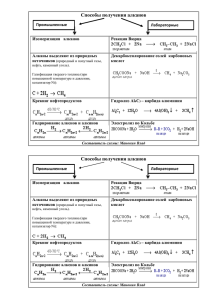
Лабораторный практикум по химии и технологии органических
advertisement

МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования «НАЦИОНАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ ТОМСКИЙ ПОЛИТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ» ________________________________________________________________ Т. Н. Волгина, А. И. Татаркина ЛАБОРАТОРНЫЙ ПРАКТИКУМ ПО ХИМИИ И ТЕХНОЛОГИИ ОРГАНИЧЕСКИХ ВЕЩЕСТВ Рекомендовано в качестве учебного пособия Редакционно-издательским советом Томского политехнического университета Издательство Томского политехнического университета 2014 УДК 547+666.7(075.8) ББК 24.23+30.50/78я73 В67 В67 Волгина Т. Н., Татаркина А. И. Лабораторный практикум по химии и технологии органических веществ / Т. Н. Волгина; Томский политехнический университет. – Томск: Изд-во Томского политехнического университета, 2014. – 71 с. В авторской редакции Учебное пособие представляет собой руководство для выполнения лабораторных работ по синтезу ряда органических веществ, выпускаемых промышленостью основного органического синтеза. Приведенные методики лабораторных работ сгруппированы по типу химических процессов, изложенных в курсе химия и технология органических веществ. Подготовлено на кафедре технологии органических веществ и полимерных материалов и предназначено для студентов ИДО, обучающихся по направлению 240100 «Химическая технология». УДК 547+666.7(075.8) ББК 24.23+30.50/78я73 Рецензенты Кандидат технических наук, ведущий инженер отдела главного технолога ЗАО Сибкабель Л. Н. Бочкарева Кандидат химических наук, доцент ТПУ О. С. Кукурина © ФГБОУ ВПО НИ ТПУ, 2014 © Волгина Т. Н., 2014 © Обложка. Издательство Томского политехнического университета, 2014 2 ОГЛАВЛЕНИЕ ПРЕДИСЛОВИЕ................................................................................................................. 4 1. ОБОРУДОВАНИЕ ДЛЯ ПРОВЕДЕНИЯ ЛАБОРАТОРНЫХ РАБОТ..................... 7 2. ИСХОДНЫЕ ВЕЩЕСТВА ДЛЯ ОСНОВНОГО ОРГАНИЧЕСКОГО СИНТЕЗА 11 2.1Очистка фракций жидких продуктов пиролиза ................................................... 11 3. ПРОЦЕССЫ ОКИСЛЕНИЯ ....................................................................................... 15 3.1. Получение синтетических жирных кислот ........................................................ 16 3.2. Получение адипиновой кислоты окислением циклогексанола ..... 19 3.3. Эпоксидирование нефтеполимерных смол ........................................................ 21 4. ПРОЦЕССЫ ХЛОРИРОВАНИЯ ................................................................................ 24 4.1. Хлорирование непредельных соединений ......................................................... 25 5. ПРОЦЕССЫ ГИДРОЛИЗА ......................................................................................... 29 5.1. Получение глицерина ........................................................................................... 29 6. ПРОЦЕССЫ ДЕГИДРАТАЦИИ ................................................................................. 32 6.1. Получение дибутилового эфира .......................................................................... 33 6.2. Получение пропилена .......................................................................................... 34 7. ПРОЦЕССЫ ЭТЕРИФИКАЦИИ ................................................................................ 36 7.1. Получение этилацетата ........................................................................................ 37 7.2. Получение бутилацетата ...................................................................................... 39 7.3. Получение дибутилфталата ................................................................................. 40 7.4. Получение лактида ............................................................................................... 42 8. ПРОЦЕССЫ ПРИСОЕДИНЕНИЯ И КОНДЕНСАЦИИ ПО КАРБОНИЛЬНОЙ ГРУППЕ ............................................................................................................................ 46 8.1. Получение дифенилолпропана ............................................................................ 47 8.2. Получение пентаэритрита .................................................................................... 49 8.3. Получение окиси мезитила .................................................................................. 51 8.4. Получение 4-аминодифениламина ..................................................................... 53 8.5. Получение капролактама ..................................................................................... 56 9. ПРОЦЕССЫ СУЛЬФИРОВАНИЯ И СУЛЬФАТИРОВАНИЯ ............................... 59 9.1. Получение n-толуолсульфокислоты ................................................................... 59 10. ПРОЦЕССЫ ДЕГИДРИРОВАНИЯ.......................................................................... 62 10.1. Получение α-метилстирола дегидрированием изопропилбензола ................ 63 11. ПРОЦЕССЫ АЛКИЛИРОВАНИЯ ........................................................................... 65 11.1. Получение изопропилбензола ........................................................................... 66 ЛИТЕРАТУРА .................................................................................................................. 69 ПРИЛОЖЕНИЕ 1. Образец титульного листа.......................................................... 70 3 ПРЕДИСЛОВИЕ Промышленность основного органического синтеза базируется на получение органических соединения, большие масштабы производства которых определяют широкое распространение высокоэффективных технологических процессов, характеризующихся непрерывностью, высоким уровнем автоматизации и высокопродуктивным оборудованием. Синтетические работы, выполненные в лаборатории, являются важным обучающим моментом в подготовке технических кадров, способных удовлетворять высоким запросам предприятий химической отрасли. Лабораторный практикум предназначается в качестве руководства студентам старших курсов для выполнения практических работ по дисциплине «Химия и технология органических веществ». Целью лабораторных занятий является освоение студентами лекционного материала и изучение классических технологических процессов получения продуктов основного органического синтеза путем выполнения экспериментальных работ на лабораторных установках. Изучение специальной литературы и другой научно-технической документации, а также достижений отечественной и зарубежной науки и техники позволит студентам расширить кругозор в области производства крупнотоннажных продуктов нефтехимии, приобрести навыки обработки полученных экспериментальных данных, и выработать определенные умения анализа и разработки технологических схем. Данное пособие составлено на основе лабораторного практикума по химии и технологии органических веществ (авторов Сухорословой М. М., Новикова В. Т. и Бондалетова В. Г.), изданного в Томском политехническом университете в 2002 году и лабораторного практикума по химии и технологии основного органического и нефтехимического синтеза (автора Одабашяна Г. В.), вышедшего в издательстве «Химия» в 1982 году. Так же в ряде разделов пособия описаны методики синтеза органических продуктов, разработанные при выполнении научноисследовательских работ на кафедре технологии органических веществ и полимерных материалов Томского политехнического университета. В учебном пособии изложены методические указания по проведению лабораторных работ, классифицированных по типу химических процессов и охватывающих все разделы теоретического курса «Химия и технология органических веществ». Каждая глава начинается с краткого теоретического введения, посвященного свойствам, методам получения и областям применения получаемого продукта, а также физико4 химическим основам и технологическим особенностям процесса. Во всех методиках, помимо указания цели и описания хода работы, приводится схема-эскиз установки. Проведение конкретной лабораторной работы возможно только после изучения теоретической части, хода работы, методов анализа, сбора элементов и монтажа экспериментальной установки. Отчет по выполненной лабораторной работе оформляется один на группу студентов в соответствии с требованиями СТО ТПУ 2.5.01-2011 по следующему плану: Цель работы Введение 1. Теоретическая часть 1.1. Методы получения продукта 1.2. Области применения продукта 1.3. Физико-химические свойства исходных реагентов 2. Экспериментальная часть 2.1. Схема установки 2.2. Ход работы 2.3. Физико-химические методы анализа 3. Результаты и их обсуждение 3.1. Химия процесса 3.2. Экспериментальные данные 3.3. Материальный баланс 3.4. Блок-схема процесса Выводы Список литературы Во введении должны быть кратко описаны общие сведения о получаемом продукте. В теоретической части приводится описание существующих промышленных методов синтеза получаемого продукта и области его применения, а также дается краткое обоснование физико-химических характеристик исходного сырья и продукта. В экспериментальной части обязательно должны быть вычерчены схемы лабораторных установок с указанием названия элементов из которых они состоят. В данном разделе также указываются, проводимые физикохимические методы анализа и дается подробное их описание и методики очистки и анализа исходных и вспомогательных веществ (в случае они производились). В экспериментальной части представляются полу5 ченные результаты эксперимента с необходимыми разъяснениями. При серии параллельных опытов цифровой материал лучше отображать в виде таблиц или графиков. В разделе результаты и их обсуждение должны быть представлены уравнения основных и побочных реакций с указанием условий их протекания. А также приведены конечные результаты по всем параллельным опытам, полученные физико-химические характеристики и выход от теоретического полученного продукта и даны выводы о факторах, влияющих на выход и чистоту полученного продукта. На основании полученных результатов и проведенного анализа составляется материальный баланс процесса и приводится блок-схема промышленного процесса получения данного продукта с описанием. В выводах следует кратко показать достигнутые в ходе проделанной работы результаты. В конце отчета приводится список литературы, который был использован при подготовке к выполнению и зищите лабораторной работы. Отчет, содержащий вышеперечисленные разделы должен быть оформлен и распечатан на бумаге формата А4. Титульный лист оформлен согласно приложению 1. Авторы выражают благодарность доценту Бочкареву В. В. за участие в разработке и редактирование учебного пособия. 6 1. ОБОРУДОВАНИЕ ДЛЯ ПРОВЕДЕНИЯ ЛАБОРАТОРНЫХ РАБОТ Дистиллятор применяется для производства дистиллированной воды и используется в различных лабораториях, медицинских учреждениях и других организациях. Дистиллятор производит дистиллированную, очищенную воду в соответствии со статьей ФС 42-2619-97 Госфармакопеи XI издания. Данный дистиллятор может производить как холодную, так и горячую (+80°С) очищенную воду, что особенно необходимо для приготовления высоконасыщенных растворов. Колбонагреватель применяется для нагрева жидкостей и твердых веществ, проведения синтеза и перегонки, контроля фракционного состава, определения содержания воды по действующим стандартам и других задач, предусматривающих нагревание при температурах до +600°С. Аналитические весы являются многофункциональными электронными весами, позволяющие решать любые задачи взвешивания в различных областях науки и техники. Весы оснащены микропроцессором. Принцип действия основан на электромагнитной компенсации. Дверцы ветрозащитной витрины открываются плавно и максимально широко, что удобно для осуществления загрузки и выгрузки взвешиваемого образца. Данные весы, также как и другие аналитические точные весы, устанавливают в лабораториях только на специальных столах (столах для весов). Механическая мешалка предназначена для перемешивания средне- и высоковязких сред, гомогенизации, приготовления взвесей и др. Скорость мешалки регулируется в широком диапазоне от 40 до 200 об/мин. Магнитная мешалка идеально подходит для перемешивания сред с низкой и средней вязкостью. Перемешивающее устройство совмещает в себе две функции в одном приборе: нагрев до 500 оС и перемешивание жидкостей. 7 Прибор для определения температуры плавления обеспечивает быстрое и точное определение точек плавления и кипения в ручном режиме в температурном диапазоне от комнатной температуры до 400 °С. Рефрактометр – прибор, предназначенный для определения показателя преломления жидкостей с высокой или низкой вязкостью. Отделение для призмы рефрактометра имеет шланговый разъем для термостатирования. Для измерения температуры применяется цифровой термометр. Измерения проводятся как в передаваемом, так и в полностью отраженном свете. С контактной жидкостью данным прибором можно измерять твердые вещества. ИК фурье-спектрометр предназначен для регистрации в ближней и средней ИК области спектров поглощения твердых, жидких и газообразных веществ (в том числе наркотиков, лаков и красок, нефтепродуктов, взрывчатых веществ, фармакологических препаратов) с их последующей идентификацией, а также для качественного и количественного анализа смесей, содержащих несколько компонентов. Хроматограф – прибор, предназначенный для анализа состава, структуры и качества жидких и газовых проб в органических и неорганических соединениях. С помощью газовой хроматографии можно проанализировать вещества с молекулярной массой меньше 400, удовлетворяющие определенным требованиям, главные из которых – летучесть, термостабильность, инертность и легкость получения. Количественный анализ можно провести только в том случае, если вещество термостойко, т. е. испаряется в дозаторе воспроизводимо и элюируется из колонки без разложения. Вещество не должно образовывать устойчивых сольватов при растворении в неподвижной жидкой фазе и реагировать с материалами, из которых изготовлены детали хроматографа. 8 Спектрофотометр – прибор, предназначенный для измерения отношений двух потоков оптического излучения, один из которых – поток, падающий на исследуемый образец, другой – поток, испытавший то или иное взаимодействие с образцом. Позволяет производить измерения для различных длин волн оптического излучения, соответственно в результате измерений получается спектр отношений потоков. Обычно используется для измерения спектров пропускания или спектров отражения излучения. Роторный испаритель (ротационный испаритель) – это устройство для быстрого удаления жидкостей отгонкой их при пониженном давлении. Роторные испарители оснащены отдельной кнопкой включения нагревательной бани, выносной панелью управления для безопасного и удобного контроля за испарителем, даже в закрытом вытяжном шкафу. Широко применяется в химических лабораториях для упаривания растворителей из смесей веществ, а также для разделения жидкостей. Высокоэффективная жидкостная хроматография (ВЭЖХ) – один из эффективных методов разделения сложных смесей веществ. Основой хроматографического разделения является участие компонентов разделяемой смеси в сложной системе Ван-дер-Ваальсовых взаимодействий (преимущественно межмолекулярных) на границе раздела фаз. Принцип жидкостной хроматографии состоит в разделении компонентов смеси, основанном на различии в равновесном распределении их между двумя несмешивающимися фазами, одна из которых неподвижна, а другая подвижна (элюент). Отличительной особенностью ВЭЖХ является использование высокого давления (до 400 бар) и мелкозернистых сорбентов (обычно 3– 5 мкм, сейчас до 1,8 мкм). Это позволяет разделять сложные смеси веществ быстро и полно (среднее время анализа от 3 до 30 мин). 9 Метод ВЭЖХ находит широкое применение в таких областях, как химия, нефтехимия, биология, биотехнология, медицина, пищевая промышленность, охрана окружающей среды, производство лекарственных препаратов и во многих других. Гель-проникающий хроматограф представляет собой вариант жидкостной хроматографии, в котором разделение происходит за счет распределения молекул между растворителем, находящимся внутри пор сорбента, и растворителем, протекающим между его частицами, т.е. неподвижной фазой служит гель, а различное удерживание веществ обусловлено различием в размерах молекул веществ, их форме и способности проникать в поры неподвижной фазы. Принципиальной особенностью гель проникающей хроматографии является возможность разделения молекул по их размеру в растворе в диапазоне практически любых молекулярных масс – от 102 до 108, что делает его незаменимым для исследования синтетических высокомолекулярных веществ и биополимеров. Хромато-масс-спектрометр – прибор, представляющий собой комбинацию газового хроматографа и масс-спектрометра, который предназначен для обнаружения и определения веществ и соединений, входящих в состав медицинской, пищевой, сельскохозяйственной и других видов продукции, лекарственных, психотропных и наркотических средств, биологических систем, контроля экологической обстановки. 10 2. ИСХОДНЫЕ ВЕЩЕСТВА ДЛЯ ОСНОВНОГО ОРГАНИЧЕСКОГО СИНТЕЗА Исходными продуктами основного органического синтеза служат парафиновые, олефиновые, нафтеновые и ароматические углеводороды, ацетилен, оксид углерода и водород (рис. 1). Индивидуальные низкомолекулярные парафины выделяют из природных и попутных газов, а из определенных фракций нефти получают смеси высокомлекулярных парафинов. Процессы изомеризации позволяют превращать нормальные парафины в изопарафины. Для получения индивидуальных ароматических и олефиновых углеводородов, ацетилена, оксида углерода и водорода применяют процессы пиролиза, каталитического крекинга, каталитического риформинга и конверсии. Выбор метода деструктивной переработки углеводородов зависит от состава исходного углеводородного сырья и от продуктов, которые желают получить из этих углеводородов. Все деструктивные методы переработки углеводородов требуют применения высоких температур. В таких условиях углеводороды подвергаются разнообразным и весьма сложным химическим превращениям. 2.1. Очистка фракций жидких продуктов пиролиза Жидкие продукты пиролиза получают на этиленовых установках при пиролизе углеводородных газов, бензинов, дизельной фракции или их смесей в качестве побочного продукта (рис. 2). 11 полиэтилен попутные газы дихлорэтан этанол этилен пластификаторы волокна гликоли окись этилена винилхлорид пластмассы растворители синтетический каучук полистирол стирол полипропилен продукты газоконденсатных месторождений окись пропилена пропилен димер нафта тетрамер додецилбензоил кумол ацетон фенол н-бутены моющие средства спирты оксосинтеза пластификаторы акрилонитрил пластмассы полиизобутилен изобутилен нефть синтетический каучук изопрен бутилкаучук метилэтилкетон растворитель бутадиен синтетический каучук бутадиен бензол фенолформальдегидные смолы фенол кумол циклогексан адипиновая кислота капролактам стирол толуол полистирол пластмассы, взрывчатые вещества Фталевый ангидрид изофталевая кислота терефталевая кислота ксилолы пластификаторы волокна кислоты парафины волокна спирты моющие средства пластификаторы ɑ-олефины Рисунок 1. Продукты переработки углеводородного сырья 12 Рисунок 2. Принципиальная схема материальных потоков (т/год) нефтехимического комплекса олефинового типа Фракции представляют собой жидкий продукт со специфическим запахом, состоящий из смеси непредельных, парафиновых, нафтеновых и ароматических углеводородов – бензола, толуола, стирола, ксилола и др., который может быть использован как сырье для получения моторного топлива, растворителей, ароматических углеводородов и нефтеполимерных смол (НПС). НПС – особый класс низкомолекулярных синтетических смол, обладающих уникальными физико-химическими свойствами. Цель работы: провести очистку дициклопентадиенсодержащей фракции от смолистых соединений и продуктов окисления. Реактивы: фракция жидких продуктов пиролиза бензина. Оборудование: колба круглодонная, холодильник прямой, насадка для перегонки, аллонж, термометр, приемники, стакан химический, цилиндр, весы. Проведение опыта: Разгонку нефти по фракциям проводят на лабораторной установке, приведенной на рисунке 3. 13 5 4 6 7 3 âî äà âî äà âàêóóì 2 8 1 Рисунок 3. Схема установки для прямой перегонки жидкостей: 1 – электроплитка; 2 – баня (водная, масляная или песчаная); 3 – колба круглодонная; 4 – насадка для перегонки, 5 – термометр; 6 – прямой холодильник; 7 – алонж; 8 – приемник Прежде чем преступить непосредственно к разгонке, необходимо взвесить круглодонную колбу для перегонки и пронумерованных приемников. После в колбу, помещают исследуемую фракцию жидких продуктов пиролиза и постепенно нагревают содержимое колбы. В приемник № 1 собирают компоненты фракции, которые выкипают до 130 °С. В диапазоне температур 130–180 °С в приемник № 2 собирают целевую фракцию. По окончании перегонки измеряют массу выделенных фракций и кубового остатка, составляют материальный баланс процесса и идентифицируют состав целевой фракции на газовом хроматографе. 14 3. ПРОЦЕССЫ ОКИСЛЕНИЯ Одно из ведущих мест в промышленности основного органического синтезе занимают процессы окисления, которые позволяют получать большое количество кислородсодержащих соединений, являющихся промежуточными продуктами органического синтеза, растворителями, мономерами и исходными веществами для производства полимерных материалов, пластификаторов и т.д. Главное место среди промышленных окислителей занимает доступный и дешевый кислород воздуха. Процессы окисления проводят в жидкой или газовой фазе с использованием инициаторов (ультрафиолетовый свет, органические гидропероксиды и др.) или катализаторов (металлы, оксиды металлов и их смеси, соли металлов переменной валентности). К реакциям окисления способны все классы углеводородов и многие их производные. Окисление парафинов может быть проведено по трем направлениям: окисление в газовой фазе (получение низших спиртов и альдегидов), термическое окисление в жидкой фазе (синтез высших вторичных спиртов), каталитическое окисление в жидкой фазе (получения карбоновых кислот). Последнее имеет наибольшее практическое значение. В общем виде окисление углеводородов может быть описано схемой, представленной на рисунке 4. Окислением нафтенов получают циклические спирты, кетоны и насыщенные дикарбоновые кислоты. Окисление циклопарафинов протекает по аналогии с окислением парафинов. В результате окислительной деструкции цикла в газовой фазе происходит образование низших кислородсодержащих соединений, двуокиси углерода и воды. 15 Рисунок 4. Схема окисления парафина Жидкофазное окисление воздухом в присутствии органических солей кобальта (при небольшой степени конверсии углеводородов – 10– 20 %) ведет к образованию спиртов и кетонов и практически исключает развитие процессов деструктивного окисления. 3.1. Получение синтетических жирных кислот В результате реакции окисления насыщенных углеводородов образуются синтетические жирные кислоты (СЖК), которые в промышленности получают продуванием потока воздуха через расплавленный парафин. Окислению подвергают углеводороды, содержащие от 12 до 20 атомов углерода в молекуле. На направление химической реакции и состав конечных продуктов влияет катализатор – перманганат калия. 16 СЖК используются в качестве замены натуральных жиров при производстве парфюмерии и бытовой химии. Цель работы: провести окисление твердого парафина и определить изменение кислотного числа. Реактивы: твердый парафин, перманганат калия, 0,1 н спиртовый раствор гидроксида калия, фенолфталеин, ацетон. Оборудование: воздуходувка, колонка с хлористым кальцием, реактор окисления, термометр до 200 °С, ловушка Дина-Старка, обратный холодильник, глицериновая баня, электроплитка, колба коническая емкостью 150 мл (4 шт.), пипетки для отбора проб. Проведение опыта: Синтез СЖК из твердого парафина проводят на лабораторной установке, представленной на рисунке 5. Парафин 25–50 г (по заданию преподавателя) загружают в реактор, расплавляют и добавляют КMnО4 (0,05–0,1 г в 0,4–0,75 мл воды). Регулируя подачу с помощью тройника, при температуре 180 °С и со скоростью 60 л на 1 кг парафина в 1 ч, через колонку с хлористым кальцием подают воздух в нижнюю часть реактора под пористую пластинку. Реактор может быть снабжен электрообогревом, либо помещен в глицериновую баню. Пройдя через реактор, воздух уходит через обратный холодильник в атмосферу. Вода, образующаяся в процессе окисления, собирается в ловушке Дина-Старка, а уносимые воздухом капли парафинов и легколетучие продукты реакции, конденсируются и возвращаются в реактор. Небольшая часть легких продуктов уносятся вместе с воздухом. Процесс проводят в течение 4 часов, отбирая каждый час пробу оксидата и определяя кислотное число. Отбор проб в предварительно взвешенные колбы ведут пипеткой. Для определения кислотных чисел пробы растворяют в нейтральном ацетоне. При проведении опыта температуру в реакторе и расход воздуха поддерживают на постоянном уровне и отмечают объем воды, собирающейся в ловушке Дина и Старка. 17 6 5 4 2 3 1 8 Рисунок 5. Схема лабораторной установки получения синтетических жирных кислот: 1 – воздуходувка, 2 – осушитель, 3 – баня глицериновая, 4 – термометр, 5 – реактор, 6 – ловушка Дина-Старка, 7 – обратный холодильник, 8 – электроплитка Навеску оксидата (0,5–1,0 г) помещают в коническую колбу на 150 мл и растворяют в 20–25 мл нейтрального ацетона. Содержимое колб нагревают на водяной бане для лучшего извлечения карбоновых кислот из оксидата. Пробы оттитровывают спиртовым раствором КОН (0,1 н) в присутствии фенолфталеина и рассчитывают кислотное число по формуле: а · Т · 𝑘 · 1000 Кислотное число = , мг КОН/г S 18 где а – объем 0,1 н раствора КОН, израсходованного на титрование навески, мл; Т – титр 0,1 н раствора КОН, г/мл; k – поправка к титру; S – навеска образца, г. Результаты эксперимента и вычислений переносят в таблицу и строят график зависимости кислотных чисел оксидата от продолжительности реакции. Проводят обсуждение полученных результатов и формулируют выводы по работе. 3.2. Получение адипиновой кислоты окислением циклогексанола Адипиновая кислота – является важнейшей в техническом отношении насыщенной дикарбоновой кислотой, которая производится в крупных заводских масштабах и применяется для получения полиамидных, полиэфирных и полиуретановых смол, а также для синтеза пластификаторов и ряда других продуктов. Наиболее экономичными методами синтеза адипиновой кислоты являются окислительные технологии, в которых используется наиболее дешевое сырье – циклогексанол, циклогексанон и циклогексан: O2 O OH O2 HOOC - (CH2)4 - COOH + H2O O2 Цель работы: получить адипиновую кислоту путем окисления циклогексанола и оценить факторы, влияющие на ее выход и чистоту. Реактивы: циклогексанол (0,1 моль), азотная кислота (51,7% -ная, 0,43 моля), ванадат аммония (0,02 г) Оборудование: колба круглодонная трехгорлая, холодильник обратный, мешалка механическая, стакан химический, термометр, капельная воронка, фильтр Шотта, колба Бунзена, баня водяная, электроплитка. Проведение опыта: Перед началом выполнения лабораторной работы необходимо рассчитать требуемое количество реагентов, согласно выданному заданию. 19 В круглодонную трёхгорлую колбу (рис. 6), снабжённую механической мешалкой, обратным холодильником, термометром и капельной воронкой, заливают азотную кислоту и добавляют катализатор (реакцию можно проводить без катализатора, в этом случае 5 температура реакции должна быть 6 85–90 °С). Колбу нагревают до 55 °С на водяной бане и при энергичном 4 перемешивании (ОСТОРОЖНО!) приливают из капельной воронки несколько капель спирта. Реакция начинается через 5 мин (после ко3 роткого периода индукции). В результате реакции происходит выделением бурых оксидов азота. Температуру в колбе (около 55– 2 60 °С) поддерживают путём охлаждения реакционной массы в бане 1 со льдом и приливания с соответствующей скоростью циклогексанола (в течение 1–1,5 ч). К концу реакции, когда в капельной воронке останется небольшое количеРисунок 6. Схема лабораторной установки: 1 – электроплитка, 2 – баня, 3 – ство циклогексанола, охлаждение круглодонная колба, 4 – термометр, 5 – прекращают и для поддержания температуры 55–60 °С слегка покапельная воронка, 6 – мешалка догревают колбу на водяной бане до окончания прибавления остатка циклогексанола. Нагревание смеси на кипящей водяной бане при непрерывном перемешивании продолжают и после прибавления всего количества циклогексанола до прекращения выделения оксидов азота. Жидкость в горячем состоянии переливают в стакан и оставляют кристаллизоваться в прохладном месте. Выпавший кристаллический осадок адипиновой кислоты отфильтровывают, промывают 10–12 мл очень холодной воды и сушат на воздухе. 20 Далее определяют массу и температуру плавления полученной адипиновой кислоты и составляют материальный баланс процесса. Обсуждают результаты опыта и формулируют выводы о выполненной работе. 3.3. Эпоксидирование нефтеполимерных смол Нефтеполимерные смолы (НПС) – низкомолекулярные термопластичные полимеры, получаемые полимеризацией жидких продуктов пиролиза нефтепродуктов, которые используются для производства синтетических олиф, масляно-смоляных лаков, алкидных смол, а также в качестве добавок в различные лакокрасочные композиции. Основными достоинствами НПС является их способность к пленкообразованию, а также водостойкость и высокая температура размягчения. Но наряду с достоинствами, НПС имеют и недостатки, главные из которых – низкая адгезия и высокая окисляемость. Улучшить качественные характеристики НПС можно путем введения различных функциональных групп в их структуру. Окислительные способы, в частности эпоксидирование, являются наиболее распространенными методами функционализации. Эпоксидные смолы стойки к действию галогенов, некоторых кислот, щелочей; обладают высокой адгезией к металлам. Из эпоксидных смол готовят различные виды клея, пластмасс, электроизоляционные лаки. Эпоксидирование относится к группе окислительных реакций, в которых сохраняется углеродный скелет, при этом под действием надкислот (по реакции Н. А. Прилежаева), кислорода воздуха в присутствии серебряного катализатора, хромовой кислоты и др. идет превращение двойной связи олефинов в эпоксидное кольцо. Ациклические и циклические алкены при взаимодействии с перкислотами (надкислотами) RCOOOH в неполярной, индифферентной среде образуют эпоксиды: R CH CH2 + R' C O O OH эфир, бензол CH2Cl2 O R CH CH2 + R' C OH O В соответствие с современной номенклатурой ИЮПАК – трехчленный цикл с одним атомом кислорода носит название оксиран. 21 Цель работы: изучить теоретические основы процесса образования α-оксидов под действием окислительной системы Н2О2+СН3СООН в присутствии каталитических количеств минеральных кислот. Реактивы: нефтеполимерная смола (С9), ледяная уксусная кислота, пероксид водорода, толуол, вода. Оборудование: колба круглодонная трехгорлая (250 мл), капельная воронка, баня водяная, мешалка, электроплитка, термометр, делительная воронка, центрифуга, чашки Петри. Проведение опыта: Перед началом опыта готовят 50 %-ный раствор НПС в толуоле, рассчитывают количество реагентов необходимых для проведения реакции, соблюдая следующие условия: мольное соотношение СН3СООН : Н2О2 равно 0,5, массовое соотношение Н2О2 : НПС также равно 0,5, количество серной кислоты составляет 0,7 % от массы окислительного комплекса. Исследование процесса эпоксидирования НПС С9 надуксусной кислотой, образующейся «in situ», проводят на установке изображенной на рисунке 6. Раствор НПС помещают в колбу (объем колбы должен в 5 раз превышать объем раствора смолы) и при перемешивании добавляют ледяную уксусную кислоту из капельной воронки. Затем смесь нагревают до 55 °С и прикапывают смесь пероксида водорода и серной кислоты. После полного добавления окислителя реакционную массу выдерживают при постоянном перемешивании при 75 °С в течение 1,5 часов, охлаждают и нейтрализуют кислоты многократной промывкой реакционной массы водой до рН=7. При помощи центрифуги отделяют органический слой от водного и сушат его на воздухе при 20–25 °С в чашках Петри. Полученные опытным путем образцы эпоксидированной НПС подвергают физико-химическим исследованиям. Структуру исходного и окисленных образцов смолы анализируют с помощью ИК-Фурье спектрометра в диапазоне длин волн 600–4000 см-1. ИК-спектры регистрируют в виде твердых таблеток, полученных холодным прессованием порошка НПС и предварительно прокаленного бромида калия. Молекулярную массу образцов определяют методом гельхроматографии, используя жидкостный хроматограф AGILENT 1200. В качестве элюента берут хлороформ, а в качестве неподвижной фазы – силикагель. Берем навеску массой 1,5 мг и растворяет в 1 мл хлороформа. По достижении полного растворения образца в растворителе, наби22 рают микрошприцем 30 мкл раствора смолы. Вводят содержимое микрошприца в петлю до упора, впрыскивают раствор смолы и снимают с прибора показания среднечисловой, среднемассовой молекулярной массы и степень полидисперсности. Определение бромного числа проводят в соответствии с методикой, описанной в разделе 3.1. Эпоксидное число (ЭЧ) определяют метод кислотно-основного титрования. В коническую колбу вместимостью 200 мл вносят 0,1–0,5 г, анализируемого продукта и растворяют в 5 мл 1,4-диоксана пипеткой добавляют 10 мл ацетонового раствора соляной кислоты (1 мл соляной кислоты и 40 мл ацетона). Выдерживают при комнатной температуре в течении 30 минут. В колбу вносят 2-3 капли метилового красного (спиртовой раствор) и титруют 0,1 н раствором гидроксида натрия до исчезновения красной окраски. Контрольный опыт проводится в тех же условиях, но без анализируемого вещества. Далее, исходя из щелочи, пошедшей на титрование, рассчитывают эпоксидное число по формуле: (𝑉1 − 𝑉0 ) ∙ 0,0043 ∙ 100 𝑥= 𝑚 где V1 – объем 0,1 н раствора гидроксида натрия, израсходованного на титрование пробы, мл; V0 – объем 0,1 н раствора гидроксида натрия, израсходованного на контрольную пробу, мл; m – масса навески, г. 23 4. ПРОЦЕССЫ ХЛОРИРОВАНИЯ Все процессы, в результате которых в органические соединения вводятся атомы галогена называют галогенированием. В зависимости от вида галогена различают реакции фторирования, хлорирования, бромирования и йодирования. Процессы хлорирования могут протекать по следующим направлениям: замещение различных атомов (или групп атомов) на атомы хлора и присоединение хлора по ненасыщенным связям или атомам, находящихся в низшем валентном состоянии. Основными промышленными процессами являются: хлорирование парафинов, олефинов и ароматических углеводородов, хлоргидринирование олефинов, гидрогалогенирование непредельных соединений, расщепление хлорпроизводных, окислительное хлорирование парафинов и олефинов: хлорирование Сl2 температура Сl . + Сl . Сl . + RH R . + HCl R . + Cl2 RCl + Cl . хлоргидринирование CH2 CH CH3 + Сl2 + H2O гидрогалогенирование R CH CH2 + НСl расщепление Сl3C-CCl3 CH2 C CH2 Cl + HCl OH R CH CH3 Cl Сl2C=CCl2 + Сl2 В качестве галогенирующих агентов в промышленности используют свободные галогены, галогенводороды, галогенангидриды неорганических кислот, соли галогенводородных кислот. Среди реакций галогенирования ведущее место занимают реакции хлорирования, позволяющие в крупных промышленных масштабах производить многочисленные галогенпроизводные: хлористый винил, хлоропрен, тетрафторэтилен, монохлортрифторэтилен, хлористый ви24 нилиден, фтористый винилиден – это хлорорганические мономеры и сырье для получения высокомолекулярных соединений; а также гексахлорциклогексан, гексахлорпентадиены и др. – галогенорганические ядохимикаты; хлористый метилен, четыреххлористый углерод, хлорэтилены – хлорорганические растворители; фреоны – хлорфторпроизводные парафинов (используемые как хладоагенты, средства пожаротушения, вспениватели и т.д.); хлористые этил, метил, аллил, хлорбензол, хлоргидрины, дихлорэтан и др. – промежуточные продукты органического синтеза. 4.1. Хлорирование непредельных соединений Процессы хлорирования проводят в широком интервале температур (20–500 °С), как в присутствии, так и в отсутствии катализаторов и инициаторов. Условия процесса выбирают таким образом, чтобы выход целевого хлорорганического соединения из исходного углеводорода был максимальным. Процессы присоединения и замещения при хлорировании олефинов в газовой фазе являются двумя независимыми параллельными реакции, которые протекают по радикально-цепному механизму. Когда продукт реакции или растворитель находится в жидкой фазе, то хлорирование идет по ионному механизму через промежуточное образование π–комплекса и иона карбония: H2C CH2 + Cl2 H2C CH2 Cl Cl + Cl H2C CH2 + Cl - Cl H2C CH2 Cl Основные продукты, которые получают присоединением галогенов по двойной связи – дихлорэтан (используемый для синтеза хлористого винила, винилиденхлорида, этилендиамина), 1,2-дихлорпропан (используемый как растворитель и фумигант), дихлорбутены (используемый для синтеза адипиновой кислоты и гексаметилендиамина). Цель работы: изучить теоретические основы присоединительного галогенирования и провести реакцию присоединения хлора к ненасыщенным соединениям. Реактивы: перманганат калия, соляная кислота концентрированная, нефтеполимерная смола, ксилол, хлороформ, раствор брома, ледяная уксусная кислота, этанол, тиосульфат натрия, йодистый калий, крахмал. 25 Оборудование: колба круглодонная трехгорлая (250 мл), капельная воронка, баня водяная, колба коническая (250 мл), барботер, мешалка, электроплитка, термометр, стеклянная трубка, стаканы (50 мл), аналитические весы. Проведение опыта: Перед началом работы, в соответствии с заданием преподавателя, следует рассчитать количество реагентов, которое необходимо подать на реакцию. Для этого на первом этапе определяют бромное число исследуемого образца смолы. Бромным числом (б.ч.) называется число граммов брома, присоединяющееся к 100 г исследуемого вещества. Оно характеризует содержание в нем непредельных соединений: С6Н5СН=СН2 + Br2 → С6Н5СНBr–СН2Br В два стакана ёмкостью по 50 мл помещают навески исследуемой смолы массой 0,1 г. Взвешивают навески на аналитических весах с точностью до 0,0001 г. В стаканы добавляют по 10 мл хлороформа, содержимое перемешивают и охлаждают в ледяной бане до 5 °С. Затем в каждую колбу приливают такое количество 0,1 н раствора брома в ледяной уксусной кислоте, чтобы содержимое колб оставалось желтым от избытка брома и после охлаждения. Полученные смеси охлаждают до 5 °С и при такой температуре выдерживают в тёмном месте в течение 5 минут. После этого добавляют по 5 мл 10 % раствора иодида калия и 1 мл крахмала (индикатор). Выделяющийся йод сразу титруют 0,1 н. раствором тиосульфата натрия при энергичном помешивании. Титрование продолжают до исчезновения синей окраски растворов: Br2 + 2KJ → 2КBr + J2 J2 + 2Na2S2O3 → 2NaJ + Na2S4O6 Параллельно проводят титрование смеси 10 мл хлороформа, 1 мл 0,1 н. раствора брома и 5 мл 10 %-го раствора иодида калия без анализируемого вещества (холостой опыт). Бромное число вычисляют по формуле: 26 б.ч. а б 0,008 К 100 g где а – объём 0,1 н раствора Na2S2O3, пошедший на титрование в холостом опыте, мл; б – объём 0,1 н раствора Na2S2O3, пошедший на титрование исследуемого образца, мл; g – навеска исследуемого образца, г; 0,008 – число граммов брома, соответствующее одному мл 0,1 н раствора Na2S2O3; К – поправка на 0,1 н раствора Na2S2O3. Далее рассчитывают количество хлора, необходимое для насыщения 20 %-ного раствора НПС массой 150 г по формуле: 𝑁= б. ч.· 𝑚 · 𝑀𝐶𝑙 100 · 𝑀𝐵𝑟 N – количество хлора, которое нужно подать на реакцию, г б.ч. – бромное число для заданного образца НПС, г m – масса смолы, г MCl – мольная масса Cl2, г/моль MBr – мольная масса Br2, г/моль Исходя из уравнения реакции, определяют требуемое количество реагентов (KMnO4 и HCl): 2KMnO4 + 16HCl → 2MnCl2 + 8Н2О + 5Cl2↑ + 2КCl М.м., г/моль 157,9 36,5 126 18 71 74,5 В трехгорлую колбу (рис. 7) помещают расчетное количество перманганата калия, в капельную воронку наливают расчетное количество концентрированной соляной кислоты. 27 4 5 6 7 3 8 2 1 Рисунок 7. Установка для насыщения раствора нефтеполимерной смолы хлором: 1 – электроплитка с мешалкой; 2 – баня водяная; 3 – трехгорлая колба; 4 – капельная воронка; 5 – термометр; 6 – стеклянная трубка (силиконовый шланг); 7 – барботёр; 8 – приемник с раствором НПС Скорость выделения хлора регулируют скоростью приливания соляной кислоты. Процесс проводят при интенсивном перемешивании, при температуре 40–45 °С. Выделяющийся хлор через трубку поступает в емкость с 20 %-ным раствором нефтеполимерной смолы в ксилоле, насыщая его. В результате реакции раствор НПС окрашивается в черно-зеленый цвет. Далее насыщенный хлором раствор переосаждают в этаноле и сушат при 20 ºС. Затем определяют б.ч., составляют материальный баланс процесса и делают выводы по работе. 28 5. ПРОЦЕССЫ ГИДРОЛИЗА Реакциями гидролиза называют процессы замещения или двойного обмена, протекающие под действием воды или щелочей. Чаще всего в технологии основного органического синтеза гидролизу подвергают хлорпроизводные, в молекулах которых при гидролизе может происходить не только замещение хлора, но и щелочное дегидрохлорирование с образованием ненасыщенных соединений или α-оксидов. Поскольку гидролиз водой идет очень медленно и обратимо, то обычно в качестве гидролизующих агентов используют растворы NaOH, Ca(OH)2 или Na2CO3. В этом случае реакция становится необратимой, происходит замещение атомов хлора на гидроксил или отщепление хлористого водорода: H3C CH CH2 Cl + HO OH H3C CH CH2 OH + Cl OH H3C CH CH2 + Cl + H2O O Дегидрохлорирование проводят с непрерывной отгонкой целевого продукта из реакционной массы. Процесс всегда ведут при атмосферном давлении и температуре, обеспечивающей кипение смеси и отгонку продукта. Наиболее легко отщепляется HCl от хлоргидринов, труднее – от полихлоридов этана. 5.1. Получение глицерина Глицериин (1,2,3-триоксипентан, пропантриол-1,2,3) – простейший представитель трёхатомных спиртов, который используют в производстве моющих и косметических средств, сельском хозяйстве, в производстве пластмасс, лакокрасочной промышленности и других отраслях. Большинство синтетических методов получения глицерина основаны на использовании пропилена в качестве исходного продукта. Хлорированием пропилена при 450–500 °С получают аллилхлорид, при присоединении к последнему хлорноватистой кислоты образуются хлоргидрины, которые при омылении щёлочью превращаются в глицерин. Завершающей стадией при получении глицерина по хлорному методу является гидролиз эпихлоргидрина, протекающий с образованием эфиров глицерина (полиглицеринов) в качестве побочных продуктов: 29 H2C CH CH2Cl O CH2 CH CH2 OH OH Cl CH2 CH CH2 OH O H2O CH2 CH CH2 OH OH Cl CH2 CH CH2 NaCl OH OH OH H2O CH2 CH CH2 NaCl OH O CH 2(OH)CH(OH)CH2OH CH2 CH CH2OCH2 CH CH2 OH OH OH OH Для повышения селективности процесса гидролиз эпихлоргидрина ведут карбонатом натрия в избытке воды. При концентрации глицерина в гидролизате 10–15 % его выход достигает 90–95 %. В промышленности гидролиз ведут непрерывно под давлением при 120–150 ℃ в двух последовательно работающих реакторах. Первую стадию процесса проводят в реакторе с мешалкой для максимальной гомогенизации реакционной массы, а вторую в трубчатом реакторе. Цель работы: изучение реакции гидролиза эпихлоргидрина в глицерин в присутствии гидроксида натрия. Реактивы: эпихлоргидрин (18,5 г), гидроксид натрия 10 %-ный водный раствор (90 мл), соляная кислота 10 %-ная. Оборудование: колба круглодонная трехгорлая, холодильник обратный, холодильник прямой, мешалка механическая, стакан химический, термометр, капельная воронка, фильтр Шотта, колба Бунзена, баня водяная, электроплитка, приемники. Гидролиз эпихлоргидрина в глицерин проводят на установке, схема которой приведена на рисунке 3. Перед началом опыта проверяют правильность сборки и герметичность всех соединений установки. Проведение опыта: В реактор загружают 90 мл 10%-ного водного раствора гидроксида натрия, 18,5 г эпихлоргидрина, подают холодную воду в обратный холодильник, включают мешалку и обогрев реактора. Для хорошего эмульгирования реакционной массы тщательно следят за скоростью вращения вала мешалки. При достижении в реакторе температуры 70 °С продолжают перемешивание реакционной массы в течение 1,5 ч. По истечению времени обогрев реактора прекращают, реакционную массу 30 охлаждают до комнатной температуры, и постепенно приливают из градуированной капельной воронки 10 %-ный раствор соляной кислоты до установления по лакмусовой бумаге нейтральной среды в реакторе. После нейтрализации продукты реакции выливают в колбу Кляйзена емкостью 250 мл и отгоняют воду до уменьшения объема реакционной массы на три четверти (рис. 3). Оставшуюся в колбе массу охлаждают до комнатной температуры, выпавшие кристаллы хлорида натрия и жидкие продукты разделяют на фильтре и перемещают в колбу Кляйзена. Фильтрат перегоняют в вакууме при 10 мм рт. ст. Выделяют головную фракцию, выкипающую до 150 oC, и фракцию глицерина с пределами кипения от 150 до 170 oC. Определяют показатель преломления и массу полученного глицерина и составляют материальный баланс процесса. Обсуждают результаты опыта и формулируют свои выводы о выполненной работе. 31 6. ПРОЦЕССЫ ДЕГИДРАТАЦИИ Процессы дегидратации имеют важное практическое значение. Этими методами в крупных масштабах получают многочисленные ненасыщенные соединения, простые эфиры, ангидриды и др. Дегидратацию осуществляют двумя основными методами: в жидкой и газовой фазах. Жидкофазную дегидратацию проводят при температуре 100– 200 °С при атмосферном давлении в присутствии кислых катализаторов (серная кислота, фосфорная кислота, катиониты) и используют в тех случаях, когда продукт или исходные реагенты недостаточно стабильны при повышенных температурах, например для получения хлорекса, диоксана и морфолина. Температура газофазной дегидратации колеблется от 225 до 720 °С используют для получения. В качестве катализатора используется фосфорная кислота на пористых носителях, оксид алюминия, кислы и средние фосфаты кальция или магния. Продуктами газофазной дегидратации являются стирол, изопрен, уксусный ангидрид и др. Большее значение процессы дегидратации имеют в качестве одной из стадий какого-либо производства. Например, стирол получают дегидратацией метилфенилкарбинола: HO CH CH3 CH CH2 + H2O Это Халкон-процесс, в котором кроме стирола в качестве товарного продукта получают оксид пропилена. Метод синтеза изопрена из изобутилена и формальдегида также связан с последовательной дегидратацией диола и ненасыщенного спирта. А межмолекулярной дегидратацией этанола на гетерогенном катализаторе при 250 ºС получают диэтиловый эфир: 2 H3C CH2 OH H3C CH2 O CH2 CH3 + H2O Некоторые двухатомные спирты в присутствии кислотного катализатора способны к замыканию цепи с образованием пяти- и шестичленных циклов. Таким путем из диэтиленгликоля получают диоксан, из бу32 тандиола – тетрагидрофуран и другие кислородсодержащие циклические продукты, являющиеся хорошими растворителями. 6.1. Получение дибутилового эфира В промышленности дибутиловый эфир получают преимущественно дегидратацией н-бутилового спирта в присутствии серной кислоты в качестве катализатора: C4H9OH H2SO4 (C4H9)2O + H2O Применяют дибутиловый эфир в комплексе кремнийфторорганических производств в качестве сырья и как растворитель для органических синтезов. Цель работы: получить дибутиловый эфир путём гидратации н-бутилового спирта, определить выход и чистоту продукта. 7 6 Реактивы: н-бутиловый спирт (20 г), серная кислота с плотностью 1,84 г/мл (5,2 г), 3 н 4 гидроксида натрия (2 мл), насы5 щенный раствор хлористого кальция (15 мл), безводный хлористый кальций 3 Оборудование: электроплитка, баня песчаная, термометр, насадка Дина-Старка, обратный 2 холодильник, прямой холодильник, круглодонная колба, насадка, 1 алонж, приемники, делительная воронка, дефлегматор. Проведение опыта: Дибутиловый эфир получают на лабораторной установке, схема Рисунок 8. Схема лабораторной установки: 1 – электроплитка, 2 – баня, 3 – которой приведена на рисунке 8. В круглодонная колба, 4 – термометр, 5 – круглодонную колбу, снабженную насадка Дина-Старка, 6 – мешалка, 7 – мешалкой, устанавливают ловушку холодильник 33 Дина-Старка, обратный холодильник и наливают 20 г н-бутилового спирта. Далее, осторожно, при перемешивании добавляют концентрированную серную кислоту, вносят «кипелки» и смесь осторожно кипятят. Нагревание прекращают, когда количество воды в ловушке перестанет увеличиваться и будет примерно равное расчетному значению, определенного предварительно по уравнению реакции. К содержимому колбы при охлаждении и перемешивании приливают 12 мл 3 н раствора гидроксида натрия, переносят в делительную воронку и промывают до щелочной реакции промывных вод. После добавляют 15 мл воды и 12 мл насыщенного раствора хлористого кальция и промывают до нейтральной реакции по лакмусу. Полученный продукт сушат безводным хлоридом кальция. Реакционную массу, после отмывки переносят в перегонную колбу с высоким дефлегматором, фильтруя ее через бумажный складчатый фильтр. Далее ведут перегонку, отбирая фракцию, кипящую до 135 °С. Затем слегка охладив, меняют дефлегматор на более низкий и собирают целевую фракцию, кипящую при 140–142 °С. (Эфиры перегонять досуха нельзя, так как они содержат перекиси, которые при перегонке досуха могут взорваться!). По окончании перегонки снимают показатель преломления, считают выход и составляют материальный баланс процесса. 6.2. Получение пропилена Пропилен представляет собой газообразное вещество с низкой температурой кипения. Используется для производства оксида пропилена, получения изопропилового спирта и ацетона, для синтеза альдегидов, для получения акриловой кислоты и акрилонитрила, полипропилена, пластмасс, каучуков, моющих средств, компонентов моторных топлив, растворителей. Одним из способов получения пропилена является реакция дегидратации изопропилового спирта, которая происходит при нагревании спирта в присутствии кислотных катализаторов. Цель работы: получение пропилена реакцией дегидратацией изопропилового спирта. Реактивы: изопропиловый спирт, вода, оксид алюминия Оборудование: реактор, перистальтический насос, поглотительная склянка, барботер, газометр. Проведение опыта: 34 Получение пропилена ведут на лабораторной установке, схема которой приведена на рисунке 9. Перед началом работы собранную стендовую установку следует проверить на герметичность. Затем включить электрообогрев реактора и после того как температура в печи достигнет 350 °С, дозирующим насосом начинают подавать в реактор изопропиловый спирт. Температура в печи не должна превышать 400 °С. Выходящий из реактора пропилен собирают в газометр. По объему воды вытесненной из газометра определяют количество образовавшегося пропилена. При расчетах также используют замеренное количество воды, выделившейся при дегидратации и сконденсировавшейся в поглотительной склянке. 2 3 âî äà 5 âî äà 6 1 4 Рисунок 9. Схема лабораторной установки дегидратации спирта: 1 – емкость для спирта, 2 – перистальтический насос, 3 – реактор, 4 – поглотительная склянка, 5 – поглотительная склянка, 6 - газометр В заключении в соответствии с проведенными расчетами определяют практический выход газа, составляют материальный баланс и схему процесса дегидратации изопропилового спирта. 35 7. ПРОЦЕССЫ ЭТЕРИФИКАЦИИ Сложные эфиры – продукты замещения атома водорода в карбоксильной группе карбоновых кислот алкильными (арильными) радикалами. В результате этерификации многоосновные кислоты могут давать продукты полного замещения и продукты неполного замещения – кислые эфиры. Из всевозможных реакций образования сложных эфиров реакции карбоновых кислот и их ангидридов со спиртами и олефинами имеют большое промышленное значение. Наиболее распространенной является этерификация при обратимом взаимодействии карбоновых кислот со спиртами. Для двух- и многоатомных спиртов возможно образование неполных и полных эфиров, что определяется соотношением реагентов: CH2 OH + RCOOH CH OH - H2O CH2 OH CH2 O C(O)R CH OH CH2 OH - H2O + RCOOH CH2 O C(O)R + RCOOH CH O C(O)R - H2O CH2 O C(O)R CH2 O C(O)R CH OH CH2 O C(O)R В случае двухатомных спиртов получаются так называемые кислые и средние эфиры, выход которых также зависит от соотношения компонентов: HOOC-R'-COOH + ROH - H2O HOOC-R'-COOR + ROH - H2O ROOC-R'-COOR Если и кислота, и спирт бифункциональны, реакция протекает с образованием высокомолекулярных соединений – полиэфиров. Этот процесс идет необратимо. Этерификацию спиртов карбоновыми кислотами можно осуществлять в отсутствие катализаторов, но в таких условиях реакция протекает медленно, и для достижения достаточной скорости необходима температура 200–300 ºС. Кислотные катализаторы (серная, соляная, катиони36 ты) позволяют вести этерификацию при 70–150 ºС. В большинстве случаев этот процесс проводят в жидкой фазе. Сложные эфиры, образованные при взаимодействии низших карбоновых кислот и простейших спиртов – бесцветные летучие жидкости часто с приятно фруктовым, или фруктово-ягодным запахом, которые используются для приготовления искусственных фруктовых эссенций в пищевой промышленности, в парфюмерии, а также в качестве экстрагентов и растворителей в различных отраслях промышленности. Высококипящие сложные эфиры хорошо совместимы со многими полимерами и широко применяются как пластификаторы. Большое применение получили также сложноэфирные смазочные масла и гидравлические жидкости, имеющие низкую температуру застывания. 7.1. Получение этилацетата Этилацетат – бесцветная жидкость с приятным сладковатым запахом. Широко используется как растворитель, из-за низкой стоимости и малой токсичности, а также приемлемого запаха. Применяется как компонент фруктовых эссенций. В промышленности этилацетат получают взаимодействием уксусной кислоты и этилового спирта: H3C C O H + H3C CH2 OH H3C C O + O CH2 CH3 H2O Цель работы: Изучение реакции этерификации карбоновых кислот на примере взаимодействия уксусной кислоты с этиловым спиртом. Реактивы: этиловый спирт (50 г), концентрированная серная кислота (17 мл), ледяная уксусная кислота (50 мл), 10%-ый раствор бикарбоната натрия, хлористый кальций кристаллический. Оборудование: двухгорлая колба, прямой холодильник, капельная воронка, термометр, баня, электроплитка, насадка, алонж, приемник, делительная воронка. Проведение опыта: В круглодонную двухгорлую колбу (рис. 10) на 250 мл, снабженную прямым холодильником с приемником и капельной воронкой заливают 8 мл этилового спирта и 8 мл концентрированной серной кислоты (охлаждая извне водой со льдом), а затем нагревают на песочной бане. При достижении температуры 140 °С начинают постепенно прибавлять из капельной воронки смесь, состоящую из 42 мл этилового спирта и 37 42 мл уксусной кислоты. Скорость прикатывания этой смеси регулируют так, чтобы происходила нормальная отгонка образующегося уксусноэтилового эфира. Реакцию заканчивают, когда отгонка эфира прекратится. 5 6 4 7 3 2 8 1 Рисунок 10. Схема лабораторной установки получения этилацетата: 1 – плитка, 2 – баня, 3 – реакционная колба, 4 – термометр, 5 – капельная воронка, 6 – холодильник, 7 – аллонж, 8 – приемник. Для нейтрализации примеси уксусной кислоты, отогнанной вместе с этилацетатом, погон встряхивают с 10%-ым раствором соды, добавляя последний малыми порциями в колбу до тех пор, пока верхний слой не перестанет окрашивать синюю лакмусовую бумажку в красный цвет. Затем оба слоя разделяют в делительной воронке и для удаления примесей этилового спирта встряхивают эфирный слой с раствором хлористого кальция (33 г соли в 33 мл воды). Снова разделяют оба слоя на делительный воронке, сушат верхний (эфирный) слой прокаленным хлористым кальцием в течение 0,5–1 ч в колбе с обратным холодильником при нагревании на водяной бане и охлаждают до комнатной температуры. 38 Высушенный уксусноэтиловый эфир перегоняется из колбы Вюрца при температуре 76–78 °С. Замеряют вес уксусноэтилового эфира, выход на взятый этиловый спирт, плотность и показатель преломления. Сравнивают полученные значения с литературными данными и делают выводы. 7.2. Получение бутилацетата Бутиловый эфир уксусной кислоты – это бесцветная или слегка желтоватая жидкость с характерным эфирным запахом. Является хорошим растворителем хлоркаучука, нитроцеллюлозы, глифталевых смол и других плёнкообразующих веществ, часто используемых в лакокрасочной промышленности. В промышленности бутилацетат получают этерификацией уксусной кислоты бутанолом в присутствии серной кислоты при 100–110 °С по непрерывной схеме: H2SO4 CH3COOH + C4H9OH O CH3 C O C4H10 + H2O Цель работы: Получить сложный эфир из бутилового спирта и уксусной кислоты и провести его идентификацию. Оценить факторы, влияющие на выход и чистоту получаемого продукта. Реактивы: уксусная кислота (14 мл), бутанол (23 мл) концентрированная серная кислота (0,5 мл), вода ледяная (75 мл), 10%-ый раствор бикарбоната натрия, хлористый кальций кристаллический. Оборудование: колба круглодонная, прямой холодильник, капельная воронка, термометр, баня, электроплитка, насадка, алонж, приемник, делительная воронка. Проведение опыта: Смесь 14 мл уксусной кислоты и 23 мл бутилового спирта помещают в круглодонную колбу с обратным холодильником (рис. 11). Прибавив 2–3 капли серной кислоты, выдерживают реакционную массу, пока пройдёт бурная экзотермическая реакция, и нагревают ее на кипящей водяной бане в течение 2 часов. После синтеза, охладив установку, содержимое колбы выливают в 75 мл ледяной воды, отделяют верхний эфирный слой в делительной воронке, затем нейтрализуют его раствором соды, промывают водой и сушат сульфатом натрия. Далее эфир очищают перегонкой, взвешива39 ют, определяют показатель преломления и сравнивают с литературными данными. Находят выход эфира в % от теоретического и составляют материальный баланс процесса. 3 4 7.3. Получение дибутилфталата 2 1 Рисунок 11. Схема установки получения бутилацетата: 1электроплитка; 2-песчаная баня; 3-обратный холодильник; 4термометр; 5- колба круглодонная CO Дибутиловый эфир фталевой кислоты – бесцветная маслянистая жидкость. Как и большинство других диалкилфталатов применяют в основном в качестве пластификатора полимерных материалов и, в частности, поливинилхлорида. В промышленности дибутилфталат получают преимущественно этерификацией фталевого ангидрида н-бутиловым спиртом в присутствии концентрированной серной кислоты или тетрабутоксититана. Присоединение первой молекулы спирта к фталевому ангидриду протекает относительно быстро и необратимо с образованием неполного эфира фталевой кислоты: COOC4H9 O + C4H9OH COOH CO Вторая молекула спирта реагирует с неполным эфиром фталевой кислоты более медленно и обратимо: COOC4H9 + C4H9OH COOH COOC4H9 COOC4H9 40 Для смещения равновесия в сторону образования полного эфира добавляют избыток спирта и образовавшуюся воду отгоняют в виде азеотропной смеси с н-бутиловым спиртом. Цель работы: Получить дибутилфталат взаимодействием фталевого ангидрида с н-бутиловым спиртом, определить выход и физические константы полученного дибутилфталата. Реактивы: фталевый ангидрид (12,3 г), н-бутиловый спирт (45,7 мл), серная кислота концентрированная (0,2 мл), карбонат натрия 5 %-ный водный раствор (33,3 мл), хлорид кальция безводный (5 г), бензол (33,3 мл). Оборудование: колба круглодонная, насадка Дина-Старка, холодильник обратный, воронка делительная, стакан, мешалка, баня, электроплитка, дефлегматор, термометр, колба коническая. Проведение опыта: Дибутилфталат получают на установке, схема которой приведена на рисунке 6. В реактор загружают 12,3 г фталевого ангидрида, 45,7 мл н-бутилового спирта, 0,2 мл концентрированной серной кислоты и опускают несколько небольших кусочков керамики. Реактор присоединяют к штативу, включают обратный холодильник и осуществляют обогрев реакционной массы с помощью плитки. Температуру процесса поддерживают таким образом, чтобы реакционная масса кипела умеренно, а скорость стекания конденсата из обратного холодильника в сепаратор составляла 1–2 капли в секунду. С момента начало закипания реакционной массы засекают время. В процессе этерификации образуется вода, которая отгоняется с н-бутиловым спиртом в виде азеотропной смеси и накапливается в нижней части сепаратора. Конец реакции определяют по прекращению выделения воды из конденсата, стекающего в ловушку Дина-Старка из обратного холодильника, и по объёму воды, скопившейся в ловушке. По окончании опыта обогрев реактора снимают, реакционную массу охлаждают до комнатной температуры, переливают в делительную воронку, добавляют 33 мл бензола и последовательно промывают 33 мл 5%-ного раствора карбоната натрия и 33 мл воды дают хорошо отстояться после каждой промывки реакционной массы. Органический слой отделенный от воды, сливают в сухую коническую колбу и сушат над безводным хлоридом кальция. После фильтрования содержимое колбы помещают в предварительно взвешенную на технических весах колбу Кляйзена, определяют массу фильтрата, опус41 кают в колбу кипелки и отгоняют из реакционной массы вещества, выкипающие до 150 °С. Остаток представляет собой достаточно чистый дибутилфталат. Его охлаждают до 40–50 °С, переливают в предварительно взвешенную на технических весах колбу, определяют массу, показатель преломления. Вычисляют выход дибутилфталата по фталевому ангидриду и потери. Составляют материальный баланс процесса и делают выводы по работе. 7.4. Получение лактида В последние десятилетия очень интенсивно ведутся разработки в области применения полимерных биоматериалов в хирургии, трансплантологии и фармакологии. Биополимеры обладают способностью к биоразложению и биологической совместимости, поэтому, по отношению к искусственным полимерам, их использование является более предпочтительным, а в ряде случаев даже незаменимым. Наиболее распространенным и перспективным синтетическим биоразлагаемым полимером является полимолочная кислота (ПМК, PLA). ПМК, синтезируемая путем поликонденсации молочной кислоты имеет низкую молекулярную массу (<20000) и не может быть использована в медицинских целях. Поэтому для получения высокомолекулярного полимера, в качестве исходного мономера, вместо молочной кислоты рекомендуется использовать изомеры лактида: CH3 O O CH3 O O O O L-лактид CH3 O O CH3 CH3 O O CH3 O O D, L-лактид D-лактид L-изомер ПМК является аморфно-кристаллическим и его биодеградация может протекать больше двух лет, а аморфного DL-изомера около 16 месяцев. При деградации ПМК, за счет разрыва эфирной связи, происходит образование молочной кислоты, которая является естественной составляющей организма человека (в обычных условиях молочная кислота формируется при распаде глюкозы). Общая схема синтеза полилактида может быть проиллюстрирована следующим образом: 42 Первой стадией синтеза лактида является концентрирование промышленных водных растворов молочной кислоты. Для этой цели используют простую и вакуумную перегонку, а также азеотропную отгонку воды с различными азеотропообразователями. При простой и вакуумной перегонке наблюдается заметный унос молочной кислоты (до 3 %), в зависимости от условий процесса. Более выгодно использовать гетероазеотропы (бензол, толуол, хлорбензол, изопропилбензол и т.п.), так как в этом случае азеотропы легко разделяются и азеотропообразователь без дополнительной очистки может быть снова использован в процессе концентрирования растворов молочной кислоты. Цель работы: провести дегидратацию 2-гидроксипропионовой (молочной) кислоты Реактивы: молочная кислота, оксид цинка, этилацетат. Оборудование: ротационный вакуумный испаритель, вакуумный насос, ловушка с силикагелем, вакуум-сушильный шкаф Memmert, весы, прибор для определения температуры плавления Melting Point M 560, высокоэффективный жидкостный хроматограф YL9100 HPLC System, газовый хроматограф Хромос ГХ-1000. Проведение опыта: В данной работе используется молочную кислоту 80%-й концентрации производителей CKИMК (L-D-изомеры) и PURAC (L-изомеры) в количестве 30 мл. Концентрирование водных растворов молочной кислоты осуществляется в течении 1,5 ч на ротационном вакуумном испарителе Heidolph при температуре 100 – 140 °С и постепенном увеличении вакуума до 40-50 мбар (рис. 12). Исходный раствор подается в круглодонную колбу испарителя, где при вращении ротора он распре43 деляется в виде пленки на внутренней поверхности колбы. За счет образования большой поверхности происходит интенсивное испарение воды с поверхности пленки. 6 5 4 7 1 3 2 8 Рисунок 12. Роторный испаритель: 1 – круглодонная колба, 2 – баня, 3 – двигатель, 4 – штатив, 5 – лапка, 6 – обратный холодильник, 7 – кран для быстрого сброса вакуума в системе или для впуска в систему инертного газа, 8 – колбаприёмник Получаемый дистиллят (вода с примесями карбоновых кислот) охлаждался в холодильнике водой, конденсируется и собирается в приемной колбе. Несконденсировавшиеся пары, отсасываемые мембранным вакуумным насосом LABOPORT N 820.3. FT.18 , проходят последовательно ловушку, наполненную прокаленным силикагелем и далее сбрасываются под тягу. Через 1,5 часа в колбу добавляют катализатор олигомеризации (окись цинка) в количестве 1,5% от массы олигомера. 44 И проводят процесс олигомеризации еще 1,5-2 часа, пока реакционная смесь не загустеет. Синтез лактида проводится в круглодонной колбе, снабженной термометром, электромагнитной мешалкой (IKA C-MAG HS7) и колбой-приемником в интервале температур 180–240 °С и вакууме 3– 8 mbar. Полученный лактид-сырец представляет собой расплав от белого до светло-желтого цвета, который кристаллизуется при комнатной температуре. Лактид-сырец, получаемый при деполимеризации олигомера молочной кислоты, обычно в основном содержит в качестве примесей исходную кислоту и олигомеры, которые значительно в дальнейшем синтезе ухудшают свойства полимеров и сополимеров. Полученный лактид-сырец очищают методом перекристаллизации из осушенного этилацетата в соотношении 1:3 (лактид:этилацетат), и далее высушивают в вакуум сушильном шкафу при 55 °С с применением неглубокого вакуума (порядка 300 mbar). Высушенный до постоянного веса лактид исследуют методом ИК-спектроскопии (ИК-Фурье спектрометр Nicolet 5700) в диапазоне длин волн 1000–3000 см-1. Та часть лактида, которая была растворена в маточнике может быть выделена после отгонки этилацетата. Маточник лактида помещают в круглодонную колбу установки прямой пергонки (рис. 3) и постепенно нагревают до 77 °C. В процессе отгонки растворителя берут пробу из колбы на чашку Петри, если из пробы выпадают кристаллы, то процесс отгонки останавливают. Из колбы отгоняется примерно 20 % этилацетата, при этом постоянно необходимо брать пробу. Насыщенный раствор лактида сливают в стакан, охлаждают, отфильтровывают осадок и высушивают. Рекомендуется проводить процесс выделения лактида, используя маточники после второй и третьей перекристаллизации лактида-сырца, так как при этом получают более чистое вещество. Регенерированный этилацетат можно после осушки снова использовать для очистки лактида. На последнем этапе работы определяют суммарный выход лактида и сравнивают физико-химические характеристика продукта с литературными данными. 45 8. ПРОЦЕССЫ ПРИСОЕДИНЕНИЯ И КОНДЕНСАЦИИ ПО КАРБОНИЛЬНОЙ ГРУППЕ Альдегиды и кетоны благодаря своей высокой реакционной способности и доступности нашли широкое применение в промышленности основного органического синтеза для производства большого числа весьма ценных продуктов (капролактама, изопрена, пентаэритрина, дифенилолпропана, бутандиола-1,4, аминов и т.д.). Многочисленные реакции, в которые вступают альдегиды и кетоны, можно условно разделить на два основных типа. К первому типу относятся реакции с веществами, обладающими π-электронами и неподеленными электронными парами на гетероатоме (органические основания, спирты, олефины, ароматические соединения и др.). Эти реакции обычно катализируются протонными кислотами (серной, соляной): + O + H R1 C H R1 C + + OH R1 C H R1 C + OH H + OH C H R1 OH + H CH R1 + OH + H + H CH R1 OH Ко второму типу относятся реакции с слабыми кислотами и псевдокислотами, т.е. реакции с веществами, содержащими подвижный атом водорода (синильная кислота, ацетилен, карбонильные соединения и др.). Эти реакции катализируются основаниями и протекают по схеме: 46 O _ + OH H3C C H H2C O C H + O H3C CH CH2 C H3C C H2C O H O + H2O C H O H3C CH CH2 C OH H3C CH CH2 C O + H2O H O H O _ + OH H Когда взаимодействуют два различных карбонильных соединений, то каждое из них может быть как метиленовой, так и карбонильной компонентой. Обычно метиленовой компонентой становится то карбонильное соединение, у которого атомы водорода в α-метиленовой группе более подвижны, а карбонильной – то соединение, у которого способность карбонильной группы к присоединению больше. Например, роль метиленовой компоненты при взаимодействии альдегида с кетоном, как правило, выполняет кетон, а при конденсации двух различных альдегидов – альдегид с более длинной и разветвленной цепью углеродных атомов. Реакционная способность карбонильных соединений зависит от величины частичного положительного заряда на атоме углерода карбонильной группы. Чем больше этот заряд, тем реакционная способность карбонильного соединения выше. Вследствие того, что алкильные группы оказывают положительный индуктивный эффект, реакционная способность карбонильных соединений снижается с увеличением длины и с разветвлением углеродной цепи алкильной группы. По этой причине кетоны значительно менее реакционноспособны, чем альдегиды, так как в кетонах с карбонильным атомом углерода связаны две алкильные группы. 8.1. Получение дифенилолпропана Дифенилолпропан [2,2-бис(n-оксифенил)-пропан] в больших количествах используют для получения эпоксидных фенолоформальдегидных смол и поликарбонатов. Дифенилолпропан синтезируют из фенола и ацетона при катализе кислотами: 47 O 2 OH + CH3 C CH3 + H OH CH3 C CH3 OH + H2O Основными побочными продуктами являются 2,4дифенилолпропан, окись мезитила и высшие фенолы. Высшие фенолы образуются при взаимодействии окиси мезитила с фенолом: 2 CH3COCH3 _ H2O CH3 CO CH C (CH3)2 + C6H5OH высшие олефины В качестве катализаторов реакции конденсации фенола с ацетоном применяют серную и соляную кислоты, ионообменные смолы и др. При сернокислотном катализе образование фенолсульфокислоты ограничивает допустимую концентрацию катализатора и температуру. В промышленности обычно применяют 70 – 74% - ную серную кислоту с добавкой тиогликолевой кислоты в качестве промотора. Цель работы: Изучение реакции конденсации фенола с ацетоном при катализе серной кислотой и влияния промотора (тиогликолевой кислоты) на выход целевого продукта. Реактивы: фенол (94 г), ацетон (29 г), 74%-ная серная кислота (240 мл), тиогликолевая кислота (0,5 г), хлорбензол (120 мл), 0,25%-ный водный раствор аммиака (500 мл), толуол (6 мл). Оборудование: колба круглодонная, холодильник обратный, капельная воронка, воронка делительная, стакан, мешалка, баня, электроплитка, термометр, колба коническая, мешалка. Проведение опыта: Конденсацию фенола с ацетоном проводят на установке, схема которой приведена на рисунке 6. Установку собирают в вытяжном шкафу с хорошо действующей тягой. Перед началом опыта проверяют правильность сборки и герметичность всех соединений установки. Убеждаются в надежной работе механической мешалки и реле-регулятора. Готовят необходимые растворы серной кислоты и аммиака. Реактор изготовлен из термостойкого стекла и представляет собой круглодонную четырехгорлую колбу емкостью 500 мл. Проводят два опыта при температуре 40 °С и мольном соотношении фенол:ацетон:серная кислота, равном 1:0,5:3, с промотором (тиогликолевой кислотой) и без него, но при одинаковой интенсивности перемешивания и продолжительности реакции. Большой избыток серной 48 кислоты необходим для предотвращения затвердевания реакционной массы, добавка небольшого количества толуола способствует образованию более крупных кристаллов дифенилолпропана и тем самым облегчает его выделение и очистку. В реактор загружают 47 г фенола, 120 мл 74%-ной серной кислоты, 3 мл толуола и 0,5 г тиогликолевой кислоты (если опыт проводится с промотором). Осторожно включают мешалку, обогрев водяной бани и продают воду в обратный холодильник. По достижении заданной температуры увеличивают интенсивность перемешивания и из капельной воронки подают в реактор 14,5 г ацетона в течение 10–15 мин. Момент включения подачи ацетона принимают за начало опыта. В ходе подачи ацетона реакционную массу охлаждают, но интенсивность охлаждения регулируют таким образом, чтобы температура в реакторе не превышала 40 °С. Через 30–40 мин после окончания подачи ацетона в реактор добавляют 40 мл хлорбензола, так как в процессе конденсации фенола с ацетоном образующийся дифенилолпропан выпадает в осадок и затрудняет перемешивание реакционной массы. Продолжительность опыта 2 ч. После эксперимента реакционную массу охлаждают до 15 °С и образовавшийся осадок отфильтровывают на воронке Бюхнера, промывают его последовательно холодной водой (1 л), 0,25 %-ным раствора аммиака (50 мл) и еще раз холодной воды. В термостойкий стакан переносят осадок, приливают хлорбензола (40 мл) и при перемешивании стеклянной палочкой нагревают на электроплитке с закрытой спиралью до 100 °С и выдерживают 30 мин при постоянной температуре. По истечении времени содержимое стакана охлаждают до 15 °С, выпавшие кристаллы дифенилолпропана отфильтровывают на воронке Бюхнера и промывают холодным хлорбензолом (20 мл), переносят массу на фильтровальную бумагу и сушат в вакуум-сушильном шкафу при 70 °С. После сушки до постоянного веса определяют массу и температуру плавления полученного продукта. Составляют материальный баланс опыта, обсуждают результаты и формулируют свои выводы о выполненной работе. 8.2. Получение пентаэритрита Пентаэритрин представляет собой четырехатомный спирт и применяется для производства синтетических смол, пластификаторов и взрывчатых веществ. Пентаэритрин получают конденсацией ацетальде49 гида с формальдегидом в присутствии щелочных катализаторов (растворов гидроксидов калия, кальция и др.). Процесс протекает в две стадии. На первой стадии в результате реакции альдольной конденсации ацетальдегида с формальдегидом образуется триметилолацетальдегид: СН3–СНО + 3НСНО + ОН− → (НОСН2)3С–СНО На второй стадии, образовавшийся триметилолацетальдегид реагирует с формальдегидом по реакции Кониццаро, т.е. формальдегид восстанавливает триметилолацетальдегид в пентаэритрит, а сам окисляется до муравьиной кислоты: (НОСН2)3С–СНО + 0,5 Ca(OH)2 → (НОСН2)3ССН2ОH + 0,5(HCOO)2Ca Основным побочным продуктом процесса является дипентаэритрит (НОСН2)3ССН2ОСН2С(СН2ОН)3, выход которого достигает 10 %. Образуются и другие побочные продукты более высокой степени конденсации. В промышленности конденсацию альдегидов проводят в водном растворе (содержание альдегидов 12–15 %) при мольном отношении ацетальдегида к формальдегиду, равном 1:5, температуре 15 °С и атмосферном давлении. В качестве катализатора используют гидроксид кальция (известковое молоко), содержащий 0,1 моль/л Са(ОН)2. Цель работы: Изучение реакции конденсации ацетальдегида с формальдегидом, определение чистоты полученного пентаэритрина и составление материального баланса опыта. Реактивы: 25%-ный водный раствор ацетальдегид (41 мл), 35%ный водный раствор формальдегид (100 мл), оксид кальция (10 г), 20%ная серная кислота (100 мл), 96%-ный этиловый спирт (70 мл), соляная кислота концентрированная (4 мл), бензальдегид (5 мл), дистиллированная вода (200 мл). Оборудование: колба круглодонная, холодильник обратный, капельная воронка, воронка делительная, стакан, мешалка, баня, электроплитка, термометр, колба коническая, мешалка. Проведение опыта: Конденсацию ацетальдегида с формальдегидом проводят на установке, схема которой приведена на рисунке 6. Установку собирают в вытяжном шкафу с хорошо действующей тягой. Перед началом опыта проверяют герметичность всех соединений установки и правильность 50 сборки, надежность работы механической мешалки. Готовят необходимые растворы ацетальдегида и формальдегида. В реактор загружают 10 г оксида кальция и 50 мл дистиллированной воды, включают холодильник, мешалку и тщательно перемешивают содержимое колбы. Полученное известковое молоко охлаждают до 15 °С на водяной бане и приливают к нему 35%-ный водный раствор формальдегида (100 мл). Реакционную массу вновь нагревают до 15 °С и постепенно добавляют 41 мл 25%-ного водного раствора ацетальдегида из капельной воронки. В процессе прибавления ацетальдегида постоянно поддерживают температуру и энергично перемешивают реакционную массу. После полного добавления ацетальдегида включают обогрев водяной бани и доводят температуру реакционной массы до 45 °С. Выдерживают при данной температуре в течение 1 ч. После реакционную массу охлаждают до комнатной температуры и нейтрализуют 20%-ной серной кислотой до нейтральной реакции по лакмусовой бумаге. Жидкие продукты реакции отделяют от осадка в колбу Кляйзена через фильтр. Осадок промывают на фильтре дистиллированной водой (20 мл), а из фильтрата с помощью вакуума, создаваемом водоструйным насосом, отгоняют воду. Процесс отгонки воды ведут при 70 °С в атмосфере азота до образования мягкого осадка. После этого давление в колбе повышают до атмосферного, водоструйный насос выключают и к осадку приливают 50 мл горячего этилового спирта. Спиртовый раствор пентаэритрита выливают в стакан (объемом 150 мл), охлаждают до 15 °С и выдерживают при данной температуре 2 ч. Выпавший осадок отфильтровывают на воронке Бюхнера, отжимают между несколькими слоями фильтровальной бумаги и сушит на воздухе. Определяют массу и температуру плавления полученного продукта. Обсуждают результаты опыта и формулируют свои выводы о выполненной работе. 8.3. Получение окиси мезитила Окись мезитила – ненасыщенный кетон алифатического ряда – бесцветная жидкость с сильным запахом мяты. Применяют как растворитель эфиров целлюлозы и поливиниловых смол, а также в качестве полупродукта в производстве лекарственных средств, инсектицидов, высыхающих масел и как экстрагент для актиноидов (торий, уран и др.) и при флотации руд. В промышленности окись мезитила получают де51 гидратацией диацетонового спирта в присутствии иода, минеральных кислот, хлоридов и бромидов железа при 100–120 °С и 100 кПа с выходом 95 % в две стадии: а) реакция конденсации ацетона в диацетоновый спирт: OH 2 H3C C CH3 H3C C O O CH2 C CH3 CH3 б) дегидратация диацетонового спирта в окись мезитила: O OH H3C C O CH2 C CH3 CH3 H3C _H O 2 C CH C CH3 CH3 Цель работы: Изучение реакции конденсации ацетона в диацетоновый спирт и дегидратации диацетонового спирта в окись мезитила, определение физических констант полученного продукта. Реактивы: ацетон (58 г), гидроксид кальция (0,25 г), щавелевая кислота (2,7 г), изопропилбензол (25 мл). Оборудование: колба круглодонная, холодильник обратный, капельная воронка, воронка делительная, стакан, мешалка, баня, электроплитка, термометр, колба коническая, мешалка. Проведение опыта: Получение окиси мезитила из ацетона в две стадии осуществляют на установке, схема которой приведена на рисунке 3. Перед работой необходимо проверить правильность сборки и герметичность всех соединений установки. Убеждаются в надежной работе механической мешалки и готовят ледяную баню вместо водяной бани. В реактор загружают 58 г ацетона, охлаждают его до 5 °С на ледяной бане, добавляют 0,25 г тонкоизмельченного гидроксида кальция и выдерживают реакционную массу при этой температуре, энергично перемешивая ее в течение 1,5 часа. После синтеза нейтрализуют гидроксид кальция щавелевой кислотой до кислой реакции по лакмусовой бумаге. Ледяную баню нейтрализуют электронагревателем, на место обратного холодильника устанав52 ливают нисходящий холодильник, приливают 15 мл изопропилбензола и отгоняют непрореагировавший ацетон. После отгонки ацетона к остатку добавляют 2,7 г тонкоизмельченной щавелевой кислоты и поднимают температуру реакционной массы до 110 °С и выдерживают при этой температуре 1 час. По окончании реакции массу охлаждают, фильтруют в куб ректификационной колонки, осадок на фильтре промывают 10 мл изопропилбензола и перегоняют. Выделяют головную фракцию, выкипающую до 128 °С, и фракцию окиси мезитила с пределами кипения от 128 до 131 °С. Определяют массу окиси мезитила, показатель преломления, анализируют его на содержание карбонильной группы и олефина. Составляют материальный баланс опыта, обсуждают результаты опыта и формулируют свои выводы о выполненной работе. 8.4. Получение 4-аминодифениламина 4-Аминодифениламин (4-АДФА) широко применяют в качестве промежуточного продукта при получении алкилированных производных 4-АДФА, используемых в качестве антиозонантов, антиоксидантов и стабилизаторов мономеров и различных полимерных материалов. Известные способы промышленного получения 4-АДФА базируются в основном на гидрировании 4-нитрозодифениламина (4нитрозоДФА) или 4-нитродифениламина (4-нитроДФА). Синтез 4-нитрозоДФА или 4-нитроДФА включает, как правило, несколько технологически сложных стадий, сопровождающихся получением ряда побочных продуктов, органических и неорганических отходов, подлежащих уничтожению, что является одной из самых серьезных проблем данного способа получения. Экологически и экономически предпочтительные альтернативные способы получения промежуточных соединений 4-АДФА включают прямое взаимодействие анилина и нитробензола. В настоящее время предложен способ получения 4-АДФА с использованием на стадии конденсации полимерного катализатора, содержащего четвертичную аммонийную группу ~N(CH3)3+. Применение данного типа катализатора исключает стадию его отделения от реакционной массы, снимает жесткие температурные и 53 концентрационные ограничения на последующих стадиях проведения процесса получения 4-АДФА, дает высокий выход продуктов: Цель работы: изучение реакции конденсации анилина с нитробензолом в щелочной среде с использованием в качестве катализатора сильноосновного анионита. Реактивы: нитробензол, диметилсульфоксид (ДМСО), гидроксид натрия марки «чда», анионит АВ-17-8 ГОСТ 20301-74, толуол, уксусная кислота, хлорид тетраметиламмония, анилин. Оборудование: колба круглодонная трехгорлая, холодильник обратный, мешалка механическая, стакан химический, термометр, капельная воронка, фильтр Шотта, колба Бунзена, баня, электроплитка. 54 Проведение опыта: Синтез 4-нитро- и 4-нитрозодифениламинов конденсацией анилина с нитробензолом проводят на установке представленной на рисунке 6. В трехгорлую колбу, снабженную капельной воронкой, механической мешалкой и термометром загружают 6,99 мл 40%-го водного раствора NaОН, 34,6 мл анилина и добавляют катализатор хлорид тетраметиламмония 13,69 г. Включают механическую мешалку, смесь нагревают до 60 °С и начинают дозировку нитробензола в количестве 9,75 мл. Продолжительность дозирования нитробензола 90 минут, продолжительность синтеза – 6 ч. Контроль над процессом конденсации осуществляется при помощи тонкослойной хроматографии (пластинки Silufol UV 254, элюент гексан-этиловый спирт в объемном соотношении 2:1) и УФ спектроскопии. После проведения синтеза, всю реакционную массу отфильтровывают на фильтре Шотта от механических примесей, добавляют к ней 800 мл дистиллированной воды. Реакционную смесь подкисляют до pH 7,0–7,5 концентрированной HCl, отгоняют 300–350 мл азеотропной смеси вода-анилин-нитробензол, охлаждают до 5–10 °С и отфильтровывают осадок 4-нитрозодифениламина (температура плавления 140–142 °С). Фильтрат подкисляют до pH 2,5–3,0, отфильтровывают осадок 4нитродифениламина (температура плавления 131–133 °С). Определяют выходы и температуры плавления и проводят спектрофотометричсекий и хроматографический анализ. Хроматографическое обнаружение 4-нитрои 4нитрозодифениламинов. При смешении анилина с нитробензолом в присутствии щелочи (NaOH) и высокоосновного анионита (АВ-17-8) наблюдается окрашивание раствора в бурый цвет. Появление интенсивной окраски реакционного раствора указывает на образование нитро- и нитрозосоединений. Кроме анилина (он взят с 4-х кратным избытком по отношению к нитробензолу) в реакционной смеси присутствуют еще два вещества с Rf 0,66 (п-нитроДФА) и 0,73 (пнитрозоДФА) соответственно. Спектрофотометрическое обнаружение 4-нитрои 4нитрозодифениламинов. Исходные реагенты анилин и нитробензол имеют полосы поглощения в ультрафиолетовой области. 4нитродифениламин и 4-нитрозодифениламин имеют характерные полосы поглощения в видимой области (410 и 440 нм соответственно). Поэтому за ходом реакции можно следить по изменению поглощения в видимой области спектра в диапазоне длин волн 400–500 нм. 55 8.5. Получение капролактама Каполактам – лактам ɛ-Аминокапроновой кислоты представляет собой бесцветное кристаллическое вещество, маслянистое на ощупь. Практически весь вырабатываемый капролактам идёт на синтез полиамида-6, который применяется для получения капроновых волокон. Для получения капролактама предложено несколько промышленных способов, где в качестве сырья могут быть использованы такие углеводороды, как бензол, циклогексан, толуол, ацетилен. Первой стадией синтеза капролактама, исходя из циклогексанона, является превращение циклогексанона в циклогексаноноксим. Это превращение осуществляется действием водного раствора сернокислого гидроксиламина: 2 O + (NH2OH)2H2SO4 + H2O NOH H2SO4 +2 NH3 NOH + (NH4)2SO4 На второй стадии под действием олеума происходит изомеризация циклогексаноноксима в капролактам (бекмановская перегруппровка): + N + H + N OH2 H2C + N _H O 2 H2C CH2 C + CH2 CH2 + H2O H2C H2C CH2 C O NH CH2 CH2 H2C H2C CH2 C OH N CH2 CH2 H2C _ + H H2C N CH2 C + OH2 N CH2 CH2 При этом происходит расширение цикла с внедрением NH-группы между двумя атомами углерода. Цель работы: Получить из циклогексанола капролактам и оценить факторы, влияющие на его выход и чистоту. Получение циклогексаноноксима Реактивы: циклогексанон ( 16,3 г), сернокислый гидроксиламин (28 г), углекислый натрий, безводный (32 г), дистиллированная вода – 80 мл. 56 Оборудование: колба круглодонная трехгорлая, холодильник обратный, мешалка механическая, стакан химический, термометр, капельная воронка, фильтр Шотта, колба Бунзена, баня, электроплитка. Проведение опыта: В круглодонной трёхгорлой колбе (рис. 6) растворяют 28 г серногокислого гидроксиламина и приливают 80 мл дистиллированной воды. При быстром перемешивании прикапывают из капельной воронки в течение 60 мин 16,3 г циклогексанона. Во время прикапывания с помощью водяной бани поддерживают температуру смеси равной 40 °С. Затем смесь осторожно нейтрализуют до pH=4 по универсальному индикатору 20 %-ным раствором углекислого натрия. При нейтрализации циклогексаноноксим выпадает в осадок, который отфильтровывают на воронке Бюхнера. Осадок оксима сушат на воздухе или в вакуум-сушильном шкафу при 40 °С. После полного высыхания определяют массу и температуру плавления полученного циклогексаноноксима, считают выход и составляют материальный баланс опыта. Получение капролактама Реактивы: циклогексаноноксим (19 г), 98-%-ная серная кислота (64 г), водный раствор аммиака, хлороформ (107 мл), лед (83 г). Оборудование: колба круглодонная трехгорлая, холодильник обратный, мешалка механическая, стакан химический, термометр, капельная воронка, фильтр Шотта, колба Бунзена, баня, электроплитка. Проведение опыта: Перед началом опыта в химический стакан, емкостью 100 мл, наливают 19 мл серной кислоты, стакан помещают в ледяную баню и небольшими порциями при непрерывном перемешивании очень осторожно прибавляют 19 г циклогексаноноксима. ОСТОРОЖНО! (Возможен выброс в результате перегрева). В результате получают суспензию циклогексаноноксима в серной кислоте, которую заливают в капельную воронку. В трехгорлую колбу (рис. 6) приливают 14 мл концентрированной серной кислоты. Содержимое колбы нагревают на масляной бане при постоянном перемешивании до 120 °С и выдерживая температуру прикапывают раствор циклогексаноноксима в серной кислоте из капельной воронки в течение 60 мин. При низкой температуре процесса капролактам образуется медленно, поэтому в колбе накапливаются исходные реагенты, которые при 57 нагревании могут быстро прореагировать, что приведет к быстрому подъему температуры и вызовет сильное осмоление. Следовательно при температуре реакционной массы меньше 115 °С добавление раствора циклогексаноноксима прекращают до тех пор, пока температура в колбе не поднимется до 120 °С. После прибавления всего раствора циклогексаноноксима нагревание при непрерывном перемешивании и 120–130 °С продолжают еще 20 мин, затем охлаждают. Холодную реакционную смесь выливают на измельченный лед (83 г). Стакан помещают в баню, заполненную охлаждающей смесью (лед и поваренная соль), нейтрализуют до pH=7 концентрированным водным раствором аммиака, поддерживая температуру при нейтрализации не выше 20 °С. В результате получают водный раствор капролактама, из которого его экстрагируют в делительной воронке энергичным встряхиванием с хлороформом (4 раза по 27 мл хлороформа). Экстракт сушат хлористым кальцием, хлороформ затем отгоняют под небольшим вакуумом, а капролактам при 12 мм рт.ст. и 140 °С. Отогнанный продукт выливают в предварительно взвешенную фарфоровую чашку, охлаждают до комнатной температуры и кристаллизации. Определяют его массу и температуру плавления и составляют материальный баланс опыта. 58 9. ПРОЦЕССЫ СУЛЬФИРОВАНИЯ И СУЛЬФАТИРОВАНИЯ Основными продуктами процессов сульфирования и сульфатирования являются поверхностно-активные вещества, которые широко применяют в промышленности и в быту. Эти процессы используют также для получения других весьма ценных многотоннажных продуктов: фенола и его производных, спиртов, ионообменных смол и т.д. Сульфированию подвергают ароматические и парафиновые углеводороды, а сульфатированию – олефины и спирты. Сульфирующими и сульфатирующими агентами служат концентрированная серная кислота, олеум, и пероксид серы (серный ангидрид). Кроме того, для сульфатирования спиртов используют органические комплексы триоксида серы с диоксаном и пиридином, хлорсульфоновую и сульфаминовую кислоты: ClSO2OH ROH SO3 NH2SO2OH ROSO2OH + HCl ROSO2OH ROSO2ONH4 Для сульфирования парафиновых углеводородов используют реакцию сульфохлорирования или сульфоокисления, так как перечисленные выше сульфирующие реагенты с парафиновыми углеводородами не взаимодействуют: RH + SO2 + Cl2 → RSO2Cl + HCl RH + SO2 + 0,5 O2 → RSO2OH 9.1. Получение n-толуолсульфокислоты n-Толуолсульфокислоту применяют для синтеза n-крезола, в качестве более мягкого, чем серная кислота, кислотного катализатора, при анализе аминов и т.д. Ее получают обычно прямым сульфированием толуола серной кислотой: 59 CH3 + H2SO4 HOSO2 CH3 + H2O Параллельно с n-толуолсульфокислотой образуется некоторое количество о-толуолсульфокислоты, так как метильная группа является заместителем первого рода. Для предотвращения образования ди- и полисульфокислот сульфирование ведут в избытке толуола и при хорошем перемешивании реакционной массы, а для смещения равновесия реакции в сторону образования сульфокислоты реакционную воду в процессе сульфирования отгоняют в виде азеотропной смеси с толуолом. Цель работы: Изучение реакции сульфирования толуола серной кислотой, выделение n-толуолсульфокислоты, определение температуры плавления, кислотного числа и составление материального баланса опыта. Реактивы: толуол (46,1 г), серная кислота концентрированная (13,3 г), соляная кислота концентрированная (120 мл), гидроксид калия (30 г) Оборудование: электроплитка, баня, круглодонная колба, термометр, насадка Дина-Старка, мешалка, холодильник. Проведение опыта: Сульфирование толуола серной кислотой ведут на установке, схема которой приведена на рисунке 6. Перед началом опыта проверяют правильность сборки и герметичность всех соединений установки. Убеждаются в надежной работе мешалки. Важно! Опыт проводят в резиновых перчатках и защитных очках при соблюдении правил работы с агрессивными веществами! В реактор, помещенный в песочную баню, осторожно загружают 46,1 г толуола и 13,3 г концентрированной серной кислоты. Устанавливают термометр, включают обратный холодильник, мешалку и электроплитку. Реакционную массу нагревают при энергичном перемешивании до умеренного кипения. Опыт продолжают до прекращения отгонки воды в насадку Дина-Старка, примерное время реакции на массу загруженного сырья составляет 5 ч. После выделения максимально возможного количества воды реакционную массу охлаждают до комнатной температуры, добавляют дистиллированной воды (3 мл). Выпавшие кристаллы nтолуолсульфокислоты отфильтровывают с помощью фильтра Шотта. 60 Побочно образовавшаяся о-толуолсульфокислота удаляется с фильтратом. Выделенные кристаллы n-толуолсульфокислоты подвергают очистке. Для этого их растворяют в 20 мл горячей воды, охлаждают до комнатной температуры, приливают 50 мл концентрированной соляной кислоты и охлаждают на ледяной бане. Выпавшие кристаллы отделяют на воронке со стеклянным фильтром и промывают холодной концентрированной соляной кислотой в объеме 10 мл. Очистку проводят дважды. После второй очистки кристаллы хорошо отжимают на фильтре с помощью стеклянной пробки, переносят в предварительно взвешенную чашку и сушат в эксикаторе над твердым гидроксидом калия до постоянной массы. После сушки определяют массу полученного продукта, его температуру плавления и составляют материальный баланс эксперимента. 61 10. ПРОЦЕССЫ ДЕГИДРИРОВАНИЯ Химические процессы, связанные с отщеплением атомов водорода от органического соединения называют дегидрированием. Повышение температуры и понижение давления, а также разбавление реакционной смеси способствует смещению равновесия в сторону дегидрирования. Катализаторами дегидрирования являются металлы (никель, платина, палладий, медь, серебро) и полупроводниковые оксиды (Fe2O3, Cr2O3, ZnO, MoO3). Наиболее типичные реакции можно классифицировать по виду связей между атомами, от которых отщепляется водород: H3C CH2 CH2 CH3 - H2 H2C CH CH2 CH3 C6H5C2H5 RCH2OH RCH2OH RCH2NH2 - H2 H2C CH CH CH2 C6H5 CH CH2 + H2 R1 C O + H2 O R2CO + H2 RCN + 2 H2 Важное значение имеют процессы дегидроциклизации, дегидроконденсации и окислительного дегидрирования: C6H14 + H2 - H2 + H2 2 CH2 CH2 + 2 H2 62 2СH3OH + 0,5O2 ↔ 2HCOH + H2O + H2 В промышленном органическом синтезе процессы дегидрирования занимают одну из ведущих ролей. Так например, дегидрированием спиртов получают: формальдегид, ацетон, метилэтилкетон, циклогексанон; дегидрированием алкилароматических соединений – стирол, αметилстирол, винилтолуол, дивинилбензол; дегидрированием парафинов получают: пропилен, бутилен, изобутилен, изопентен, высшие олефины, бутадиен и изопрен. 10.1. Получение α-метилстирола дегидрированием изопропилбензола Большое количество стирола идет на производство пластмасс, обладающих хорошими электроизоляционными свойствами и большой химической стойкостью. α-Метилстирол используют для производства синтетических каучуков, но в производстве пластических масс его используют в ограниченном количестве. Дегидрирование этилбензола или изопропилбензола является в настоящее время основным способом получения стирола: H2C CH3 HC CH2 H3C H3C HC CH3 C CH2 - H2 - H2 При этом протекают побочные реакции: H3C HC CH3 H2C CH3 + H2 + H2 H2C CH3 + H2 + CH4 + C2H6 HC CH2 CH3 + CH4 + CH4 CH3 + C2H4 + C2H4 63 Цель работы: Получить α-метилстирол дегидрированием изопропилбензола и рассмотреть факторы, влияющие на выход продукта. Реактивы: Изопропилбензол, раствор бромистого натрия и брома в метаноле, 10 %-ный водный раствор йодистого калия, 0,1 н раствор тиосульфата натрия, крахмал. Оборудование: реактор, насос, поглотительная склянка, конические колбы, бюретка, холодильник обратный, газометр. Проведение опыта: Дегидрирование кумола проводят на специально собранной стендовой установке (рис. 9), которую перед началом работы проверяют на герметичность. После включают электрообогрев печи реактора и доводят температуру в реакционной зоне до 550–600 ºС, после чего устанавливают подачу кумола в реакторе со скоростью 1 мл/мин. Объем кумола идущего на 1 опыт составляет примерно 50 мл. В процессе синтеза замеряют объем вытекающей воды, собирающейся в мерной склянке. Количество образовавшейся воды соответствует количеству выделяющегося при реакции газа. После того, как весь объем кумола будет добавлен в реактор, выдерживают температуру реакции еще в течение 15 минут и после отключают обогрев печи. Замеряют объем выделившегося водорода, который будет равен объему вытесненной из газометра воды. Осторожно, стравливая газы через специальный шланг в тягу, заполняют газометр водой. Катализат из поглотительной склянки взвешивают и определяют его массу. Из двух проб катализата определяют среднее бромное число. Для этого в колбу с притертой крышкой наливают 20 мл раствора бромистого натрия и брома в метаноле, вносят 0,1– 0,2 г катализата и ставят колбу в темное место на 15 мин. После выдержки в темноте к реакционной массе добавляют 10 %-ный водный раствор KI в объеме 20 мл. Выделившийся в процессе реакции йод оттитровывают раствором тиосульфата натрия (0,1 н) в присутствии крахмала. Параллельно ставят холостой опыт. Бромное число (б.ч.) рассчитывают по формуле приведенной в разделе 4.1. После анализа проводят вычисления, замеряют объем газа, полученного в процессе дегидрирования и составляют материальный баланс процесса. 64 11. ПРОЦЕССЫ АЛКИЛИРОВАНИЯ Алкилированием называют процессы, в которых происходит введение алкильных групп в молекулы органических и неорганических соединений. Среди этих реакций большое практическое значение имеют процессы алкилирования ароматических соединений в ядро. Также важно проведение алкилирования изопарафинов, меркаптанов, аминов и др. Процессы алкилирования часто являются промежуточными стадиями в производстве мономеров, моющих веществ и т.д. В процессах алкилирования в качестве алкилирующих агентов используются: ненасыщенные соединения – олефины и ацетилен (у которых происходит разрыв связи между атомами углерода), хлорпроизводные с достаточно подвижным атомом хлора (способным замещаться под влиянием различных агентов), спирты, простые и сложные эфиры (например оксиды олефинов, у которых при алкилировании разрывается углерод-кислородная связь). Этилен, пропилен, бутилен и высшие имеют первостепенное значение в качестве алкилирующих агентов. Одна из классификаций процессов алкилирования основана на типе вновь образующейся связи, т. е. на том, с каким атомом связывается алкильная группа. По этой классификации различают: а) С-алкилирование парафиновых и ароматических углеводородов (замещение на алкильную группу атома водорода, находящегося при углеродном атоме): + RCl R1 + HCl б) О-алкилирование (связывание алкильной группы с атомом кислорода): OH + RCl NaOH O R1 + NaCl + H2O в) N-алкилирование (замещение атомов водорода в аммиаке или аминах на алкильные группы): NH3 + RCl → RNH2 + HCl NH3 + ROH → RNH2 + H2O 65 К продуктам алкилирования, которые производятся в очень крупных масштабах относятся: этилбензол, изопарафиновый алкилат, высшие алкилбензолы, гликоли и другие продукты превращения α-оксидов. Данные продукты являются не только промежуточными, но и широко используются в качестве компонентов топлив, растворителей, пластификаторов, присадок к маслам и других целях. 11.1. Получение изопропилбензола Изопропилбензол (кумол) – ароматическое соединение, которое в промышленности получают алкилированием бензола пропиленом. При этом протекает ряд побочных реакций заключающихся в последовательном введении алкильных радикалов в образовавшийся изопропилбензол: CH3 + + C H + R1 C CH3 H CH3 C H + R1 + H R1 H CH3 C H + R1 Катализатором процесса является хлоралюминиевый каталитический комплекс (АlСl3 с НСl и алкилароматическим углеводородом). Процесс проводят при 100–130 °С и мольном соотношении бензол:пропилен равным 3:3,5. За рубежом используют фосфорнокислотные катализаторы на твердом носителе. Процесс осуществляют при температуре около 200 °С и давлении 2,8–4,2 мПа. Для уменьшения выхода побочных продуктов процесс ведут при мольном соотношении бензол:пропилен 10:1. При это выход кумола достигает 96–97 %. Применяют изопропилбензол для производства фенола, ацетона, αметилстирола, и в качестве добавки к авиационным бензинам. Цель работы: Изучить реакцию алкилирования бензола пропиленом в присутствии хлористого алюминия и рассмотреть факторы, влияющие на выход продуктов алкилирования, определить константы скорости реакции. Реактивы: пропилен (1 моль), бензол (80–90 г), треххлористый алюминий (2–15 %), 5 %-ная соляная кислота. 66 Оборудование: реактор, насос, поглотительная склянка, конические колбы, бюретка, холодильник обратный, газометр. Проведение опыта: Бензол, катализатор и 1-2 капли воды (для активации катализатора) загружают в реактор и включают мешалку (рис. 13). Используемый бензол должен быть осушен с помощью CaCl2 и перегнан. Пропилен, необходимый для алкилирования получают путем дегидратации изопропилового спирта, на установке, описанной в разделе 5.2. Расход пропилена на алкилирование устанавливают 240–250 мл/мин. Момент включения подачи пропилена принимают за начало опыта. Выходящий из реактора непрореагировавший пропилен собирают в газометр. 2 5 3 âî äà âî äà 6 1 4 7 Рисунок 13. Схема лабораторной установки получения изопропилбензола: 1 – емкость для спирта, 2 – перистальтический насос, 3 – реактор, 4 – поглотительная склянка, 5 – поглотительная склянка, 6 – газометр, 7 – реактор дегидрирования Алкилирование ведут при температуре 25–50 °С, поддерживая температуру подачей холодной воды в рубашку реактора. На протяжении всего процесса проводят отбор трех проб по 25–30 мл через 30, 45 и 60 мин после начала опыта. Через час подачу спирта прекращают, и остаточный пропилен стравливают через тягу в атмосферу. Каждую 67 пробу для удаления AlCl3 промывают до нейтральной реакции 20–30 мл 5%-ной соляной кислоты, затем водой, сушат над хлористым кальцием, взвешивают и разгоняют. При перегонки выделяют фракции до 78 °С – бензола с водой в виде азеотропа (может отсутствовать), 78–85 °С – бензол, 85–155 °С – изопропилбензол, выше 155 °С – остаток, принимаемый за полиалкилбензолы. У отогнанных фракций определяют массу и показатели преломления, сравнивают с табличными значениями, делают выводы и составляют материальный баланс процесса. Далее определяют мольный состав каждой из проб и мольное содержание каждого компонента, допуская, что продукт, кипящий выше 155 °С, является полиалкилбензолом. По данным расчета строят график зависимости мольного состава от времени реакции и находят величину константы скорости первой и второй стадии алкилирования. 68 ЛИТЕРАТУРА Обязательная литература 1. Лебедев Н. Н. Химия и технология основного органического и нефтехимического синтеза: учебник. – М.: Альянс, Изд. IV, 2013. – 592 с. 2. Мозговой И. В. Технология органических веществ (курс лекций): учебное пособие. – Омск: Изд-во ОмГТУ, 2006. – 184 с. 3. Тимофеев В. С., Серафимов Л. А.. Принципы технологии основного органического и нефтехимического синтеза: учебное пособие для вузов, 2-е изд., перераб. – М.: Высшая школа, 2003. – 536 с. 4. Сухорослова М. М., Новиков В. Т., Бондалетов В. Г. Лабораторный практикум по химии и технологии органического синтеза. – Томск: Изд. ТПУ, 2002. – 132с. 5. Одабашян Г. В. Лабораторный практикум по химии и технологии основного органического и нефтехимического синтеза: учебное пособие. – М.: Химия, 1992. – 239 с. 6. Фитерер Е. П., Троян А. А., Новиков В. Т. Техника лабораторных работ: Учебное пособие. – Томск: Изд-во ТПУ, 2013. – 159 c. 7. Гутник С. П. Примеры и задачи по технологии органического синтеза: учебное пособие. – М.: Химия, 1984. – 192 с. 8. Гутник С. П., Сосонко В. Е., Гутман В. Д. Расчеты по технологии органического синтеза: учебное пособие. – М.: Химия, 1988. – 272 с. Дополнительная литература 9. Сорока Л. С., Волгина Т. Н. Промышленная органическая химия. Основной органический синтез: учебное пособие для вузов. – Томск: Издво ТПУ, 2006. – 163 с. 10. Мозговой И. В. Технология нефтехимического синтеза. Мономеры: учебное пособие. – Омск: Изд-во ОмГТУ, 2008. – 279 с. 11. Столяров В. А., Поконова Ю. В. Сырье и продукты промышленности органических и неорганических веществ. – 2005 г. – 1141 с. 12. Волгина Т. Н., Сорока Л. С., Мананкова А. А. Лабораторный практикум по промышленной органической химии. – Томск: Изд. ТПУ, 2009. – 100 с. 13. Фитерер Е. П., Троян А. А., Новиков В. Т. Техника лабораторных работ: Учебное пособие. – Томск: Изд-во ТПУ, 2013. – 159 c. 14. Химическая промышленность сегодня: научно-технический журнал. – М.: «Химпром Сегодня». 15. Химическая технология: ежемесячный производственный, научнотехнический журнал. – М.: ООО «Наука и технология». 69 ПРИЛОЖЕНИЕ 1. Образец титульного листа МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ Федеральное государственное автономное образовательное учреждение высшего образования «НАЦИОНАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ ТОМСКИЙ ПОЛИТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ» Институт природных ресурсов Направление Химическая технология АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ ОЛЕФИНАМИ: ПОЛУЧЕНИЕ ИЗОПРОПИЛБЕНЗОЛА Отчёт по лабораторной работе № 1 Вариант 1 по дисциплине: Химия и технология органических веществ Исполнитель: студент группы ____________ Иванов Иван Иванович 01.02.2014 группа Ф.И.О. студента дата _____________ _______________________ _____________ звание, должность Ф.И.О. преподавателя дата Руководитель: преподаватель Томск – 2014 70 Учебное издание ВОЛГИНА Татьяна Николаевна Химия и технология органических веществ Учебное пособие Научный редактор доктор филологических наук, профессор З.И. Резанова В авторской редакции Верстка Л.А. Егорова Отпечатано в Издательстве ТПУ в полном соответствии с качеством предоставленного оригинал-макета Подписано к печати Формат 60×84/16. Бумага «Снегурочка». Печать Xerox. Усл. печ. л. 5,46. Уч.-изд. л. 4,95. Заказ . Тираж экз. Национальный исследовательский Томский политехнический университет Система менеджмента качества Издательства Томского политехнического университета сертифицирована NATIONAL QUALITY ASSURANCE по стандарту BS EN ISO 9001:2008 . 634050, г. Томск, пр. Ленина, 30. Тел./факс: 8(3822)56-35-35, www.tpu.ru 71