидного клубочка). Кроме того, снижение перфузии почечной паренхимы приводит к увеличению секреции ренина юкстагломерулярными клетками. При длительном применении этих препаратов в организме задерживаются натрий и вода. Глава 20. Средства, применяемые при артериальной гипертензии… 485 В норме тонус кровеносных сосудов регулируют нейрогуморальные механизмы. Нейрональная регуляция осуществляется симпатической нервной системой (стимуляция постсинаптических α1-адренорецепторов повышает тонус сосудов). Гуморальные системы регуляции тонуса сосудов многочисленны. Вазоспастическим действием обладают циркулирующие моноамины (адреналин, норадреналин), ангиотензин II, вазопрессин. Понижают тонус сосудов эндотелиальный релаксирующий фактор (NO), натрийуретические пептиды (А, В и С) и др. (табл. 20.1). Таблица 20.1. Вазоактивные эндогенные вещества Сосудосуживающие эндогенные вещества Сосудорасширяющие эндогенные вещества Адреналин Эндотелиальный релаксирующий фактор (NO) Норадреналин Натрийуретические пептиды (А, В, С) Вазопрессин Ацетилхолин Эндотелин Брадикинин Ангиотензин II Гистамин Нейропептид Y Аденозин, АТФ Простагландин F2б Простациклин Тромбоксан Адреномедуллин В зависимости от механизмов снижения тонуса сосудов антигипертензивные средства миотропного действия классифицируют на следующие группы: ● блокаторы кальциевых каналов; ● активаторы калиевых каналов; ● донаторы оксида азота (NO); ● прочие. 20.3.1. Блокаторы кальциевых каналов В качестве антигипертензивных средств применяют селективные блокаторы кальциевых каналов II класса — призводные дигидропиридина. К ним относят нифедипин, амлодипин (норваск♠), фелодипин (плендил♠), нитрендипин, исрадипин, лацидипин (лаципил♠) и др. Ионы кальция играют важную роль в регуляции тонуса сосудов. При деполяризации мембраны ангиомиоцитов открываются потенциалзависимые кальциевые каналы L-типа, и ионы кальция поступают в клетку. Это приводит к повышению концентрации цитоплазматического кальция, 486 Часть II. Частная фармакология который в комплексе с кальмодулином активирует киназу легких цепей миозина. В результате повышается фосфорилирование легких цепей миозина, что способствует взаимодействию актина с миозином и сокращению гладких мышц. Блокаторы кальциевых каналов из группы производных дигидропиридина избирательно блокируют потенциалзависимые кальциевые каналы L-типа в гладкомышечных клетках кровеносных сосудов. При этом нарушается вход ионов кальция в клетки, что приводит к снижению тонуса сосудов, уменьшению ОПСС и АД. В настоящее время предпочтение отдают препаратам пролонгированного действия. При их применении наблюдают более длительное антигипертензивное действие и менее выраженные побочные эффекты со стороны гемодинамики. Амлодипин (норваск♠) при приеме внутрь хорошо всасывается, биодоступность 64–80%. Максимальная концентрация в плазме крови достигается через 6–12 ч, а равновесная концентрация — через 7–8 дней. Метаболизируется в печени с образованием неактивных метаболитов, выводится почками в неизмененном виде (10%) и в виде метаболитов (60%); t½ составляет 35–50 ч, что соответствует режиму назначения 1 раз в сутки. Применяют для систематического лечения артериальной гипертензии и при стенокардии. Препарат хорошо переносим. Побочные эффекты — рефлекторная тахикардия, отеки лодыжек, головокружение, головная боль, сонливость, тошнота. Аналогичными свойствами обладают фелодипин (плендил♠), лацидипин (лаципил♠). Нифедипин отличается быстрым и относительно непродолжительным (6–8 ч) антигипертензивным действием. При сублингвальном применении антигипертензивное действие возникает через 15 мин. В связи с этим короткодействующие формы нифедипина (коринфар♠, фенигидин♠) применяют под язык для купирования гипертензивных кризов. Для систематического лечения артериальной гипертензии рекомендуют пролонгированные формы нифедипина (коринфар ретард♠, кордипин ретард♠, кордафлекс ретард♠, адалат СЛ♠), которые назначают внутрь 2 раза в сутки. 20.3.2. Активаторы калиевых каналов Препараты этой группы представлены миноксидилом (лонитен♠) и диазоксидом (гиперстат♠). Эти препараты способствуют открытию калиевых каналов в мембранах гладких мышц кровеносных сосудов, что приводит к выходу ионов калия из клеток и развитию гиперполяризации мембраны. Гиперполяризация Глава 20. Средства, применяемые при артериальной гипертензии… 487 препятствует открытию потенциалзависимых кальциевых каналов, вследствие чего уменьшается внутриклеточная концентрация ионов кальция. В результате снижается тонус сосудов, ОПСС и АД. Миноксидил (лонитен♠) эффективен при приеме внутрь, препарат расширяет главным образом резистивные сосуды, антигипертензивный эффект сохраняется до 24 ч (рис. 20.9). Миноксидил применяют при тяжелых формах гипертензии, резистентных к другим антигипертензивным средствам (назначают, как правило, в комбинации с α-адреноблокаторами и диуретиками). Побочные эффекты — рефлекторная тахикардия, увеличение секреции ренина, задержка натрия и воды, головная боль, увеличение роста волос (гипертрихоз, гирсутизм). Рис. 20.9. Химические структуры миноксидила и диазоксида Диазоксид (гиперстат♠) расширяет главным образом резистивные сосуды (см. рис. 20.9). После внутривенного введения гипотензивное действие развивается через 1 мин, максимальный эффект достигается через 2–5 мин. Длительность гипотензивного действия достигает 12 ч. Диазоксид используют для купирования гипертензивных кризов. Вводят внутривенно быстро, так как более 90% препарата связывается с белками плазмы. В качестве побочных эффектов вызывает рефлекторную тахикардию, увеличение секреции ренина (в связи с чем задерживает в организме натрий и воду), гипергликемию и гиперурикемию. 20.3.3. Донаторы оксида азота Натрия нитропруссид (ниприд♠) (рис. 20.10) по механизму сосудорасширяющего действия сходен с нитроглицерином. Высвобождающийся из препарата оксид азота (NO) проникает в гладкомышечные клетки сосудов, активирует цитозольную гуанилатциклазу, вызывая повышение содержания циклического гуанозинмонофосфата, что приводит к снижению концентрации ионов кальция в клетке и снижению тонуса сосудов. При применении препарата расширяются как резистивные, так и емкостные сосуды. Расширение резистивных сосудов (артерий) при- 488 Часть II. Частная фармакология водит к снижению ОПСС и АД. Расширение емкостных сосудов (вен) приводит к снижению венозного давления, венозного возврата к сердцу и уменьшению преднагрузки на сердце. Продолжительность действия натрия нитропруссида при внутривенном введении не превышает 1–2 мин, в связи с чем его вводят внутривенно капельно при острой сердечной недостаточности (для разгрузки сердца), при хирургических операциях (для управляемой гипотензии) и при гипертензивном кризе. Выводится препарат почками. Побочные Рис. 20.10. Химическая струк- эффекты — рефлекторная тахикардия, головная боль, мышечные подергивания и др. тура натрия нитропруссида 20.3.4. Разные миотропные препараты Гидралазин (апрессин♠) (рис. 20.11) хорошо всасывается из ЖКТ (около 90%), но метаболизируется при первом прохождении через печень. Скорость метаболизма гидралазина (активность метаболизирующего фермента) обусловлена генетически и значительно варьирует. Поэтому биодоступность гидралазина при приеме внутрь составляет около 30% у медленных ацетиляторов и 10% — у быстрых ацетиляторов. Максимальный эффект при приеме препарата внутрь достигается через 1–2 ч. Метаболизируется в печени с образованием нескольких активных метаболитов. Продолжительность действия препарата обычно составляет 6–8 ч. Механизм сосудорасширяющего действия неясен. Гидралазин расширяет преимущественно резистивные сосуды (артериолы и мелкие артерии), в результате чего снижаются ОПСС и АД. Препарат принимают внутрь и вводят внутривенно. Из побочных эффектов отмечают рефлекторную тахикардию, задержку натрия и воды, отеки, диарею, анорексию, тошноту, рвоту, головную боль. При передозировке возможно развитие синдрома, подобного системной красной волчанке. Сходный с гидралазином дигидралазин входит в состав таблеток адельфан-эзидрекс К♠. В настоящее время ведут активные поиски новых миотропных сосудорасширяющих средств. К перспективным препаратам относят аналоги натрийуретических пептидов, антагонисты эндотелина и антагонисты вазопрессина. Незиритид — препарат рекомбинантРис. 20.11. Химическая струкного натрийуретического пептида типа В. тура гидралазина Глава 20. Средства, применяемые при артериальной гипертензии… 489 Натрийуретические пептиды вырабатываются в ответ на повышение АД или объема циркулирующей крови. Предсердия вырабатывают натрийуретический пептид типа А, желудочки — натрийуретический пептид типа В и эндотелиоциты — натрийуретический пептид типа С. Помимо натрийуретического действия указанные пептиды обладают сосудорасширяющим эффектом. При стимуляции специфических рецепторов типов А и В в гладкомышечных элементах сосудистой стенки увеличивается продукция циклического гуанозинмонофосфата, и тонус гладких мышц сосудов уменьшается. Незиритид связывается с рецепторами (типа А и В) натрийуретического пептида в эндотелиоцитах и ангиомиоцитах. При его применении расширяются как емкостные, так и резистивные сосуды (препарат относят к сбалансированным артериовенозным вазодилататорам). Это приводит к снижению пред- и постнагрузки. Применение незиритида не сопровождается существенной рефлекторной тахикардией и повышенным уровнем норадреналина. Помимо расширения сосудов большого круга кровообращения применение препарата приводит к расширению коронарных сосудов и диуретическому эффекту (натрийуретическое действие). Вводят внутривенно. Препарат хорошо переносится и вызывает мало побочных эффектов (отмечаются лишь гипотензия и головная боль). К антагонистам вазопрессина относят толваптан и кониваптан. Вазопрессин — гормон задней доли гипофиза. Стимулируя V 1Aрецепторы ангиомиоцитов, он вызывает вазоконстрикцию; стимулируя V2-рецепторы собирательных трубок, увеличивает реабсорбцию воды. Кониваптан неизбирательно блокирует V1A-рецепторы и V2-рецепторы вазопрессина. Толваптан избирательно блокирует V2-рецепторы. Препараты проходят III фазу клинических испытаний. К антагонистам эндотелина относят тезозентан (эндотелин-пептид, состоящий из 21 аминокислотного остатка). Обладает выраженным сосудосуживающим (вазоконстрикторным) эффектом, оказывает провоспалительное действие. Эндотелин участвует в патогенезе многих заболеваний, сопровождающихся нарушением функции сердца, спазмом сосудов и ишемией. В частности, уровень эндотелина резко повышается при инфаркте миокарда, что обусловливает сужение коронарных сосудов и ухудшение функций миокарда. Тезозентан блокирует рецепторы эндотелина (ЕТ-А и ЕТ-В) в ангиомиоцитах и эндотелиоцитах. Препарат вводят внутривенно. Тезозентан характеризуется коротким периодом полужизни, что обеспечивает легкую титрацию доз. Препарат проходит III фазу клинических испытаний. 490 Часть II. Частная фармакология Аналоги натрийуретических пептидов, антагонисты эндотелина и вазопрессина рекомендованы к применению при острой сердечной недостаточности. Прямое миотропное действие на гладкие мышцы сосудов оказывают также бендазол (дибазол♠) и магния сульфат. Препараты расширяют сосуды, снижают АД, но их гипотензивный эффект непродолжителен и выражен умеренно. Бендазол (дибазол♠) применяют обычно в комбинации с другими антигипертензивными средствами, а сульфат магния используют для внутримышечного введения при гипертензивных кризах (при внутривенном введении существует риск угнетения дыхательного центра). 20.4. МОЧЕГОННЫЕ СРЕДСТВА (ДИУРЕТИКИ) В фармакотерапии артериальной гипертензии диуретики применяют как для снижения АД (в качестве антигипертензивных средств), так и в комбинации с другими антигипертензивными средствами. Антигипертензивное действие диуретиков обусловлено снижением объема циркулирующей крови и сосудорасширяющим (вазодилатирующим) действием (сосудорасширяющее действие имеет определяющее значение). Оно обусловлено способностью диуретиков выводить избыток натрия. Это уменьшает «перегрузку» ангиомиоцитов натрием и снижает трансмембранный транспорт кальция. Это приводит к уменьшению гиперреактивности сосудистой стенки, а расширение кровеносных сосудов — к снижению ОПСС и АД. Диуретические средства (тиазиды) могут использоваться как средство монотерапии на ранних стадиях гипертонической болезни. Применение диуретиков в комбинации с другими антигипертензивными средствами обусловлено тем, что практически все антигипертензивные средства задерживают в организме воду и электролиты. Задержка воды и электролитов при применении антигипертензивных средств имеет компенсаторный характер. Одна из систем, участвующая в этом процессе, — ренин-ангиотензин-альдостероновая система. При медикаментозном понижении АД уменьшается почечный кровоток происходит увеличение секреции ренина активация образования ангиотензина II стимуляция АТ1-рецепторов надпочечников увеличение секреции альдостерона задержка натрия и воды. Вторая система, приводящая к задержке воды, — «система контроля объема». При медикаментозном снижении АД возбуждаются рецепторы, реагирующие на уменьшение давления («сенсоры низкого давления»), распо- Глава 20. Средства, применяемые при артериальной гипертензии… 491 ложенные в предсердиях и легочных сосудах. Афферентные импульсы от них поступают через продолговатый мозг в гипоталамус. Это приводит к увеличению выделения нейрогипофизом антидиуретического гормона вазопрессина, который задерживает воду (а также оказывает вазоспастическое действие). Кроме того, снижается выделение натрийуретических пептидов типов А, В и С). Это приводит к задержке реабсорбции натрия и воды (а также к вазоспастическому действию). В фармакотерапии артериальной гипертензии часто используют тиазидные и тиазидоподобные диуретики, петлевые диуретики, антагонисты альдостерона. Индапамид (арифон♠, индап♠) — сульфамидное нетиазидное производное (рис. 20.12). При приеме внутрь быстро и полно абсорбируется, максимальная концентрация в плазме крови достигается через 1–2 ч, t½ составляет 18 ч. Отличительной особенностью препарата является то, что при приеме в дозе 2,5 мг/сут он практически не оказывает диуретического действия, однако вызывает снижение ОПСС и АД. Увеличение дозы не приводит к усилению антигипертензивного действия, однако приводит к возникновению диуретического эффекта. В качестве побочных эффектов вызывает гипокалиемию и ортостатическую гипотензию. При систематическом лечении артериальной гипертензии широко применяют тиазидный диуретик гидрохлоротиазид (дихлотиазид♠, гипотиазид♠). Препарат часто сочетают с другими антигипертензивными средствами, он входит в состав комбинированных антигипертензивных средств (адельфан♠, трирезид К♠). Петлевой диуретик фуросемид (лазикс♠) также применяют для систематического лечения артериальной гипертензии. При внутривенном введении он приводит к быстрому снижению АД (это связано не с диуретическим, а с прямым венодилатирующим действием). Применение диуретиков в комплексной терапии артериальной гипертензии не только уменьшает задержку воды, вызываемую антигипертензивными средствами, но и увеличивает эффективность этих препаратов (в немалой степени за счет нормализации электролитного баланса). Глав- Рис. 20.12. Химическая структура индапамида 492 Часть II. Частная фармакология ный побочный эффект петлевых, тиазидных и тиазидоподобных диуретиков — способность вызывать гипокалиемию и гипомагниемию. В связи с этим их часто применяют в комбинации с препаратами калия и магния или с калий-магний сберегающими диуретиками: триамтереном, антагонистом альдостерона спиронолактоном (верошпирон♠, альдактон♠). Комбинированные антигипертензивные препараты фиксированного состава: ● адельфан♠ — таблетки, содержащие резерпина 0,0001 г и дигидралазина 0,01 г; ● адельфан-эзидрекс♠ — содержит резерпина 0,0001 г, дигидралазина 0,01 г и гидрохлоротиазида 0,01 г; — содержит те же ингредиенты, что и адель● адельфан-эзидрекс-К ♠ фан-эзидрекс , а также калия хлорида 0,6 г в одном драже; ● драже кристепина♠ — содержит резерпина 0,0001 г, дигидроэрготоксина 0,0005 г, клопамида 0,005 г; ● трирезид К♠ — таблетки, содержащие резерпина 0,0001 г, дигидралазина сульфата 0,01 г, гидрохлоротиазида 0,01 г, калия хлорида 0,35 г. Вопросы и задания для самоконтроля 1. Нейротропные антигипертензивные средства: а) моксонидин; б) доксазозин; в) нифедипин; г) атенолол; д) небиволол. 2. Средства, угнетающие ренин-ангиотензиновую систему: а) лозартан; б) эналаприл; в) пропранолол; г) гидрохлоротиазид; д) доксазозин. 3. Миотропные антигипертензивные средства: а) доксазозин; б) амлодипин; в) лозартан; г) гидралазин; д) нитропруссид. Глава 20. Средства, применяемые при артериальной гипертензии… 493 4. Средство, применяемое для систематического лечения артериальной гипертензии. Назначается 1 раз в сутки. Стимулирует имидазолиновые I1-рецепторы в ядре солитарного тракта. В качестве побочного эффекта вызывает сухость во рту: а) гуанфацин; б) моксонидин; в) клонидин; г) резерпин; д) карведилол. 5. Длительно действующее нейротропное антигипертензивное средство. Снижает АД за счет расширения кровеносных сосудов. В качестве побочных эффектов вызывает постуральную гипотензию и умеренную рефлекторную тахикардию. Облегчает мочеотделение при аденоме предстательной железы: а) дилтиазем; б) верапамил; в) доксазозин; г) амлодипин; д) небиволол. Глава 21 СРЕДСТВА, ПОВЫШАЮЩИЕ АРТЕРИАЛЬНОЕ ДАВЛЕНИЕ (ГИПЕРТЕНЗИВНЫЕ СРЕДСТВА) Препараты, повышающие АД, применяют при лечении артериальной гипотензии (снижение систолического АД менее 105 мм рт.ст.). Гипотензия может быть хронической и острой (шок, коллапс, острая кровопотеря). Хроническая гипотензия может быть первичной (идиопатической) и вторичной (при которой необходима лекарственная терапия). Основные причины развития гипотензии — снижение сердечного выброса и/или снижение ОПСС. Для лечения артериальной гипотензии применяют следующие препараты. ● Средства, повышающие сердечный выброс и тонус периферических сосудов: —α-, β-адреномиметики: эпинефрин (адреналин♠). ● Средства, повышающие преимущественно тонус периферических сосудов: —α-адреномиметики: норэпинефрин (норадреналина гидротартрат♠), фенилэфрин (мезатон♠); —ангиотензинамид, гипертензин♠. Адреномиметики применяют при острой артериальной гипотензии, вводят внутривенно (в виде инфузии). Фенилэфрин вводят также подкожно или внутримышечно. Эпинефрин — высокоэффективное средство при анафилактическом шоке (см. раздел «Адреномиметические средства»). Ангиотензинамид — амид ангиотензина II, стимулирует ангиотензиновые рецепторы артериол, оказывая выраженное и быстрое сосудосуживающее действие (по активности превосходит эпинефрин). Наиболее выражено сужение сосудов внутренних органов, кожи и почек. Стимулирующее влияние ангиотензинамида на продукцию альдостерона приводит к задержке в организме натрия и воды, увеличению объема вне- Глава 21. Средства, повышающие артериальное давление… 495 клеточной жидкости, повышению АД. Под влиянием ангиотензинамида увеличивается выделение адреналина из мозгового вещества надпочечников; препарат также стимулирует сосудодвигательный центр и симпатические ганглии. Препарат применяют при острой артериальной гипотензии; вводят внутривенно, действует он кратковременно. Поскольку 50% введенной дозы, по-видимому, разрушается за один кругооборот крови, препарат вводят инфузионно с определенной скоростью для обеспечения необходимого АД. Побочные эффекты — аллергические реакции, брадикардия, головные боли. В определенных случаях можно применять кардиотонические средства — сердечные гликозиды, допамин (см. главу «Кардиотонические средства»). При гиповолемической форме гипотензии (например, при кровопотере) применяют переливание крови, инфузию крове- и плазмозаменителей или изотонического раствора натрия хлорида (см. главу «Плазмозамещающие и дезинтоксикационные средства»). При хронической гипотензии возможно применение кофеина, никетамида (см. раздел «Психостимуляторы», главу «Аналептики»), эфедрина (см. раздел «Симпатомиметические средства»), препаратов общетонизирующего действия (препараты лимонника, элеутерококка, женьшеня и др.). Вопросы и задания для самоконтроля 1. При гипотензии, вызванной фентоламином, дальнейшее снижение АД вызовет: а) ангиотензинамид; б) никетамид; в) кофеин; г) эпинефрин; д) норэпинефрин. 2. Препараты, вызывающие брадикардию: а) эпинефрин; б) фенилэфрин; в) кофеин; г) норэпинефрин; д) ангиотензинамид. Глава 22 СРЕДСТВА, УВЕЛИЧИВАЮЩИЕ СОКРАТИМОСТЬ МИОКАРДА. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ Сократимость (инотропизм) — главная функция рабочих кардиомиоцитов, благодаря которой сердце выполняет насосную функцию и доставляет к периферическим тканям необходимое для их нормального функционирования количество крови. Снижение насосной функции сердца проявляется сердечной недостаточностью (острой и хронической). Для стимуляции сократимости миокарда применяют кардиотонические средства. 22.1. КАРДИОТОНИЧЕСКИЕ СРЕДСТВА Кардиотонические средства (средства, увеличивающие сократимость миокарда, средства с положительным инотропным эффектом) действуют непосредственно на рабочие кардиомиоциты, стимулируя их сократительную активность. Сокращение миоцитов — результат взаимодействия сократительных белков — актина и миозина (рис. 22.1). Их ассоциации препятствует тропонин С. Ионы Са2+ ингибируют тропониновый комплекс и тем самым увеличивают ассоциацию актина и миозина (таким образом, сократимость кардиомиоцитов — кальцийзависимый процесс). Движение ионов через клеточные мембраны происходит двумя основными способами: ● через ионные каналы (по электрохимическому градиенту); ● посредством активного транспорта (симпорт или антипорт независимо от электрохимического градиента). Ионы Са2+ входят в кардиомиоциты через кальциевые каналы (рецепторозависимые или потенциалозависимые), а выводятся из кардиомиоци- Рис. 22.1. Механизмы действия кардиотонических средств разных групп Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 497 498 Часть II. Частная фармакология тов при помощи кальций-натриевого антипорта (3 Na+ в обмен на 1 Са2+). В цитоплазме кардиомиоцитов Са2+ образует комплекс с кальмодулином. Кроме того, при помощи АТФ-зависимого транспортера Са2+ проходит через мембрану саркоплазматического ретикулума, где депонируется (секвестируется), связываясь с белком кальсеквестрином. Саркоплазматический ретикулум может секвестрировать значительные количества кальция. Десеквестрация Са2+ из саркоплазматического ретикулума происходит через так называемые рианодиновые рецепторы (кальциевые каналы мембраны саркоплазматического ретикулума). Этот процесс стимулируют поступающие извне ионы Са2+. Процесс сокращения кардиомиоцитов происходит следующим образом. Деполяризация мембран кардиомиоцитов (открытие натриевых каналов и вход ионов Na+ в клетку) инициирует последующее открытие потенциалзависимых кальциевых каналов. Ионы Са2+ входят из межклеточного пространства в кардиомиоциты и, стимулируя рианодиновые рецепторы саркоплазматического ретикулума, вызывают десеквестрацию Са2+. В цитоплазме ионы Са2+ связываются с тропонином С тропонин-тропомиозинового комплекса кардиомиоцитов и, изменяя конформацию этого комплекса, устраняют его тормозное влияние на взаимодействие актина и миозина. Это приводит к увеличению ассоциации актина и миозина. В результате этого взаимодействия кардиомиоцит сокращается. Среди существующих в настоящее время кардиотонических средств можно выделить: сократимость миокарда за счет увели● препараты, увеличивающие чения концентрации Са2+ в цитоплазме кардиомиоцитов (сердечные гликозиды, β1-адреномиметики, ингибиторы фосфодиэстеразы III типа); ● препараты, увеличивающие сократимость миокарда за счет увеличения чувствительности тропонина к ингибирующему действию Са2+ — сенситайзеры кальция (левосимендан). Традиционно кардиотонические средства подразделяют на две группы: ● сердечные гликозиды (кардиотонические средства гликозидной структуры); ● кардиотонические средства негликозидной структуры. 22.1.1. Сердечные гликозиды Эта группа препаратов введена в медицинскую практику в 1785 г. Уильямом Уитерингом. За более чем двухсотлетнюю историю применения сердечных гликозидов отношение к ним менялось от восторженного до скептического. До наших дней эта группа средств не утеряла значи- Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 499 мости, хотя в настоящее время сердечные гликозиды не расценивают в качестве основной группы средств, применяемых при сердечной недостаточности. Сердечные гликозиды — средства растительного происхождения, обладающие выраженным кардиотоническим действием (рис. 22.2). Их получают из растительного лекарственного сырья — наперстянки (пурпуровой, ржавой и шерстистой), строфанта (гладкого, Комбе), ландыша майского, горицвета весеннего, морского лука и др. В связи с этим сердечные гликозиды принято классифицировать по источникам получения. (дилакор♠, диланацин♠, ланикор♠), ● Гликозиды наперстянки: дигоксин ♠ ацетилдигоксин В (новодигал ), ланатозид С (цедигалан♠), дигитоксин. Г♠). ● Гликозиды строфанта Комбе: уабаин (строфантин ♠ ● Гликозиды ландыша майского: коргликон , конваллотоксин. Из перечисленных препаратов в настоящее время применяют только гликозиды наперстянки — дигоксин, ацетилдигоксин В и ланатозид, а также гликозид строфанта Комбе уабаин. Рис. 22.2. Химические структуры некоторых сердечных гликозидов 500 Часть II. Частная фармакология Молекула сердечного гликозида состоит из гликона (сахаристой части) и агликона, или генина (несахаристой части). Гликон состоит из моносахаридов, которых у разных гликозидов может быть от 1 до 4 (1 — у конваллотоксина, 2 — у строфантина, по 3 — у дигоксина и дигитоксина и 4 — у целанида♠). При попадании в организм происходит последовательное отщепление сахаридов от гликона и образование вторичных гликозидов, также обладающих фармакологической активностью. Этим обусловлена высокая продолжительность циркуляции сердечных гликозидов в системном кровотоке (даже самых короткодействующих). Таким образом, гликон определяет преимущественно фармакокинетические свойства сердечных гликозидов (способность к кумуляции, всасывание, экскреция). Агликон (несахаристая часть, генин) имеет стероидную структуру. Он состоит из циклопентанопергидрофенантрена, соединенного с ненасыщенным лактоновым кольцом. Эта часть сердечных гликозидов (благодаря высокой липофильности) способна легко преодолевать гистогематические барьеры (в том числе ГЭБ), вызывая различные фармакологические эффекты. Таким образом, агликон определяет преимущественно фармакодинамические свойства сердечных гликозидов. Воздействуя на сердце, сердечные гликозиды вызывают положительный инотропный (кардиотоническое действие), отрицательный хронотропный, отрицательный дромотропный, а также положительный батмотропный эффекты. Положительный инотропный эффект (от греч. inos — волокно, мускул, tropos —направление), кардиотоническое действие — увеличение силы сердечных сокращений (усиление и укорочение систолы). В кардиомиоцитах после возбуждения клетки для восстановления потенциала покоя транспортный фермент — магнийзависимая К+-,Na+-АТФаза — осуществляет активный обменный транспорт (антипорт) Na+ и К+ в соотношении 3:2 (Na+ выводится из кардиомиоцита, а К+ активно транспортируется в кардиомиоцит). Сердечные гликозиды связываются с тиоловыми группами К+-,Na+-АТФазы, нарушая ее транспортную функцию. К+ перестает доставляться в кардиомиоциты, и его концентрация в клетке снижается (гипокалийгистия). Na+ перестает выводиться из кардиомиоцитов, и его концентрация увеличивается (гипернатрийгистия). Увеличение концентрации Na+ в кардиомиоцитах вызывает деполяризацию мембран кардиомиоцитов и приводит к открытию потенциалзависимых кальциевых каналов. Наряду с этим нарушается функция Na+-, Са2+-антипорта, что способствует накоплению Са2+ в цитоплазме кардиомиоцитов (гиперкальцийгистия). Гиперкальцийгистия стимулирует десеквестрацию Са2+ из саркоплазматического ретикулума и ингибирование тропонина С. Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 501 Увеличивается ассоциация актина и миозина, т.е. повышается сила сердечных сокращений. Положительный инотропный эффект приводит к усилению и укорочению систолы, в результате чего увеличивается сердечный выброс. Отрицательный хронотропный эффект (греч. сhronos — время) — брадикардия (урежение ЧСС). Этот эффект (наряду с укорочением систолы в результате положительного инотропного эффекта) приводит к удлинению диастолы. При этом создаются условия, благоприятствующие восстановлению энергетических ресурсов миокарда, и удлиняется период коронарного кровотока. Таким образом, комбинация положительного инотропного и отрицательного хронотропного эффектов устанавливает более экономный режим работы сердца (с минимальным увеличением потребления миокардом кислорода). Отрицательный дромотропный эффект (от греч. dromos — дорога) — снижение проводимости возбуждения. Под действием сердечных гликозидов скорость проведения возбуждения (по интернодальным пучкам) от синусового к атриовентрикулярному узлу снижается. Кроме того, существенно сокращается и проведение через атриовентрикулярное соединение (вплоть до полной атриовентрикулярной блокады). Механизмы отрицательного хронотропного и отрицательного дромотропного эффектов сходны и в значительной степени зависят от способности сердечных гликозидов повышать тонус блуждающих нервов. Механизм ваготонического действия сердечных гликозидов включает 3 компонента. ● Сердечные гликозиды проникают через ГЭБ и прямо стимулируют ядро блуждающего нерва. ● Сердечные гликозиды инициируют кардио-кардиальный рефлекс. Стимулируя чувствительные окончания блуждающего нерва в миокарде, увеличивают афферентацию в продолговатый мозг и повышают тонус ядра блуждающего нерва. ● Сердечные гликозиды инициируют барорецепторный прессорнодепрессорный рефлекс. Ввиду оказываемого гликозидами положительного инотропного эффекта увеличивается сердечный выброс. Это приводит к стимуляции барорецепторов дуги аорты и каротидных клубочков. Увеличивается афферентация в ядро солитарного тракта, приводящая к увеличению тонуса ядра блуждающего нерва. Повышение тонуса ядра блуждающего нерва (ваготоническое действие) приводит к увеличению нисходящих тормозных холинергических влияний (через М2-холинорецепторы) на узлы проводящей системы. Снижение автоматизма синусового узла («водитель ритма первого порядка») 502 Часть II. Частная фармакология приводит к отрицательному хронотропному действию. Снижение проводимости через атриовентрикулярное соединение проявляется отрицательным дромотропным действием. Влияние сердечных гликозидов на автоматизм сердца неоднородно. Так, автоматизм водителей ритма сердечные гликозиды угнетают (за счет ваготонического действия). Автоматизм желудочков (типичных кардиомиоцитов и волокон Пуркинье) сердечные гликозиды повышают за счет прямого действия (гипокалийгистия, гиперкальцийгистия). Повышение автоматизма желудочков при передозировке сердечных гликозидов может приводить к образованию эктопических очагов и возникновению желудочковых экстрасистол. Влияние сердечных гликозидов на возбудимость также неоднородно и зависит от доз. В малых дозах сердечные гликозиды снижают порог возбудимости миокарда (увеличивают возбудимость миокарда — положительный батмотропный эффект, от греч. bathmos — порог). В больших дозах сердечные гликозиды понижают возбудимость миокарда. Диуретическое действие — важный экстракардиальный эффект сердечных гликозидов. Оно связано с ингибированием К+-Na+-АТФазы базальных мембран эпителиоцитов почечных канальцев. В результате этого снижается реабсорбция натрия и эквивалентных количеств воды. Кроме того, диурез может повышаться в виду уменьшения застойных явлений в почечной паренхиме и нормализации ее функционирования. В целом применение сердечных гликозидов при сердечной недостаточности положительно отражается на гемодинамике: снижается застой в периферических тканях, уменьшаются периферические отеки, преднагрузка на сердце и одышка. Общие показания к применению сердечных гликозидов — сердечная недостаточность (хроническая и острая) и тахисистолическая форма мерцательной аритмии. Более детальные показания к применению сердечных гликозидов обусловлены инидивидуальными свойствами препаратов. Так, уабаин целесообразно применять при острой сердечной недостаточности, поскольку его вводят только внутривенно, он имеет короткий латентный период (2–10 мин). Дигоксин можно применять как при острой сердечной недостаточности (внутривенное введение, латентный период 5–30 мин), так и при хронической сердечной недостаточности (прием внутрь, латентный период 30–120 мин), а также при тахисистолической форме мерцательной аритмии (прием внутрь). К побочным эффектам сердечных гликозидов относят: ● желудочковые экстрасистолы; ● атриовентрикулярную блокаду; Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 503 ● тошноту; ● рвоту; ● диарею; ● расстройства зрения (в том числе ксантопсия — изменение цветоощущения в желтом и зеленом спектрах); ● расстройства психики (возбуждение, галлюцинации); ● нарушения сна; ● головные боли. Поскольку сердечные гликозиды — средства растительного происхождения, в процессе производства лекарственных препаратов проводят многократную биологическую стандартизацию как растительного сырья, так и препаратов. Это обусловлено тем, что растительное сырье содержит ферменты, превращающие гликозиды друг в друга (например, первичные «генуинные» гликозиды превращаются в более стойкие вторичные). Таким образом, активность разных гликозидов из одного и того же растительного сырья может существенно различаться. При биостандартизации проводят оценку активности сырья или препарата в сравнении со стандартным препаратом. Обычно активность препаратов определяют в опытах на лягушках и выражают в «лягушачьих ЕД». Одна лягушачья ЕД (ЛЕД) соответствует минимальной дозе стандартного препарата, в которой он вызывает остановку сердца в систоле у большинства подопытных лягушек. Так, 1 г листьев наперстянки должен содержать 50–66 ЛЕД; 1 г семян строфанта — 2000 ЛЕД; 1 г дигитоксина — 8000–10 000 ЛЕД; 1 г целанида♠ — 14 000–16 000 ЛЕД; а 1 г строфантина — 44 000–56 000 ЛЕД. Реже для стандартизации используют кошачьи и голубиные ЕД. Дигоксин (ланикор ♠ ) — гликозид наперстянки шерстистой (Digitalis Lanata). Среди сердечных гликозидов его наиболее широко применяют в клинической практике. Препарат хорошо всасывается из ЖКТ (степень и скорость всасывания из таблеток, выпускаемых различными фирмами, варьирует). Биодоступность дигоксина при введении внутрь составляет 60–85%, связь с белками плазмы крови — 25–30%. Метаболизируется дигоксин только в небольшой степени и в неизмененном виде (70–80% от принятой дозы) выводится почками; t½ 32–48 ч. У больных с хронической почечной недостаточностью почечный клиренс дигоксина снижается (необходимо уменьшение дозы). Дигоксин назначают внутрь при хронической сердечной недостаточности и тахисистолической форме мерцательной аритмии, а также внутривенно при острой сердечной недостаточности. Кардиотонический эффект при приеме внутрь развивается через 1–2 ч и достигает максимума в течение 8 ч. При внутривенном введении действие наступает через 20–30 мин и достигает максимума через 3 ч. 504 Часть II. Частная фармакология Действие после прекращения приема препарата при нормальной функции почек продолжается 2–7 дней. Ввиду длительного t½ и способности связываться с белками плазмы при применении дигоксина существует риск материальной кумуляции и интоксикации. Ацетилдигоксин В (новодигал♠) — ацетилированное производное дигоксина. По фармакодинамике и основным фармакокинетическим параметрам мало отличается от дигоксина. Всасывается в тонкой кишке. В процессе прохождения через кишечную стенку практически полностью деацетилизируется, достигает системного кровотока уже в виде дигоксина. Ацетильная группа выполняет функцию переносчика и увеличивает резорбцию препарата. Ланатозид С (целанид♠) — первичный (генуинный) гликозид из листьев наперстянки шерстистой (Digitalis Lanata), сходен с дигоксином. При введении внутрь имеет меньшую, чем дигоксин, биодоступность (30–40%). Связывается с белками плазмы крови на 20–25%. В ходе метаболических изменений ланатозида образуется дигоксин. Выводится почками в неизмененном виде, в виде дигоксина и метаболитов, t½ составляет 28–36 ч. Применяют по тем же показаниям, что и дигоксин. Оказывает более «мягкий» эффект (лучше переносится пожилыми больными). Дигитоксин — гликозид, содержащийся в листьях наперстянки пурпуровой (Digitalis purpurea). До недавнего времени его широко применяли в клинической практике. Представляет собой липофильное неполярное соединение, поэтому полностью всасывается из ЖКТ (биодоступность 95–100%). С белками плазмы крови связывается на 90–97%. При приеме внутрь латентный период составляет 2–4 ч, максимальный эффект развивается через 8–12 ч, продолжительность действия после однократно введенной дозы — 14–21 дней. Дигитоксин метаболизируется в печени и в виде метаболитов выводится с мочой. Кроме того, частично экскретируется с желчью в кишечник, где подвергается энтерогепатической рециркуляции (снова реабсорбируется и поступает в печень); t½ составляет 4–7 дней. Применяют внутрь при хронической сердечной недостаточности и наджелудочковых тахиаритмиях. Вследствие перечисленных выше фармакокинетических особенностей (высокая степень связывания с белками плазмы, медленный метаболизм, длительная циркуляция в системном кровотоке) препарат обладает выраженной способностью к материальной кумуляции. В связи с этим при применении дигитоксина риск возникновения интоксикации гораздо выше, чем при применении других гликозидов наперстянки. Уабаин (строфантин Г♠) — гликозид из семян строфанта гладкого (Strophantus gratus) и строфанта Комбе (Strophantus Kombe), полярное со- Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 505 единение, практически не всасывается из ЖКТ, в связи с чем его вводят внутривенно. Имеет короткий латентный период (действие проявляется через 2–20 мин, достигая максимума через 30–120 мин, и продолжается 1–3 дней). Связь с белками плазмы составляет около 40%, практически не метаболизируется в организме и выводится почками в неизмененном виде. Препарат применяют при острой сердечной недостаточности (или при хронической сердечной недостаточности III—IV функционального классов) и тахисистолической мерцательной аритмии в качестве средства скорой помощи. Вводят внутривенно медленно в растворе глюкозы♠. Ввиду относительно непродолжительной циркуляции в системном кровотоке уабаин меньше, чем гликозиды наперстянки, кумулирует и создает риск возникновения интоксикации. Коргликон♠ — препарат, содержащий ряд гликозидов из листьев ландыша (Сonvallaria majalis). По характеру действия и фармакокинетическим свойствам близок к строфантину♠. Оказывает несколько более продолжительное действие. Применяют при острой сердечной недостаточности. Вводят внутривенно медленно (в растворе глюкозы). Вследствие выраженной способности сердечных гликозидов к материальной кумуляции и небольшой широты их терапевтического действия при применении этой группы кардиотоников высок риск интоксикации. Гликозидная интоксикация проявляется кардиальными и экстракардиальными нарушениями. К кардиальным проявлениям относят желудочковую экстрасистолию и атриовентрикулярную блокаду. Желудочковые экстрасистолы возникают в результате повышения автоматизма, вызываемого гипокалийгистией и гиперкальцийгистией. Частота их возникновения увеличивается при гипокалиемии и гипомагниемии, которые может вызвать применение петлевых и тиазидных диуретиков. Желудочковые экстрасистолы могут протекать по принципу бигеминии (экстрасистола после каждого нормального сердечного сокращения) или тригеминии (экстрасистола после каждых двух нормальных сокращений сердца). Атриовентрикулярная блокада (частичная или полная) — результат отрицательного дромотропного действия, обусловленного усилением вагусных влияний на сердце. Наиболее частая причина смерти при интоксикации сердечными гликозидами — фибрилляция желудочков (беспорядочные несинхронные сокращения отдельных пучков мышечных волокон с частотой 450–600 в минуту, быстро приводящие к асистолии — остановке сердца). Наиболее эффективные средства при интоксикации гликозидами наперстянки — препараты антител к дигоксину. При желудочковых экстрасистолиях применяют блокаторы натриевых каналов класса IB (фенитоин, лидокаин), поскольку они действуют избирательно на желудочки, 506 Часть II. Частная фармакология снижая автоматизм и не снижая сократимость, атриовентрикулярную проводимость. При атриовентрикулярной блокаде применяют атропин, который блокирует М2-холинорецепторы сердца, препятствуя тормозному действию блуждающего нерва на атриовентрикулярную проводимость. Для восполнения дефицита ионов калия и магния применяют калия хлорид и комбинированные препараты калия и магния — панангин♠ и аспаркам♠. Ионы магния активируют К+-,Na+-АТФазу, способствуя транспорту ионов калия в кардиомиоциты. Для связывания ионов кальция в системном кровотоке внутривенно вводят динатриевую соль этилендиаминтетрауксусной кислоты (трилон Б♠). Для восстановления активности К+-,Na+-АТФазы применяют донатор сульфгидрильных групп унитиол♠, который благодаря наличию сульфгидрильных (тиоловых) групп связывается дисульфидными мостиками с гликозидом, освобождая тем самым тиоловые группы АТФазы (это приводит к восстановлению ее транспортной функции). 22.1.2. Кардиотонические средства негликозидной структуры Эта группа средств появилась в клинической практике в 80-е годы прошлого века. В течение некоторого времени на эту группу возлагались надежды как на группу препаратов, способную заменить сердечные гликозиды (с этой целью были созданы негликозидные кардиотоники для приема внутрь — дофаминомиметик ибопамин, ингибитор фосфодиэстеразы-III милринон♠). Однако в тех дозах, в которых применяют негликозидные кардиотоники, их побочные и токсические эффекты выражены ярче и проявляются чаще, чем у сердечных гликозидов. При длительном применении негликозидные кардиотоники повышают летальность. В связи с этим в настоящее время группа кардиотонических средств негликозидной структуры применяется в качестве средств скорой помощи (кратковременно) при острой (декомпенсированной) сердечной недостаточности. По механизму действия их классифицируют на 3 группы. ● Агонисты β1-адренорецепторов: —β1-адреномиметики — добутамин; —дофаминомиметики — допамин. ● Ингибиторы фосфодиэстеразы III типа — милринон♠. — левосимендан. ● Сенситайзеры кальция Добутамин (добутрекс♠) — β1-адреномиметик (рис. 22.3). Стимулируя β1-адренорецепторы миокарда, активирует GS-белки, которые увеличивают активность аденилатциклазы (см. рис. 22.1). В результате увеличивается образование цАМФ из АТФ. Накапливающийся в кардиомиоцитах цАМФ Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 507 активирует цАМФ-зависимые протеинкиназы, способствующие открытию кальциевых каналов. Это повышает вход Са2+ в кардиомиоциты и усиливает десеквестрацию Са2+ из саркоплазматического ретикулума. Увеличивается сила сердечных сокращений (при этом ЧСС, автоматизм и проводимость увеличиваются в меньшей степени). Добутамин вводят внутривенно капельно (или с использова- Рис. 22.3. Химическая структура нием инфузионного насоса) со скоростью добутамина 2,5–10 (но не более 40) мкг/кг/мин. Доза и скорость инфузии зависят от степени гемодинамических расстройств. Препарат обладает быстрым и коротким действием (начинает действовать через 1–2 мин, а максимальный эффект развивается через 10 мин). Метаболизируется с образованием 3-О-добутамина и быстро выводится через почки (t½ 2 мин). Кроме того, добутамин увеличивает коронарный кровоток, повышая доставку кислорода к миокарду, а также снижает ОПСС и сосудистое сопротивление в малом круге кровообращения, не понижая существенно системное АД. Основная сфера применения — острая сердечная недостаточность или острая декомпенсация хронической сердечной недостаточности. Из побочных эффектов возможны тахикардия, аритмии (как желудочковая, так и наджелудочковая). Допамин — дофаминомиметик, предшественник норадреналина. Его кардиотоническое действие, как и у добутамина, обусловлено стимуляцией β1-адренорецепторов. Однако в отличие от добутамина, мало влияющего на тонус периферических сосудов, допамин в небольших дозах расширяет сосуды почек и брыжейки (за счет стимуляции дофаминовых рецепторов), а в высоких дозах повышает тонус периферических сосудов, оказывая прессорное действие (стимуляция α-адренорецепторов). Сочетание кардиотонического и прессорного эффектов обусловливает применение допамина при острой сердечно-сосудистой недостаточности, кардиогенном (а также послеоперационном, инфекционно-токсическом, анафилактическом) шоке. Вводят допамин внутривенно капельно со скоростью 4–10 (но не более 20) мкг/кг/мин. Препарат обладает быстрым, но коротким действием (5–10 мин). Среди побочных эффектов отмечают сужение периферических сосудов, тахикардию, аритмию, тошноту, рвоту. Милринон♠ (примакор♠) — ингибитор фосфодиэстеразы кардиомиоцитов (фосфодиэстераза-III). Угнетение фосфодиэстеразы-III приводит к увеличению внутриклеточной концентрации цАМФ и активации 508 Часть II. Частная фармакология цАМФ-зависимых протеинкиназ, открывающих кальциевые каналы. За счет этого увеличивается концентрация ионов Са2+ в кардиомиоцитах и усиливаются сокращения миокарда. За счет неизбирательного угнетения фосфодиэстеразы ангиомиоцитов милринон оказывает сосудорасширяющее действие, снижая ОПСС и постнагрузку. Препарат применяют внутривенно для кратковременной терапии острой сердечной недостаточности, резистентной к другим кардиотоникам. Вначале вводят нагрузочную дозу 50 мкг/кг (в течение 10 мин), затем — поддерживающую дозу 0,375–0,75 мкг/кг/мин. В качестве побочных эффектов отмечают аритмогенное действие, ангинальные боли, тромбоцитопению. Левосимендан (симдакс♠) — производное пиридазинон-динитрила, относят к группе негликозидных кардиотонических средств, повышающих чувствительность миофибрилл сердца к ионам кальция (сенситайзерам) Несколько раньше был разработан пимобендан, использующийся в настоящее время только в Японии. Эта группа отличается от описанных выше групп кардиотонических средств тем, что не увеличивает внутриклеточную концентрацию Са2+ и, таким образом, в меньшей степени способствует возникновению аритмий, обусловленных гиперкальцийгистией (поздняя постдеполяризация). Левосимендан связывается с N-концевой частью тропонина С, повышая его аффинитет к ионам Са2+. При этом ингибирование тропонина С и увеличение сократительной активности миофиламентов происходят без увеличения концентрации кальция в кардиомиоцитах. Взаимодействие левосимендана с тропонином С осуществляется только в период систолы. Таким образом, левосимендан, повышая интенсивность систолы, не препятствует полному расслаблению желудочков в диастолу. В связи с этим потребность миокарда в кислороде при применении левосимендана возрастает в меньшей степени, чем при применении других негликозидных кардиотоников. Не исключено, что левосимендан обладает способностью ингибировать фосфодиэстеразу-III, однако эта способность проявляется в дозах, значительно превышающих терапевтические. Ценное свойство левосимендана — способность активировать АТФ-зависимые калиевые каналы ангиомиоцитов. Это приводит к расширению сосудов большого круга кровообращения (снижение постнагрузки и преднагрузки на сердце), а также к расширению коронарных сосудов (увеличение оксигенации миокарда). Эти эффекты чрезвычайно важны при сердечной недостаточности. Левосимендан при введении в системный кровоток связывается с белками плазмы на 97–98%. Препарат почти полностью метаболизируется (5% составляет активный метаболит), t½ составляет около 1 ч. Левосимендан обладает быстрым и непродолжительным действием, Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 509 в связи с чем его применяют внутривенно капельно. Вначале вводят нагрузочную дозу 24 мкг/кг в течение 10 мин, затем поддерживающую дозу 0,1 мкг/кг/мин в течение 24 ч. Среди побочных эффектов отмечают только гипотензию и головную боль. 22.2. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ Сердечная недостаточность обусловлена нарушением насосной функции сердца и ухудшением гемодинамики периферических органов и тканей, проявляющихся явлениями застоя. При этом недостаточность насосной функции сердца может быть результатом систолической или диастолической дисфункции. ● Систолическая дисфункция (уменьшение фракции выброса левого желудочка) возникает в результате первичного уменьшения сократительной активности миокарда. Снижение сократимости при этом может быть обусловлено коронарогенными поражениями сердца (инфаркт миокарда, миокардиодистрофии), миокардитами, кардиомиопатиями. ● Диастолическая дисфункция обусловлена вторичной перегрузкой миокарда (увеличением преднагрузки или постнагрузки на сердце) при относительно сохранной сократимости миокарда. Это можно наблюдать при клапанных пороках сердца, гипертонической болезни, артериовенозных шунтах. Позже присоединяется систолическая дисфункция. Симптоматика сердечной недостаточности зависит от преимущественной локализации процесса. Так, недостаточность левых отделов сердца приводит к застойным явлениям в малом круге кровообращения (острая левожелудочковая недостаточность проявляется острым отеком легких), а недостаточность правых отделов сердца приводит к застойным явлениям в большом круге кровообращения и гипоксии периферических тканей. При этом у пациентов возникают акроцианоз (цианоз кожи и слизистых оболочек), гипостатические периферические отеки. Кроме того, застой в большом круге кровообращения приводит к повышению давления в легочных капиллярах и нарушению газообмена в легких. В результате возникает одышка. Распространенность сердечной недостаточности в популяции составляет 1,5–2%, с возрастом этот показатель увеличивается. Так, у людей старше 65 лет она встречается уже в 6–10% случаев. 510 Часть II. Частная фармакология По длительности течения сердечной недостаточности различают хроническую (застойную) сердечную недостаточность и острую сердечную недостаточность. 22.2.1. Средства фармакотерапии хронической застойной сердечной недостаточности Хроническая (застойная) сердечная недостаточность обычно характеризуется прогрессирующим течением, при котором усугубляются общие застойные явления, а также возникает изменение геометрии левого желудочка, обозначаемое термином «ремоделирование» (гипертрофия стенки, дилатация камеры, регургитация через митральный клапан). Ремоделирование, в свою очередь, приводит к еще большему увеличению гемодинамической нагрузки на миокард, еще большему снижению насосной функции сердца и дальнейшему нарастанию явлений застоя. Существуют различные классификации хронической (застойной) сердечной недостаточности. Наиболее распространена классификация по степени тяжести (функциональная) Нью-Йоркской ассоциации сердца (NYHA): ● I функциональный класс — бессимптомное течение при обычной физической нагрузке, появление симптомов при значительной нагрузке; ● II функциональный класс — появление симптомов при умеренной нагрузке; ● III функциональный класс — появление симптомов при легкой (незначительной) физической нагрузке; ● IV функциональный класс — появление симптомов в покое. В основе прогрессирования сердечной недостаточности лежит активация нейрогуморальных систем — симпато-адреналовой, ренинангиотензин-альдостероновой, системы эндотелина, вазопрессина, натрийуретического пептида и др. Наиболее взаимосвязаны из них — симпато-адреналовая и ренин-ангиотензиновая системы, поскольку они оказывают друг на друга взаимное активирующее влияние (стимуляция β1-адренорецепторов юкстагломерулярного аппарата стимулирует секрецию ренина, а ангиотензины повышают тонус симпатической нервной системы) (рис. 22.4). Застой в большом круге кровообращения снижает перфузию почечной паренхимы. Снижение давления в приносящем сосуде юкстагломерулярного аппарата приводит к усилению выделения ренина. Ренин, поступая в системный кровоток, превращает ангиотензиноген в ангио- Рис. 22.4. Механизмы прогрессирования сердечной недостаточности и некоторые препараты, применяемые для ее лечения Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 511 512 Часть II. Частная фармакология тензин I, который под действием АПФ переходит в ангиотензин II. Ангиотензин II играет существенную роль в прогрессировании сердечной недостаточности. Стимулируя АТ1-рецепторы резистивных сосудов, он повышает их тонус и увеличивает постнагрузку на сердце (способствуя вторичной перегрузке сердца и его ремоделированию). Стимуляция АТ1-рецепторов коры надпочечников ангиотензином II увеличивает выделение в кровь альдостерона (вторичный гиперальдостеронизм), вызывающего задержку Na+ и воды. Это, в свою очередь, способствует возникновению отеков, увеличению преднагрузки и ремоделирования сердца. В миокарде ангиотензин II может превращаться в ангиотензин III, который стимулирует фибротические процессы и тем самым усугубляет ремоделирование. Кроме того, ангиотензин II повышает тонус симпатической нервной системы и активирует симпато-адреналовую систему. Это приводит к стимуляции адренореактивных структур в сердечно-сосудистой системе. Стимуляция α-адренорецепторов резистивных сосудов (так же как и стимуляция АТ1-рецепторов) повышает их тонус и увеличивает постнагрузку. Стимуляция β1-адренорецепторов миокарда приводит к аритмиям, увеличению потребности миокарда в кислороде (и, таким образом, усилению гипоксии миокарда, что содействует ремоделированию), гибернации сократительных кардиомиоцитов (уменьшение сократимости вследствие гипоксии). Стимуляция β1-адренорецепторов юкстагломерулярного аппарата приводит к активации выделения ренина и стимуляции ренин-ангиотензин-альдостероновой системы. Существенный вклад в прогрессирование хронической застойной сердечной недостаточности вносят другие регуляторные системы. Так, увеличение объема внеклеточной жидкости и уменьшение сердечного выброса приводят к нарушению функционирования так называемых сенсоров объема. Недостаточная импульсация «сенсоров высокого давления» приводит к увеличению продукции вазопрессина (суживает сосуды и задерживает воду). Кроме того, снижается выработка натрийуретических пептидов (расширяют сосуды и выводят Na+ и воду). В результате происходят повышение тонуса сосудов и задержка воды и электролитов. Это еще больше усугубляет диастолическую перегрузку миокарда и способствует прогрессированию сердечной недостаточности. Основная стратегия в фармакотерапии хронической застойной сердечной недостаточности — замедление прогрессирования заболевания ЛС, влияющими на разные звенья его патогенеза. Прямая стимуляция сократимости миокарда кардиотоническими средствами при этом играет далеко не ведущую роль. Средства, применяемые в комплексной терапии Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 513 хронической застойной сердечной недостаточности, представлены следующими группами препаратов: ● ингибиторы АПФ; ● диуретики; ● β-адреноблокаторы; ● сердечные гликозиды; ● вазодилататоры. Кроме того, в комплексной терапии хронической застойной сердечной недостаточности можно применять антиагреганты, антикоагулянты, антиаритмические средства, препараты витаминов и др. Ингибиторы АПФ назначают всем больным сердечной недостаточностью, связанной с систолической дисфункцией (фракция выброса ≤35–40%). Наиболее часто при сердечной недостаточности назначают эналаприл и лизиноприл (возможно также применение фозиноприла и периндоприла). При этом следует учитывать, что симптоматический эффект ингибиторов АПФ проявляется медленно (иногда спустя несколько недель или месяцев). Эффективность этих препаратов при хронической застойной сердечной недостаточности обусловлена тем, что они прерывают один из главных механизмов прогрессирования заболевания — ингибируя АПФ, они нарушают образование ангиотензина II. Уменьшение воздействия ангиотензина II на сосуды приводит к снижению постнагрузки на сердце. Уменьшение воздействия ангиотензина II на надпочечники снижает явления вторичного гиперальдостеронизма (это способствует снижению преднагрузки на сердце). Уменьшение вторичной перегрузки сердца уменьшает процесс ремоделирования и, таким образом, замедляет прогрессирование заболевания. Установлено, что применение ингибиторов АПФ уменьшает смертность больных. При назначении ингибиторов АПФ больным с сердечной недостаточностью применяют тактику «титрования доз». Она предполагает назначение препарата в низкой дозе (например, 2,5 мг эналаприла или лизиноприла 2 раза в сутки) с постепенным повышением дозы (дозу удваивают каждые 3–7 дней) до терапевтической. Побочные эффекты, вызываемые ингибиторами АПФ, можно условно разделить на 2 группы: ● связанные с подавлением образования ангиотензина II (гипотензия, ухудшение функции почек и задержка калия); ● связанные с накоплением кининов (ангионевротический отек и сухой кашель) — развиваются у 5–15% лиц, принимающих ингибиторы АПФ, не купируются противокашлевыми средствами, не проходят самопроизвольно и являются основанием для отмены препаратов. 514 Часть II. Частная фармакология Помимо ингибиторов АПФ при хронической застойной сердечной недостаточности возможно назначение блокаторов ангиотензиновых рецепторов (лозартан). Теоретически эти препараты способны более полно, чем ингибиторы АПФ, «выключать» ренин-ангиотензин-альдостероновую систему, поскольку ангиотензин II может образовываться не только в системном кровотоке под влиянием АПФ, но и в тканях, откуда выделяется в системный кровоток в готовом виде. Однако в настоящее время не существует убедительных данных, свидетельствующих о преимуществе блокаторов ангиотензиновых рецепторов перед ингибиторами АПФ. В связи с этим блокаторы рецепторов ангиотензина II целесообразно назначать в случае непереносимости больными ингибиторов АПФ (например, тем больным, у которых ингибиторы АПФ вызывают сухой кашель). В отличие от ингибиторов АПФ диуретики оказывают очень быстрый (в течение нескольких дней и даже часов) симптоматический эффект. Диуретики обеспечивают адекватный контроль водного баланса (выведение избытка воды приводит к снижению отеков и массы тела). Кроме того, нормализуя электролитный баланс, диуретики создают предпосылки для успешного применения других групп средств. Монотерапия сердечной недостаточности диуретиками не эффективна. Выбор диуретика и его доза зависят от степени задержки жидкости. Назначение диуретиков начинают с низких доз (20–40 мг фуросемида), и при необходимости дозу увеличивают под контролем снижения массы тела (0,5–1 кг/сут). Основная опасность применения петлевых и тиазидовых диуретиков состоит в их способности вызывать гипокалиемию и гипомагниемию, что может усилить проаритмическое действие сердечных гликозидов. Для компенсации гипокалиемии возможно использование препаратов калия и магния. Однако более целесообразно применение калий-магний-сберегающих диуретиков (лучше переносятся больными, чем препараты калия и магния). Особого внимания заслуживает использование антагонистов альдостерона. Антагонист альдостерона спиронолактон традиционно относят к медленно действующим диуретикам с низкой диуретической эффективностью. Однако в условиях вторичного гиперальдостеронизма при хронической застойной сердечной недостаточности этот препарат может действовать быстро и эффективно. Кроме того, ценное свойство спиронолактона — калий-магний-сберегающее действие. Как показали статистические исследования, применение спиронолактона снижает смертность и риск повторных госпитализаций. В целом порядок назначения диуретиков при хронической застойной сердечной недостаточности может быть следующим. При значительной задержке жидкости назначают петлевой диуретик фуросемид в комбинации с антагонистом альдостерона Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 515 спиронолактоном. Избыток жидкости эффективно выводится фуросемидом. За это время начинает проявляться диуретический эффект спиронолактона, после чего петлевой диуретик может быть отменен. β-Адреноблокаторы. Применение этой группы препаратов при сердечной недостаточности может показаться парадоксальным, учитывая тот факт, что одно из свойств β-адреноблокаторов — отрицательный инотропный эффект. Однако статистические мультицентровые исследования показали, что применение β1-адреноблокаторов (метопролол), а также применение α-, β-адреноблокатора карведилола приводит к значительному снижению смертности и риска повторных госпитализаций больных с сердечной недостаточностью (по некоторым данным, высокая эффективность метопролола определяется наличием у него свойств инверсированного агониста β-адренорецепторов). Следует особо отметить, что β-адреноблокаторы не применяют при выраженных признаках декомпенсации, а также при IV функциональном классе сердечной недостаточности. У пациентов с сердечной недостаточностью I функционального класса β-адреноблокаторы снижают риск нарастания нарушений кровообращения. Наиболее выраженное улучшение состояния наблюдают при назначении β-адреноблокаторов пациентам с сердечной недостаточностью II и III функциональных классов (со снижением фракции выброса <35–40%). Эффективность β-адреноблокаторов при сердечной недостаточности обусловлена их способностью устранять активацию нейрогуморальных систем прогрессирования заболевания. Здесь возможны несколько механизмов: ● блокада β1-адренорецепторов юкстагломерулярного аппарата приводит к подавлению активности ренин-ангиотензин-альдостероновой системы — одной из главных систем, вовлеченных в прогрессирование сердечной недостаточности; ● расширение периферических сосудов (за счет уменьшения их стимуляции ангиотензином II) приводит к уменьшению нагрузки на сердце; ● блокада β1-адренорецепторов миокарда препятствует избыточной стимуляции сердца циркулирующим адреналином и норадреналином в области синаптических окончаний; это снижает риск возникновения аритмий; ● блокада β1-адренорецепторов миокарда приводит к «дегибернации» сократительных кардиомиоцитов. В условиях гипертрофии стенки желудочков кардиомиоциты пребывают в состоянии гипоксии (рост коронарных сосудов отстает от увели- 516 Часть II. Частная фармакология чения массы кардиомиоцитов). Продолжительный дисбаланс доставки/ потребления кислорода приводит к обратимой диссинергии, при которой морфологически неповрежденные кардиомиоциты перестают сокращаться. Возникают участки «спящего» миокарда (гибернация). Гибернация приводит к уменьшению сердечного выброса. Применение β1-адреноблокаторов (особенно в комбинации с ингибиторами АПФ) восстанавливает баланс доставки/потребления. Сократительная активность «спящих» (гибернированных) кардиомиоцитов восстанавливается. Этим объясняется (парадоксальная на первый взгляд) способность β-адреноблокаторов увеличивать фракцию выброса, уменьшая функциональный класс сердечной недостаточности. Как видно из перечисленного, позитивные свойства β-адреноблокаторов — результат блокады β1-адренорецепторов. Применение неселективных блокаторов (пропранолол) нежелательно ввиду их способности блокировать β2-адренорецепторы и повышать ОПСС. Теоретически преимущество карведилола состоит в его способности блокировать α2-адренорецепторы и понижать постнагрузку на сердце. Однако реальные преимущества карведилола перед метопрололом и бисопрололом не доказаны. Назначение β-адреноблокаторов лицам с сердечной недостаточностью II–III функциональных классов проводят крайне осторожно — только после полной компенсации водно-электролитного баланса и по принципу «титрования» доз, начиная с очень малых (1,25 мг бисопролола в сутки; 12,5 мг метопролола в сутки; 3,125 мг карведилола дважды в сутки). При хорошей переносимости дозы β-адреноблокаторов удваивают каждые 2–4 нед. В качестве побочных эффектов β-адреноблокаторов отмечают гипотензию (наиболее возможна при применении карведилола за счет блокады α2-адренорецепторов), задержку жидкости и нарастание сердечной недостаточности в начале терапии (задержка жидкости возникает на 3–5 сут, явления сердечной недостаточности могут нарастать в первые 1–2 нед), брадикардию и атриовентрикулярную блокаду. Противопоказания к назначению β-адреноблокаторов при хронической застойной сердечной недостаточности — признаки декомпенсации, атриовентрикулярная блокада II–III степени, выраженная брадикардия, выраженная задержка жидкости, бронхоспазм. Сердечные гликозиды в настоящее время не рассматривают как ведущую группу препаратов для лечения хронической застойной сердечной недостаточности. Их применение не увеличивает выживаемость пациентов. Дигоксин уменьшает симптомы сердечной недостаточности, улучшает качество жизни и повышает толерантность к физической нагрузке, заметно стабилизирует состояние больных (особенно при признаках де- Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 517 компенсации или мерцании предсердий). Назначение дигоксина лицам со скрытой дисфункцией левого желудочка или с сердечной недостаточностью I функционального класса нецелесообразно. Препарат применяют главным образом в комбинации с ингибиторами АПФ, диуретиками и β-адреноблокаторами. При этом не используют насыщающих схем дигитализации. Как начальная, так и поддерживающая дозы дигоксина достаточно низкие и обычно составляют 0,25 мг/сут. При таком способе назначения уровень дигоксина в плазме крови не поднимается выше 2 нг/мл, в связи с чем побочные эффекты отмечают крайне редко. Вазодилататоры (изосорбида динитрат и гидралазин) оказывают фармакотерапевтическое действие при хронической застойной сердечной недостаточности за счет уменьшения преднагрузки и постнагрузки на сердце. При этом не исключено, что изосорбида динитрат ингибирует патологический рост кардиомиоцитов, замедляя ремоделирование, а гидралазин обладает некоторыми антиоксидантными свойствами. Применение вазодилататоров при хронической застойной сердечной недостаточности не имеет самостоятельного значения. Их назначают только в случае невозможности применения ингибиторов АПФ (например, при выраженной почечной недостаточности или гипотензии). 22.2.2. Средства фармакотерапии острой сердечной недостаточности Острая сердечная недостаточность может возникать либо в результате декомпенсации хронической сердечной недостаточности, либо в результате значительных морфологических поражений сердца (острый инфаркт миокарда, операции на сердце). Острая левожелудочковая недостаточность проявляется отеком легких. Повышение давления в легочных капиллярах приводит к транссудации жидкости в альвеолы. Транссудат вспенивается за счет дыхательных экскурсий грудной клетки. Пена препятствует нормальному газообмену в альвеолах. В результате возникает гипоксия, которая может быстро привести к смерти. Острая сердечная недостаточность требует экстренных терапевтических мероприятий. При этом применяют средства, вводимые внутривенно, с быстрым и мощным действием. Основные мероприятия при острой сердечной недостаточности: ● поддержание работы сердца; ● разгрузка кругов кровообращения; ● профилактика осложнений. 518 Часть II. Частная фармакология При острой сердечной недостаточности применяют: ● кардиотонические средства; ● вазодилататоры; ● диуретики; ● средства симптоматической терапии. Из кардиотонических средств при острой сердечной недостаточности применяют быстродействующие негликозидные кардиотоники (добутамин, левосимендан), а также сердечные гликозиды, вводимые внутривенно (дигоксин). Если в фармакотерапии хронической сердечной недостаточности применение вазодилататоров имеет второстепенное значение («разгрузки» сердца достигают применением ингибиторов АПФ), то при острой сердечной недостаточности (когда ингибиторы АПФ нецелесообразны ввиду длительного латентного периода их действия) применение вазодилататоров имеет основополагающее значение. Ценность вазодилататоров при острой сердечной недостаточности состоит не только в их способности снижать нагрузку на сердце. Не менее значима их способность снижать давление в легочных капиллярах. Это приводит к уменьшению одышки и транссудации, что особенно необходимо при фармакотерапии отека легких (клиническое проявление острой левожелудочковой недостаточности). При острой сердечной недостаточности применяют нитроглицерин и нитропруссид натрия. Оба препарата вводят внутривенно капельно. Они расширяют как емкостные (снижая преднагрузку), так и резистивные сосуды (снижая постнагрузку). Основной недостаток донаторов NO — их способность вызывать толерантность. Кроме того, значительное расширение емкостных сосудов приводит к резкой гипотензии и рефлекторной тахикардии. Ввиду того, что вазодилататоры — одна из важнейших групп средств, применяемых при острой сердечной недостаточности, постоянно ведут поиски новых вазодилататоров. Незиритид — препарат рекомбинантного натрийуретического пептида типа В. Он связывается с рецепторами (типа А и В) натрийуретического пептида в эндотелиоцитах и ангиомиоцитах. При этом в гладкомышечных элементах сосудистой стенки увеличивается продукция циклического гуанозинмонофосфата, а тонус ангиомиоцитов уменьшается. Незиритид расширяет как емкостные, так и резистивные сосуды, снижая как пред-, так и постнагрузку. Кроме того, препарат оказывает коронарорасширяющее и диуретическое (натрийуретическое) действия. При внутривенном введении хорошо переносится и вызывает мало побочных эффектов (отмечают лишь умеренную рефлекторную тахикардию, гипотензию и головную боль). Глава 22. Средства, увеличивающие сократимость миокарда. Средства, применяемые… 519 Кроме того, вазодилатирующее действие могут оказывать антагонисты эндотелина (тезозентан♠, блокатор ЕТА и ЕТВ рецепторов эндотелина) и антагонисты вазопрессина (толваптан♠, блокирующий V2-рецепторы, и кониваптан♠, блокирующий V1A- и V2-рецепторы). Из диуретиков при острой сердечной недостаточности наиболее часто используют фуросемид (средство выбора при отеке легких), обладающий быстрым и эффективным мочегонным действием. Быстрое выведение избытка воды приводит к снижению преднагрузки и уменьшению гидратации легочной паренхимы. Также фуросемид обладает прямым венодилатирующим действием. Расширение емкостных сосудов приводит к снижению преднагрузки на сердце. Кроме того, происходит снижение давления в легочных капиллярах и уменьшается транссудация. Среди средств симптоматической терапии следует отметить: ● противоаритмические средства; ● морфин (уменьшает преднагрузку и давление в легочных капиллярах, что важно при отеке легких); ● этиловый спирт (при ингаляционном введении уменьшает вспенивание транссудата при отеке легких). В числе немедикаментозных мероприятий проводят кислородотерапию (с целью коррекции гипоксемии). Вопросы и задания для самоконтроля 1. При хронической сердечной недостаточности применяют: а) ингибиторы АПФ; б) диуретики; в) β-адреноблокаторы; г) кардиотонические средства; д) сосудосуживающие средства. 2. Сердечные гликозиды применяют при: а) хронической сердечной недостаточности; б) острой сердечной недостаточности; в) атриовентрикулярной блокаде; г) тахисистолической форме мерцательной аритмии. 3. Повышает в кардиомиоцитах концентрацию ионов кальция. Оказывает положительное инотропное, отрицательное хроно- и дромотропное действия. Применяется при сердечной недостаточности и тахисистолической форме мерцательной аритмии: 520 Часть II. Частная фармакология а) добутамин; б) левосимендан; в) дигоксин; г) эналаприл; д) фуросемид. 4. Оказывает кардиотоническое действие за счет повышения чувствительности тропонина к ионам кальция. Снижает нагрузку на сердце и увеличивает доставку кислорода к миокарду. Действует непродолжительно. Применяется путем внутривенной капельной инфузии при острой сердечной недостаточности. Редко вызывает аритмии: а) эналаприл; б) добутамин; в) дигоксин; г) левосимендан; д) милринон. 5. Расширяет артериальные сосуды и снижает постнагрузку на сердце. Уменьшает секрецию альдостерона и снижает преднагрузку на сердце. Действует путем угнетения биосинтеза ангиотензина. Применяется при хронической застойной сердечной недостаточности и артериальной гипертензии. Повышает уровень брадикинина. Вызывает сухой кашель и ангионевротический отек: а) бисопролол; б) спиронолактон; в) лозартан; г) эналаприл; д) дигоксин. Глава 23 СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ НАРУШЕНИИ МОЗГОВОГО КРОВООБРАЩЕНИЯ Различают острые и хронические нарушения мозгового кровообращения. ● Острые нарушения мозгового кровообращения могут возникать вследствие спазма, тромбоза или эмболии сосудов мозга (ишемический инсульт), например на фоне атеросклеротического поражения сосудов (особенно стеноза сонных и позвоночных артерий). Другой формой острого нарушения мозгового кровообращения считают кровоизлияния в мозг (геморрагический инсульт), причиной которых могут быть артериальная гипертензия, разрыв аневризмы и др. При продолжительной и выраженной ишемии развивается некроз ткани мозга. ● Хронические нарушения мозгового кровообращения обычно связаны с возрастными изменениями, в том числе с атеросклерозом сосудов, артериальной гипертензией, нарушениями метаболических процессов в тканях мозга. При этом наблюдают постепенное расстройство памяти, нарушения интеллекта, другие изменения в психической сфере, двигательные нарушения. Один из основных способов профилактики и лечения хронических ишемических нарушений мозгового кровообращения — применение средств, расширяющих сосуды мозга (увеличивают мозговой кровоток), в меньшей степени влияя на системную гемодинамику. Эти препараты могут также использовать для лечения остаточных явлений после перенесенных острых нарушений мозгового кровообращения (в основном после ишемических инсультов). К таким препаратам относят: ● некоторые блокаторы кальциевых каналов (нимодипин, циннаризин); ● производные алкалоидов барвинка (винпоцетин); ● производные ГАМК (никотиноил-ГАМК); ● некоторые производные алкалоидов спорыньи (ницерголин); ● производные никотиновой кислоты (ксантинола никотинат) и др. 522 Часть II. Частная фармакология Из этих препаратов преимущественное (более избирательное) действие на сосуды мозга оказывают нимодипин, винпоцетин и производные ГАМК. Ницерголин, ксантинола никотинат, циннаризин расширяют также периферические сосуды, их применяют при нарушениях периферического кровообращения (болезнь Рейно, диабетическая ангиопатия и др.). Некоторые препараты могут улучшать мозговое кровообращение не только за счет расширения кровеносных сосудов, но и за счет улучшения реологических свойств крови, как, например, циннаризин, винпоцетин, пентоксифиллин и др. 23.1. БЛОКАТОРЫ КАЛЬЦИЕВЫХ КАНАЛОВ Нимодипин (нимотоп♠) и циннаризин (стугерон♠) блокируют поступление ионов Са2+ в гладкомышечные клетки сосудов мозга, что приводит к снижению тонуса сосудов. Нимодипин (рис. 23.1) — блокатор кальциевых каналов, производное 1,4-дигидропиридина, преимущественно расширяет артериолы мозга и увеличивает мозговой кровоток без значительного снижения системного АД. Может предотвращать или устранять спазм сосудов мозга. Нимодипин применяют для профилактики и лечения нарушений мозгового кровообращения (после перенесенной острой ишемии мозга, при хронической ишемии мозга). Нимодипин — единственный препарат этой группы, который можно использовать для уменьшения неврологических нарушений после перенесенного субарахноидального кровоизлияния. Препарат назначают внутрь (быстро и полностью всасывается из ЖКТ, максимальная концентрация в крови достигается через 0,5–1 ч), а при острой ишемии мозга внутривенно капельно. Рис. 23.1. Химическая структура нимодипина Глава 23. Средства, применяемые при нарушении мозгового кровообращения 523 Наиболее частые побочные эффекты нимодипина — снижение системного АД (при внутривенном введении), рефлекторная тахикардия. Реже отмечают чувство жара и гиперемию лица, желудочнокишечные расстройства, повышение активности трансаминаз печени и снижение функции почек. Препарат практически не влияет на сократимость миокарда. Поливинилхлорид хорошо абсорбирует нимодипин, поэтому его можно хранить только в упаковке из полиэтилена. Срок хранения инфузионного раствора при рассеянном дневном свете или искусственном освещении не более 10 ч. Циннаризин (рис. 23.2) — блокатор кальциевых каналов, имеющий тропность к артериолам головного мозга, снижает реакцию гладкомышечных клеток сосудов на эндогенные сосудосуживающие вещества (катехоламины, брадикинин, ангиотензин, вазопрессин и др.), умеренно расширяет периферические сосуды. Реологические эффекты циннаризина заключаются в повышении способности эритроцитов к деформации и уменьшению повышенной вязкости крови. Это приводит к улучшению микроциркуляции в ишемизированных участках мозга. Рис. 23.2. Химическая структура циннаризина Циннаризин применяют при остаточных явлениях после инсульта и черепно-мозговых травм, для профилактики и лечения нарушений периферического кровообращения. Помимо влияния на мозговой кровоток циннаризин проявляет умеренную антигистаминную активность. Препарат уменьшает возбудимость вестибулярного аппарата, вследствие чего его применяют для профилактики морской и воздушной болезни (болезни движения). Назначают по 25 мг за 30 мин до предстоящей поездки, при необходимости — повторно через 6 ч. Побочные эффекты — сухость во рту, сонливость, диспептические явления. При длительном применении рекомендуют проведение контрольного исследования функции печени, почек, картины периферической крови. 524 Часть II. Частная фармакология 23.2. ПРОИЗВОДНЫЕ АЛКАЛОИДОВ БАРВИНКА Винпоцетин (кавинтон♠) — полусинтетическое производное алкалоида винкамина, содержащегося в растении барвинке (Vinca minor L. и Vinca erecta), расширяет преимущественно сосуды мозга. Миотропное спазмолитическое действие препарата связывают с ингибированием фосфодиэстеразы, что способствует накоплению в гладкомышечных клетках цАМФ. Винпоцетин не вызывает феномена «обкрадывания». Не влияя на кровоснабжение интактных областей, он усиливает кровоснабжение ишемизированных участков мозга. Препарат улучшает мозговое кровообращение (микроциркуляцию) не только вследствие расширения сосудов мозга, но и за счет повышения способности эритроцитов к деформации, что приводит к снижению повышенной вязкости крови, так как агрегация тромбоцитов уменьшена. Кроме того, винпоцетин повышает устойчивость мозга к гипоксии, повышает утилизацию кислорода, усиливает поглощение и метаболизм глюкозы с переключением на энергетически более выгодный, аэробный путь окисления. Винпоцетин показан при хронической недостаточности мозгового кровообращения, состояниях после инсульта, при неврологических и психических нарушениях у больных с цереброваскулярной недостаточностью, при сосудистых заболеваниях глаз и снижении остроты слуха сосудистого или токсического генеза. Назначают внутрь в течение длительного времени. При необходимости вводят внутривенно капельно. Побочные эффекты — снижение АД, тошнота и головокружение. При быстром внутривенном введении возможны тахикардия, экстрасистолия, замедление внутрижелудочковой проводимости. 23.3. ПРОИЗВОДНЫЕ АЛКАЛОИДОВ СПОРЫНЬИ Ницерголин (сермион ♠) — синтетическое производное алкалоидов спорыньи, с присоединенным бромзамещенным остатком никотиновой кислоты. Сосудорасширяющий эффект препарата связан с α-адреноблокирующим и прямым миотропным действиями. Расширяет артериолы головного мозга и периферические сосуды. Улучшает мозговой, легочный, почечный кровоток и кровоснабжение в конечностях. Применяют при нарушениях мозгового кровообращения на фоне атеросклероза и тромбоэмболии, при расстройствах периферического кро- Глава 23. Средства, применяемые при нарушении мозгового кровообращения 525 вообращения и др. Вводят внутрь и парентерально. При приеме внутрь в терапевтических дозах не влияет на АД, после внутривенного введения возможен гипотензивный эффект. Из побочных эффектов также возможны головокружение, зуд, диспептические расстройства. 23.4. ПРОИЗВОДНЫЕ НИКОТИНОВОЙ КИСЛОТЫ Никотиновая кислота оказывает выраженное сосудорасширяющее действие как на периферические сосуды, так и на сосуды мозга. Никотиновую кислоту плохо переносят (см. главу «Средства, применяемые при атеросклерозе»), поэтому при нарушениях мозгового кровообращения используют ее производные с менее выраженными побочными эффектами. Ксантинола никотинат (компламин♠) в своей структуре имеет элементы никотиновой кислоты и теофиллина. Препарат расширяет периферические сосуды и сосуды мозга, улучшает как периферическое, так и мозговое кровообращение, уменьшает явления гипоксии мозга, несколько снижает агрегацию тромбоцитов. Препарат применяют при нарушениях периферического и мозгового кровообращения, связанных с атеросклерозом сосудов мозга. Никотиноил-ГАМК (пикамилон♠) относят к группе производных ГАМК, сочетает структуру ГАМК и никотиновой кислоты, преимущественно расширяет сосуды мозга. 23.5. ПРОИЗВОДНЫЕ КСАНТИНА Пентоксифиллин (трентал♠, агапурин♠) по химической структуре близок к теофиллину, блокирует аденозиновые рецепторы и, будучи ингибитором фосфодиэстеразы, повышает содержание цАМФ в гладкомышечных клетках сосудов, с чем связывают его умеренное сосудорасширяющее действие. Препарат уменьшает агрегацию тромбоцитов (см. раздел «Средства, снижающие агрегацию тромбоцитов»), повышает эластичность оболочки эритроцитов, снижает вязкость крови, улучшает микроциркуляцию и доставку кислорода к тканям. При внутривенном введении пентоксифиллин дополнительно приводит к усилению коллатерального кровообращения с увеличением объема протекающей крови через единицу сечения и возрастанию содержания АТФ в клетках головного мозга. 526 Часть II. Частная фармакология Пентоксифиллин показан при ишемических формах нарушения мозгового, коронарного и периферического кровообращения. Препарат принимают внутрь после еды 3 раза в сутки. Пролонгированные формы назначают 2 раза в сутки. Таблетки, покрытые кишечнорастворимой оболочкой, проглатывают целиком, запивая небольшим количеством воды. При необходимости препарат вводят внутривенно капельно. Побочные эффекты: тошнота, рвота, чувство жара и гиперемия кожи лица и верхней части грудной клетки, кровотечения из сосудов кожи и слизистых оболочек, аллергические реакции, при внутривенном введении — снижение АД. Передозировка пентоксифиллина вызывает снижение АД, повышение температуры тела, потерю сознания, судороги, желудочно-кишечное кровотечение. Лечение передозировки: промывание желудка с активированным углем, поддержание дыхания и АД. При ишемических нарушениях мозгового кровообращения применяют комбинированный препарат интестенон♠, в состав которого входят гексобендин (оказывает спазмолитическое и коронарорасширяющее действие), этамиван (стимулирует ЦНС, оказывает аналептическое действие), этофиллин (гидроксиэтилтеофиллин, улучшает мозговой кровоток). Для предупреждения нарушений мозгового кровообращения, связанных с тромбозом сосудов мозга, в том числе для профилактики ишемического инсульта, можно назначать антиагреганты — ацетилсалициловую кислоту, дипиридамол, тиклопидин, клопидогрел, а также антикоагулянты (см. главу «Средства, влияющие на гемостаз и тромбообразование»). Однако эти препараты могут вызвать внутричерепные кровоизлияния, поэтому они и противопоказаны при опасности возникновения геморрагического инсульта. Другим направлением в терапии ишемии мозга считают применение нейропротекторов — препаратов, повышающих устойчивость нейронов к гипоксии. К нейропротекторам относят вещества различных фармакологических групп с различными механизмами действия. Так, натрия оксибат♠ повышает устойчивость мозга к гипоксии (оказывает влияние на метаболизм мозга). Нейропротекторное действие оказывают вещества, устраняющие эффекты возбуждающих аминокислот (антагонист NMDA-рецепторов — дизоцилпин). Блокаторы кальциевых каналов не только улучшают мозговое кровообращение, но и оказывают нейропротекторное действие, связанное с уменьшением входа ионов кальция в клетки мозга. Есть данные о нейропротекторных свойствах винпоцетина. Глава 23. Средства, применяемые при нарушении мозгового кровообращения 527 23.6. СРЕДСТВА, ПРИМЕНЯЕМЫЕ ПРИ МИГРЕНИ Мигрень проявляется периодическими приступами односторонней пульсирующей головной боли (продолжительностью в среднем от 4 до 72 ч), при этом могут возникать тошнота, рвота, зрительные и слуховые нарушения и другие характерные симптомы. Приступы могут повторяться в течение многих лет с периодичностью 1–4 раза в месяц. Заболевание связывают с нарушением регуляции тонуса сосудов мозга; природа заболевания окончательно не выяснена. Получены данные о роли серотонинергической системы в патогенезе мигрени. Для устранения боли при приступах мигрени применяют неопиоидные анальгетики (парацетамол), а также ацетилсалициловую кислоту, ибупрофен и некоторые другие НПВС. Из веществ, влияющих на мозговое кровообращение, для купирования острых приступов мигрени используют суматриптан и алкалоиды спорыньи. Суматриптан (имигран♠) — синтетическое производное триптамина, селективный агонист центральных 5-НТID-рецепторов, локализованных преимущественно в сосудах головного мозга. Со стимуляцией этих рецепторов связывают вазоконстрикторное действие препарата в отношении сосудов мозга. Суматриптан уменьшает чрезмерную пульсацию мозговых сосудов и связанную с этим головную боль. Кроме того, стимулируя пресинаптические 5-НТ-рецепторы, суматриптан уменьшает высвобождение вещества Р, с чем также может быть связано уменьшение болевых ощущений. Препарат используют для купирования приступов мигрени, в особенности у больных, не реагирующих на ненаркотические анальгетики. Препарат вводят внутрь, подкожно и интраназально. Биодоступность при введении внутрь составляет 14%, эффект развивается через 30 мин, при подкожном введении — через 10 мин; продолжительность действия — около 12 ч. Суматриптан может вызвать спазм коронарных сосудов, в связи с чем его не рекомендуют назначать при ишемической болезни сердца. Из других побочных эффектов отмечают тошноту, рвоту, нарушение вкуса, головокружение, утомляемость. Препарат противопоказан при заболеваниях печени, беременности и лактации, в детском (до 18 лет) и пожилом (более 65 лет) возрасте. Для купирования приступов мигрени применяют алкалоиды спорыньи — эрготамин и их дигидрированные производные (дигидроэрготамин). Дигидроэрготамин (дигидергот♠) стимулирует 5-НТ-рецепторы, в особенности 5-НТID-рецепторы. Используется в виде назального спрея 528 Часть II. Частная фармакология для быстрого купирования приступа мигрени. Для устранения рвоты, возникающей при мигрени, назначают противорвотные средства (метоклопрамид). Для профилактики приступов мигрени применяют антагонист 5-НТ2-рецепторов метисергид♠, а также β-адреноблокаторы (пропранолол), трициклические антидепрессанты (амитриптилин), противоэпилептические средства (карбамазепин, клоназепам). Вопросы и задания для самоконтроля 1. Блокатор кальциевых каналов: а) циннаризин; б) суматриптан; в) пентоксифиллин; г) винпоцетин. 2. Полусинтетическое производное алкалоида барвинка: а) нимодипин; б) ницерголин; в) ксантинола никотинат; г) винпоцетин. 3. Производное алкалоидов спорыньи: а) нимодипин; б) винпоцетин; в) ниц