Теория фазового соответствия
advertisement

ТЕОРИЯ ФАЗОВОГО
СООТВЕТСТВИЯ
Под фазовым соответствием мы будем
понимать закономерности сопряженного
изменения составов сосуществующих минералов
при воздействии на них температуры, давления и
других факторов равновесия.
На общей теории фазового соответствия
базируется современная термометрия,
барометрия, рН-метрия, определение fO2
активностей компонентов во флюидах и т.п. по
минеральным равновесиям в магматических и
метаморфических породах из различных
геодинамических обстановок. Но достигается ли
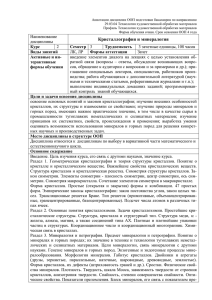
в них равновесие? Рассмотрим пример.
Grt
A
Opx
Grt
300
0
мкм
Концентрация элементов, усл. ед.
A
40
Mg
30
Mg
Mg
20
Grt
Opx
10
Grt
Fe
Fe
Fe
Ca
0
0
Ca
Ca
100
200
300
400
Расстояние, мкм
Распределение Сa, Fe и Mg в сосуществующих Орх и Grt из
ксенолита в кимберлитовой трубке Сан Жуан, штат Юта, США
Межфазовое распределение компонентов.
Обменное равновесие между фазами и :
2 + 1 = 2 + 1
(247)
Его константа равна:
lnKP = ln (a1a2/a1a2) = (G0T/RT )P (248)
a1 = X11
(249)
a2 = X22
(250)
a1= (X1 1)
(251)
a2=( X22)
(252)
где a – активность соответствующего компонента,
а - коэффициент его активности.
G0T = G02 + G01 – G02 – G01
(253)
Подставляем уравнения (249) - (252) в (248) и находим:
lnKР = ln[(X1/X2)(X2 /X1)] (12/12)
=(G0T/RT )P
В соответствии с (239)
(X1 /X2) (X2 /X1) = KD
(254)
(239)
KD - коэффициент распределения. В случае
идеального распределения при постоянных Т и Р
коэффициент распределения, KD– величина постоянная
и графически может быть представлена в виде
диаграмм фазового соответствия:
диаграмм фазового соответствия:
=
KD
2
=
1
0.
5
K D=0. 2
2
2
4
6
X1/(1 - X1 )
8
D
K
=
D
0.2
=
0.4
D
K
=
K
0.5
=
KD
4
D
0.6
6
K
D
0.8
10
=5
D
K
=1
KD =
2
KD =5
8
X 1
K
X1 /(1 - X1 )
10
0.2
10
Рис.1.
.05
0
=
KD
0.2
0.4
0.6
0.8 X1
Подставим (239) в (254):
lnKD = (G0/RT)P+ln(21/21)
или
RTlnKD = G0 +RTln(21/21)
Согласно (198)
RTlni= Gie
И далее, RTlnKD=
=RTln2+RTln1–RTln1–RTln2+ G0
(258)
RTlnKD=
= (G1e)+(G2e)–(G2e)–(G1e)+G0=Ge+G0
KD=exp[(G0+Ge)/RT],
где
G0=G0(247) = H0(247) – TS0(247)
(255)
(256)
(257)
(259)
(260)
(261)
Эти же соотношения можно вывести из условия
равенства в равновесии парциальных молярных
свободных энергий Гиббса, т.е. химических
потенциалов каждого изоморфного компонента в
двух фазах (см. уравнения 220 и 221).
Домашнее задание
Продолжить вывод КD, начиная с (227) и (228)
при условии Ge=0.
Температурная зависимость KD
или
(lnKD/T )P = Ho(247) /RT2
{lnKD/(1/T)}P=Ho(247) /R
(262)
(263)
(G0/T)P = So(247)
(265)
Барическая зависимость KD:
(lnKD/P)T = Vo(247)/RT
(264)
Уравнения (262)-(265) описывают вариации KD
и G в случае идеального распределения, т.е.
:
e
когда G =0 и постоянных Р и Т величина
КD=const при любых составах равновесных фаз
(см. рис.1).
Пример идеального распределения.
В интервале 1000-1300 К для реакции
Fe2SiO4 + Mg2Si2O6 = Fe2Si2O6+Mg2SiO4
величина G 0.
(266)
По справочникам находим энергии образования
фаялита, форстерита, энстатита и ферросилита.
Вычисляем G0T реакции (266) при каждой данной
температуре и Р=1 бар:
G0=lnKD=(XFe/XMg)Opx/(XFe/XMg)Ol =
=G0Fa+G0En–G0Fo–G0Fs (267)
Далее строим график зависимости lnKD от 1/T (рис.2).
1400 1300 1200 1100
1000
o
900 K
lnKD
lnKD
0.50
-0.44
0.45
0.40
-0.42
1б
ар
-0.46
0.35
0.30
0.25
0.20
-0.40
-0.38
0.7 0.8
3
0.9
10 / T
1
1.1
11
00
10
00
13
00
Рис.2
0.15
0.10
0.05
0
10 20 30 40 50 60 70 80 90 100
P, кбар
В случае неидеального распределения
(lnKD /T)P =H/RT2 = Ho+He/RT2
(RTlnKD/P)T=V=Vo+Ve
(268)
(269)
lnKD = lnKDP + lnKDT
(270)
lnKD(T)= (H/RT2)PT
(271)
lnKD(P) = (V/RT)TP
(272)
и далее
lnKD =(H/RT2)PT+ (V/RT)TP =
– {[Ho+(He1–He2)T,P+(He2–He1)T,P]/RT2}PT+
{[Vo+(Ve2–Ve1)T,P+(Ve1–Ve2)T,P]/RT}TP
(273)
Аналогичным образом решается задача для обменного
равновесия двух пироксенов
CaMgSi2O6+Fe2Si2O6= Mg2Si2O6+CaFeSi2O6
Результаты расчётов показаны на диаграммах фазового
соответствия рис.3.
Cpx
Cpx
XMg
XMg
0.8
K
0.8
0.4
=1
KD
0.4
0.6
K
0.6
D
K
K
00
1 0 2 00 0 K K
0
1
14 60 0
1
1 0 80 6 00
00 0 K K
K
80
0K
=1
60
0
(a)
0.2
1.0
0.8
(б)
0.6
X
0.4
Opx
Mg
0.2
0
0.2
0.4
X
0.6
Ol
Mg
0.8
0.2
1.0
Рис.3.
В случае неидеального распределения
( ln KD/T)P = Ho+ He /RT2 = H/RT2
или
(RTlnKD/P)T = V = Vo+ Ve
lnKD = lnKDP + ln KDT
lnKDT = (H/RT2)PT
lnKDP = (V/RT)TP
и далее
(268)
(269)
(270)
(271)
(272)
lnKD=(H/RT2)PT+(V/RT)TP =
=–{[Ho+(He1–He2)T,P + (He2–He1)T,P] /RT2}PT+
+{[Vo+(Ve2–Ve1)T,P+(Ve1–Ve2)T,P]/RT}TP (273)
где
Ho=Ho1+Ho2–Ho2–Ho1
Vo = Vo2 + Vo1 –Vo1 –Vo2
(274)
(275)
С учетом уравнений (173) и (174) равенство
(274) можно переписать так:
lnKD={[(Ho + (He/X1)T,P +
(He/X2)T,P)/RT2]T}+{[(Vo +
+(Ve/X2)T,P+ (Ve/X1)T,P)/RT]P}
(276)
На диаграмме рис.4 приведены изотермы
неидеального распределения компонентов 1 и
2 между минералами и β при Р=const и t6 >
t5>…> t1.
Рис.4
Рассмотрим случай распределения компонентов в
многофазных системах с участием твердых растворов.
Пусть при t′ сосуществуют минералы α′+δ′+ε′ (см.
рис.5), а распределение компонентов между ними будет
идеальным. При повышении температуры от t′ до t′′
произойдет перераспределение компонентов между
этими минералами так, как это показано на рис.5 и 6.
Причём в фазовом треугольнике (рис.5) положение
точки валового состава системы не изменилось, а
количественные соотношения минералов остались
прежними. Следовательно, число степеней свободы n
равно
n=k+1-=3+1-3=1.
3
3
Рис.5
A
A
t
1
2
=1
0.2
t
0.2
0.8
D
K
0.4
X2
X2
0.4
X2
1
2
t
0.4
t
t
0.8
t
K
0.2
=1 0.4
D
0.4
X2
X2
Рис.6
t
t
K
0.2
=1
D
0.4
X2
ВЛИЯНИЕ СОСТАВА ФАЗ И ФАЗОВЫХ ЕРЕХОДОВ
НА ИЗОТЕРМИЧЕСКОЕ РАСПРЕДЕЛЕНИЕ
КОМПОНЕНТОВ. ЭКСТРЕМАЛЬНЫЕ
СОСТОЯНИЯ.
Если в равновесии находятся два раствора, в которых
наблюдается переменная концентрация трех
компонентов, то величина lnKD для компонентов 1 и 2
при прочих равных условиях (Р, Т, Х1 и Х1) зависит от
концентрации третьего компонента. Это положение
легко доказать из принципа химического равновесия:
парциальные молярные свободные энергии, или же
химические потенциалы всех компонентов системы при
постоянных Р и Т должны быть равны. Например, для
системы с трёхкомпонентными твердыми растворами
должно выполняться следующее условие:
G1 = G 1
G2 = G2
G3 = G 3
(277)
И далее:
(Go1)+RTlna1 = (Go1)+RTlna1
(Go2)+RTlna2 = (Go2)+RTlna2
(Go3)+RTlna3 = (Go3)+RTlna3
(278)
Подставляем вместо RTlnai его значение из формул (197) и (198)
и после преобразований находим:
RT ln(X1X2/X1X2)=(Go1) – (Go2)+(Go2) –
– (Go1) + (Go3) – (Go3) + (Ge1) – (Ge2) + (Ge2) –
– (Ge1) + (Ge3) – (Ge3) +RTln(X3/X3),
(279)
где
Х1+Х2+Х3=1
(280)
или же KD компонентов 1 и 2 зависит от соотношения
мольных долей компонента 3 в фазах и :
RTlnKD (239) =G0+Ge+RTln(X3/X3)
(281)
Из этого уравнения следует, что даже в случае Ge
= 0, распределение компонентов 1 и 2 между фазами
и не остается идеальным. Оно зависит (при Т=сonst)
от соотношения X3/X3. Но для идеальных растворов
X3/X3 =const и при Т=const lnKD (239) =const.
В случае фазового перехода в одном из равновесных
твердых растворов на графиках зависимости G=f(T) и lnKD=
f (1/T) должен появиться излом (рис.7, 8), поскольку резко
изменяется величина H обменной реакции.
2
+
lnKD
+
1
G обменной реакции
2
+
+ 1
T1
T
T1
1/T
Рис.7. Фазовый переход в фазе β на диаграммах ΔG-T и
lnKD-1/Т для обменного равновесия β1+2= β1+2
12
3
1.2
1.1
10
Число образцов
10 /T K
1.3
8
6
4
2
-15
0
+15
T1 -To
1.0
0.8
0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 2.6 2.8
lnKD (Crd-Grt)
Рис.8. Зависимость lnKD от обратной температуры
для обменного равновесия кордиерит - гранат
В ряде случаев изотермы (изобары) распределения на
диаграммах фазового соответствия могут пересекать
линию равного распределения изоморфных компонентов.
Это значит, что при данных Т и Р составы равновесных
фаз одинаковы, а точка равных составов соответствует
минимуму или максимуму одного из этих внешних
параметров системы. В таком случае говорят, что система
находится в экстремальном состоянии, а сама точка носит
название экстремальной (Рис. 9). Подход к этой точке от
конечных членов твердых растворов зависит от влияния
на нее Т или Р. Если в системе появляется Tmax,то ему
соответствовует Pmin. Отсюда и различная триангуляция на
диаграммах состав-парагенезис: на Рис. 9 вершины
треугольников составов направлены в противоположные
стороны.
Рис.9. Экстремумы Р-Т параметров в трехкомпонентной
трехфазной системе с двумя фазами переменного состава
1
T1=min
P1=max
X1
T2 =min
P2=max
T3=min
P3=max
2
X1
1
Рис.10. Смещение экстремальных точек на диаграмме
фазового соответствия при систематическом возрастании в
системе компонента-примеси (3).
МЕТОДЫ ВЫВОДА ДИАГРАММ
ФАЗОВОГО СООТВЕТСТВИЯ
1. Экспериментальные:
а) Прямое изучение равновесий двух фаз
переменного состава при разных Р и Т.
б) Изучение обменных равновесий типа “минерал водный раствор”.
Например:
Ab+(KCl)aq = (NaCl)aq+ San
Ne+(KCl)aq = (NaCl)aq+Ks
Ab+Ks = San+Ne
2. Расчетные, основанные на термодинамике
минералов: используются формулы (268) и (269)).
3. Эмпирические, основанные на анализе природных
минеральых парагенезисов (рис.11).
1
X
0.8
T2
0.6
0.4
T1
0.2
0.2
0.4
0.6
0.8
X
1
Рис.11. Эмпирический вывод диаграммы фазового
соответствия на основе известных геотермометров
ОБЩИЕ ЗАКОНОМЕРНОСТИ
РАСПРЕДЕЛЕНИЯ
КОМПОНЕНТОВ МЕЖДУ
СОСУЩЕСТВУЮЩИМИ
МИНЕРАЛАМИ
Прежде, чем приступить к решению этой проблемы
необходимо доказать, что в природных минеральных
ансамблях достигается химическое равновесие. Это
следует доказать двумя путями. Путь первый: доказать,
что при постоянстве Т и Р составы сосуществующих
фаз гомогенны. Иногда это удается (рис. 12). Но в
большинстве случаев они зональны. Обычно
зональность возникает потому, что скорость изменения
внешних параметров системы (Т, Р и др.) выше
скорости диффузии компонентов в кристаллических
решетках минералов. Согласно принципу локального
равновесия Д.С.Коржинского зональность в каждом из
сосуществующих минералов должна быть
закономерной.
Grt
A
Opx
Grt
300
0
мкм
Концентрация элементов, усл. ед.
A
40
Mg
30
Mg
Mg
20
Grt
Opx
10
Grt
Fe
Fe
Fe
Ca
0
0
Ca
Ca
100
200
300
400
Расстояние, мкм
Рис.12. Распределение Сa, Fe и Mg в сосуществующих Орх и Grt из
ксенолита в кимберлитовой трубке Сан Жуан, штат Юта, США
К доказательству этого положения можно придти
только после рассмотрения общих закономерностей
распределения компонентов между сосуществующими
минералами. Понять эти закономерности можно
исключительно путем анализа химических и
структурных свойств сосуществующих минералов.
§1. Сравнительная сила кислотных и основных
компонентов.
Способность элементов отдавать с внешней орбиты
электрон или принимать его определяет основные или
кислотные свойства данного элемента. Поэтому
относительная сила отрыва (или присоединения)
электрона служит мерой кислотности - основности
атомов. Она носит название электроотрицательности
(по Л.Полингу).
0= 0.399(ZЭФ/r2)+ 0.744 э.в.
r – ковалентный радиус, ZЭФ – эффективный заряд ядра
атома.
Величины элекктрооотрицательности некоторых
элементов (по К. Дею и Д. Селбину, 1971)
Элемент
F
O
№
9
8
о
4.1
3.5
Cl
Ni
Si
Fe
17
28
14
26
2.83
1.75
1.74
1.64
Mn
25
1.6
Элемент
Cr
Al
№
24
13
о
1.56
1.47
Ti
Mg
Ca
Na
84
12
58
11
1.44
1.23
1.08
1.01
K
19
0.91
№ - порядковый номер в периодической таблице Менделеева
(282)
Коэффициент ионизации
= amM+ /aaqMmOn
(283)
определяет сравнительную основность окислов при
их ионизации в водном растворе. Для основных
окислов справедлива такая реакция ионизации:
MmOn + 2nH+ = mM(2n/m)+ + nH2O
(284)
KP = (aH2O)n/(aH )+2n =exp(G(284)/RT )
(285)
и, так как aH2O=1,
KP =(aH+)–2n
(286)
В качестве стандартного состояния можно принять
твердый окисел (Sol). Тогда формула (283) обретет
вид:
= (aM+)m/(aMmOn)Sol = (aM+)m
(287)
=(aM+)m = KP(aH+)+2n
(288)
lg = mlgaM+= (G(284)/2.303RT) - 2n(pH)
(289)
Кислотные окислы ионизируют с образованием
анионов:
MmOn+ rH2O = m(MO(n+r):m)-2r + 2rH+
(290)
KP={[(aMO)(n+r):m]-2r).m(aH+)2r}/{aMmOn(aH2O)r} =
= exp(G(290)/RT)
(291)
KP =(aH+)2r=exp(G(290)/RT)
(292)
lg=mlg(aMO)-2r =(G(290)/2.303RT)+2r(pH)
(293)
или
откуда
Таблица 3. Коэффициенты ионизации некоторых
наиболее важных петрогенных соединений*
Окисел
25 оС
1000 оС
K2O
Na2O
35.2
26.5
7.4
3.8
CaO
MgO
FeO
Fe2O3
18.5
7.6
-0.3
-21.8
-6.7
-11.4
-14.0
-30.4
Al2O3
-6.7
-21.0
Окисел,
кислота
SiO2
CO2
HF
HCl
SO3
Al2O3
25 оС
1000 оС
14.3
13.2
9.1
4.4
21.5
27.3
46.4
2.8
4.7
10.2
12.4
9.8
*Перчук Л.Л. Зависимость изоэлектрических точек ионизированных амфотерных окислов от
температуры и их значение для геохимии // Геохимия. 1964. № 11.
§2. Правило Соболева
В тройной взаимной системе при малой величине
поляризации (Zэф) устойчива та пара солей, где
сочетается слабая кислота со слабым основанием и
сильная кислота с более сильным основанием. При
значительной величине поляризации аниона
соотношения могут измениться на обратные. К
числу таких анионов относятся S-2, F-, Cl- и др.
Из правила, в частности, следует, что окислы
всегда более железисты, чем сосуществующие с
ними силикаты. Например,
MgO + FeSiO3 FeO +MgSiO3
(295)
2MgFe2O4+Fe2SiO4 2Fe3O4+Mg2SiO4
(296)
§3 Правило Рамберга
Оно полностью соответствует правилу Соболева, но
распространяется только на кислородные соединения:
обменные равновесия смещены в сторону образования
соединений радикалов сильных кислот с сильными
основаниями плюс соединения радикалов слабых
кислот со слабыми основаниями
(Ме2)A + (Me1)B (Me2)B + (Me1)A
(297)
где
Me1 - сильное основание
Me2 - слабое основание
А
- сильная кислота
В
- слабая кислота
(см. таблицу 2)
Таблица 2. Логарифмы констант обменных реакций сульфатов и карбонатов
lnKD
Обменная реакция
298 oC
lnKD
1000 oC
Обменная реакция
298 oC
1000 oC
MgCO3 + Li2SO4 = Li2CO3 + MgSO4
-5,88
-1,60
FeCO3 + K2SO4= K2CO3+ FeSO4
-88,1
-26,7
MgCO3+ K2SO4 = K2CO3 + MgSO4
-18,01
-5,02
CaCO3+ Na2SO4= CaSO4 + Na2CO3
-5,56
-1,49
MgCO4+Na2SO4= Na2CO3+MgSO4
-12,87
-3,38
BaCO3+ Na2SO4=BaSO4 + Na2CO3
-0,9
-0,22
MgCO3 +CaSO4= CaCO3+MgSO4
-7,28
-1,8
MnCO3+ Na4SO4=MnSO4 + Na2CO3
-15,2
-4,05
MgCO3 + BaSO4= BaCO3+MgSO4
-11,95
-3,35
FeCO3+ Na2SO4= FeSO4+Na2CO3
-82,3
-24,15
MnCO3+ MgSO4= MgCO3+ MnSO4
-2,32
-0,68
BaSO4+ MnCO3= MnSO4 +ВаСO4
-14,3
-4,02
FeCO3 + MgSO4= MgCO3 +FeSO4
-67,6
-19,4
CaSO4+ MnCO3= MnSO4 +CaCO3
-9,63
-2,56
MnCO3 +Li2CO3 = Li2SO4+ MnSO4
-8,8
-2,8
CaSO4+ FeCO3= FeSO4 + CaCO3
-75,7
-22,6
FeCO3 + Li2SO4 = Li2CO3 + FeSO4
-75,5
-22,8
BaSO4 + FeCO3=FeSO4+ BaCO3
-76,5
-24,1
Na2CO3+ K2SO4 = K2CO3+ Na2SO4
-6,11
-2,62
MnSO4+FeCO3= FeSO4+ MnCO3
-66,8
-20,02
CaCO3+ K2SO4 = K2CO3+ CaSO4
-11,65
-4,11
Li2SO4+BaCO3=Li2CO3+ BaSO4
+0.79
+0. 01
BaCO3+ K2SO4= K2CO3 + BaSO4
-7,0
-2,64
Li2SO4+ Na2CO3=Li2CO3 + Na2SO4
+5.45
+1. 41
MnCO3+K,SO4= K2CO3 + MnSO4
-21,8
-6,66
Li2SO4+ K2CO3 =Li2CO3 + K2SO4
+6.35
+1.15
Большинство реакций в таблице 2 подчиняются правилу
Рамберга. Но есть и исключения. Например:
BaCO3 + Li2SO4 => Li2CO3 + BaSO4
(298)
lgKP,298 = 5.45
(299)
Na2CO3 + Li2SO4 => Li2CO3 + Na2SO4
(300)
lgKP,298 = 0.79
(301)
Хотя правило Рамберга относится только у
кислородным соединениям, он в ряде случаев применимо
и к другим солям, Так, оно полностью выполняется в
случае равновесий галогенидов Fe и Mg, но не
выполняется для галогенидов K и Na.
Рамберг считал, что сила электростатических связей в
таком случае важнее электроотрицательности. Реакции в
таких случаях смещены в сторону образования
соединений типа:
крупный катион+крупный анион и мелкий
катион+мелкий анион.
Например: KF + NaCl NaF + KCl
(302)
Значительная часть силикатных равновесий
подчиняется правилу Соболева - Рамберга.
Силикаты – соли поликремниевых кислот (H2SiO3,
H4SiO4, H2Si2O5 и т.д.). Мера сравнительной силы
этих кислот – эффективная электроотрицательность
кислорода. Для атомов О она тем выше (в связке
МеО), чем больше мостиковых связей SiOSi, так
как Si наименее электроположительный металл.
Ряд повышающейся кислотности
гипотетических кремнекислот по Рамбергу
выглядит так:
H4SiO4 <H6Si2O7<H2SiO3< H12+nAlnSi8-nО22<
H2+nAlnSi2-nO5< HnAlnSim-nO2m
Отсюда можно наметить кристаллохимический
ряд повышения силы кремнекислотного радикала:
Ортосиликат < диортосиликат <цепочечный
метасиликат<кольцевой метасиликат<
<ленточный метасиликат <слоистый
диметасиликат< каркасный силикат
Один из факторов, снижающих эффективную
электроотрицательность атомов кислорода вхождение Al в тетраэдр. Но об этом чуть позже.
Хорошо известные примеры выполнения правила
Рамберга для равновесий силикатов - ряды
сравнительной “калиевости”, “магнезиальности” и т.п.
Так, например, магнезиальность нарастает в ряду:
глиноземистые породы:
гранат <ортопироксtн < биотит < хлорит <кордиерит
Са-содержащие породы:
гранат < ортопироксен (или клинопироксен) <
амфибол < хлорит
§4. Общий принцип фазового
соответствия
Он определяет закономерности влияния
температуры и давления на характер
перераспределения изоморфных компонентов
между двумя и более сосуществующими
минералами.
Прежде, чем перейти к формулировке основных
правил и самого принципа, рассмотрим
некоторые диаграммы распределения элементов
между природными минералами (рис. 13 и 14).
Mg/(Mg+Fe+Mn) в Cpx
1.0
0.8
=1
D
K
0.6
0.4
0.2
0
0.4
0.5
0.6
0.7
0.8
0.9
Mg/(Mg+Fe+Mn) в Bt
Рис.13. Распределение Mg и Fe между биотитом и
клинопироксеном из вулканитов ( ), нефелиновых сиенитов и
фенитов ( ) и карбонатитов ( )
Grt
X Mg
Op
x
X Mg
0.8
0.8
0.6
0.6
0.4
0.4
0.2
0.2
0.0
0.2
0.4
0.6
0.8
Grt
XMg
Bt
X Mg
0.0
0.4
0.6
l
0.8X Hb
0.2
0.4
0.6
0.8
Mg
Cp
x
XMg
0.8
0.8
0.6
0.6
0.4
0.4
0.2
0.2
0.0
0.2
0.2
0.4
0.6
0.8
Hb
l 0.0
XM
g
Hb
l
XM
g
Рис.14. Распределение Mg и Fe между некоторыми минералами из
комплексов различных метаморфических фаций
Из анализа этих и подобных диаграмм для равновесных
природных минералов вытекает следующее
эмпирическое правило:
c возрастанием температуры Mg
перераспределяется из водосодержащих
минералов в безводные, а Fe из безводных в
водосодержащие.
Это означает, что обменные равновесия типа
AMg·nH2O + BFe=BMg + AFe·nH2O
(303)
с возрастанием температуры смещаются, либо имеют
тенденцию к смещению вправо. Объясним это явление.
Сравним эффективную электроотрицательность
кислорода в водосодержащих и в безводных силикатах
с одинаковым кремнекислородным радикалом.
В соответствии с правилом Соболева - Рамберга такие
силикаты, как St и Grt, Cld и Grt, Cld и St, Spr и Ol, Grt
и Ol должны иметь одинаковую магнезиальность. Но
это не так. В умеренном интервале Т железистость
[Fe/(Fe+Mg)] граната выше, чем у сосуществующих с
ним силикатов. Изменение прочности химической связи
Si–O (от одного радикала к другому), приводит к
изменению прочности связи Ме–О. Это значит, что чем
сильнее связь Ме–О, тем выше эффективная
электроотрицательность кислорода и тем более прочная
ионная связь в звене Ме–О–Si. С переходом от
ортосиликатов к каркасным силикатам тип связи
меняется по схеме:
2(Ме–О–Si–O) (Si–O–Si–O) + 2(Me–O)
Металл попадает во все более тесное окружение
атомами кислорода с усилением степени ионности
связи за счет увеличения эффективной электроотрицательности кислорода. И чем она выше, тем
более электроположительный металл стремиться
войти в это окружение. Возрастает упорядоченность
структуры в последовательности ортосиликаты=>
диметасиликаты (слоистые и ленточные)=>
каркасные силикаты. Действительно, в
ортосиликатах (например, оливин и гранат)
неизвестен распад твердых растворов. Появляется он в
метасиликатах (например, амфиболы и пироксены), и
уже четко проявлен в слоистых и каркасных силикатах
(например, полевые шпаты, нефелин).
Итак, еще одно правило:
Увеличение концентрации мостиковых кислородов,
т.е. связок Si–O–Si, приводит к “изолированности”
тетраэдров в структуре силикатов, снижению их
атомной энтропии.
В водосодержащих силикатах появляются
“бруситоподобные”, “корундоподобные” и т.п. слои, что,
несомненно, свидетельствует об увеличении
эффективной электроотрицательности кислорода в
тетраэдре и степени ионности связи (Ме–О) и снижению
ее в связке (Si–O).Отсюда следует, что в октаэдрических
позициях водосодержащих силикатов степень ионности
связи всегда выше, чем у безводных силикатов того же
кристаллохимического класса.
Сравните St или Cld c Ol и/или Grt, - все они ортосиликаты. В
метасиликатах тетраэдры образуют цепи благодаря связкам
типа Si–O–Si – по две связки из четырех на каждый тетраэдр, у
турмалина хорошо выражены “бруситоый” и «тальковый» слои.
“ Тальковый” слой
Y6(Si,Al)6O20(OH) 4 {
“Бруситовый“
cлой Y6(OH)6
{
атомы кислорода
c sin
{
{
{
сетка
тетраэдро в
гидроксил
гидроксил+кислород
ионы группы Y
(Si, Al)
слой
октаэдров
сетка
тетраэдро в
Ось Х
а
Рис. 15. Проекция структуры хлорита на плоскость (010) (McMurchi, 1934)
Правило:
Водосодержащие силикаты обладают более высокой
эффективной электроотрицательностью кислорода
по сравнению с безводными.
Повысим температуру. Связи ослабятся за счет
разупорядочения структур силикатов, т.е. за счет
возрастания энтропии. Снижается относительное
число связок Si–О–Si , уменьшается эффективная
электроотрицательность кислорода, т.е. свойства
силикатов как бы нивелируются.
При этом нивелируется и различие в кислотно-основных
свойствах компонентов, что приводит к ослаблению
прочности связей типа “сильное основание + сильная
кислота” и усиливается прочность связей полярного
типа (“сильный + слабый”). Т.е. реакция (297) с ростом
температуры смещается влево.
Если два изоморфных металла отличаются по
величине электроотрицательности, в равновесиях их
силикатов прочность связей более
электроположительного металла с гипотетическими
поликремнекислотами с ростом температуры
увеличивается (а менее электроположительного
уменьшается) в следующей последовательности:
каркасный силикат => слоистый диметасиликат =>
ленточный метасиликат => цепочечный метасиликат
=>диортосиликат.
Эта последовательность строго противоположна
правилу Рамберга.
Можно предположить, что относительное
изменение прочности связей в сосуществующих
силикатах при изменении температуры на некоторую
величину ΔT = Т2-Т1 будет тем больше, чем дальше
отстоят в выведенном выше ряду силикаты.
Следовательно, и большей должна быть величина
энтропии ΔS обменных реакций. Это можно показать
на примерах, однако прежде рассмотрим возможность
систематизации силикатов на основе трех признаков:
1) водный или безводный (химический признак)
2) кремнекислотный радикал (кристаллохимический
признак)
3) сингония (категория симметрии).
В природных силикатах и алюмосиликатах наиболее
развиты два типа изовалентного изоморфизма: Mg<=>Fe
и Na<=>К. Распределим эти минералы по
перечисленным выше трем признакам.
В табл.4, составленной для Fe-Mg-силикатов, минералы
размещены в соответствующих клетках в зависимости
от (1) принадлежности их к тому или иному
кристаллохимическому подклассу, (2) от содержания в
них воды и (3) от категории их симметрии. По первому
признаку они разбиты на две большие группы – водные
и безводные минералы.
Табл.4. Фазовое соответствие железо-магнезиальных минералов
Таблица 5. Систематика K-Na силикатов
В пределах каждой группы они систематизированы по
кристаллохимическим типам (табл. 4 и 5) – от каркасных
силикатов до ортосиликатов (группа безводных минералов
представлена ортосиликатами и цепочечными
метасиликатами). Эффективная электроотрицательность
кислорода уменьшается от первой графы до последней.
Вправо таблица раскрывается в сторону возрастания
категорий симметрии минералов. В зависимости от этих
показателей каждый породообразующий силикат помещен
в соответствующую клетку табл. 4 и 5 и ему присвоен
условный порядковый номер. Его полиморфным и
политипным разновидностям дан тот же номер, но с
соответствующим числом штрихов. Если два минерала
принадлежат к одной сингонии и к одному подклассу,
последовательность их расположения в таблице
определяется по категориям симметрии их полиморфных
разновидностей.
Например, в табл. 4 Chl и Bt – оба водные слоистые
диметасиликаты моноклинной сингонии. Но у Chl имеется
триклинная полиморфная разновидность, тогда как
полиморфы (точнее политипы) Bt более симметричные,
вплоть до гексагональных. То же касается Cld и St –
водных ортосиликатов.
Пустые клетки в таблицах могут заполняться как
новыми минералами, так и полиморфными разновидностями соседних по таблицам силикатов. В соответствии с
расположением минералов закономерно изменение их
физических свойств. Так, плотность Fe-Mg минералов
(табл.4) изменяется не только от низших сингоний к
высшим, но и в пределах каждой сингонии в соответствии
с возрастанием номера силиката в каждой колонке.
Например, плотность моноклинных силикатов (в г/cм3)
во второй колонке изменяется так: Srp – 2.5-2.6, Bt – 2.73.0; Na-Am – 3.04-3.5; Сpх – 3.21-3.56; Spr – 3.4-3.6.
Изменение плотности ромбических силикатов (исключая
полиморфные разновидности моноклинных фаз) также
вполне закономерное: Сrd – 2.52-2.78; Hbl – 2.98-3.6; Орх
– 3.21-3.96; 0l – 3.2-4.38.
Из рассмотренных закономерностей перераспределения
Fe и Mg между силикатами на основе табл. 4 следует
общий вывод: чем дальше отстоят в ней минералы друг
от друга, т. е. чем больше разность порядковых номеров,
тем больше эффект перераспределения при изменении
температуры.
Разность плотностей миналов
1.4
, см 3
Grt+Crd
1.2
1.0
Grt+Chl
Grt+Bt
0.8
Grt+Cpx
0.6
0.4
0.2
0
Grt+Hbl
Grt+Opx
Grt+Cld
Ol+Opx
Grt+St
Opx+Cpx
2
4
6
8
10
12
14
Разность номеров в табл.4
Рис.16. Корреляция Δρ с разностью условных номеров в табл.4
А на рис.17 показана корреляция разности номеров
минералов в табл.4 с разностью молярных объемов Feи Mg-миналов участников обменных реакций.
НОМЕР МИНЕРАЛА В ТАБЛИ ЦЕ 4.
18
16
298 К
14
12
безводные
силикаты
10
8
водные силикаты
6
4
2
0
0
0.10
0.20
0.30
V(Fe) - V(Mg), дж/бар
Рис.17. Корреляция
номера минерала в табл. 4
с разностью мольных
объемов некоторых
миналов водосодержащих
и безводных Fe и Mg
силикатов и алюмосиликатов
Разность номеров в табл. 4
20
18
Grt+Crd
16
14
Grt+Hbl
}
12
Grt+Chl
Grt+Bt
10
8
6
4
Grt+Cld
Grt+St
Grt+Cpx
Ol+Opx
2
Ol+Cr
0
Opx+Cpx
2
4
6
8
10
12
14
S , э. е.
Рис.18. Корреляция усредненной величины энтропии обменной
реакции (ΔS, кал/моль) с разностью условных номеров в табл.4
Из рассмотрения таблицы 4 для Fe-Mg
силикатов и алюмосиликатов следуют три
четких правила:
Правило 1. Если данный Fe-Mg минерал имеет
более симметричные полиморфы, то с ростом T
в них перераспределяется Mg – более
электроположительный металл, чем Fe; если
же данный минерал равновесен с любым из
своих полиморфов более низких сингоний, то
с ростом Т он обогащается Mg – более
электроположительным металлом – за счет
своих полиморфов. Это правило находит
замечательное экспериментальное
подтверждение на примере группы оливина.
Оливин – ромбический ортосиликат, а его
полиморфная разновидность рингвудит –
кубическая (см.табл.4).
.
В табл. 4 оливин и рингвудит располагаются под
номерами 17 и 17‘, соответственно. Из правила
следует, что с ростом температуры Mg должен
перераспределяться из оливина в рингвудит,
причем энтропийный эффект обменной реакции:
OlMg+RinFe=RinMg+OlFe
должен быть незначительный. И это действительно так: согласно экспериментальным данным
(Akimoto S. & Fujisawa H. Olivine – ringwoodite
solid solution. Journal of Geophysical research.
1968. V.73. P.467-480) влияние температуры на
эту реакцию очень слабое (см. рис. 19).
Mg2SiO4 в Rin
0.8
0.6
0.4
0.2
0
0.2
0.4
0.6
Mg2SiO4 в Ol
0.8
1.0
Рис.19. Распределение Mg и Fe между оливином и
рингвудитом при 800о и 1200 оС
Правило 2. С ростом температуры Mg как
наиболее электроположительный металл
стремится вытеснить Fe из силикатов высших
категорий симметрии в силикаты низших
симметрий.
Правило 3. С ростом температуры Mg
перераспределяется из водосодержащих
силикатов в безводные - это правило мы уже
формулировали.
Все три правила достаточно универсальны и
могут быть распространены на обменные
равновесия любых алюмосиликатов и силикатов
с изовалентным изоморфизмом.
Теперь можно сформулировать общий принцип фазового
соответствия:
Если два изоморфных катиона различаются по
величине электроотрицательности, то при прочих
равных условиях степень перераспределения их
между сосуществующими силикатами и/или
алюмосиликатами в зависимости от температуры
тем выше, чем больше разница в силе
кремнекислотных радикалов и в категориях
симметрии этих минералов; наиболее сильные
эффекты перераспределения достигаются в
равновесиях водных и безводных минералов.
“Исключения” из этого принципа:
1. Резкие отклонения твердых растворов
равновесных минералов от идеальности
(таковы обменные равновесия Ne – Kfs, Ol –
Opx, может быть, Opx – Ged).
2. Влияние концентрации Al в тетраэдрической
координации на эффективную
электроотрицательность кислорода (ЭЭО).
Рассмотрим второй случай. Это поможет нам
убедиться в фундаментальности принципа
фазового соответствия (потому он и назван
«общим»)
В ряду ортосиликаткаркасный силикат нарастает ЭЭК
и при этом облегчается возможность вхождения Al в
тетраэдр, в связку Si–O–Si, т.к. ХAl =1.47 э.в., XSi = 1.74э.в.
Это снижает ЭЭК в связках Ме–О (например, Mg–O–Si, Ca–
O–Si и т.п.). В результате нарастает “концентрация” связей
слабых катионов с кислородом. Такие равновесия смещены
в сторону образования пар соединений “сильных” металлов
со “слабыми” кислотными радикалами и соединений
“слабых” (менее основных) металлов с “сильными”
кислотными радикалами. Температура стремится сместить
такие равновесия в противоположную сторону. Таково,
например, распределение Mg и Fe между глинозёмистым
хлоритом и амфиболом (рис.20).
100.Mg/(Mg+Fe) в хлорите, мольный %
60
I
40
II
20
0
20
40
60
80
100
100.Mg/(Mg+Fe) в амфиболе, мольный %
Рис.20. Соотношение магнезиальностей хлорита и
амфибола в зеленых сланцах (I) и хлорит-эпидотовых
амфиболитах (II) (Добрецов и др., 1970, стр. 416)
ДИАГРАММЫ ФАЗОВОГО СООТВЕТСТВИЯ
Термометр Барта.
В 1951 году норвежский ученый Барт предложил
использовать в качестве геотермометра
температурную зависимость коэффициента
разделения альбитового компонента между
сосуществующими полевыми шпатами. Этот
коэффициент легко получить из равенства
химических потенциалов альбитового
компонента в плагиоклазе (Pl) и калишпате (Kfs):
GAb(Pl) = GAb(Kfs)
И далее
(304)
m
m
GoAb(Pl) -GoAb(Kfs) +GAb(Pl)
-GAb(Kfs)
=0
Так как
GoAb(Pl) = GoAb(Kfs)
(305)
(306)
то можно приравнять
Pl
Kfs
aAb
= aAb
(307)
и тогда
Pl
Kfs
RTln(X Ab
/X Ab
)=GeAb(Kfs) - GeAb(Pl) =ΔGe
æD G e ÷
ö
÷
K Ab = exp çç
÷
çè RT ø
÷
P
(308)
(309)
Из уравнения (309) видно, что в случае идеальности обоих
полевых шпатов распределение Аb между ними было бы
одинаковым, т.е. K¯Ab = 1. В действительности же Pl всегда
богаче альбитом, чем равновесный Kfs.
Экспериментальные данные показывают, что
распределение Ab между этими фазами лучше
описывается выражением
KDAb = [XAb/(1 – XAb)]Pl·[(1 – XAb)/XAb]Kfs
(310)
чем величиной K Ab . Таким образом,
распределение Аb между полевыми шпатами
полностью определяется величинами их
избыточных функций.
Сейчас имеется множество экспериментальных
и теоретических данных для вывода диаграмм
фазового соответствия. Расчетные варианты таких
диаграмм приведены на рис.21.
0.7
KAb
5 50
0.8
0.6
0.7
0.6
0.5
0.5
0.4
0.4
Kfs(low)+Pl
Kfs( high)+Pl
0
0.2
0.4
0.6
XAb в Kfs
=1
C
o
50
0
40 0o
45 0o
3 00 o
=1
o
6 00 o
Xab в Pl
0.8
0.9
XAb в Pl
0.9
1.0
b
500 C 6 0 0 C
OC
0
0
7
o
C
0
0
8o C
00
9
1 00 0o
o
o
KA
1.0
0.8
1.0
0
0.2
0.4
0.6
0.8 1.0
XAb в Kfs
Рис.21. Распределение альбитового компонента между
равновесными Pl и Kfs разной степени упорядочения
НЕФЕЛИН-ПОЛЕВОШПАТОВЫЙ ТЕРМОМЕТР
Обменное равновесие Ness с Kfs описывается
гипотетической обменной реакцией:
NaAlSiO4+ KAlSi3O8=NaAlSi3O8+ KAlSiO4
(311)
Отсюда находим выражение для lnKD:
RTlnKD = RTln[(XNaKfs(1–X NaNe))/((1–XNaKfs)XNaNe)] =
= Go+Ge
(312)
где
Go = GoNe +GoKAlSi3O8 – GoNaAlSi3O8– GoKs
(313)
Ge = GeNe +GeKAlSi3O8 – GeNaAlSi3O8 – GeKs
(314)
Теоретически величины Gо и Ge зависят от степени
упорядочения Ness и Kfs. Но практически – очень слабо.
Экспериментальное изучение этого равновесия проводилось
методом раздельного изучения таких обменных реакций:
Ne + [KCl]aq = [NaCl]aq +Ks
Abhigh + [KCl]aq = [NaCl]aq + San
Ablow + [KCl]aq = [NaCl]aq + Mic,
(315)
(316)
(317)
где [KCl]aq и [NaCl]aq – водные (aq) растворы (при t < 800оC)
хлоридов щелочей. Для этих равновесий справедливо
выражение коэффициента распределения в виде
Min
X aq
X
K
Na
=K D
aq
Min
X Na X K
Результаты экспериментального изучения обменных
реакций (315)-(317) с участием как водных растворов, так
и хлоридных расплавов, приведены на рис. 22 и 23,
соответственно.
Ne ss +Ks ss +(K,Na) Claq
0.6
0.4
K, N
a )Cl aq
0.8
Ksss +
(
o
T = 70 0 C
P = 1000 бар
Nes +
(K, Na a q
)Cl
( )
KC l:(KCl+NaCl) в водном растворе aq
1.0
aq
l
Kfs +(K,Na)C
high
0.2
0.0
0.0
0.2
0.4
0.6
0.8
K:(K+Na) в минерале
1.0
Рис.22. Изотермы распределения K и Na в равновесии
твердых растворов нефелина и разупорядоченного
калишпата с водным раствором хлоридов щелочей
1.0
NeSS+KsSS+(K,Na)Cl
( K,N
a )C
l
me
lt
0.6
0.4
me lt
l
, Na)C
K
(
+
K fs
lo w
0.2
0.0
m elt
melt
NeSS+KsSS+(K,Na)Cl
0.6
Ne +
(K, Na m el t
)C l
KCl:(KCl+NaCl) в расплаве
melt
o
T=900 C
P=6 бар
0.8
KsS
S+
0.8
(K,N m
a)C e lt
l
T = 900 C
P = 6 бар
KSs
S +(K
, Na
) Cl
o
Nes+s
KCl :(KCl+ NaCl ) в расплаве
1.0
0.4
hi gh
Kfs
l
+ (K, Na)C
melt
0.2
A
0.0
0.2
0.4
0.6
0.8
K:(K+Na) в минерале
B
1.0
0.0
0.0
0.2
0.4
0.6
0.8
1.0
K:(K+Na) в минерале
Рис.23. Изотермы распределения K и Na в равновесии
твердых растворов нефелина и Kfs разной степени
упорядочения c расплавом KCl-NaCl
В отличие от Kfs, нефелин содержит избыточный
нормативный кварц в форме NaAlSi3O8. Причем его
растворимость в твердом растворе нефелина есть
функция температуры: чем выше Т, тем больше альбита
растворяется в натровой части Ness. Это приводит к
смещению изотерм распределения щелочей между Ness и
сосуществующими фазами в сторону нефелинового
минала. Это видно на рис. 25 – 27: повышение Qtz в
твердом растворе нефелина приводит к смещению
изотерм в натровую область Ness. Этот очень важный
эффект необходимо учитывать при термометрии
нефелин-полевошпатовых интрузивных и вулканических
пород. В противном случае цифры температур
равновесий в фонолитах окажутся сильно заниженными.