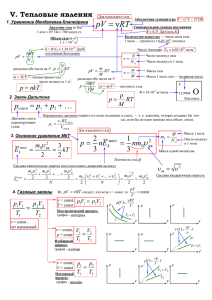
Законы термодинамики и имическое равновесие
advertisement

ФЕДЕРАЛЬНОЕ АГЕНСТВО ПО ОБРАЗОВАНИЮ КЕМЕРОВСКИЙ ТЕХНОЛОГИЧЕСКИЙ ИНСТИТУТ ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ Л.И. Холохонова Е.В. Короткая ЗАКОНЫ ТЕРМОДИНАМИКИ И ХИМИЧЕСКОЕ РАВНОВЕСИЕ Учебное пособие для студентов вузов Кемерово 2007 2 УДК: 536 (075) ББК. 22.317я7 Х73 Рецензенты: Т.Г. Черкасова, д-р. хим. наук, профессор, зав. кафедрой химии и технологии неорганических веществ КемГТУ; М.М. Колосова канд. хим. наук, доцент, зав. кафедрой химии Кемеровского государственного сельскохозяйственного института Рекомендовано редакционно-издательским советом Кемеровского технологического института пищевой промышленности Холохонова Л.И., Короткая Е.В. Х73 Законы термодинамики и химическое равновесие: Учебное пособие. – / Кемеровский технологический институт пищевой промышленности. – Кемерово, 2007. – 119 с. ISBN – Пособие освещает основные теоретические положения и закономерности химической термодинамики и учения о химическом равновесии, рассматривает методы решения типичных задач. Учебное пособие составлено в соответствии с программой дисциплины «Физическая химия» и предназначено для студентов технологических специальностей всех форм обучения. УДК: 536 (075) ББК. 22.317я7 ISBN – © Л.И. Холохонова, Е.В. Короткая, 2007 © КемТИПП, 2007 3 Введение В пособии рассматриваются основные теоретические положения и закономерности химической термодинамики и учения о химическом равновесии. Инженер любой специальности должен обладать достаточными знаниями в этой области, студенту химическая термодинамика необходима для успешного изучения последующих общенаучных и специальных дисциплин. При изучении теории (гл. 1, 2) рекомендуем Вам воспользоваться следующими советами. • При первом чтении особенно не задерживайтесь на математических выводах, старайтесь получить общее представление об изложенных вопросах. Внимательно прочитайте текст, выделенный жирным шрифтом, рассмотрите схемы, рисунки, графики. • При повторном чтении нужно усвоить все теоретические положения, математические зависимости и их выводы. Старайтесь понять сущность, а не запоминать отдельные факты. • Задания, которые предлагается выполнять при изучении теоретического материала, очень важны: это лучший способ закрепления опорных знаний и самоконтроля. Знания и умения, которые Вы получите при изучении гл. 3, помогут овладеть техникой термодинамических расчетов. В этой главе рассматриваются методики решения расчетных задач с подробным описанием хода решения и анализом результатов. Решая задачи самостоятельно, примите к сведению наши советы. • Некоторые задачи требуют длительных расчетов, причем ответы, полученные на отдельных стадиях, далее входят в заключительную стадию вычислений, поэтому не делайте арифметических ошибок, следите за размерностью и порядком величин, не нарушайте правил округления. • Научитесь анализировать полученные численные значения величин. Полезно пытаться предсказывать результаты расчетов и затем сравнивать с полученными. • Не ограничивайтесь заданиями, входящими в обязательный учебный план; старайтесь решить как можно больше задач. Это позволит Вам полнее раскрыть свой творческий потенциал и добиться успехов в профессиональной деятельности. 4 1. ОСНОВЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ 1.1. Что такое химическая термодинамика Слово «термодинамика» состоит из двух греческих слов «терме» – «теплота» и «динамос» – «сила». Термодинамика возникла как наука о превращениях тепла в механическое движение, в работу. В процессе своего развития термодинамика необычайно расширилась и приобрела характер фундаментальной физической науки. В основе термодинамики – три закона, которые служат такой же основой для построения «строгого и изящного здания» классической термодинамики, какой являются, например, законы Ньютона в классической механике. Законы термодинамики являются постулатами, справедливость которых подтверждается всем накопленным в результате человеческой деятельности опытом. В отличие от ньютоновской механики или евклидовой геометрии, принятая в термодинамике система постулатов является единственной, пригодной для изучения взаимосвязи материи и энергии. Хотя альтернативные постулаты и могут быть в принципе предложены, в настоящее время они не известны. Химическая термодинамика изучает, используя термодинамический метод, химические и физико-химические процессы; одна из её основных задач – предсказание направления химической реакции и расчет равновесного состава системы в зависимости от исходного состава реакционной смеси, её температуры и давления. Теоретическая база химической термодинамики – три основополагающих закона термодинамики и их следствия, которым в химической термодинамике придаётся форма, наиболее удобная для решения химических проблем. Огромный вклад в химическую термодинамику внесен Г.И. Гессом (основной закон термохимии, 1840 г), Дж. Гиббсом (метод термодинамических потенциалов 1876 – 78 г.), Г. Гельмгольцем (применение второго закона термодинамики к химическим реакциям, 1882 г.), Я. Вант-Гоффом (термодинамика химических реакций и растворов 1883 – 90 г.), А. Ле Шателье (принцип смещения равновесия 1883 – 88 г.), Б. Нернстом (третий закон термодинамики, 1906 г.), Г. Льюисом (метод активностей) и другими исследователями. Основные экспериментальные методы химической термодинамики - калориметрия, измерение изменения давления газовых систем при протекании в них химических процессов, измерение электродвижущих сил электрохимических элементов. Для практических расчетов используются таблицы термодинамических свойств индивидуальных веществ и ионов, в которых обычно приводятся значения теплот образования, энтропии, теплоемкости и некоторых вспомогательных функций. Методы химической термодинамики широко используются не только в химии, но и в биологии, геологии, металлургии. 5 1.2. Основные понятия и определения 1.2.1. Параметры и функции состояния системы Термодинамической системой называется тело или группа тел, находящихся во взаимодействии и условно обособленных от окружающей среды. В зависимости от рассматриваемого явления система может быть выбрана различного размера и сложности, но всегда она должна состоять из большого числа частиц, т.е. быть макроскопической. Только для макроскопических систем можно оперировать такими понятиями, как температура, давление, теплота и некоторыми другими. Система называется закрытой, если она не обменивается с окружающей средой веществом, т.е. не теряет и не получает вещество извне. В дальнейшем мы будем говорить только о закрытых системах. Состояние системы есть совокупность физических и химических свойств, характеризующих данную систему. Для характеристики состояния системы обычно используют следующие величины: 1) температура системы параметры 2) объем системы состояния системы 3) давление в системе 4) внутренняя энергия системы 5) энтальпия системы 6) энтропия системы 7) изохорно-изотермический потенциал системы 8) изобарно-изотермический потенциал системы Определение Пример функции состояния системы Внутренней энергией системы (U) называется совокупность всех видов энергии, которой обладает система, за исключением кинетической энергии системы в целом и ее потенциальной энергии положения. ∗∗∗ Пусть термодинамическая система представляет собой сосуд, в котором находится 1 моль (т.е. 6⋅1023 молекул) двухатомного газа. 6 В такой системе можно отметить следующие виды движения: 1) поступательное движение молекул газа, которому соответствует энергия Uпост., 2) вращательное движение молекул газа, мерой вращательного движения является энергия Uвращ., 3) колебательное движение атомов в молекулах, мерой этого движения является энергия Uколеб., 4) движение электронов вокруг атомов с энергией Uэл., 5) взаимодействия внутри атомов, характеризующиеся энергией Uяд. Внутренняя энергия системы, согласно определению, равна: Uсистемы = Uпост. + Uвращ. + Uколеб. + Uэл.+ Uяд. Если рассматриваемую в нашем примере термодинамическую систему (баллон с газом) поднять над землей на высоту h , то общая энергия системы увеличится на величину mgh. E полная энергия системы = U внутренняя энергия системы + mgh потенциальная энергия ситемы в целом Если термодинамическая система (баллон с газом) будет двигаться со скоростью υ, то общая энергия системы увеличится на величину m⋅υ2/2 E полная энергия системы = U внутренняя энергия системы + m ⋅υ 2 2 кинетическая энергия ситемы в целом Однако в соответствии с определением, ни кинетическая энергия системы в целом, ни ее потенциальная энергия положения не включается в понятие внутренней энергии системы. ∗∗∗ Определение Энтальпией системы (H) называется функция состояния системы, которая определяется выражением: H = U + P⋅⋅V (1.1) 7 Определение Энтропией системы называется функция состояния системы, которая характеризует хаотичность системы и определяется выражением S=R⋅⋅lnW , (1.2) где R – константа Больцмана; W – термодинамическая вероятность (число способов, которыми можно построить систему). Пример ∗∗∗ Пусть термодинамическая система представляет собой 1 моль (т.е. 6⋅1023 атомов) кристаллического, простого вещества. Рассмотрим два состояния системы: 1-е состояние – 2-е состояние – идеальное кристаллическое кристаллическое вещество, вещество, находящееся при находящееся при Т = 10 К (в Т = 0 К (в этом случае атотаком кристалле будет мы не колеблются, и потому какое-то количество атонет никаких нарушений кримов, покинувших свои мессталлической решетки вета и занявших места в щества) междоузлиях решетки) Такую систему можно построить только одним способом. Задание Такую систему можно построить большим числом способов. W >> 1 W=1 Таким образом, из наших рассуждений ясно, что система во втором состоянии (при Т = 10 K) более хаотична и W – является мерой хаотичности системы. (Чем больше W, тем хаотичнее система). Энтропия системы, связанная с W (термодинамической вероятностью системы) соотношением S=R⋅lnW, следовательно, также является мерой хаотичности системы. ∗∗∗ Ответить на вопросы: что больше а) энтропия 1 моля жидкой воды или энтропия 1 моля льда? б) энтропия 1 кг железа при Т = 300 К или энтропия 1 кг железа при Т = 800 К? ∗∗∗ 8 Определение Изохорно-изотермическим потенциалом системы (или энергией Гельмгольца) называется функция состояния системы, которая определяется выражением: F = U – T⋅⋅S Определение (1.3) Изобарно-изотермическим потенциалом системы (или энергией Гиббса) называется функция состояния системы, которая определяется выражением: G = H – T⋅⋅S (1.4) Величина функций состояния системы (U, H, S, F, G) зависит только от состояния системы, но не зависит от того, каким путем система пришла в это состояние, поэтому изменение функций состояния при переходе системы из состояния 1 в состояние 2 не зависит от пути процесса (см. рис.1.1). Рис.1.1. Схема термодинамического процесса U 2 − U1 = ∆U H 2 − H1 = ∆H S2 − S1 = ∆S не зависят от пути процесса F2 − F1 = ∆F G2 − G1 = ∆G Единицами измерения величин U, H, F, G являются единицы энергии (Дж, кДж), энтропия системы (S) измеряется в Дж/К, кДж/К. 1.2.2. Теплота и работа термодинамического процесса Определение Термодинамическим процессом называется всякое изменение в системе, связанное с изменением хотя бы одного из свойств системы. 9 Пусть термодинамическая система из состояния 1, характеризующегося параметрами P1, V1, T1 и функциями состояния U1, H1, S1, F1, G1 перешла в состояние 2, характеризующееся параметрами P2, V2, T2 и функциями состояния U2, H2, S2, F2, G2, благодаря протекающему в системе термодинамическому процессу (см. рис. 1.2) Рис. 1.2 Схема термодинамического процесса С точки зрения термодинамики суть любого процесса в том, что при его протекании в системе увеличивается или уменьшается интенсивность движения. Определение Увеличить интенсивность движения в системе можно 2-мя путями: 1) заставить каждую от- 2) вызвать движение знадельную молекулу двигать- чительных масс вещества. ся быстрее. Мерой передаваемого таким Мерой передаваемого таким путем движения является путем движения является величина, которая называ- величина, которая называется теплотой процесса. ется работой процесса. Пример ∗∗∗ Пусть термодинамическая система представляет собой 1 моль газа, находящегося в цилиндре с движущимся поршнем. Чтобы увеличить интенсивность движения в этой системе можно поступить 2-мя путями: 1) закрепить поршень и на2) сжать газ с помощью греть газ. В этом случае кажпоршня и заставить все модая молекула газа начнет двилекулы двигаться в одну стогаться быстрее и движение в рону. системе в целом станет инВ этом случае говорят, что тенсивнее. над системой совершена раВ этом случае говорят, что бота. система получила какое-то количество теплоты. ∗∗∗ 10 Теплота процесса Итак, любой термодинамический процесс характеризуется двумя величинами: теплотой и работой. Обе величины являются мерой передаваемого в результате данного процесса движения, т.е. мерой передаваемой энергии. Полезно помнить следующие сведения о теплоте процесса: 1. теплота процесса, приводящего к значительным изменениям в системе (макропроцесс), обозначается Q; приводящего к незначительным изменениям в системе (элементарный процесс), обозначается δ Q, 2. теплота измеряется в единицах энергии: Дж, кДж, 3. принято считать, что Q > 0, если система получает тепло из окружающей среды, Q < 0, если система отдает тепло, 4. теплота процесса, приводящего к изменению температуры системы от Т1 до Т2, может быть вычислена из соотношений: δ Q = С⋅⋅(T2 – T1) = С⋅⋅dT: T2 Q = ∫ C ⋅ dT : (1.5) (для элементарных процессов) (1.6) (для макропроцессов), T1 где С – теплоемкость системы; Работа процесса о работе процесса: 1. работа процесса, приводящего к значительным изменениям в системе, обозначается A, работа процесса, приводящего к незначительным изменениям в системе обозначается δ A, 2. работа процесса измеряется в Дж, кДж, 3. принято считать, что А > 0, если система совершает работу, А < 0, если над системой совершают работу, 4. виды работы весьма разнообразны: работа перемещения заряженных частиц в электрическом поле (электрическая работа), работа перемещения частиц в поле силы тяжести, работа сил поверхностного натяжения и др. Работу любого вида можно представить как произведение двух величин: фактора интенсивности на изменение фактора емкости; так электрическая работа это произведение величины перенесенного заряда (q) на разность потенциалов электрического поля (φ2 – φ1); работа перемещения частиц в поле сил тяжести произведение силы тяжести (m·g) на перемещение (h2 – h1) и т.д. В курсе химической термодинамики чаще всего рассмат- 11 риваются системы, которые не совершают какой-либо работы, кроме работы расширения газа. Пусть газ помещен в цилиндр с поршнем и его объем меняется от V1 до V2, тогда работа расширения определяется выражениями: δ A = P⋅⋅(V2 – V1) = P⋅⋅dV: V2 A = ∫ P ⋅ dV : (1.7) для элементарного процесса (1.8) для макропроцесса, V1 где P – давление газа под поршнем Задание Решение ∗∗∗ Пусть в системе, представляющей собой 100 г жидкой воды, находящейся при нормальном атмосферном давлении (P0) и температуре 300 K, некоторое количество воды (v моль) испарилось и превратилось в водяной пар, объем системы при этом увеличился от V1 до V2. Для протекания процесса потребовалось некоторое количество теплоты Q. Определить знак теплоты и работы процесса. ∗∗∗ Работа процесса испарения воды, протекающего в данных условиях – величина положительная и может быть рассчитана из соотношений: A= V2 ∫ p ⋅ dV = p ⋅ (V2 − V1 ) = vRT > 0 0 V1 Для преодоления сил межмолекулярного притяжения в жидкой фазе необходимо некоторое количество энергии. Температура системы не меняется, следовательно, необходимое количество теплоты (Q) поступило из окружающей среды. Таким образом, теплота процесса испарения воды, протекающего в данных условиях – величина положительная. Q = v ⋅ Qисп.H 2 O > 0 , где Qисп.H 2 O - мольная теплота испарения воды. 12 1.3 Формулировка закона Первый закон термодинамики Пусть в системе протекает процесс, в результате которого система переходит из состояния 1, характеризующегося внутренней энергией – U1, в состояние 2, характеризующееся внутренней энергией – U2. тогда справедливы следующие соотношения: для макропроцесса: для элементарного процесса (т.е. приводящего к бесконечно малым изменениям): U2 – U1 = δQ – δA U2 – U1 = Q – A ∆U = Q – A (1.9) dU = δQ – δA (1.10) где Q, δQ – теплота процесса, A, δA – работа процесса. Приведенные соотношения являются математическим выражением важнейшего закона естествознания – первого начала термодинамики. Первое начало термодинамики является частным случаем закона сохранения энергии, в соответствии с которым энергия не исчезает и не возникает, но только переходит из одного вида в другой. Известно несколько равноценных формулировок первого начала термодинамики; если одну из них рассматривать, как исходную, другие получаются из неё как следствия∗. Применяя первое начало термодинамики к химическим процессам, можно сделать ряд выводов и доказать важные законы, имеющие практическое применение. Раздел химической термодинамики, изучающий тепловые эффекты реакций и их зависимость от параметров состояния системы называется термохимией. ∗ Подробно об этом смотри в учебниках [1, 2, 3]. 13 1.4. Применение первого закона термодинамики к химическим процессам Пусть термодинамическая система представляет собой ν1 моль вещества А и ν2 моль вещества В, и пусть исходные вещества А и В в результате необратимой химической реакции: ν1A + ν2B → ν3C + ν4Д, превратились в продукты С и D. При протекании химического процесса разрываются химические связи между атомами исходных молекул А и В (на это нужно потратить некоторое количество энергии), и образуются новые химические связи между атомами, образующими молекулы С и D (энергия при этом выделяется). Если «новые» химические связи прочнее «старых», то химический процесс сопровождается освобождением энергии. Такие реакции идут с выделением теплоты в окружающую среду или приводят к повышению температуры системы, если она изолирована от окружающей среды (экзотермические реакции). И наоборот, система либо охлаждается, либо поглощает тепло из окружающей среды, если «старые» связи прочнее «новых» (эндотермические реакции). Описанное химическое превращении иллюстрирует рисунок 1.3: Рис. 1.3 Изменение термодинамических свойств системы, в которой происходят химические изменения. Термодинамический процесс, в результате которого система перешла из начального состояния 1 в конечное состояние 2, есть химическая реакция: ν1A + ν2B → ν3C + ν4Д, которая может протекать в одну стадию (один путь) или несколько стадий (другие пути). Химическая реакция, как и всякий термодинамический процесс характеризуется теплотой (Q) и работой (А). Из схемы ясно, что изменение внутренней энергии (∆U =U2 – U1) и энтальпии (∆Н = Н2 – Н1) системы в процессе химического превращения веществ в соответствии с первым законом термодинамики не зависит от пути , по которому протекает реакция, а 14 лишь от химической природы и состояния начальных и конечных веществ .Теплота процесса (Q), напротив, в общем случае зависит от пути, другими словами, химическое превращение: ν1A + ν2B → ν3C + ν4Д, можно осуществить так, что выделится значительное количество теплоты, но не совершится никакой работы, а можно так, что теплота окажется незначительной, зато система совершит работу. Однако, по крайней мере, в двух простейших случаях теплота термодинамического процесса (в том числе и химической реакции) не зависит от его пути. Первый случай: процесс протекает в системе, объем которой не меняется V1 = V2 = V = const, и при этом не совершается ни электрической, ни каких других видов работы δA = 0. Второй случай: процесс протекает при постоянном давлении P1 = P2 = P = const, и не совершается никакой работы, кроме работы расширения газа: δA = p·dV. В данных частных случаях выполняется закон Гесса. 1.4.1. Закон Гесса Формулировка закона Гесса Если из данных веществ можно получить заданные конечные продукты различными путями, то суммарная теплота процесса на одном пути равна суммарной теплоте процесса на любом другом пути. ДоказательМожно доказать, что при Р = const теплота процесса, не соство вершающего никакой работы кроме работы расширения, равна закона Гесса изменению энтальпии системы QP = H2 – H1 = ∆H (1.11) Поскольку Н2 и Н1 определяются только начальным и ко- 15 нечным состоянием системы и не зависят от пути протекания процесса, то и теплота Q не должна зависеть от пути перехода из начального состояния в конечное. Соотношение (1.11) легко получить, применяя 1-й закон термодинамики: (1) dU = δQ – P⋅dV ⇐ 1-й закон термодинамики (2) H = U + P⋅V ⇐ определение энтальпии (3) dH = dU + P⋅dV+ V⋅dP ⇐ математическое преобразо- для системы, которая совершает только работу расширения (δ A = P⋅dV) (4) dH = δQ – P⋅dV + P⋅dV+ V⋅dP (5) dH = δQ + 0 2 2 1 1 ∫ dH =∫ δQ H2 – H1 = QP вание (дифференцирование выражения (2)) ⇐ математическое преобразование (сложение (1) и (3)) ⇐ так как P = const, то dP = 0 ⇐ что требовалось доказать Легко доказать, что при V = const, теплота реакции равна изменению внутренней энергии системы: QV = U 2 − U 1 = ∆U Связь QP и QV (1.12) Следовательно, если реакция протекает при постоянном объеме, ее теплота тоже зависит только от начального и конечного состояния системы, но не от пути процесса. Соотношение (1.12) легко получить из 1-го закона термодинамики (доказательство самостоятельно). Соотношения (1.11) и (1.12) позволяют сделать еще один важный вывод: Теплота реакции, протекающей при постоянном объеме (QV = ∆U ) не всегда равна теплоте той же реакции, протекающей при постоянном давлении (QV = ∆H ) . Можно доказать, что: 1) если в реакции участвуют только твердые и жидкие вещества, то ∆H ≈ ∆U и QP ≈ QV; (1.13) 16 2) если в реакции участвуют газообразные вещества (причем условия таковы, что газы можно считать идеальными), то ∆H = ∆U + ∆n⋅⋅R⋅⋅T или QP = QV + ∆n⋅⋅R⋅⋅T (1.14) где R – газовая постоянная, Т – абсолютная температура, ∆n – разность числа молей газообразных продуктов реакции и газообразных исходных веществ (в соответствии со стехиометрическим уравнением химической реакции). Пример ∗∗∗ Для реакции сгорания жидкого гептана: C7H16(ж) + 11O2(газ) → 7CO2(газ) + 8H2O(ж) QP = QV + ∆n⋅R⋅T, Стандартное состояние вещества ∗ см. гл.3 стр. 72. ∆n = 7 – 11 = – 4 т.е. при сгорании 1 моль жидкого гептана в реакторе, где сохраняется постоянное давление (P = const), выделится на 4RT тепла больше, чем в закрытом сосуде, объем которого остается неизменным (V = const). ∗∗∗ Закон Гесса позволяет вычислить теплоты процессов, для которых отсутствуют экспериментальные данные, комбинируя стехиометрические уравнения и известные теплоты других реакций∗; необходимо только сравнивать теплоты реакций, протекающих в одних и тех же условиях, и следить, чтобы участники всех используемых в расчетах химических процессов находились в одинаковых (например, в некоторых заранее оговоренных стандартных) состояниях. В связи с этим возникла необходимость введения понятий «стандартного состояния вещества» и «стандартного теплового эффекта химической реакции». Стандартным состоянием индивидуального газообразного вещества принято считать гипотетическое (воображаемое) состояние, при котором газ находится при стандартном давлении P0 = 1 атм. = 1,013·105 Па, подчиняется законам идеальных газов, а его энтальпия остается равной энтальпии реального газа. За стандартное состояние чистого жидкого или кристаллического вещества принимают его наиболее устойчивое физическое состояние при данной температуре и нормальном атмосферном давлении P0 = 1 атм. 17 Например, при температуре t = 25°С H2O в стандартном состоянии – жидкость (H2O(ж)), а не лед; S в стандартном состоянии – кристаллическое вещество, имеющее ромбическую (Sромб), а не гексагональную кристаллическую решетку; C в стандартном состоянии – графит (Cграфит), а не алмаз. Стандартный Стандартным тепловым эффектом (∆H°) реакции: тепловой эффект ν1A + ν2B → ν3C + ν4Д, называют теплоту процесса превращения ν1 молей A и ν2 молей B в ν3 моль C и ν4 моль D при условии, что 1) при этом не совершается никакой работы кроме работы расширения; 2) вещества A ,B, C и D находятся при одинаковой температуре; 3) процесс химического превращения проходит при постоянном стандартном давлении P0 = 1 атм. = 1,013·105 Па Из закона Гесса вытекает ряд следствий, два из которых используют для вычисления тепловых эффектов реакций по справочным данным. 1.4.2. Следствия закона Гесса. Определение Пример Стандартной теплотой образования химического соединения называют теплоту, которая могла бы выделяться (или поглощаться) при образовании одного моль соединения из соответствующих простых веществ, взятых в своих стандартных состояниях, при стандартном давлении P0 = 1атм. = 1,013·105 Па. Стандартная теплота образования химического соединения обозначается ∆H 0f . Для многих веществ ∆H 0f определены прямыми калориметрическими измерениями или косвенными методами и приведены в справочной литературе. ∗∗∗ Стандартной теплотой образования кристаллического сульфата меди ( ∆H f , 298,CuSO4 ) в соответствии с определением следует считать тепловой эффект реакции: CuКР. + SКР. РОМБ. + 2O2 ГАЗ → CuSO4 КР., 18 при давлении 1 атм. и температуре 298 К ∗∗∗ Стандартные теплоты образования химических соединений при комнатной температуре (Т = 298 К) являются справочными величинами. Зная теплоты образования химических соединений, можно посчитать тепловой эффект любой реакции, в которой участвуют эти соединения. Для этого следует воспользоваться первым следствием закона Гесса. 1-е следствие закона Гесса Пример Тепловой эффект реакции равен разности между суммой стандартных теплот образования продуктов реакции и суммой стандартных теплот образования исходных веществ, с учетом стехиометрических коэффициентов. ∗∗∗ Тепловой эффект реакции: 2 NO + O2 = 2 NO2 при стандартных условиях можно вычислить из соотношения: 0 ∆H реакции = 2 ⋅ ∆H 0f , NO2 − ∆H 0f ,O2 − 2∆H 0f , NO Определение. Пример ∗∗∗ Стандартной теплотой сгорания химического соединения называется тепловой эффект реакции сгорания 1-го моля вещества до конечных продуктов сгорания (H2O, CO2, N2, SO2) при стандартных условиях. Стандартная теплота сгорания обозначается ∆H сгор ∗∗∗ Стандартной теплотой сгорания C6H5NH2 следует называть тепловой эффект реакции: C6H5NH2 + 7,75 O2 → 6 CO2 + 3,5H2O + 0,5 N2 ∗∗∗ Стандартные теплоты сгорания многих химических соединений 0 приведены в справочниках. Зная ∆H сгор соединений, участвующих в химической реакции, можно вычислить тепловой эффект этой реакции. Для этого следует воспользоваться вторым следствием закона Гесса. 2-е следствие Тепловой эффект реакции равен разности между суммой закона стандартных теплот сгорания исходных веществ и суммой Гесса стандартных теплот сгорания продуктов реакции, с учетом стехиометрических коэффициентов. 19 1.4.3. Теплоемкость Определение Теплоемкостью вещества называется количество тепла, которое необходимо для нагревания определенного количества вещества на 1 градус. Количество тепла, которое необходимо для нагревания 1-го грамма вещества, называется удельной теплоемкостью вещества и обозначается "с". Удельная теплоемкость измеряется в Дж/(г⋅К). Количество тепла, которое необходимо для нагревания 1-го моля вещества называется мольной теплоемкостью вещества и обозначается "С". Мольная теплоемкость измеряется в Дж/(моль⋅К). Экспериментальное определение удельной (с) или мольной (С) теплоемкости вещества заключается в измерении теплоты Q, поглощенной при нагревании известной массы вещества на T2 – T1 = ∆T градусов при тех или иных условиях (например, при V = const или P = const). При этом определяют средние теплоемкости C в интервале T2 – T1. ( ) C= Q , T2 − T1 (1.15) где, Q – количество тепла, требуемое для нагревания 1 моля вещества от T1 до T2. В настоящее время методы измерения теплоемкостей достигли такого уровня, что есть возможность уменьшить экспериментальную величину (T2 – T1) до нескольких сотых градуса; измеренную в таком узком температурном интервале теплоемкость считают истинной теплоемкостью вещества при данной температуре T (T ≈ T2≈ T1). Для истинной теплоемкости согласно определению справедливо соотношение: C= δQ dT (1.16) где, δQ – количество тепла, требуемое для нагревания 1 моля вещества на T2 – T1 = dT градусов. Теплоемкость вещества зависит от условий, при которых происходит нагревание. Например, для того, чтобы при постоянном давлении на- 20 греть 1 моль газа на 1 градус (условие можно выполнить, если нагревать газ в цилиндре с поршнем) потребуется больше тепла чем для нагревания 1-го моля того же газа при постоянном объеме в закрытом сосуде (см. рис.1.4 и 1.5). В связи с этим принято различать теплоемкость вещества при постоянном давлении (СP) и постоянном объеме (СV). Поскольку при P = const δQ = dH, а при V = const δQ = dU, для изобарной (СP) и изохорной (СV) теплоемкости вещества выражение (1.4) можно записать в виде: CP = dH dT CV = (1.17) dU dT (1.18) Рис 1.4 Нагревание газа в цилиндре с Рис. 1.5 Нагревание газа в закрытом сосуде характеризует теплопоршнем характеризует тепемкость при постоянном лоемкость вещества при пообъеме стоянном давлении dU dH CV = CP = dT dT Связь изобарной и изохорной теплоемкости идеального газа Можно доказать, что для идеальных газов CP = CV + R, (1.19) где R – газовая постоянная. (1) H = U + P⋅V Доказательство: ⇐ определение энтальпии (2) P⋅V = R⋅T ⇐ закон Клапейрона-Менделеева, справедливый для идеальных газов (3) H = U + R⋅T ⇐ из (1) и (2), (4) dH/dT = dU/dT + R ⇐ дифференцирование равенства (3), 21 (5) CP = CV + R ⇐ так как CP = dH dU и CV = dT dT Для твердых и жидких веществ изобарная (CP) и изохорная (CV) теплоемкости отличаются мало∗ и при расчетах принимается, что CP тв ≈ CV тв CP ж ≈ CV ж (1.20) Теплоемкость вещества зависит от температуры Например, для того, чтобы нагреть 1 моль твердого вещества от 10 до 11 K потребуется меньше тепла, чем для нагревания 1-го моля того же вещества от 310 до 311 K. Теплоёмкость Экспериментальные и теоретические исследования позвокристалличе- лили доказать, что ских 1. теплоемкость кристаллических веществ изменяется с ростом веществ температуры следующим образом: а) при абсолютном нуле теплоемкость вещества равна 0: при Т = 0 CV = 0, б) при температурах, близких к абсолютному нулю, теплоемкость мала, но быстро растет с ростом Т: при Т → 0 CV = α⋅Т3, в) при температурах выше 30 К рост теплоемкости с увеличением температуры замедляется, г) при комнатных температурах атомная теплоемкость всех кристаллических веществ согласно правилу Дюлонга-Пти Дж . составляет примерно 3R 25 моль ⋅ К Графическая зависимость теплоемкости кристаллических веществ от температуры в общем случае имеет вид, изображенный на рис 1.6. Изменение 2. При фазовых переходах (из одной кристаллической модифитеплоёмкости кации в другую, из твердого состояния в жидкое, и т.п.) теплопри фазовых емкость меняется скачкообразно, при этом для большинства переходах веществ CV жидкого вещества при температуре плавления не- сколько больше CV кристаллического (рис.1.7). ∗ Это можно строго доказать, см., например, А.Г.Стромберг, Д.П.Семченко «Физическая химия», 1999г, стр.73. 22 Рис. 1.6. Зависимость теплоемкости кристаллических веществ от температуры. Рис. 1.7. Зависимость теплоемкости HCl от температуры T TФ.П. – температура фазового перехода, TПЛ. – температура плавления, TКИП. – температура кипения. 3. теплоемкость газообразных и жидких веществ обычно растет с повышением температуры (рис. 1.8). Рис. 1.8 Зависимость теплоемкости газов от температуры: а) если кривая при невысоких T имеет большую кривизну, чем при высоких, её предпочтительнее описывать эмпирическим степенным рядом вида: CP = a + в⋅T + c’/T2 б) кривую б степенным рядом вида: CP = a + в⋅T + c⋅T2. 23 Зависимость теплоемкости веществ от температуры в интервале от 298 до T принято описывать для неорганических веществ для органических веществ эмпирическим уравнением: эмпирическим уравнением: CP = a + в⋅⋅T + c’/T2 Задание CP = a + в⋅⋅T + c⋅⋅T2 (1.21) (1.22) Коэффициенты а, в, с, с', экспериментально определены и приведены в справочной литературе (см., например, [4]). ∗∗∗ Согласно справочным данным зависимость изобарной молярной теплоемкости кислорода от T в интервале 298 – 3000 K описывается уравнением: C P = a + bT + O2 Дж , моль ⋅ K c' T2 где a = 31,46, b = 3,39·10-3, c’ = -3,77·105. Рассчитать C P400 - молярную теплоемкость O2 при P = const и T = 400 K, CV400 - молярную теплоемкость O2 при V = const и T = 400 K, 1.4.4. Зависимость теплового эффекта реакции от температуры. Закон Кирхгофа По закону Гесса можно вычислить тепловой эффект реакции при той температуре (обычно это 298 K), при которой известны стандартные теплоты образования или сгорания реагентов. Если процесс протекает при иной температуре, его теплота при T = отличается от стандартного теплового эффекта ∆H 298 298 K. ∗∗∗ При образовании 2 молей СО и 2 молей H2 из 1 моля СH4 и 1 моля СO2 при Т = 298 К поглощается 247,4 кДж теплоты: ( Пример CН4 + СO2 = 2 СO2 + 2 Н2 ) ∆Н°300 = 247,4 кДж/моль, а при образовании 2 молей СО и 2 молей H2, из 1 моля СН4 и 1 моля СO2 при Т = 1000 К поглощается 260,9 кДж теплоты, т.е. на 13,5 кДж больше: CН4 + СO2 = 2 СO2 + 2 Н2 ∗∗∗ ∆Н°1000 = 260,9 кДж/моль. 24 Формулировка закона Кирхгофа Согласно закону Кирхгоффа (1858 г): Зависимость теплового эффекта реакции ν1A + ν2B = ν3C + ν4Д, (1) от температуры описывается уравнениями: d∆ H = ∆C P dT d∆ U = ∆CV dT или (1.23) (1.24) если реакция протекает при если реакция протекает при постоянном объеме постоянном давлении V = const P = const Те же уравнения в интегральной форме имеют вид: T + ∆C dT (1.25) ∆H T = ∆H 298 ∫ P T + ∆U T = ∆U 298 298 ∫ ∆CV dT (1.26) 298 где ∆H T ( ∆U T ) – тепловой эффект реакции при температуре Т. ( ∆U ) – тепловой эффект реакции при стандартной ∆H 298 298 ∆C P ∆CV температуре Т = 298 К. = ν 3 ⋅ C P , C + ν 4 ⋅ C P , Д − ν 2 ⋅ C P, В − ν 1 ⋅ C P , А – разность тепло= ν 3 ⋅ CV , C + ν 4 ⋅ CV , Д − ν 2 ⋅ CV , В − ν 1 ⋅ CV , А емкостей продуктов реакции и веществ, вступивших в реакцию. Доказательство Пусть HА, HВ, HС, HД, – энтальпии реагентов А, В, С, Д, отнесенные к 1-му моль вещества. Тогда тепловой эффект реакции (1) при постоянном давлении (QP) равен: (1) QP = ∆H° = ν3·HС + ν4·HД – ν1·HА – ν2·HВ (2) (3) d∆H dT =ν3 dH C dT +ν 4 dH Д dT − ν1 dH А dT −ν 2 dH В ⇐ дифференци- руем (1) по температуре, считая P = const. dT d∆H = ν 3 ⋅ CPC + ν 4 ⋅ CPД − ν 1 ⋅ CPА − ν 2 ⋅ CPВ dT ⇐ так как dH i CPi = dT при P = const 25 d ∆H = ∆C P , (4) dT ⇐ что и требовалось доказать Аналогично можно доказать соотношение (1.24) для реакций, протекающих при постоянном объёме. Из дифференциальной формы уравнения Кирхгоффа (1.23) видно что, влияние температуры на тепловой эффект обусловлено знаком величины ∆СP: 1) если ∆СP < 0, то первая производная d∆H < 0 , следовательно, dT функция ∆H° = f(T) – убывающая (рис. 1.9). 2) если ∆СP > 0, то первая производная d∆H > 0 , следовательно, dT функция ∆H° = f(T) – возрастающая (рис. 1.10). Рис. 1.9 Зависимость ∆H° от T, если Рис. 1.10 Зависимость ∆H° от T, если ∆СP > 0: ∆СP < 0: а) тепловой эффект эндотермичеа) тепловой эффект эндотермической реакции растёт с ростом T ской реакции убывает с ростом (при высоких T поглощается T (при высоких T поглощается больше тепла); меньше тепла); б) тепловой эффект экзотермичеб) тепловой эффект экзотермической реакции с ростом T станоской реакции с ростом T становится менее отрицательным (при вится более отрицательным (при высоких T выделяется меньше высоких T выделяется больше тепла). тепла). d∆H 3) если ∆СP = 0, то = 0 , и тепловой эффект не зависит от dT температуры (рис. 1.11). 4) если величина ∆СP при высоких и низких температурах имеет разный знак, то зависимость ∆H° от температуры имеет более сложную зависимость: кривая ∆H° = f(T) – проходит через мак- 26 симум или минимум (рис. 1.12). Рис. 1.11 Зависимость ∆H° от T, если ∆СP = 0. Как пользоваться интегральной формой закона Кирхгоффа Рис. 1.12 Зависимость теплового эффекта от температуры: 1) 3/2H2 + 1/2N2 → NH3 2) C + 1/2O2 → CO Для расчета тепловых эффектов реакций при температурах отличных от стандартной пользуются интегральной формой уравнений Кирхгоффа (1.25) и (1.26). В нешироком температурном интервале (десятки градусов) можно пренебречь зависимостью теплоемкостей реагентов от температуры и считать ∆СP постоянной величиной, не зависящей от T, тогда для теплового эффекта при температуре T получаем: T + ∆H T = ∆H 298 ∫ ∆CP dT = ∆H 298 + ∆CP (T − 298) 298 где T – не слишком отлична от 298 К. При интегрировании уравнения Кирхгоффа в широком интервале температур (298 – T) пользуются температурной зависимостью теплоёмкости веществ в виде степенных рядов (1.21) и (1.22), справедливых в данном температурном интервале. В общем случае, если в реакции участвуют и органические и неорганические вещества для разности теплоёмкостей конечных и начальных веществ получится выражение вида: ∆CP = ∆a + ∆в⋅T + ∆c⋅T2 + ∆c’/T2 Тогда для теплового эффекта при высокой температуре T имеем: + ∆H T = ∆H 298 ∆c ' 2 ∆a + ∆в ⋅ T + ∆c ⋅ T + 2 dT T 298 T ∫ Примеры расчетов тепловых эффектов реакций по законам Гесса и Кирхгоффа приведены в 3-й главе. 27 1.5. Второй закон термодинамики При исследовании любой системы возникают важные вопросы: • будет ли система оставаться неизменной во времени или следует ожидать, что её свойства (T, P, V, U и др.) будут меняться? • как предсказать какие изменения в данной системе окажутся естественными процессами, приближающими её к устойчивому (равновесному) состоянию, а какие нет? Ответы на эти вопросы можно получить, применяя к исследуемой системе второй закон термодинамики. Это – общий закон природы, действующий в любых системах, состоящих из достаточно большого∗ числа частиц. Для формулировки второго закона термодинамики введем некоторые новые понятия. 1.5.1. Самопроизвольные и несамопроизвольные процессы Определение Пример 1 Процессы, протекающие сами собой без воздействия внешней силы и приближающие систему к равновесию, называются самопроизвольными. Процессы, которые сами собой протекать не могут, называются несамопроизвольными. ∗∗∗ Пусть термодинамическая система представляет собой смесь жидкой воды и льда, находящуюся при t = 200 С. При t = 200 С система не находится в равновесии: в такой системе самопроизвольно протекает процесс таяния льда. Таким образом, в данной системе при данных условиях процесс таяния льда: H2Oкр → H2Oж есть самопроизвольный процесс; а кристаллизация воды: H2Oж → H2Oкр есть несамопроизвольный процесс. Направление процесса в этом случае мы определяли, опира∗ частиц должно быть достаточно для того, чтобы их поведение подчинялось законам статистики. 28 ясь на практический опыт. Пример 2 Термодинамическая система представляет собой смесь N2 и H2, находящуюся при t = 6000 С. Система не находится в равновесии: в системе будет протекать процесс образования NH3. 3N2 + H2 → 2NH3 Этот процесс прекратится не тогда, когда весь N2 или Н2 вступит в реакцию, а когда концентрации N2, Н2 и NH3 примут определенные значения, причем эти равновесные концентрации будут связаны уравнением: 2 c NH KC = 3 3 cN 2 ⋅ cH 2 В этом случае направление процесса и условие равновесия мы определили, опираясь на знания свойств N2, Н2 и NH3, известные нам из общей химии. ∗∗∗ Задание Опираясь на собственный опыт, укажите какие из перечисленных процессов, являются самопроизвольными: а) диффузия газов в воздушной среде; б) передача тепла от холодного тела к горячему; в) растворение этилового спирта в воде при комнатной температуре; г) горение топлива в печи. ∗∗∗ Самопроизвольный процесс, в результате протекания которого система переходит из начального состояния (1) в конечное состояние (2) в разных условиях может протекать по-разному. Обратимые и Различают обратимые процессы (протекающие равновеснеобратимые ным путем) и необратимые (протекающие неравновесным пупроцессы тем). Необратимыми называют процессы, после протекания которых нельзя и систему, и окружающую среду вернуть в первоначальное состояние, останутся некоторые изменения – «следы» необратимого процесса. Все реальные процессы в той или иной степени необратимы, однако можно представить некий идеальный процесс, идущий под воздействием бесконечно малых движущих сил, так что в любой момент времени система, по существу, находится в равновесии. Под воздействием противоположно направленных движущих сил систему можно вернуть в первоначальное положение через те же бесконечно близкие к равновесию 29 Пример 1 состояния, при этом не остается никаких изменений во всех участвующих в процессе телах и окружающей среде. Такие идеальные процессы, протекающие равновесным путем и в прямом и в обратном направлении, называются обратимыми. ∗∗∗ Процесс расширения газа можно провести равновесным путем (обратимо), если поместить газ под поршень и расширять его так, чтобы в каждый момент времени внешнее давление (PВН) на поршень было на бесконечно малую величину меньше давления газа. При этом совершается работа, которую можно аккумулировать, например, превращая её в энергию сжатой пружины (рис. 1.13). Рис. 1.13 Модель процесса обратимого расширения газа Пример 2 Процесс можно провести в обратном направлении, если бесконечно-медленно увеличивать внешнее давление на поршень, используя энергию сжатой пружины. Если поршень невесом и движется без трения, то запасенной энергии пружины в точности хватит для проведения процесса сжатия газа и возвращения системы в первоначальное состояние, при этом никаких изменений ни в окружающей среде, ни в системе не будет. На рис. 1.14 изображен процесс равновесного испарения жидкости при постоянной температуре и постоянном давлении пара (P, T = const). Рис.1.14 Модель процесса обратимого испарения жидкости при P, T = const. Внешняя температура отличается от температуры системы на бесконечно малую величину dT, поэтому процесс испарения происходит медленно, так что давление пара под поршнем возрастает настолько медленно, что его разность с внешним дав- 30 Задание лением бесконечно мала. Таким образом, система в каждый момент времени находится в состоянии очень близком к равновесному. Равновесный процесс протекает бесконечно медленно, при этом система совершает максимально возможную работу. ∗∗∗ Работа любого необратимого процесса (A) всегда меньше работы процесса, протекающего равновесным путем (Amax). Чем меньше работа по сравнению с максимальной, тем более необратим процесс. Процессы, которые протекают без совершения работы (A = 0) называются полностью необратимыми. Второй закон термодинамики формулирует принцип, позволяющий различать самопроизвольные и несамопроизвольные процессы в любой системе. Предложено значительное количество формулировок второго закона термодинамики; все они равноценны: если одну из них выбрать в качестве постулата, остальные вытекают из неё как следствия. ∗∗∗ Прочитайте в учебниках [1, 2] формулировки второго закона термодинамики Клаузиуса, Томсона, Оствальда, принцип Каратеодори, интерпретацию второго закона с точки зрения теории вероятности. ∗∗∗ Мы приведем математическую формулировку второго закона термодинамики в форме, удобной для исследования химических и физических превращений в системе. Формулировка второго закона термодинамики Рис. 1.15 Схема термодинамического превращения Если термодинамическая система переходит из состояния 1, характеризующегося параметрами P1, V1, T1 и функциями состояния U1, H1, S1, F1, G1 в состояние 2, характеризующееся параметрами P2, V2, T2 и функциями состояния U2, H2, S2, F2, G2, самопроизвольно 31 равновесным путем, то δQ S 2 − S1 = T dS неравновесным путем, то (1.27) δQ S 2 − S1 > T dS (1.28) где δQ – теплота процесса, протекающего в системе. Второй закон термодинамики является постулатом, сформулированным на основе накопленного человечеством опыта; доказательством его справедливости служит то, что все выводы, вытекающие из него, находят экспериментальное подтверждение, и до сих пор не произошло ничего, что «запрещает» второй закон термодинамики. Из второго закона термодинамики путем несложных математических преобразований можно получить ряд следствий, которыми удобно пользоваться для определения направления процесса и условий, при которых в системе наступает равновесие. 1.5.2. Условие самопроизвольного протекания процесса в изолированной системе, т.е. при U = const и V = const. Термодинамическая система называется изолированной, если её объём постоянен, и она не обменивается с окружающей средой энергией и веществом. Если обратиться к схеме, приведенной на рис. 1.15, то для изолированной системы: V1 = V2 = V = const U1 = U2 = U = const Критерием самопроизвольного протекания процесса в изолированной системе является энтропия. Следствие В изолированной системе могут самопроизвольно 2 –го закона протекать только те процессы, которые приводят к возрастермодинамики танию энтропии в системе. Когда энтропия достигнет макдля симального при данных U и V значения, все процессы преизолированных кратятся и система достигнет равновесного состояния. систем 32 Итак если U = const и V = const, если U = const и V = const, то при самопроизвольном то при равновесии процессе, протекающем неравновесным путем S2 – S1 > 0 S2 – S1 = 0 dS > 0 ∆S > 0 dS = 0 ∆S = 0 Доказательство. (1) dU = δQ – P⋅dV (2) dS ≥ δQ T ⇐ 1-й закон термодинамики для систем, которые совершают только работу расширения против внешних сил ⇐ 2-й закон термодинамики для систем, в которых идут самопроизвольные процессы Объединив (1) и (2),получим: (3) dU P ⋅ dV ≤ T ⋅ dS − 0 0 ⇐ объединенное выражение 1-го и 2-го законов, справедливое для самопроизвольных процессов. Из (3) ясно, что если U = const и V = const, то dU = 0, dV = 0 и для самопроизвольного процесса, протекающего неравновесным путем: T⋅dS > 0 т.е dS > 0 и S2 – S1 > 0, а при равновесии: T⋅dS = 0 т.е dS = 0 и S2 – S1 = 0. 1.5.3. Условия самопроизвольного протекания процессов в системе, находящейся при постоянном давлении и температуре (Р = const, Т = const) и не совершающей никакой работы кроме работы расширения газа (δA = P⋅dV). Многие физические и химические процессы протекают при постоянном давлении и температуре, если они проводятся в открытых термостатированных реакторах. О направлении процессов и равновесии в таких системах судят по изменению изобарно-изотермического потенциала (свободной энергии) системы (G). Следствие В системе, не совершающей никакой работы, кроме 2 –го закона работы расширения, и находящейся при постоянных термодинамики температуре и давлении (Р = const, Т = const), самопроиз- 33 при P, T = const вольно могут протекать только те процессы, которые приводят к уменьшению изобарно-изотермического потенциала системы (G). Когда G достигнет минимального значения при данных Р и Т, система приходит в равновесие и процесс прекращается. Обозначим величины так, как на рис. 1.15, тогда если 1) δA = P⋅⋅dV если 1) δA = P⋅⋅dV 2) Р = const, Т = const 2) Р = const, Т = const то при самопроизвольном то при равновесии процессе, протекающем неравновесным путем G2 – G1 = 0 G2 – G1 < 0 dG = 0 ∆G = 0 dG < 0 ∆G < 0 Доказательство (1) dU = δQ – P⋅dV (2) dS ≥ δQ ⇐ 1-й закон термодинамики для систем, которые совершают только работу расширения, ⇐ 2-й закон термодинамики (справедлив, если в системе процесс протекает самопроизвольно), T Объединив (1) и (2),получим: (3) dU ≤ T ⋅ dS − P ⋅ dV ⇐ объединенное выражение 1-го и 2-го законов термодинамики (справедливо для самопроизвольного процесса). (4) G = H – T⋅S = U + P⋅V – T⋅S ⇐ определение изобарноизотермического потенциала, дифференцируя (4) получаем: (5) dG = dU + P⋅dV + V⋅dP – T⋅dS – S⋅dT объединяя (3) и (5), имеем (6) dG ≤ V ⋅ dP S ⋅ dT − 0 0 ⇐ объединенное выражение 1-го и 2-го законов термодинамики (справедливо для самопроизвольных процессов). 34 Из (6) ясно, что если Р = const, Т = const, то dP = 0, dT = 0. Следовательно, при самопроизвольном протекании процесса dG < 0, или G2 – G1 < 0 или G2 < G1, т.е. свободная энергия системы уменьшается. При равновесии dG = 0 или ∆G = 0 при этом G2 = G1 принимает минимальные значения. 1.5.4. Условие самопроизвольного протекания процесса в системе, объём и температура которой остаются постоянными Процессы протекают при постоянном объеме и температуре, если их проводят в закрытых, термостатированных аппаратах, например, в автоклавах. Критерием протекания процесса в этом случае служит изменение изохорно-изотермического потенциала (энергии Гельмгольца) системы. Опираясь на второй закон термодинамики, легко доказать следующее правило. Следствие Если система не совершает никакой работы, и её объем и 2 –го закона температура остаются постоянными (V = const, Т = const), термодинамики то в такой системе самопроизвольно могут протекать при V, T = const только те процессы, которые приводят к уменьшению изохорно-изотермического потенциала системы (F). Когда F достигнет минимального при данных V и Т значения, система приходит в равновесие и процесс прекращается. Итак если 1) δA = P⋅dV + … = 0 если 1) δA = P⋅dV + … = 0 2) V = const, Т = const 2) V = const, Т = const то при самопроизвольном то при равновесии процессе F2 – F1 < 0 F2 – F1 = 0 dF < 0 ∆F < 0 Задание dF = 0 ∆F = 0 Доказательство (самостоятельно). ∗∗∗ Термодинамическая система представляет собой смесь разбавленного раствора KCl в воде и кристаллов KCl, находящуюся при нормальном атмосферном давлении и комнатной температуре. Р =1 атм., T = 298 К Система неравновесна, в ней будет самопроизвольно протекать процесс растворения кристаллов соли KCl в воде: 35 KClКР. → KClРАСТВОР Задание Определить, как при этом изменяется (увеличивается или уменьшается) каждая из функций состояния системы. ∗∗∗ С помощью преобразований, аналогичных приведенным выше (стр. 33) получить выражение объединенного 1-го и 2-го законов термодинамики в форме: dH ≤ TdS + VdP Задание для самопроизвольных процессов, не совершающих никакой работы, кроме работы расширения системы. Показать, что при S = const, и P = const критерием направления процесса могло бы служить изменение энтальпии. Сформулировать соответствующее правило. ∗∗∗ Химическая реакция, протекающая в открытых или закрытых реакторах не совершает никакой работы кроме, быть может, работы расширения системы. Однако, если химическое превращение осуществляется в особых устройствах – гальванических элементах, то за счет химической реакции совершается работа другого вида – электрическая (δ Аэл , А эл ). Причем, если гальванический элемент работает в условиях близких к равновесным, совершаемая им электрическая эл работа будет максимальной (δ Аmax ) (см. стр. 30). С помощью преобразований, аналогичных приведенным на стр. 33, доказать, что для токообразующей реакции v1A + v2B → v3C + v4Д, протекающей в гальваническом элементе при P, T = const равновесным путем, справедливо соотношение: эл G2 – G1 = – Аmax Задание эл ∆G = – Аmax ∗∗∗ Некоторые жидкости можно охладить ниже их температуры замерзания. Такие жидкости называют переохлажденными. В переохлажденных жидкостях процесс кристаллизации может протекать самопроизвольно даже в изолированных системах. Как объяснить, почему этот процесс протека- 36 Задание ет, несмотря на то, что кристаллы имеют меньшую энтропию, чем жидкость, находящаяся с ними в равновесии. Почему следствие второго закона термодинамики для изолированных систем в данном случае не нарушается? ∗∗∗ Какую величину следует использовать в качестве критерия направления процесса в следующих случаях: а) необходимо определить возможность протекания реакции 2NOгаз + O2 → 2NO2 газ в герметичном, термостатированном реакторе; б) определить возможность получения алмаза из графита Cграфит → Cалмаз при давлении 120000 атм. и T = 3000 K. 37 1.6. Постулат Планка. Абсолютные энтропии Формулировка постулата Планка Энтропия индивидуального кристаллического вещества с идеально построенной кристаллической решеткой при абсолютном нуле равна нулю. ST =0 K = 0 для идеального кристаллического вещества Замечание Энтропия стеклообразного (некристаллического) вещества и твердых растворов не равна 0 и при Т = 0 К. Постулат Планка не вытекает из 1-го и 2-го законов термодинамики и не может быть доказан в рамках классической термодинамики, его считают третьим законом термодинамики. С помощью постулата Планка можно вычислить абсолютную величину энтропии∗ индивидуального вещества при любой температуре. Эта величина называется абсолютной энтропией вещества и обозначается ST0. Абсолютные энтропии большинства веществ рассчитаны и приведены в справочной литературе (см. [4]). Как Для вычисления абсолютной энтропии газообразного верассчитать щества при температуре T необходимо знать: абсолютную 1) теплоемкость этого вещества в кристаллическом состоянии от энтропию температуры плавления (TПЛ.) и до температур близких к абсовещества лютному нулю; Измерение теплоемкостей веществ в области низких температур является сложной задачей. В настоящее время методами низкотемпературной калориметрии исследованы многие вещества и получены их низкотемпературные теплоемкости. 2) теплоты фазовых переходов вещества (плавления, испарения и др.) при стандартном давлении; 3) температурные зависимости теплоемкости вещества в жидком состоянии в интервале от TПЛ. до TКИП. и газообразного вещества при температурах выше TКИП.. Предположим 1 моль кристаллического вещества, находящегося при абсолютном нуле (T = 0 K), превращается при постоянном давлении P° = 1 атм. в газообразное вещество при температуре T. При этом происходят следующие процессы: 1) нагревание кристаллического вещества от 0 до TПЛ.; 2) плавление вещества при постоянной температуре (TПЛ.) и стандартном давлении 1 атм.; ∗ Абсолютную величину U°, H°, F°, G° в рамках классической термодинамики рассчитать невозможно. 38 3) нагревание жидкого вещества от TПЛ. до TКИП.; 4) испарение жидкости при TКИП. и P = 1 атм.; 5) нагревание газообразного вещества от TКИП. до T (см. рис.1.16). Т=0K TПЛ. TКИП. T нагревание нагревание нагревание от 0 K до TПЛ. от TПЛ до TКИП от TКИП до T S кр. 0К S газ, T Рис. 1.16 Схема превращения «абсолютно холодного» кристалла в «горячий» газ. Можно вычислить изменение энтропии на каждом этапе превращения и рассчитать прирост энтропии S газ, T - S кр. 0К ; Как вычислить изменение энтропии вещества при нагревании изменение энтропии при нагревании При нагревании n молей вещества от T1 до Т2 при постоянном давлении изменение энтропии можно посчитать по формуле T2 CP dT T T S − S = n∫ 0 T2 0 T1 (1.29) 1 где ST02 – энтропия вещества при температуре Т2, ST01 – энтропия вещества при температуре T1, СP – мольная теплоемкость вещества при постоянном давлении. Доказательство. (1) δQ = dH при P = const (2) C P = (3) dS = dH ⇐ определение изобарной тепили dH = CP⋅dT лоемкости системы (стр. 20), dT δQ ⇐ 2-й закон термодинамики для процесса, проведенного равновесным путем. T Объединив (1) (2), (3) имеем: δQ C P ⋅ dT T T Интегрируя (4), получаем: (4) dS = ⇐ см. стр. 15, = 39 ST2 T2 ST1 1 ∫ dS = CP ∫ T dT T для n молей вещества теплоемкость системы равна n·CP, где CP – мольная теплоемкость вещества, тогда ST02 − T2 ST01 ⇐ что и требовалось доказать. CP dT T T1 = n∫ При нагревании n молей вещества от T1 до Т2 при постоянном объеме изменение энтропии можно посчитать по формуле: T2 CV dT T T S T0 − ST0 = n ∫ 2 1 (1.30) 1 где ST02 – энтропия вещества при температуре Т2, ST01 – энтропия вещества при температуре T1, СV – мольная теплоемкость вещества при постоянном объеме. Доказательство (самостоятельно). изменение энтропии при фазовых переходах Как вычислить изменение энтропии при плавлении вещества При переходе n молей вещества из твердой фазы в жидкую ТВ → Ж (т.е. при плавлении вещества) изменение энтропии можно посчитать по формуле: S Ж − SТВ =n Q ПЛ TПЛ (1.31) где S°Ж – энтропия жидкого вещества при температуре плавления, S°ТВ – энтропия твердого вещества при температуре плавления, QПЛ – мольная теплота плавления (т.е. количество теплоты, которое необходимо, чтобы расплавить 1 моль вещества), ТПЛ – температура плавления вещества, n – число молей расплавившегося вещества. Доказательство. (1) dS = δQ T ⇐ 2-й закон термодинамики для процессов, проведенных равновесным путем. 40 Интегрируя (1), получим: S Ж (2) 2 ∫ dS = ∫ 1 SТВ δQ 2 или T S Ж − SТВ = ∫ δQ 1 T Так как Т = const = ТПЛ, то 2 (3) ∫ 1 δQ T = nQПЛ TПЛ и следовательно, (4) S Ж − SТВ = n QПЛ TПЛ ⇐ что и требовалось доказать. Аналогично доказываются выражения, позволяющие вычислить изменение энтропии при других фазовых переходах. Изменение энтропии при возгонке вещества При переходе n молей вещества из твердой фазы в газообразную ТВ. → Г (т.е. при возгонке вещества) изменение энтропии можно посчитать по формуле: 0 =n S Г0 − SТВ QВОЗГ . TВОЗГ . (1.32) где QВОЗГ. – мольная теплота возгонки, TВОЗГ. – температура возгонки вещества. Изменение энтропии при испарении вещества При переходе n молей вещества из жидкой фазы в газообразную Ж→Г (т.е. при испарении вещества) изменение энтропии можно посчитать по формуле: S Г0 − S Ж0 = n Q ИСП . TКИП . где QИСП. – мольная теплота испарения, TКИП. – температура кипения вещества. (1.33) 41 Согласно (1.29), (1.31), (1.32), (1.33) изменение энтропии при превращении 1 моля кристаллического вещества, находящегося при 0 К, в газообразное вещество при температуре T, определяется выражением: 0 0 S ГАЗ ,T − S ТВ , 0 K = TПЛ ∫ 0 T T C P ТВ . Q C Q C dT + ПЛ + ∫ P Ж dT + ИСП + ∫ P ГАЗ dT (1.34) T TПЛ T T TКИП T T КИП ПЛ КИП так как согласно постулату Планка энтропия кристаллического 0 вещества при 0 К равна 0 ( S КР , 0 K = 0 ), то абсолютная энтропия 1 Задание 0 моля газа при температуре Т ( S ГАЗ ,T ) может быть легко вычислена по формуле (1.34), для этого необходимо знать теплоты плавления и испарения вещества и теплоемкость кристаллического, жидкого и газообразного вещества в интервале температур от 0 до Т К. ∗∗∗ Доказать, что изменение энтальпии и внутренней энергии при превращении 1 моля кристаллического вещества, находящегося при Т = 0 К в газообразное вещество при температуре Т можно рассчитать по формулам: H T0 − H 00K = U T0 − U 00K = TПЛ TКИП 0 TПЛ T ПЛ TКИП 0 TПЛ ∫ CP КР.dT + QP ПЛ + ∫ CV КР.dT + QV ПЛ + ∫ CP Ж dT + QP ИСП + ∫ CV Ж dT + QV ИСП + T CP ГАЗ dT T TКИП ∫ T CV ГАЗ dT T TКИП ∫ (1.35) (1.36) Почему соотношения (1.35) и (1.36) не позволяют рассчитать абсолютное значение внутренней энергии (U°T) и энтальпии (H°T) вещества? ∗∗∗ 42 1.7. Уравнения Гиббса-Гельмгольца Как было показано выше, выражение объединенного 1-го и 2-го законов термодинамики может быть записано при помощи любой из функций состояния, характеризующих энергию системы (U, H, F или G): dU = TdS − PdV (1.37) dH = TdS + VdP (1.38) dF = − SdT − PdV (1.39) dG = − SdT + VdP (1.40) Соотношения (1.37), (1.38), (1.39), (1.40) справедливы для любой равновесной системы, совершающей только работу расширения. Они равноценны и превращаются друг в друга путем несложных математических преобразований. Можно показать, что каждая из характеристических функций (U, H, F, G) обладает интересным свойством; любая из них может дать полную термодинамическую характеристику системы, если считать её функцией естественных переменных: U = f(S, V); H = f(S, p); F = f(T, V); G = f(T, p). Это означает, что все термодинамические величины, характеризующие систему, можно выразить с помощью выбранной характеристической функции (U, H, F или G) и её производных по естественным переменным. Так как энтропия неудобный параметр (её величину нельзя непосредственно измерить), то при решении практических вопросов внутренняя энергия и энтальпия редко используются как характеримтические функции, зато широкое применение нашли F (изохорно-изотермический потенциал или энергия Гельмгольца) и G (изобарно-изотермический потенциал или энергия Гиббса). Пусть необходимо выразить термодинамические свойства системы, используя функцию G = f (P, T) и F = f (V, T). Интересное свойство термодинамических потенциалов (1) G = f (P, T) (1’) F = f (V, T) дифференцируя (1), получим дифференцируя (1’), получим ∂G ∂G (2) dG = ∂P dP + ∂T dT ∂F ∂F (2’) dF = ∂V dV + ∂T dT T V T P 43 запишем объединенный 1-й и 2-й законы термодинамики в форме: запишем объединенный 1-й и 2-й законы термодинамики в форме: (3) dG = VdP – SdT (см. (1.40)) (3’) dF = – PdV – SdT (см. (1.39)) сравнивая (2) и (3) видим, что ∂G ∂P T (4) V = (4’) ∂G ∂T P ∂F P = − ∂V T ∂F ∂T V (5) S = − (5’) S = − из определений G, H, F следует: ∂G (6) H = G + T ⋅ S = G − T ⋅ ∂T P (7) сравнивая (2) и (3) видим, что U = H − PV = из определений F, H, G следует: ∂F ∂T V (6’) U = F + T ⋅ S = F − T ⋅ (7’) H = U + PV = ∂G ∂G = G −T ⋅ − P ⋅ ∂T P ∂P T ∂G ∂P T ∂F ∂F = F −T ⋅ −V ⋅ ∂T V ∂V T ∂F ∂V T (8) F = U − T ⋅ S = G − P ⋅ (8’) G = H − T ⋅ S = F − V ⋅ Таким образом, все свойства системы (V, S, H, U, F) удалось выразить через G, P и T. Таким образом, все свойства системы (P, S, H, U, G) удалось выразить через F, V и T. Рассмотренные свойства функций состояния G и F дают возможность установить связь между максимальной работой процесса, протекающего равновесно, и теплотой того же процесса, но протекающего неравновесно. Эту связь устанавливают уравнения Гиббса-Гельмгольца. Этими уравнениями в интегральной и дифференциальной форме широко пользуются в химической термодинамике. 44 Уравнения ГиббсаГельмгольца Уравнения Гиббса-Гельмгольца в интегральной форме в дифференциальной форме ∂∆G ∆G = ∆ H + T ⋅ ∂T P (1.41) или ∆G = ∆H - T·∆S ⇔ ∂ ∆G ∆H =− 2 ∂T T T (1.43) ⇔ ∂ ∆F ∆U =− 2 ∂T T T (1.46) (1.42) ∂∆F ∆ F = ∆U + T ⋅ ∂T V (1.44) или ∆F = ∆U - T·∆S (1.45) Доказательство. (1) (2) ∂G H 2 = G2 − T ⋅ 2 ∂T P ⇐ уравнение (6) на стр. 42 для ко- ∂G H1 = G1 − T ⋅ 1 ∂T P ⇐ чального состояния системы нечного состояния системы уравнение (6) на стр. 42 для на- Вычитая (2) из (1) получаем: ∂(G2 − G1 ) (3) H 2 − H 1 = (G2 − G1 ) − T ⋅ ∂T P или ∂∆G ∆H = ∆G − T ⋅ или (4) ∂T P ∂∆G (5) ∆G = ∆H + T ⋅ ∂T P ⇐ что и требовалось доказать. ∂G ∂∆G Учитывая, что S = − и ∆S = − ∂T P ∂T P уравнение Гиббса-Гельмгольца можно представить в виде: (6) ∆G = ∆H - T·∆S 45 Задание Пример Аналогично можно доказать уравнения (1.44) и (1.45). ∗∗∗ Равносильность уравнений (1.41) и (1.43), а также (1.44) и (1.46) доказать самостоятельно, пользуясь правилами дифференцирования сложной функции. ∗∗∗ Связь термодинамических характеристик процессов, протекающих равновесным и неравновесным путем, иллюстрирует следующий пример. ∗∗∗ Пусть в термодинамической системе при постоянном давлении и температуре (P, T = const) совершается химическое превращение: v1A + v2B → v3С + v4Д (∗) и система переходит из начального состояния 1 в конечное состояние 2 (см. рис. 1.17). Рис. 1.17 Изменение термодинамических свойств системы, при протекании химической реакции: а) в реакторе полностью неравновесным путем; б) в электрохимическом элементе равновесным путем. Рассмотрим два возможных пути протекания процесса: 1. термодинамическая система представляет собой реактор, в котором неравновесным путем протекает реакция (∗); в такой системе не совершается никакой работы, 46 кроме быть может работы расширения газа∗: AРАСШИР. = p · (V2 – V1) (1.47) теплота реакции (QНЕРАВН.) равна изменению энтальпии системы: QНЕРАВН. = H2 – H1 = ∆H (1.48) Соотношение (1.1,48) доказано на стр. 15, теплота реакции, протекающей в таких условиях называется «тепловым эффектом реакции». 2. Термодинамическая система – электрохимический (гальванический) элемент, который за счет реакции (∗) совершает электрическую работу (Aэл.). Если элемент работает в условиях близких к равновесным, работа процесса максимальна и её величина определяется (см. стр. 35) выражением: эл Amax = −(G2 − G1 ) = −∆G (1.49) Теплота процесса, протекающего равновесным путем (QРАВН.) в соответствии со вторым законом термодинамики (см. стр. 30) равна: QРАВН. = T · (S2 – S1) = T · ∆S. (1.50) Подставляя (1.48), (1.49), (1.50) в уравнение ГиббсаГельмгольца в форме (1.42), получаем : эл – Amax = QНЕРАВН. – QРАВН. эл Amax = QРАВН. – QНЕРАВН. (1.51) Если протекающая при постоянных p и T химическая реакция (∗) – эндотермическая (H2 – H1 > 0; ∆H > 0), то при работе гальванического элемента из окружающей среды поглощается больше тепла (QРАВН.), чем при протекании реакции неравновесным путем в обычном реакторе (QНЕРАВН. = ∆H). Часть поглощенной из окружающей среды теплоты тратится на совершение электрической ра∗ работа расширения совершается, если объем газообразных продуктов реакции отличается от объема газообразных исходных веществ. 47 боты. А часть на повышение энтальпии системы. QРАВН. = Aэл + ∆H Задание (1.52) Если химическая реакция (∗) – экзотермическая (H2 – H1 < 0; ∆H < 0), то электрическая работа совершается в гальваническом элементе за счет снижения энтальпии системы, при этом либо в окружающую среду выделяется меньше тепла (QРАВН. = T · ∆S < 0), чем при протекании реакции неравновесным путем, либо теплота вообще не выделяется, но даже поглощается из окружающей среды (QРАВН. = T · ∆S >0). ∗∗∗ В гальваническом элементе Якоби – Даниэля электрическая работа совершается за счет химической реакции: Zn + Cu2+ → Zn2+ + Cu (∗∗) Пусть элемент работает при комнатной температуре в условиях близких к равновесным. эл Рассчитать 1) максимальную электрическую работу ( Amax ), РАВН. 2) теплоту токообразующей реакции (∗∗) (Q ), если известны, рассчитанные по справочным данным [4] термодинамические характеристики окислительно-восстановительной реакции (∗∗), протекающей в водном растворе при комнатной температуре (T = 298 K): ∆H 298 = ∆H f ,Cu + ∆H f , Zn 2 + − ∆H f ,Cu 2 + − ∆H f , Zn = −220,58 кДж моль Дж 2+ 2+ ∆S 298 = S Cu + S Zn − S Cu − S Zn = −26,39 моль ⋅ K Ответить на вопросы: 1) в каком случае в окружающую среду выделяется больше тепла: а) если реакция протекает в водном растворе; б) в гальваническом элементе? 2) почему закон Гесса в данном случае не выполняется, и теплота реакции зависит от пути процесса? ∗∗∗ 48 1.8. Парциальные величины. Химический потенциал Состав многокомпонентной газовой смеси характеризуют, задавая либо молярную концентрацию (c1, c2, …cK моль/дм3), либо молярную долю (χ1, χ2, …χK) компонентов, либо парциальное давление каждого газообразного вещества в смеси (P1, P2, …PK атм.). Определение Парциальным давлением газа в смеси называют давление, которое оказывал бы газ, если бы занимал объем, предоставленный всей смеси, при отсутствии всех других компонентов. Если смесь представляет собой идеальную смесь идеальных газов, то парциальное давление каждого газа (Pi) можно вычислить из соотношений: Pi = P·χi = RT·Ci (1.53), Pi = RT·Ci (1.54) где P – общее давление смеси, [Па], χi – мольная доля iого газа в смеси. R = 8,31 [Дж/(моль·К)] – газовая постоянная, Ci - концентрация i-го газа в смеси [моль/м3], T – температура [К]. Для характеристики многокомпонентных термодинамических систем используют парциальные физические величины. Свойства термодинамической системы, как известно, характеризуются пятью функциями состояния (U, Н, S, F, G), важно знать, какая часть вклада в термодинамические функции приходится на долю каждого из веществ, составляющих систему (т.е. парциальные U i , H i , Si , Fi , Gi каждого вещества в смеси). Определение Если при P, T = const к бесконечно-большому количеству смеси определенного состава добавить 1 моль i-го вещества, то внутренняя энергия системы вырастет на U i , энтропия на H i , на S i , изохорно-изотермический потенциал на Fi , энтальпия на G i . Величины U i , H i , S i , Fi , называются соответственно парциальной внутренней энергией, парциальной энтальпией, парциальной энтропией и т.п. i-го вещества в смеси. Парциальный изобарно-изотермический потенциал i-го компонента (G i ) часто называют химическим потенциалом изобарно-изотермический потенциал этого компонента и обозначают µi ( G i = µ i ) 49 Согласно данному определению: ∂U Ui = ∂n i ∂H ;H i = ∂n P,T, состав i ; P ,T , состав ∂S Si = ∂n i ∂F ;F i = ∂n P ,T , состав i ; P ,T , состав (1.55) ∂G ∂n i P ,T , состав µi = (1.56) В соответствии с определением химический потенциал компонента смеси можно рассматривать как энергию Гиббса, приходящуюся на 1 моль этого компонента в смеси. То же касается и других парциальных термодинамических величин. Для многокомпонентной системы, состоящей из n1 моль первого компонента, n2 моль второго …, ni моль iого компонента, поэтому справедливы соотношения: G = n1µ1 + n2 µ 2 + ... + ni µi = ∑ ni µi (1.57) i U = n1U 1 + n2U 2 + ... + ni U i = ∑ ni U i (1.58) i H = n1 H 1 + n2 H 2 + ... + ni H i = ∑ ni H i и т.д. (1.59) i Можно доказать, что для парциальных термодинамических величин справедливы те же соотношения, что и для величин, характеризующих систему в целом (таблица 1.1) Таблица 1.1. Термодинамические соотношения Для системы в целом Для отдельного компонента системы H = U + PV H i = U i + Pi ⋅ V F = U – TS Fi =U i −T ⋅Si G = H – TS µi = H i −T ⋅Si ∆G = ∆H – T∆S ∆µ = H i − T ⋅ ∆ S i 50 1.9. Зависимость химического потенциала компонента от состава смеси Опираясь на законы термодинамики, можно доказать∗ следующие важные соотношения. 1. Для смеси идеальных газов выражение, описывающее зависимость химического потенциала i-го газа от парциального давления этого газа в смеси, простое и удобное: ~ µ i = µ i0 + RT ⋅ ln Pi , ~ P Pi = i P0 (1.60) где µi – химический потенциал i-го газа в смеси, µi0 – стандартный химический потенциал i-го газа, т.е. потенциал газа в смеси, где его парциальное давление равно стандартному давлению P0. T – температура, R – газовая постоянная, Pi – парциальное давление i-го газа в смеси, выраженное в атмосферах или Па, P0 – давление газа в стандартном состоянии P0 = 1 атм. = 1,013·105 Па, ~ P Pi = i – относительное парциальное давление i-го газа P0 P Pi Па ~ Pi = i = 1 атм 1,013 ⋅ 105 Па ~ Pi – безразмерная величина, её численное значение совпадает с Pi, если давление выражено в атмосферах и не совпадает, если давление газа измерено в Па, В идеальной газовой смеси µi0 равно∗∗ свободной энергии 1 моля индивидуального i-го газообразного вещества в стандарт ном состоянии ( µi0 = Gi0 ), поэтому µi определяется только свойствами идеального газа и зависит только от температуры. Метод ∗ 2. Для смеси реальных газов выражение, описывающее зависимость химического потенциала от парциального давления газа, очень громоздкое. Чтобы облегчить термодинамические выводы и не нарушать изящности математического аппарата термодинамики, Льюис предложил следующий метод. Выражение, описывающее зависимость химического по- Доказательство приведено во всех учебниках по физической химии, см., например, [2]. Это утверждение следует из вывода уравнения (II.173) см. [2] на стр.100. ∗∗ 51 Льюиса Летучесть газа тенциала от состава газовой смеси, оставляют без изменения, однако вместо Pi, ставят величину fi, которая называется "летучесть" газа и связана с Pi соотношением: fi = γi⋅Pi (1.61) где γi –коэффициент летучести, причем, чем больше смесь отличается от идеальной, тем больше γi отличается от 1 и fi от Pi. Коэффициент летучести можно измерить экспериментально или рассчитать теоретически, но уже не в рамках классической термодинамики. Таким образом, для смеси реальных газов ~ µ i = µ i0 + RT ⋅ ln f i f ~ f i = i0 f (1.62) где µi – химический потенциал i-го газа в смеси, µi0 – константа, которая зависит только от Т, fi – летучесть i-го газа, ~ f i – относительная летучесть i-го газа, f 0 = P° = 1 атм = 1,013·105 Па – летучесть в стандартном состоянии. 3. Для идеальной жидкой смеси (жидкого идеального раствора) зависимость µI = f(ci) описывается соотношением: µ i = µ i0 + RT ⋅ ln с~i с с~i = i сст (1.63) где µi – химический потенциал i-го компонента, µi0 – константа, зависящая от Т и внешнего давления, ci – молярная концентрация i-го компонента в растворе, с~i – относительная концентрация i-го компонента в растворе, cст – молярная концентрация в стандартном состоянии. 4. Для реального жидкого раствора (согласно метода Льюиса) µ i = µ i + RT ⋅ ln a~i 0 а a~i = i aст где ai – активность i-го вещества в растворе, a~i – относительная активность i-го вещества в растворе, (1.64) 52 aст – активность i-го вещества в стандартном растворе. Активность вещества (ai) связана с его концентрацией соотношением: ai = γi⋅ci (1.65) где γi –коэффициент активности, ci – концентрация i-го вещества в растворе. 5. Химический потенциал индивидуального твердого или жидкого вещества есть величина постоянная. µ тв. инд. в - ва = µ тв. инд. в - ва = const µ жид. инд. в - ва = µ жид. инд. в - ва = const Задание ∗∗∗ Вспомнить, что называется идеальным газом и идеальным раствором. ∗∗∗ 53 2. ХИМИЧЕСКОЕ РАВНОВЕСИЕ 2.1. Динамический характер химического равновесия Допустим, термодинамическая система состоит из нескольких веществ А, В, С, Д, способных реагировать друг с другом в соответствии с химическим уравнением: В реакционной смеси наряду с химическим взаимодействием между исходными веществами А и В (прямая реакция) может протекать и химическое взаимодействие между продуктами С и Д, в результате которого вновь образуются исходные вещества (обратная реакция). Если в системе условия таковы, что скорость образования С и Д в результате прямой реакции больше, чем скорость исчезновения этих веществ в результате обратной реакции, то количество продуктов С и Д в реакционной смеси со временем будет увеличиваться по сравнению с исходным состоянием. С точки зрения термодинамики это означает, что исходная реакционная смесь была неравновесна, и в ней самопроизвольно протекает процесс, описываемый уравнением прямой реакции: в результате которого система придет в равновесное состояние. Если в системе при данных условиях скорость обратной реакции больше, то со временем в системе продуктов С и Д будет становиться меньше, а исходных веществ А и В больше по сравнению с исходным. С точки зрения термодинамики: в неравновесной исходной реакционной смеси самопроизвольно протекает процесс, который описывается уравнением обратной реакции. Процесс закончится, когда в реакционной смеси наступит состояние химического равновесия. При наступлении химического равновесия число молекул (молей) веществ, составляющих систему, перестает меняться во времени при неизменных внешних условиях. Химическое равновесие носит динамический характер. Это означает, что в системе возникли такие условия, при которых скорости прямой и обратной реакций оказались одинаковыми. Если эти равновесные условия изменить, (например, повысить температуру смеси), то скорость одной из реакций станет больше, и равновесие нарушится, но, если условия станут прежними, то и система вернется в прежнее равновесное состояние. 54 2.2. Условие протекания химического процесса в закрытых системах при постоянных давлении и температуре Уравнение изотермы химической реакции Согласно 2-му закону термодинамики, при постоянных давлении и температуре любой процесс ( в том числе химическая реакция) возможен только в том случае, если это приведет к уменьшению свободной энергии системы ( G после процесса − G до процесса < 0 ). Изменение свободной энергии, которое вызовет химическая реакция, можно рассчитать по уравнению, известному как уравнение изотермы химической реакции. Пусть при постоянных давлении и температуре (P, T = const) в смеси, содержащей бесконечно большое количество идеальных газов А, В, С, Д происходит химическая реакция, в результате которой ν1 молей вещества А и ν2, молей вещества В превратились в ν1′, молей вещества С и ν2′ молей вещества Д: ν1A + ν2B → ν1′C + ν2′Д, (на составе смеси это превращение почти не скажется, так как смеси – бесконечно большое количество). Тогда справедливо уравнение: ~ν ' ~ν ' P ⋅ PД C G 2 − G1 = ∆GT0 + RT ⋅ ln ~ ν ~ ν PА ⋅ PВ ∆G 1 1 2 2 (2.1) где G2 – свободная энергия системы после реакции, G1 – свободная энергия системы до реакции, ∆GT0 – стандартный изобарный потенциал химической реакции, ~ ~ ~ ~ РА , РВ , РС , Р Д , – относительные парциальные давления газов в исходной реакционной смеси. ОтносительОтносительные парциальные давления – величины безразные мерные, их рассчитывают по формулам: парциальные ~ РА ~ РВ ~ РС ~ Р Д давления Р (2.2) А = Р ; РВ = Р ; РС = Р ; Р Д = Р газов 0 0 0 0 где PA, PB, PC, PД – парциальные давления газов в смеси, P0 – давление газа в стандартном состоянии. Задание ∗∗∗ Посчитать относительное парциальное давление газа А в смеси, где его парциальное давление РА = 1,5 атм. = 1,5·1,013·105 Па = = 1,5·760 мм рт ст. 55 Решение Парциальное давления газа может быть выражено в любых единицах: атмосферах (атм.), паскалях (Па), миллиметрах ртутного столба (мм рт ст). В стандартном состоянии давление газа принято равным нормальному атмосферному давлению Р0. Р0 = 1 атм. = 1,013·105 Па = 760 мм рт ст Легко видеть, что величина относительного парциального дав~ ления газа Р А не зависит от размерности РА и P0. ( ) ~ РА = РА Р0 = 1,5атм 1атм = 1,5 ⋅ 1,013 ⋅ 105 Па 1,013 ⋅ 105 Па = 1,5 ⋅ 760 ммртст 760 ммртст = 1,5 ∗∗∗ Стандартный Стандартный изобарный потенциал химической реакции изобарный ( ∆G 0 ) является константой, величина которой зависит только от T потенциал природы веществ, участвующих в реакции, и температуры. Можхимической но доказать, что ∆GT0 определяется формулой: реакции ∆GT = ∆H T − T ⋅ ∆ST (2.3) где ∆H T – тепловой эффект реакции при температуре Т, ∆ST0 = ν '2 S 0Д +ν '1 S С0 −ν 1 S А0 −ν 2 S В0 – разность абсолютных энтропии продуктов и исходных веществ данной реакции при температуре Т. ∆H T0 и ∆ST0 рассчитывают, пользуясь таблицами термодинамиче- ских величин индивидуальных веществ, приведенными в справочной литературе (см. [4]). Уравнение изотермы (2.2) позволяет определить направление химической реакции в реакционной смеси. Так, если мы предполагаем, что в смеси газов А, В, С, Д известного состава для достижения равновесия некоторое количество А и В должно превратиться в С и Д, т.е. должна идти реакция ν1A + ν2B → ν1′C + ν2′Д Как определить направление химической (∗), то наше предположение легко проверить. Для этого следует вычислить по уравнению (2.1) как изменяется изобарноизотермический потенциал системы при протекании в системе предполагаемой реакции. Если изобарно-изотермический потенциал уменьшается: G2 – G1 < 0 56 реакции то в смеси будет расти количество веществ С и Д, т.е. реакция протекает в прямом направлении. Если изобарно-изотермический потенциал увеличивается: G2 – G1 > 0, то реакция (∗ ∗) должна протекать в обратном направлении, т.е. количество исходных веществ А и В в реакционной смеси будет расти. А продуктов С и Д уменьшаться. Если G достиг минимального значения и G2 – G1 = 0, то смесь равновесна, состав её не меняется во времени. Уравнение изотермы химической реакции (2.1) можно доказать, используя понятие химического потенциала компонентов реакционной смеси. Доказательство уравнения (2.1). Пусть в бесконечно-большом количестве реакционной смеси при P = const и T = const протекает химическая реакция: ν1A + ν2B → ν1′C + ν2′D, Как известно, изобарно-изотермический потенциал системы зависит от состава системы, её температуры и давления. (1) где nA, nB, nC, nD – количество молей А, В, С, D в смеси. G = f (P, T, nA, nB, nC, nD), Дифференцируя (1), имеем: (2) ∂G ∂G ∂G ∂G dG= dP+ dT+ dnA + dnB + P T n ∂ n ∂ ∂ ∂ T, состав P, состав A P,T, состав B P,T, состав µ A µ B ∂G ∂G + dnC + dnD ∂ n ∂ n C P,T, состав Д T, состав P, µ C µ D Так как по смыслу dnA, dnB, – это число молей веществ А и В, которые исчезли из реакционной смеси, а dnC, dnD - число молей веществ С и D, которые в результате химической реакции появились в смеси, то (3) dnA = –ν1; dnB = –ν2; dnC = ν1′; dnD = ν2′ 57 Учитывая (3), а также то, что при P = const, dP = 0 и при T = const, dT = 0, получаем: (4) G2 – G1 = µC⋅ν1′ + µD⋅ν2′ – µА⋅ν1 – µВ⋅ν2, где µА, µВ, µC, µD⋅– химические потенциалы газообразных веществ А, В, С, D в реакционной смеси (см. (1.43). Если газы А, В, С, Д можно считать идеальными, то согласно (1.60) ~ µ А = µ А0 + RT ⋅ ln PA ~ µ В = µ В0 + RT ⋅ ln PВ (5) ~ µ С = µ С0 + RT ⋅ ln PС ~ µ D = µ D0 + RT ⋅ ln PD , ~ ~ ~ ~ где РА , РВ , РС , Р Д – относительные парциальные давления газов в исходной смеси, µ А0 , µ B0 , µ C0 , µ D0 – константы, которые зависят только от Т и определяются свойствами веществ А, В, С, Д в стандартном состоянии. Подставляя (5) в (4), получаем: (6) ( ) ( ( ) ) ~ ~ G2 − G1 = µ C0 + RT ln PC ⋅ν 1' + µ Д0 + RT ln PД ⋅ν 2' − ( ) ~ ~ − µ А0 + RT ln PА ⋅ν 1 − µ В0 + RT ln PВ ⋅ν 2 Или после преобразований ~ν ~ν P ⋅P (7) G 2 − G1 = (µ C0 ⋅ν 1' + µ 0Д ⋅ν 2' ) − (µ А0 ⋅ν 1 + µ В0 ⋅ν 2 ) + RT ln ~C ~Д PАν ⋅ PВν G исходных веществ 0 G конечных продуктов свободная энергия продуктов реакции находящихс я в стандартно м состоянии ' 1 ' 2 1 2 0 свободная энергия исходных веществ находящихс я в стандартно м состоянии 0 0 (8) Gкон − Gисх = ∆GT0 – называют стандартным изобарным потенциалом химической реакции, его величина определяется свойствами веществ А, В, С, Д в стандартном состоянии и зависит 58 только от температуры. Подставляя (8) в (7) получаем уравнение (2.1), что и требовалось доказать. Уравнение (2.3) можно доказать, опираясь на свойства парциальных термодинамических величин (см. стр. 48)*. Так как µ i = H i − TS i и µi = H i − TS i , то для ∆GT получим: ( ) ( ) ( ) ( ) Gкон. − Gисх. = HC −TSC ⋅ν1' + H Д −TS Д ⋅ν2' − HА −TSА ⋅ν1 − HВ −TSВ ⋅ν2 = ' = HC ⋅ν1' + HД ⋅ν2' − HА ⋅ν1 + HВ ⋅ν2 −T SC ⋅ν1' + S Д ⋅ν2 − SA ⋅ν1 + SB ⋅ν2 = 0 0 0 0 H S Hпрод. Sпрод. исх. исх. ( )( ( ) ( ) ( )( ) ) = Hпрод. − Hисх. −T ⋅ Sпрод. − Sисх. = ∆HT −T ⋅ ∆ST , где ∆Н T – стандартное изменение энтальпии при температуре Т, величина ∆Н T равна тепловому эффекту реакции, протекающей при Р = const и Т = const, ∆ST – изменение стандартной энтропии при температуре Т, которую можно рассчитать как разность стандартных энтропий продуктов реакции и исходных веществ, при данной температуре Т. Вопрос Ответ ∗∗∗ Можно ли определить направление химической реакции, если известна величина ∆GT ? Нет. Критерием направления процесса является измененение свободной энергии системы: ∆G = G2 – G1, а не стандартный изобарный потенциал реакции ∆GT . ∆GT может помочь определить направление реакции только в таких системах, где ∆G оказывается равным ∆GT . Например, если реакционная смесь представляет собой индивидуальные твердые или жидкие вещества, не растворимые друг в друге, или если реакция протекает в такой газовой смеси, где относительные парциальные давления всех участников реакции равен 1 ~ ~ ~ ~ ( Р А = РВ = РС = Р Д ). * или применяя уравнение Гиббса-Гельмгольца. 59 60 2.3. Условие химического равновесия в закрытых системах при постоянной температуре и давлении. Закон действующих масс Согласно 2-му закону термодинамики при P, T = const система, в которой может протекать химический процесс ν1A + ν2B ↔ ν1′C + ν2′Д, (∗) находится в равновесии, если свободная энергия системы (G) минимальна, и для реакции (∗), протекающей в равновесных условиях выполняется общее условие химического равновесия. G 2 − G1 = ∑ µ i ⋅ ν i = 0 (2.4) i Тогда в соответствии с (2.1) получаем: ~ ~ ( ) ( )равн. P ⋅ P равн. 0 = ∆G + RT ⋅ ln ~ (P )равн. ⋅ (P~ )равн. ν '1 ν '2 C T Д ν1 ν2 А В ~ ~ ( ) ( )равн. P ⋅ P равн. ∆G = − RT ⋅ ln ~ (P )равн. ⋅ (P~ )равн. ν '1 ν '2 C T (~ ) (~ ) (~ ) или Д ν1 ν2 А В (2.5) (~ ) где PАν 1 равн. , PВν 1 равн. , PСν 1 равн. , PДν 1 равн. – относительные парциальные давления идеальных∗ газов A, B, C, Д в равновесной смеси, ∆GT – стандартный изобарный потенциал химической реакции, величина которого зависит только от температуры. Так как ∆GT = const, то правая часть соотношения (2.5) тоже есть величина постоянная, следовательно, и выражение под логарифмом также является постоянной величиной. Обозначается эта постоянная величина K° и называется стандартной константой равновесия химической реакции. Стандартная Таким образом, в равновесной смеси: константа равновесия ∗ (P~ν ) (P~ν ) ( ) ( ) ~ν '2 ⋅ P Д равн. C равн. = K ~ ν2 1 А равн. ⋅ PВ равн. '1 (2.6) Если условия таковы, что газы в реакционной смеси нельзя считать идеальными, то, согласно методу Льюиса, выражения (2.1), (2.5), (2.6) по форме не меняются, но вместо парциальных давлений газов, следует поставить их летучести. 61 и выражение (2.5) можно переписать в виде ∆GT = − RT ⋅ ln K (2.7) где K° – стандартная константа равновесия химической реакции, которая зависит только от температуры. Выражение (2.6) означает, что в условиях равновесия парциальные давления всех веществ, участвующих в реакции, связаны между собой. Если изменить парциальное давление одного из них, изменятся и парциальные давления всех остальных веществ, участвующих в реакции, причем соотношение (2.6) между парциальными давлениями всех веществ, участвующих в реакции, останется строго определенным при данной температуре. Уравнение (2.6) называется законом действующих масс. Связь между равновесными парциальными давлениями (или равновесными концентрациями) веществ, участвующих в химической реакции впервые была установлена К. Гульбергом и П. Вааге (1867 г) из кинетических соображений. Термодинамический вывод закона действующих масс был дан Вант-Гоффом (1885 г). Закон действующих масс может быть выражен еще в трех формах. Можно доказать, что для реакции ν1A + ν2B ↔ ν1′C + ν2′Д, (∗) в равновесной идеальной газовой смеси (рис. 2.1) справедливы соотношения: (χ )νравн. ⋅ (χ )νравн. Kχ = (χ )νравн. ⋅ (χ )νравн. '1 '2 Д C 1 А В (P )νравн. ⋅ (P )νравн. = (P )νравн. ⋅ (P )νравн. (2.9) (C )νравн. ⋅ (C )νравн. = (C )νравн. ⋅ (C )νравн. (2.10) '1 KP '2 Д C 1 А 2 В '1 KC (2.8) 2 '2 Д C 1 А 2 В где (χi)равн. – мольные доли, (Рi)равн. – парциальные давления, (сi)равн. – концентрации компонентов в равновесной смеси. 62 Робщ., V, T Равновесная смесь газов: А, В, С, Д - количество газов в nA, моль nВ, моль nС, моль nД, моль смеси а) (χА)равн. б) (РА)равн в) (P~А )равн. (χВ)равн. (РВ)равн г) (СВ)равн (СА)равн (P~ )равн. В (χС)равн (РС)равн (χД)равн (РД)равн (P~ )равн. (P~ )равн. Д С (СС)равн (СД)равн состав смеси, выраженный разными способами G – минимальна при данных P и T Рис. 2.1 Характеристики равновесной термодинамической системы. Состав равновесной смеси газов А, В, С, Д, выраженный разными способами: а) через мольные доли компонентов ni n χi = = i n1 + n2 + n3 + n4 ∑ n б) через парциальные давления компонентов ni , Па или атм. Pi = Pобщ. n1 + n2 + n3 + n4 в) через относительные парциальные давления компонентов Pi Па ~ P P атм Pi = i = i = P0 1 атм 1,013 ⋅ 105 Па г) через концентрации ci = ni моль , V м3 Учитывая, что для идеальных газов: Рi = ci ⋅ RT = Pобщ. ⋅ χ i , (2.11) где Робщ. – общее давление газов в смеси. можно вывести уравнения, связывающие эмпирические константы Kχ, KP, KC и стандартную константу равновесия K0. Pобщ. − ∆ν Kχ = K P0 (2.12) K P = K (P0 ) (2.13) ∆ν 63 RT KC = K − ∆ν или P ∆ν P KC = K RT (2.14) где ∆ν = ν 1' + ν 2' − ν 1 − ν 2 - изменение числа молей в результате реакции, Pобщ. – общее давление в системе, P0 – стандартное давление, P0 =1 атм = 1,013·105 Па, K° – термодинамическая константа равновесия. Размерности констант равновесия Рассчитывая Kχ, KP и KC следует следить за размерностями величин. Kχ будет величиной безразмерной и одинаковой независимо от того, в каких единицах измерено давление; важно только, чтобы Pобщ. и P0 имели одинаковую размерность (см. (2.12)). ∆ν ∆ν KP измеряется в Па или атм в зависимости от размерности давления. Полезно иметь в виду, что K° и KP численно совпадают, если равновесные давления выражены в атм. (см. (2.13)). Единицы измерения KC зависят от размерности R и P0. Так Дж если принять R = 8,31 и P0 = 1,013·105 Па, то размермоль ⋅ K ность KC в соответствии с (2.14) окажется равной: Па [K C ] = Дж ⋅ К/ моль ⋅ К/ Задание Задание ∆ν н/ ⋅ моль = 2 м ⋅ н/ ⋅ м ∆ν моль = м3 ∆ν Все четыре формы закона действующих масс могут быть использованы при расчете состава равновесной смеси (см. гл. 3), теоретически или экспериментально могут быть определены численные значения K°, Kχ, KP или KC, но только одна из них – K° вычисляется по соответствующим значениям ∆G° или ∆H° и ∆S° с использованием таблиц стандартных термодинамических величин. ∗∗∗ Используя соотношение (2.11), доказать (2.12), (2.13), (2.14) самостоятельно (см. также гл. 3). ∗∗∗ Рассчитать K°, Kχ, KP и KC реакции: 2NO + O2 ↔ 2NO2, 64 Решение если температура газовой смеси 600 К и общее давление в реакторе 3 атм. Стандартный изобарный потенциал реакции при Т = 600 К рассчитан по справочным данным и составляет 0 ∆G500 = 25,19 кДж/моль . Давление и температура в системе постоянны. ∗∗∗ Стандартную константу равновесия(K°) рассчитывают по уравнению (2.7). ∆GT0 25,19 =− = −5,0521 8,31 ⋅ 10 −3 ⋅ 600 RT (1) ln K = − (2) K = e − 5,0521 = 6,40 ⋅ 10 −3 Константа равновесия, выраженная через мольные доли компонентов (Kχ) в соответствии с (2.12) равна: (3) Pобщ. − ∆ν Kχ = K P0 3 = 6,40 ⋅10 − 3 ⋅ 1 +1 ⇐ учитываем что Pобщ. = 3 атм, P0 = 1 атм., и для данной реакции ∆v = 2 – 2 – 1 = -1 Константа равновесия, выраженная через парциальные давления (KP)рассчитывается по уравнению (2.13). Величина KP численно совпадает с K°, если P измерены в атм: (4) K P = K (P0 ) ∆ν = 6,40 ⋅ 10 −3 (1атм ) = 6,40 ⋅ 10 −3 атм −1 , −1 и не совпадает, если давление газов измерены в других единицах, например, в Па. (5) K P = K (P0 ) ∆ν = 6,40 ⋅ 10 −3 (1,013 ⋅ 105 Па ) = 6,40 ⋅ 10 −8 Па −1 . −1 Константа равновесия, выраженная через концентрации газов (KC) рассчитывается по уравнению (2.14): (6) RT K C = K P = 3,15 ⋅ 10 − ∆ν моль 3 м −4 Дж ⋅К − 3 8,31 ⋅ 600 моль ⋅ К = 6,40 ⋅ 10 ⋅ = 1,013 ⋅ 105 Па −1 = 3,15 ⋅ 10 −1 дм 3 моль 65 2.4. Химическое равновесие в гетерогенных системах Химические реакции, участники которых находятся в разных фазах, называются гетерогенными. Пусть при постоянных P и T в системе протекала гетерогенная реакция: ν1A(газ) + ν2B(конд.) → ν3C(газ) + ν4Д(конд.), и установилось химическое равновесие. (Индекс «конд.» означает конденсированную фазу – твердую или жидкую). Если газообразные вещества А и С – подчиняются законам идеальных газов, а конденсированные вещества В и Д не образуют растворов (твердых или жидких), для гетерогенной системы будет справедливо соотношение: ~ ( P ) равн. = ~ , (P ) равн. ν3 K 0 C (2.15) ν1 А (~ ) (~ ) где PC равн. , PА равн. – относительные парциальные давления газов при равновесии, K0 – стандартная константа равновесия, величина которой зависит только от температуры. Как видно из уравнения (2.15) в константу равновесия гетерогенной реакции входят лишь парциальные давления газообразных веществ. Доказательство: При равновесии (1) ∑ µ i ⋅ν i = 0 или ⇐ общее условие химического равновесия (см. µ C ⋅ν 3 + µ Д ⋅ν 4 − µ А ⋅ν 1 − µ В ⋅ν 2 = 0 (2.6) на стр. 60) ~ (2) µ C = µ C0 + RT ln( PC ) равн. ⇐ химический ~ µ А = µ А0 + RT ln( PА ) равн. µ В = µ В0 = const (3) µ Д = µ 0Д = const ⇐ потенциал компонента идеальной газовой смеси, находящейся в равновесии (см. стр. 58) химические потенциалы твердых и жидких веществ являются постоянными величинами, если они не образуют растворов (см. стр. 52). Подставляя (2) и (3) в (1), после несложных математических преобразований, получаем: 66 (4) (ν 3 ⋅ µ С0 + ν 4 ⋅ µ 0Д ) − (ν 1 ⋅ µ А0 + ν 2 ⋅ µ В0 ) = − RT (ν 3 ⋅ ln(P~С ) равн. −ν 1 ⋅ ln(P~А ) равн. ) GT0 нач. прод. GT0 кон. прод. Поскольку GT кон. − GT нач. = GT , то (5) ∆ G T ~ ν ( P ) = − RT ⋅ ln (P~ )ν 3 C А 1 равн. равн. Так как ∆GT – постоянная при данных Р, Т величина, то и выражение под логарифмом есть константа: (6) (P~ )ν (P~ )ν 3 C А 1 равн. = K , что и требовалось доказать. равн. Константа равновесия гетерогенной реакции, несмотря на то, что выражается через парциальные давления только газообразных реагентов, будет зависеть от свойств всех участников реакции. Это следует непосредственно из (4): левая часть этого уравнения содержит стандартные химические потенциалы всех веществ. Рассчитать стандартный изобарный потенциал гетерогенной реакции ( ∆GT ) можно также как и для газовых реакций по уравнению: ∆GT = ∆H T − T ⋅ ∆ST Для расчета необходимо знать для каждого реагирующего вещества: стандартную теплоту образования (∆H f ) , стандартную эн- Задание тропию ( S 298 ) и температурную зависимость теплоемкости: СP = f(T). ∗∗∗ Написать выражение стандартной константы равновесия следующих гетерогенных реакций: MgCO3кр ↔ MgOкр. + CO2(газ) С + CO2(газ) ↔ 2CO(газ) ∗∗∗ 67 Пример Если в гетерогенной реакционной смеси конденсированные фазы – не индивидуальные вещества, а твердые и жидкие реальные растворы, и газы, не подчиняются законам идеальных газов (т.е. являются реальными газами), то аналогичным способом, используя общее условие химического равновесия (2.4), можно выразить константу равновесия гетерогенной реакции, протекающей в данной смеси, используя равновесные активности (аi) компонентов, находящихся в растворе и равновесные летучести (fi) реальных газов. Если в реакции участвуют практически чистые твердые или жидкие вещества (а не их растворы), то они попрежнему будут исключены из выражения для констант равновесия. ∗∗∗ Та, если в гетерогенной смеси (рис. 2.2) протекает реакция: Ар-р + Вкр + 2Сгаз → Дгаз + 2Ер-р константа равновесия определяется выражением: K f ,a = f Д ⋅ a E2 f С2 ⋅ a А , (2.16) где fД, fC – летучести реальных газов С и Д при равновесии, аА, аЕ – равновесные активности веществ А и Е в растворе. Рис. 2.2 Равновесная гете- Задание Кристаллическое вещество В не включе- рогенная смесь веществ А, В, С, Д, Е. но в выражение (2.16). ∗∗∗ Доказать, что для реакции восстановления водородом оксида железа (FeO), растворенного в жидком железе: FeO(раствор в Fe) + H2 (газ) → Fe(ж) + H2O(газ) отношение PH 2O в равновесной смеси PH 2 пропорционально содержанию FeO в расплаве. Учесть, что растворимость FeO в железе невелика; газы принять Рис. 2.3 Равновесная смесь, идеальными. содержащая Fe, FeO, H2, и H2O 68 Решение ∗∗∗ Запишем выражение для константы равновесия данной гетерогенной реакции: (1) K p ,a = a Fe ⋅ PH 2O a FeO ⋅ PH 2 , где a Fe = γ Fe ⋅ χ Fe , a FeO = γ FeO ⋅ χ FeO - активности Fe и FeO в расплаве. Мольные доли компонентов раствора Fe и FeO связаны соотношением: (2) χ Fe = 1 − χ FeO Если растворимость FeO невелика, можно принять (3) χ Fe ≈ 1 , a Fe = γ Fe ⋅ χ Fe ≈ 1 Тогда (1) можно переписать в виде: (4) K p ,a = PH O 2 χ FeO ⋅ γ FeO ⋅ PH , 2 где χ FeO – мольная доля FeO в растворе, γ FeO – коэффициент активности FeO в растворе. (5) PH 2O PH 2 = K p , a ⋅ χ FeO ⋅ γ FeO Из (5) следует, что отношение парциальных давлений газообразP ных участников реакции H 2O пропорционально мольной доле PH 2 FeO в жидком расплаве. ∗∗∗ 69 2.5 Влияние температуры на химическое равновесие Уравнение изобары химической реакции Изменение температуры равновесной реакционной смеси нарушит равновесие, состав смеси начнет меняться, и через некоторое время установится новое равновесное состояние, соответствующее изменившейся температуре. Смещение равновесия вызовет изменение величины константы равновесия химической реакции (K°). Зависимость K° от температуры описывается уравнением, которое получило название «уравнения изобары химической реакции». Согласно этому уравнению: Уравнение изобары химической реакции d ln K 0 ∆H 0 = dT RT 2 (2.17) или дифференциальная форма 1 ∆H 0 ln K = ⋅ ∫ 2 dT + J R T 0 (2.18) интегральная форма где К° – константа равновесия химической реакции, R – газовая постоянная, ∆Н° – тепловой эффект реакции, J – константа интегрирования. Доказательство Для доказательства (2.17) воспользуемся уравнением Гиббса-Гельмгольца. (1) ∂ ∆G 0 ∆H 0 =− 2 ∂T T T ⇐ уравнение Гиббса-Гельмгольца для стандартных величин (см.(1.28) на стр. 43) (2) ∆GT0 = − RT ⋅ ln K 0 ⇐ уравнение изотермы химической реакции для равновесных систем (см.(2.9) на стр. 61) Объединяя (1) и (2) , имеем ∂R ln K 0 ∆H 0 − =− 2 ∂T T или d ln K 0 ∆H 0 = , dT RT 2 что и требовалось доказать. Уравнение (2.18) получается интегрированием (2.17). 70 Интегральное уравнение изобары химической реакции в нешироком интервале температур Тепловой эффект реакции ∆H° зависит от температуры и для расчета интеграла в выражении (2.18) необходимо эту зависимость знать. Если исследуют влияние температуры (T) на константу равновесия (K°) в широком интервале температур, то пренебрегать зависимостью ∆H° от T нельзя. Если рассматривать зависимость K° от Т в нешироком интервале температур (∼100°), то тепловой эффект реакции можно считать независимым от температуры (∆H° = const). Это позволяет упростить интегральное уравнение изобары химической реакции. В нешироком интервале температур зависимость константы равновесия от температуры описывается уравнением: ∆H 0 ln K = − +J, RT 0 (2.19) где ∆Н0 – тепловой эффект реакции в данном температурном интервале, J – константа интегрирования. Доказательство выражения 2.19. Так как в нешироком интервале температур тепловой эффект реакции меняется мало с изменением температуры, то можно считать, что (1) ∆Н = const. Интегрируя уравнение (2.19) с учетом (1), получаем: (2) ∆H 1 ln K = ⋅ ∫ 2 dT + J R T (3) ln K = − или ∆H 1 ⋅ + J , что и требовалось доказать. R T Легко видеть, что в нешироком интервале температур зависимость константы равновесия от температуры в координатах lnK° – l/T линейна (рис. 2.4). угловой коэффициент прямой (a) равен тангенсу угла наклона 71 прямой к оси ox и связан с тепловым эффектом реакции соотношением: ∆H a=− R а) если реакция эндотермическая ∆Н° > 0, то K° растет с ростом T б) если реакция экзотермическая ∆Н° < 0, то K° убывает с ростом T в) если ∆Н° = 0, то K°не меняется с ростом T Рис. 2.4 Зависимость константы равновесия от температуры а) К° растет с ростом T, б) К° убывает с ростом T, в) К° не меняется с ростом T. Применение 1. Выражение (2.19) позволяет вычислить константу равновесия интегрального при одной температуре ( K ) , если известно экспериментальT2 уравнения ное её значение при другой температуре ( K T1 ) и тепловой эфизобары для термодинамифект реакции ∆Н° в данном температурном интервале. Для эточеских го следует записать уравнение (2.19) для двух температур и расчетов 72 решить систему уравнений относительно ( K T2 ) (1) (2) ∆H 0 0 +J ln K T1 = − RT1 0 ln K 0 = − ∆H + J 2 RT2 Вычитая (1) из (2), получаем ln K T02 = ln K T01 ∆H 0 1 1 − − R T2 T1 (2.22) Если будут известны значения констант равновесия при двух температурах ( K T1 и K T2 ) , то тем же способом можно рассчитать константу равновесия при некоторой третьей температуре ( K T3 ). Если T1, T2, T3 не слишком различаются (лежат в пределах неширокого интервала температур), можно записать уравнения: (1) (2) (3) ∆H 0 0 +J ln K T1 = − RT 1 ∆H 0 0 +J ln K 2 = − RT 2 ∆H 0 0 ln K 3 = − +J RT3 и решить систему уравнений относительно ( K T03 ) . 2. Уравнение изобары (2.19) позволяет вычислить тепловой эффект реакции, если экспериментально определить константы равновесия при нескольких температурах в интервале порядка 100°градусов. Расчеты удобно выполнять графически (рис. 2.4). Нужно построить график в координатах lnK° – 1/T, тангенс угла наклона прямой к оси ox связан с тепловым эффектом соотношением: ∆H 0 tgα = − . R 73 Как пользоваться дифференциальным уравнением изобары химической реакции Уравнение изобары в дифференциальной форме (2.19) позволяет предвидеть зависимость константы равновесия от температуры. Согласно (2.17), влияние температуры на K° обуславливается знаком теплового эффекта. Известно, что функция y = f(T) – возрастающая. Если перdy dy вая производная > 0 и y = f(T) – убывающая, если < 0. dx dx Из уравнения а) б) d ln K ∆H = видно, что dT RT 2 d ln K если реакция эндотермическая ∆Н > 0, то >0, dT следовательно, функция lnK° = f(T) – возрастающая d ln K < 0, dT следовательно, функция lnK° = f(T) – убывающая если реакция экзотермическая ∆Н < 0, то Рис. 2.5 Зависимость константы равновесия от температуры а) К° увеличивается с ростом T, б) К° уменьшается с ростом T. Правило Эти же выводы получаются из общего принципа смещения Ле Шателье равновесия Ле Шателье-Брауна, согласно которому если на сис- тему, находящуюся в устойчивом равновесии, оказать воздействие, изменяя какой-нибудь параметр, определяющий равновесное состояние, например, температуру, давление или др., то усилится то из направлений процесса, протекание которого ослабит оказанное воздействие. 74 3. НЕКОТОРЫЕ ПРИЛОЖЕНИЯ ЗАКОНОВ ТЕРМОДИНАМИКИ 3.1. Расчеты тепловых эффектов и других термодинамических характеристик химических процессов. Термохимические уравнения Пример Химические уравнения, для которых указан тепловой эффект, называются термохимическими. Тепловой эффект зависит от агрегатного состояния и кристаллической модификации веществ, участвующих в реакции, поэтому в термохимических уравнениях указывают состояния веществ. Условимся, жидкое состояние обозначать (ж), растворенное в воде (aq), газообразное (г), кристаллическое (кр), (если требуется, тип кристаллической решётки вещества можно уточнить, например, Sромб., Pбелый, Cграфит и т.д.). Из закона Гесса следует, что с термохимическими уравнениями, если они приведены для одних и тех же внешних условий, можно оперировать так же, как с алгебраическими. В частности их можно умножать на некоторое число, складывать или вычитать одно из другого. ∗∗∗ Например, термохимическое уравнение (1) (1) ∆H1 2A → 2D + L можно получить, если сложить уравнение (2), уравнение (3), умноженное на коэффициент 2, и уравнение (4), умноженное на коэффициент –1. (2) A + B → 2C ∆H 20 1 (3) C + 0,5A → D + K ∆H 30 2 (4) L + B → 2K ∆H 40 -1 A + B + 2C + 2·0,5A – L – B → 2C + 2D + 2K – 2K 1∆H 20 + 2∆H 30 − 1∆H 40 , после алгебраических преобразований имеем: (5) 2A → 2D + L ∆H 20 + 2 ⋅ ∆H 30 − ∆H 40 Таким образом тепловой эффект реакции (1) легко вычислить, если известны тепловые эффекты реакций (2), (3) и (4): (6) ∆H 10 = ∆H 20 + 2 ⋅ ∆H 30 − ∆H 40 75 Задание 1 ∗∗∗ Показать на примере реакции тримеризации ацетилена: 3C2H2(г) → С6H6(ж) Решение справедливость первого и второго следствий закона Гесса. ∗∗∗ Согласно 1-му следствию закона Гесса тепловой эффект данной реакции ( ∆H ) можно рассчитать по стандартным теплотам образования участвующих в реакции веществ: (1) ∆H = ∆H fC 6 H 6 (ж) − 3 ⋅ ∆H fC 2 H 2 (г ) Соотношение (1) можно доказать, если представить термохимическое уравнение (2): (2) ∆H 3C2H2(г) → С6H6(ж) как комбинацию уравнений (3) и (4) (3) 6C(графит) + 3H2(г) → C6H6(ж) ∆H 1 1 (4) 2C(графит) + H2(г) → C2H2(г) ∆H 2 -3 6С(графит) + 3H2(г) – 3·2С(графит) – 3H2(г) → С6H6(ж) – 3С2H2(г) ∆H − 3 ⋅ ∆H 1 2 После алгебраических преобразований (5) ∆H = ∆H1 − 3 ⋅ ∆H2 3C2H2(г) → С6H6(ж) По определение тепловой эффект реакции (3) есть стандартная теплота образования бензола (6) ∆H 1 = ∆H fC 6 H 6 (ж) а тепловой эффект реакции (4) – стандартная теплота образования ацетилена: (7) ∆H 2 = ∆H fC 2 H 2 (г ) Подставляя (6) и (7) в (5) получаем соотношение (1), выражаю- 76 щее 1-ое следствие закона Гесса и позволяющее рассчитать тепловой эффект реакции по стандартным теплотам образования реагентов. Согласно 2-ому следствию закона Гесса тепловой эффект реакции можно рассчитать, используя стандартные теплоты сгорания веществ: 0 0 ∆H 0 = 3 ⋅ ∆H сгор. − ∆H сгор. C 2 H 2 (г ) C6 H 6 (ж ) (8) Справедливость этого соотношения легко видеть, если представить термохимическое уравнение (2) как комбинацию уравнений (9) и (10) (9) С6H6(ж)+7,5О2(г) → 6СО2(г)+3Н2О(ж) ∆H 30 -1 (10) С2H2(г)+2,5О2(г) → 2СО2(г) +Н2О(ж) ∆H 40 3 33С2H2(г) + 3·2,5О2(г) - С6H6(ж) - 7,5О2(г) →3·2СО2(г) + 3Н2О(ж) - 6СО2(г) -3Н2О(ж) (11) 3С2H2(г) → С6H6(ж) ∆H X0 = 3 ⋅ ∆H 40 − ∆H 30 По определению тепловой эффект реакции (9) есть стандартная теплота сгорания жидкого бензола: (12) 0 ∆H 30 = ∆H сгор. C6 H 6 ( ж ) Тепловой эффект реакции (10) – стандартная теплота сгорания ацетилена: (13) 0 ∆H 40 = ∆H сгор. C2 H 2 ( г ) Подставляя (12) и (13) в (11), получаем соотношение (8), выражающее 2-ое следствие закона Гесса: 0 0 ∆H 0 = 3 ⋅ ∆H 40 − ∆H 30 = 3 ⋅ ∆H сгор. − ∆H сгор. C 2 H 2 (г ) C6 H 6 (ж ) Тепловые эффекты реакций определяют экспериментально в специальных приборах – калориметрах. Однако прямые калориметрические измерения возможны не всегда. Закон Гесса позволяет рассчитывать тепловые эффекты любого процесса, даже если он не осуществим экспериментально. 77 Задание 2 Решение ∗∗∗ Рассчитать стандартную теплоту образования карбида вольфрама WC(кр), если, согласно калориметрическим данным: (1) С(графит) + О2(г) → СО2(г) (2) WC(кр) + 2,5О2(г) → WO3(кр) + СО2(г) ∆H 0 = −1192,44 кДж/моль 298 (3) Wкр + 1,5О2(г) → WO3(кр) 0 ∆H 298 = −393,51 кДж/моль 0 ∆H 298 = −837,47 кДж/моль ∗∗∗ Согласно определению стандартная теплота образования карбида вольфрама ∆H 0f WCкр есть тепловой эффект реакции: W(кр) + С(графит) → WC(кр) Интересующее нас термохимическое уравнение можно получить, если сложить уравнения (1) и (3) и вычесть из них уравнение (2). (1) С(графит) + О2(г) → СО2(г) ∆H 10 1 (2) WC(кр) + 2,5О2(г) → WO3(кр) + СО2(г) ∆H 20 -1 (3) Wкр + 1,5О2(г) → WO3(кр) ∆H 30 1 (4) W(кр) + С(графит) → WC(кр) ∆H10 + ∆H 30 − ∆H 20 Следовательно ∆H 0f WC = ∆H 10 + ∆H 30 − ∆H 20 = −393,51 − 837,47 − (−1192,44) = −38,54 кДж/моль кр Задание 3 ∗∗∗ Закон Гесса распространяется не только на химические реакции, но и на все процессы, сопровождающиеся тепловыми эффектами, поэтому термохимические расчеты можно использовать для определения энергии химических связей, тепловых эффектов фазовых превращений, теплот растворения и т.п. ∗∗∗ Показать, что в тройной точке (т.е. в одинаковых условиях) теплота возгонки (∆Hвозг.), теплота плавления (∆Hпл.) и теплота испарения (∆Hисп.) вещества связаны соотношением: 78 Решение ∆Hвозг = ∆Hпл + ∆Hисп. ∗∗∗ Запишем термохимические уравнения процессов плавления (1), испарения (2) и возгонки (3) вещества А. Легко видеть, что термохимическое уравнение (3) есть сумма уравнений (1) и (2): (1) А(кр.) → А(ж.) ∆Hпл. 1 (2) А(ж.) → А(г.) ∆Hисп. (3) А(кр.) → А(г.) ∆Hвозг = ∆Hпл + ∆Hисп 1 Данные процессы можно представить в виде энтальпийной диаграммы изображенной на рис. 3.1. Рис. 3.1 Энтальпийная диаграмма фазовых переходов в тройной точке ∗∗∗ Комбинированием химических уравнений можно рассчитать не только тепловой эффект интересующей нас реакции (∆H°), но и другие её термодинамические характеристики, в том числе ∆G° и константу равновесия K°. Так как величина G – есть функция состояния системы, то ∆G не зависит от пути процесса. Поэтому изобарный потенциал реакции, совершающейся в несколько (одну, две, три…) стадий, равен сумме величин ∆Gi для каждой стадии, и если одно из значений ∆Gi, входящих в сумму, неизвестно, то оно может быть рассчитано. Для таких расчетов, аналогичных расчетам ∆H° по закону Гесса, необходимо, чтобы вещества, участвующие в каждой реакции, находились в одинаковом состоянии. Удобно, если все вещества находятся в стандартном состоянии, т.е. каждый Стандартное из компонентов реакции находится при стандартном давлесостояние нии P°° = 1 атм., причем твердые и жидкие вещества являются вещества чистыми фазами, а газообразные вещества – идеальными газами (для последних безразлично, находятся ли они в смеси или в индивидуальном состоянии, важно лишь, что их отно- 79 Пример ~ сительные парциальные давления Pi = 1 ), в таком случае ∆G = ∆G°. Вследствие логарифмической зависимости между ∆G°и K°, все действия сложения и вычитания для величин ∆G° при комбинировании реакций превращаются в действия умножения и деления для величин K°. ∗∗∗ Пусть известны ∆G° и K° для реакций (1) и (2): (1) CO(г) + H2O(г) → CO2(г) + H2(г) ∆G10 = − RT ln K10 (2) 2CO(г) + O2(г) → 2CO2(г) ∆G20 = − RT ln K 20 Требуется найти константу равновесия K 30 реакции (3) (3) 2H2(г) + O2(г) → 2H2O(г) ∆G30 = − RT ln K 30 при той же температуре. Проведем те же операции, что и при расчете теплоты реакции по закону Гесса: (3) = (2) – (1)·2, поэтому (4) ∆G30 = ∆G20 − 2 ⋅ ∆G10 , отсюда (5) RT ln K 30 = RT ln K 20 − 2 ⋅ RT ln K10 , следовательно (6) ln K 3 = ln K 2 − ln( K1 ) 2 (7) K 2 K3 = 2 K1 и K 2 ln K 3 = ln , тогда 2 K1 ( ) ( ) 3.1.1. Задачи для самостоятельного решения Задача 1 (оценка 1∗) Известны константы равновесия при 500K для реакций: (1) CH4 + H2O → CO + 3H2 K10 = 7,8 ⋅ 10 −11 80 (2) CH4 + CO2 → 2CO + 2H2 K 20 = 4,03 ⋅ 10 −13 (3) 2CO + O2 → 2CO2 K 3 = 1,95 ⋅ 1050 Рассчитать из этих данных константу равновесия реакции разложения водяного пара при T = 500K; 2H2O → 2H2 + O2 Задача 2 (оценка 1∗) Записать выражение, с помощью которого можно рассчитать константу равновесия реакции: 4NH3(г) + 7O2(г) → 4NO2(г) + 6H2O(г), если известны K° при той же температуре для следующих реакций: Задача 3 (оценка 1∗) (1) N2(г) + 3H2(г) → 2NH3(г) K10 (2) N2(г) + O2(г) → 2NO2(г) K 20 (3) 2H2(г) + O2(г) → 2H2O(г) K 30 Записать выражение для расчета теплового эффекта, стандартного изобарного потенциала и константы равновесия реакции Cграфит + H2O(г) → CO(г) + H2(г) при температуре T, если известны термодинамические параметры при той же температуре следующих реакций: Задача 4 (оценка 1∗) (1) CO(г) + 1/2O2(г) → CO2(г) ∆H 10 , ∆G10 , K10 (2) H2(г) + 1/2O2(г) → H2O(г) ∆H 20 , ∆G20 , K 20 (3) Cграфит + O2(г) → CO2(г) ∆H 30 , ∆G30 , K 30 Стандартная теплота сгорания жидкого пиридина (С5H5N) составляет (– 2755,16) кДж/моль. Считая, что теплоты образования продуктов реакции: CO2, H2O, N2 Вам известны из справоч- 81 ных данных, рассчитайте стандартную теплоту образования С5H5N(ж). Сравните полученную величину со справочной. Какую величину необходимо знать, чтобы рассчитать теплоту образования Задача 5 (оценка 1∗) газообразного пиридина ( ∆H f C5 H 5 N ( г) ). Имеются следующие данные: Уравнения реакций (1) Fe(кр) + 2HCl(aq) → FeCl2(aq) + H2(г) 0 ∆H 298 кДж/моль - 87,3 (2) FeCl2(кр) + aq → FeCl2(aq) - 81,0 (3) HCl(г) + aq → HCl(aq) - 72,7 (4) H2(г) + Cl2(г) → HCl(г) - 182,8 Определить стандартную теплоту образования кристаллического FeCl2. Задача 6 (оценка 1∗) Вычислить стандартную теплоту сгорания этана в озоне: C2H6 (г) + 7/3O3 (г) → 2CO2 (г) + 3H2O(ж) если ∆H сгор. C 2 H 6 ( г) в кислороде составляет (- 1561,0) кДж/моль и Задача 7 (оценка 1∗) ∆H f O3 = 142,3 кДж/моль. ( г) Вычислить количество теплоты, которое выделяется при производстве 1000 т. аммиака в сутки, если N2 (г) + 3H2 (г) → 2NH3 (г) ∆H = - 92 кДж Как используют эту теплоту на производстве? Задача 8 (оценка 1∗) Вычислить стандартную теплоту образования Fe2O3, если известно изменение энтальпии при T = 298 K в реакции: Fe2O3 (кр) + 3CO(г) → 2Fe(кр) + 3CO2 (г) ∆H = - 28,4 кДж Стандартные теплоты образования CO2 (г) и CO (г) равны соответственно (- 393,70 и (- 110,5) кДж/моль. 82 3.2. Применение законов термодинамики к идеальным газам Идеальный газ Идеальный газ – система, которая состоит из молекул, между которыми нет межмолекулярного взаимодействия, и размеры которых несопоставимо малы по сравнению со средними расстояниями между ними. Уравнение состояния идеального газа Параметры термодинамической системы, представляющей собой n моль идеального газа, связаны уравнением, которое называется уравнением состояния идеального газа: P ⋅V = n ⋅ R ⋅ T (3.1) Можно доказать, что для системы, параметры которой связаны уравнением (3.1) выполняются соотношения: ∂U =0 ∂P T и ∂U =0 ∂V T (3.2) Следовательно, U такой системы не зависит от ее объема и давления. Из этого следует важный вывод: Важное свойство идеального газа Внутренняя энергия идеального газа зависит только от температуры U = f (T) (3.3) Классическая термодинамика не может рассчитать абсолютное значение внутренней энергии системы (U) и ее теплоемкость CV CV = dU . Но для идеальных газов появляется возмож dT ность оценить U и CV, опираясь на законы молекулярнокинетической теории газов. Внутренняя энергия газовой системы складывается из энергии поступательного и вращательного движения молекул в пространстве, энергии колебательного движения атомов, энергии электронных переходов. U = Uпост + Uвращ + Uколеб + Uэл Переход электронов в молекулах на более высокие энергетиче- 83 ские уровни происходит лишь при температурах выше 2000 К, при более низких температурах Uэл можно пренебречь. Колебательное движение атомов подчиняется законам квантовой механики, и молекулярно-кинетическая теория не позволяет рассчитать Uколеб и теплоемкость газа, связанную с этим видом движения. Зато энергию поступательного и вращательного движения молекул (Uпост, Uвращ) рассчитать можно. Поступательное движение молекулы газа в пространстве в любом произвольном направлении может быть разложено по 3-м координатным осям (в этом случае говорят, что частица имеет 3 степени свободы). На каждую степень свободы движения молекулы приходится энергия, равная kТ (или в пересчете на 1 моль 2 газа RT ). Эта величина, как принято считать, не зависит от вида 2 теплового движения молекулы. Если кроме поступательного молекула совершает еще и вращательное движение, то на каждую «вращательную» степень свободы тоже приходится энергия RT в 2 расчете на 1 моль газа. Эти соображения позволяют оценить внутреннюю энергию 1-го моля одноатомного, двухатомного или трехатомного идеального газа, а затем и его теплоемкости по уравнению CV = dU . dT Таблица 3.1 Значения изохорной теплоемкости идеальных газов, рассчитанные в рамках молекулярно-кинетической теории Газ Виды движения Число степеней свободы одноатомный Ar, He и т.п. поступательное 3 двухатомный H2, HCl и т.п. поступательное вращательное 3 2 колебательное трехатомный с нелинейными молекулами поступательное вращательное колебательное U 3⋅ 3 RT 2 RT 2 2 dU CV = dT RT 3 2 2 RT 5 2 5 2 R R+C кол. оценить невозможно 3 3 6⋅ RT 2 оценить невозможно 3R + C кол. 84 Рассчитанные и измеренные экспериментальным путем теплоемкости одноатомных газов (Не, Аr, Ne) хорошо совпадают. Теплоемкости многих двухатомных газов отличаются от рассчитанной величины равной 2,5 R Теплоемкость идеальных газов Дж моль ⋅ К и зависят от температуры; теплоемкости газов, молекулы которых состоят из трех и более атомов, сильно отличаются от расчетных. Этого и следовало ожидать, так как, оценивая энергию системы, не учитывали внутримолекулярные колебания, вклад которых в энергию системы тем больше, чем сложнее молекула. Итак, полезно помнить, что теплоемкость одноатомного идеального газа при постоянном объеме можно оценить по формуле: CV = 3 CV = 5 2 R, (3.4) R (3.5) двухатомного газа: Замечание 2 Выполняя термодинамические расчеты следует учитывать, что поведение газовой системы соответствует поведению идеального газа лишь при относительно высоких температурах и низких давлениях в газовой системе, только в этом случае молекулы могут свободно перемещаться, не взаимодействуя друг с другом. Важно научиться рассчитывать термодинамические характеристики процессов, протекающих в идеальных газовых системах, с этой целью рассмотрим некоторые примеры. 3.2.1. Изобарное расширение газа Постановка задачи Изобарным называют процесс, протекающий при постоянном давлении (Р = const). Пусть моль идеального газа находится в цилиндре с поршнем, на который оказывается постоянное давление Р. Система получает некоторое количество теплоты Q, в результате чего газ расширяется и его объем меняется от V1 до V2, а температура от Т1 до Т2 (рис. 3.2). Требуется рассчитать: 1) максимальную работу (А), которую совершает расширяющийся газ; 2) количество теплоты (Q), необходимое для протекания про- 85 цесса; 3) изменение внутренней энергии (∆U = U2 – U1), энтальпии (∆H = H2 – H1) и энтропии (∆S = S2 – S1) системы. Рис. 3.2 Изобарное расширение газа Решение задачи Для решения задач подобных этой необходимо: 1• уравнение состояния системы: f (Р, V, Т, n) = 0 (т.е. уравнение, связывающее параметры данной системы: Р, V, Т, n); для идеальных газов уравнение состояния известно: РV = nRT (3.1) 2• математические выражения первого и второго законов термодинамики: Максимальная работа совершается при протекании процесса бесконечно медленно в условиях близких к равновесным, и рассчитывается (см. стр. 28) по формулам: δA = PdV или в интегральной форме V2 A = ∫ PdV . V1 Если Р = const, интеграл легко рассчитать V2 A = ∫ PdV = P ⋅ (V 2 − V1 ) V1 (3.6) 86 Изменение внутренней энергии газа, связанное с повышением его температуры от Т1 до Т2, можно рассчитать, если известна теплоемкость газа СV; согласно (1.6) dU = CV ⋅ dT , считая теплоемкость газа постоянной величиной, не зависящей от температуры (СV = const), получаем: U2 T2 U1 T1 ∫ dU = ∫ CV ⋅ dT ; U 2 − U 1 = CV ⋅ (T2 − T1 ) (3.7) Изменение энтальпии газа при повышении его температуры от Т1 до Т2 считаем тем же способом: dH = C P ⋅ dT H2 T2 H1 T1 ∫ dH = ∫ C P ⋅ dT , если CР = const, то H2 – H1 = CР ·(T2 – T1) (3.8) Количество теплоты (Q), которое необходимо подвести к системе для протекания процесса при P = const, равно изменению энтальпии: Q = H2 – H1 = ∆H (3.9) Соотношение (3.9) следует из 1-го закона термодинамики и легко выводится (см. стр. 13). Изменение энтропии считаем, опираясь на объединенный 1-й и 2-й закон термодинамики (см. стр. 31). Для равновесных процессов справедливы выражения: (1) dU = TdS − pdV ⇐ объединенный 1-й и 2-й законы (2) CV ⋅ dT = TdS − pdV ⇐ так как dU = CV ⋅ dT (3) dS = CV dT p + dV T T термодинамики ⇐ математические преобразования 87 (4) dS = S2 ⇐ CV dT R + dV T V ⇐ интегрирование T2 V2 CV R (5) ∫ dS = ∫ dT + ∫ dV S1 T1 T V1 V (6) S 2 − S1 = CV ⋅ ln так как P·V = R·T, то p T = R V уравнения (4) T2 V + R ⋅ ln 2 T1 V1 ⇐ интегрирование при условии СV = const Из уравнения состояния (3.1) следует, что при Р = const, (7) V2 V1 = T2 T1 ⇐ так как pV = RT, то ; P/ V2 P/ V1 = R/ T2 R/ T1 учитывая также, что изобарная и изохорная теплоемкости идеального газа связаны соотношением: ⇐ см. стр. 20 (8) СP = СV + R (9) S 2 − S1 = CV ⋅ ln V2 V1 + R ⋅ ln V2 V1 = ln V2 V1 ⋅ (CV + R) = C P ⋅ ln V2 V1 Итак, энтропия газа при изобарном расширении растет, ее изменение (∆S = S2 – S1) можно рассчитать по формулам: ∆S = C P ⋅ ln V2 V1 или ∆S = C P ⋅ ln T2 T1 (3.10) Полученные результаты иллюстрирует рисунок 3.3. Рис. 3.3 Изменение функции состояния идеального газа при изобарном расширении 88 3.2.2. Изотермическое расширение газа Постановка задачи Изотермические процессы протекают при постоянной температуре (Т1 = Т2 = Т = const) Пусть n моль идеального газа находится в цилиндре с поршнем. Система получает некоторое количество теплоты (Q), газ расширяется, его объем меняется от V1 до V2, а температура остается постоянной (Т = const) см. рис.3.4). Рассчитать термодинамические характеристики процесса: 1) максимальную работу, которую может совершить газ при расширении; 2) количество теплоты, которое необходимо подвести к системе, чтобы его объем изменился от V1 до V2; 3) изменение функций состояния системы (∆U, ∆Н, ∆S, ∆F, ∆G) в результате протекания данного процесса. Рис. 3.4 Изотермическое расширение газа Решение задачи Максимальную работу расширения газа: V2 A = ∫ P ⋅ dV (1) V1 легко рассчитать, если задано уравнение состояния газа f(P, V, T, n) = 0, которое позволит выразить Р как функцию V. Для идеального газа уравнение состояния известно. Согласно (3.1) для идеального газа P= (2) nRT V , тогда подставляя (2) в (1) и интегрируя, получаем: (3) A= V2 ∫ V1 nRT V V dV = nRT ⋅ lnV V2 = nRT ⋅ ln 1 V2 V1 89 Итак, максимальная работа изотермического расширения газа определяется выражением: V A = nRT ⋅ ln 2 (3.11) V1 Внутренняя энергия идеального газа зависит только от температуры. Если Т не меняется, то U2 = U1 и ∆U = 0 (3.12) Количество теплоты, необходимое для изотермического расширения газа согласно 1-му закону термодинамики равно работе процесса. Действительно, (1) Q = ∆U + А; ⇐ 1-й закон термодинамики для макропроцессов ⇐ так как ∆U = 0. (2) Q = А Изменение энтропии можно определить, опираясь на математическое выражение объединенного 1-го и 2-го законов термодинамики. Для равновесных процессов справедливы соотношения: (1) dU = TdS – PdV, (2) dS = P T dV ⇐ объединенный 1-й и 2-й законы термодинамики ⇐ так как U = const, dU = 0 Для идеальных газов согласно (3.1) (3) P= nRT V Подставляя (3) в (2) и интегрируя, получаем (4) (5) S2 V2 S1 1 ∫ dS = V nR ∫ V dV = nR ln V2 1 V S 2 − S1 = nR ln V2 V1 или в интегральной форме 90 Итак, энтропия идеального газа при изотермическом расширении растет, её изменение (∆S = S2 - S1) можно рассчитать по формуле : V ∆S = nR ln 2 (3.13) V1 Энтальпия идеального газа при протекании изотермических процессов не меняется. Действительно (1) H = U + p·V = U + nRT ⇐ определение энтальпии при условии pV = nRT; (2) dH = dU + nRdT = 0 ⇐ дифференцирование (1); (3) dH = 0 ⇐ так как T = const, dT = 0 U = const, dU = 0; (4) H2 - H1 = 0; ∆H = 0 ⇐ после интегрирования (3) Изменение изобарно-изотермического (∆G = G2 - G1) и изохорно-изотермического (∆F = F2 - F1) потенциалов легко рассчитать, опираясь на их определения: (1) G = H – T·S ⇐ определение G (2) dG = dH – TdS – SdT, ⇐ дифференцирование (1), (3) dG = – TdS ⇐ так как H = const, T = const, то dH = 0, dT = 0; (4) ∆G = −T ⋅ ∆S ⇐ после интегрирования (3); (5) V ∆G = − nRT ln 2 ⇐ так как ∆S = nR ln V1 согласно (3.13) V2 V1 Таким образом, свободная энергия системы (G) уменьшается при изотермическом расширении газа. V ∆G = nRT ln 1 V2 Подобным образом можно показать, что (3.14) 91 V ∆F = ∆G = nRT ln 1 V2 (3.15) Полученные результаты иллюстрирует рисунок 3.5. Рис. 3.5 Изменение функций состояния идеального газа при изотермическом расширении 3.2.3. Адиабатическое расширение газа Постановка задачи Процесс называется адиабатическим, если система, в которой он протекает, не получает теплоты извне и не отдает в окружающую среду (Q = 0). Пусть 1 моль идеального газа расширяется от V1 до V2 в адиабатических условиях (см. рис.3.6). Рис. 3.6 Схема адиабатического расширения газа Определить, как изменится температура и давление газа. 92 Решение Можно доказать, что при адиабатическом расширении газ охлаждается, Т2 < Т1. (1) dU = δQ − pdV ⇐ 1-й закон термодинамики (2) dU = − PdV ⇐ δQ = 0 для адиабатических процессов (3) CV dT = − ⇐ так (4) (5) RT dV V P= CV R dT = − dV T V T2 V 2 CV R ∫ T dT = − ∫ V dV T1 V1 T как dU = CV ⋅ dT и RT V ⇐ разделяем переменные ⇐ интегрируем от начального до конечного состояния, при условии, что СV = const V (6) CV ⋅ lnT T2 = − R lnV V2 (7) CV ⋅ ln (8) ln (9) ln T2 = ln T1 − 1 1 T2 V = − R ln 2 T1 V1 T2 V R =− ⋅ ln 2 T1 CV V1 V R ⋅ ln 2 CV V1 ⇐ математические вания преобразо- ⇐ математические вания преобразо- ⇐ математические вания преобразо- Таким образом, при расширении газа (т.е. если V2 > V1) его температура понижается (Т2 < Т1). Учитывая уравнение состояния идеального газа (3.1), можно преобразовать (8) и вычислить, как изменяется давление газа в системе. (10) T2 PV = 2 2 T1 PV 1 1 T2 P2 V + ln 2 (11) ln = ln T1 P1 V1 ⇐ так как для 1-го моля идеального газа RT = PV ⇐ логарифмируем (10) 93 (12) ln P2 V V R + ln 2 = − ⋅ ln 2 P1 V1 CV V1 ⇐ подставляем (11) в (8) (13) ln P2 R = −1 + P1 CV ⇐ математические преобразования (14) ln C + R V2 P2 ⋅ ln = − V P1 C V1 V ⇐ математические вания (15) ln P2 C V = − P ⋅ ln 2 P1 CV V1 ⇐ так как CP = CV + R для идеального газа (16) ln P2 C P V = ⋅ ln 1 P1 CV V2 ⇐ математические вания (17) V2 ⋅ ln V1 преобразо- преобразо- CP P2 V1 CV = P1 V 2 Давление в системе уменьшается и может быть рассчитано по уравнению (17). 3.2.4. Адиабатические химические реакции Постановка задачи Реакция может протекать, как в изотермических (Т = const), так и в адиабатических условиях (Q = 0), то есть так, что система в которой протекает химическая реакция, не получает и не отдает тепло в окружающую среду; энергия, которую бы потеряла или приобрела система в изотермических условиях, в этом случае идет на ее охлаждение или нагревание. Некоторые химические процессы, например, реакции горения и взрывы, протекают так быстро, что в их центре приблизительно сохраняются адиабатические условия. Это позволяет рассчитывать максимальные температуры пламени или взрыва по теплотам реакции при низких температурах (∆Н298) и теплоемкостям продуктов реакции. Пусть при Р = const в адиабатических условиях (рис. 3.7) протекает химический процесс: Реагенты → Продукты при Т = 298К при Т1К ( ∗) Доказать, что для адиабатической реакции (∗), протекаю- 94 щей при постоянном давлении (Q = 0, Р = const) выполняется соотношение: − ∆H 298 = T2 ∫ C P продуктов dT 298 где ∆Н298 – тепловой эффект реакции при Т = 298К, СP – теплоемкость продуктов реакции, которая складывается из мольных теплоемкостей соответствующих веществ ( C P продуктов = ∑ν i ⋅ C P ) i i Т2 – температура реакционной смеси после процесса (температура пламени). Рис. 3.7 Схема химической реакции, протекающей в адиабатических условиях Решение задачи (1) H продуктов, T2 − H реагентов, 298 = 0 T2 (2) H продуктов, T2 = H продуктов, 298 + ∫ C P пр dT 298 ⇐ так как Р = const ∆Н = Q, Q = 0, если процесс адиабатный ⇐ dН = CPdТ или в интегральной форме H T2 − H 298 = T2 ∫ CP dT 298 Подставляя (2) в (1), получаем (3) H продуктов, 298 + T2 ∫ C P пр dT − H реагентов, 298 = 0 298 95 T2 (4) H 298 − H реаг, 298 = − ∫ C P пр dT пр, 298 ∆H 298 (5) − ∆H 298 = T2 ∫ C P пр dT , ⇐ математическое преобразование что и требовалось доказать. 298 Математические преобразования можно проиллюстрировать графически (см. рис. 3.8). Рис. 3.8 Сопоставление изотермического (∆Н < 0) и адиабатического (∆Н = 0) процессов протекающих в одной и той же системе при Р = const т.А – энтальпия реагентов при Т1 = 298К, т. В – энтальпия продуктов при Т1=298К, т. С – энтальпия реагентов при температуре Т2, А → С – адиабатическая химическая реакция, А → В – изотермическая химическая реакция, В → С – нагревание продуктов реакции. 3.2.5. Задачи для самостоятельного решения Задача 1 (оценка 1*) Вычислить конечную температуру обратимого адиабатического расширения 200 г неона от 20 до 50 литров. Начальная температура газа 25°С. Составить схему процесса. Задача 2 (оценка 1*) Вычислить максимальную работу 1)изотермического и 2) адиабатического расширения 2 моль двухатомного идеального газа от 10 до 20 литров с начальной температурой 25°С. Составить схему процесса. 96 Задача 3 (оценка 1*) Клапан цилиндра, содержащего 10 л газа при давлении 25 атм. и температуре 25°С, открыт в атмосферу, давление в которой 760 мм рт ст, а температура 25°С. Определить работу процесса расширения газа, считая процесс изотермическим. Составить схему процесса. Задача 4 (оценка 1*) Доказать что параметры системы при протекании адиабатических процессов в идеальных газах связаны соотношениями: P ⋅ V γ = const и T ⋅ V γ −1 = const , где γ = Задание 5 CP CV В соответствии с выбранным вариантом (таблица 3.2) Таблица 3.2 № варианта 1 2 3 4 5 Вещество А H2 N2 CO NO O2 Температурная зависимость теплоемкости C P = a + b ⋅ T + c' / T 2 a b·103 c’·10-5 27,28 27,87 28,41 29,58 31,46 3,26 4,27 4,10 3,85 3,39 0,502 - 0,46 - 0,59 - 3,77 решить следующие задачи. Задача 5.1 (оценка 1*) Определить количество теплоты, поглощенное при изобарном нагревании 1 кг вещества А от 300 до 1000 К с учетом зависимости изобарной теплоемкости от температуры. Изобразить схему процесса. Задача 5.2 (оценка 1*) В каком из процессов работа расширения 2-х молей идеального газа А больше: а) изотермическое расширение при 300 К от 10 до 100 м3; б) изобарное расширение при повышении температуры от 300 до 500 К. Изобразить схему процессов. Задача 6 (оценка 1*) В соответствии с выбранным вариантом (таблица 3.3), рассчитать работу, совершаемую реакцией А против внешнего давления при постоянном давлении и температуре 298 К. Составить схему процесса. 97 Таблица 3.3 № варианта 1 2 3 4 5 Задача 7 (оценка 2*) Реакция А 3H2 + N2 → 2NH3 2NO2 → N2O4 2CO + O2 → 2CO2 2NO + O2 → 2NO2 4NH3 + 5O2 → 4NO + 6H2O Вычислить максимальную температуру пламени при адиабатическом горении ацетилена в кислороде: C2H2 + 2,5O2 → 2CO2 + H2O, Считая, что теплота ∆H°298 адиабатически нагревает продукты до температуры пламени Т. Необходимые данные взять из справочника [4]. Задача 8 (оценка 2*) Оценить максимальную температуру пламени в следующих процессах: а) горение H2 на воздухе; б) горение H2 в чистом кислороде; в) горение СH4 на воздухе; г) горение H2 во фторе. Необходимые данные взять из справочника [4]. Задача 9 (оценка 2*) В изолированной калориметрической бомбе находится стехиометрическая смесь водорода и кислорода: 2H2 + O2 (гремучая смесь) при T = 298 K и P = 1 атм. Оценить температуру и давление после взрыва. Теплота сгорания этана, этилена и ацетилена равна соответственно 70129, 64973 58657 кДж/м3. Объяснить, почему для сварки пользуются ацетиленом, а не этаном или этиленом. Принимая, что сгорание в ракетном двигателе происходит адиабатически, вычислить температуру продуктов сгорания, если топливом служит этан (C2H6 (г)). Вычисляя температуру принять, что полное сгорание происходит при T = 25 °С, а все тепло идет на нагревание продуктов. Задача 10 (оценка 2*) Задача 11 (оценка 2*) 98 3.3. Расчет термодинамических параметров химических реакций и определение направления процесса в системе Для выполнения расчетов необходимы величины, характеризующие термодинамические свойства веществ, участвующих в реакции: ∆H 0f – стандартные теплоты образования, 0 S 298 – стандартные энтропии, СP = f(T) – теплоемкости вещества в заданном интервале температур. Термодинамические свойства большинства индивидуальных химических соединений и ионов приведены в справочной литературе∗. Стандартный изобарный потенциал химической реакции (∆GT ), её константу равновесия (K°) и другие термодинамиче- ские параметры (∆H 298 , ∆H T , ∆ST ) рассчитывают по известным термодинамическим соотношениям: (1) ∆H 298 = ∑ ∆H f прод. − ∆H f исх. (2) ∆HT = ∆H 298 + T ∫ ∆CP dT 298 ∗ ⇐ 1-е следствие закона Гесса ⇐ закон Кирхгоффа в интегральной форме (3) ∆S 298 = ∑ S 298 прод. − S 298 исх. (4) ∆ST = ∆S298 + (5) ∆GT = ∆HT − T∆ST ⇐ уравнение ГиббсаГельмгольца (6) ∆GT = − RT ln K ⇐ уравнение изотермы реакции в равновесной системе T ∆CP dT T 298 ∫ ⇐ изменение энтропии при протекании реакции в стандартных условиях есть разность абсолютных энтропий продуктов реакции и исходных веществ В наших расчетах использован “Краткий справочник физико-химических величин” под ред. А.А. Равделя и A.M.Пономаревой – Л. Химия, 1983. 99 Пример ∗∗∗ Пусть нас интересует реакция: 2Н2 + СО → СН3ОН(ГАЗ), (∗ ) протекающая в газовой фазе при при T = 500 K. Вычисление ∆H 298 1. Рассчитаем тепловой эффект реакции (∗) при комнатной температуре T = 298 К ( ∆H 298 ). По первому следствию закона Гесса: ∆H 298 = ∆H f , CH 3OH − ∆H f , CO − 2 ⋅ ∆H f , H 2 = = −201,00 − (−110,53) − 2 ⋅ 0 = −90,47 кДж/моль Анализ результата Тепловой эффект реакции получился отрицательным, это означает, что при протекании данной реакции тепло выделяется. Если температура исходных веществ и продуктов одинакова и равна 298 K, то при образовании 1-го моля СН3ОН из 2-х моль Н2 и 1-го моль СО выделится в окружающую среду 90,47 кДж/моль тепла и энтальпия системы (Н) уменьшится на 90,47 кДж. Задание Посчитать количество тепла, которое выделится при образовании 1,5 кг газообразного СН3ОН из водорода и оксида углерода (II), если температура исходных веществ (Н2, СО) и продукта реакции (СН3ОН) - равна 298 K. Вычисление 2. Рассчитаем тепловой эффект реакции (1) при T = 500 K ∆H T ( ∆H 500 ). Чтобы посчитать тепловой эффект реакции при температуре, значительно отличающийся от стандартной, например, при Т = 500 К, следует воспользоваться интегральной формой закона Кирхгофа и учесть, что в широком интервале температур нельзя пренебречь зависимостью теплоемкости веществ от температуры. По закону Кирхгофа: ∆H 500 = ∆H 298 + 500 ∫ ∆C P ⋅ dT , 298 где ∆CP = CP,CH3OH − CP,CO − 2 ⋅ CP, H 2 - разность теплоемкостей продуктов реакции и исходных веществ. 100 Рассчитаем ∆CP = f(T) учитывая, что согласно справочным данным температурная зависимость теплоемкостей участников реакции в интервале температур 298 - 1000 K описывается уравнениями: CP,CH3OH = 15,28 + 105,20⋅10–3⋅T – 31,04⋅10–6⋅T Дж/(моль⋅К) 1 2 -1 CP,CO = 28,41 + 4,10⋅10–3⋅T – 0,46⋅105⋅T –2 Дж/(моль⋅К) -2 CP,H 2 = 27,28 + 3,26⋅10–3⋅T + 0,50⋅105⋅T –2 Дж/(моль⋅К) ∆CP = ∆ a + ∆ в ⋅ T + ∆ c′ ⋅T –2 + ∆ c ⋅T 2, где ∆ a = 15,28 – 28,41 – 2⋅27,28 = – 67,69 ∆ в = (105,20 – 4,10 – 2⋅3,26) ⋅10–3 = 94,56⋅10–3 ∆ c′ = (0 + 0,46 – 2⋅0,50) ⋅105 = – 0,54⋅105 ∆ c = –31,04⋅10–6 – 0 – 2⋅0 = – 31,04⋅10–6 Итак, зависимость ∆CP от температуры описывается уравнением: ∆C P = −67,69 + 94,58 ⋅ 10 −3 ⋅ T − 0,54 ⋅ 10 −6 ⋅ T 2 Дж/(моль⋅К) 500 Вычисляем ∫ ∆CP dT : 298 500 ∫ ( ∆a + ∆в ⋅ T + ∆c '⋅T − 2 + ∆c ⋅ T 2 ) dT = ∆a ⋅ (500 − 298) + 298 + (∆в / 2) ⋅ (500 2 − 298 2 ) − ∆c '⋅(1 / 500 − 1 / 298) + (∆c / 3) ⋅ (500 3 − 2983 ) = = −13673 + 7621,3 − 73,20 − 1019 = −7144 Дж/моль = − 7,14 кДж/моль. Тогда ∆H 500 = ∆H 298 + 500 ∫ ∆CP ⋅ dT = (−90,47 − 7,14) кДж/моль= −97,61кДж/моль 298 Типичные ошибки Следует обращать внимание на размерность величин, приведенных в справочнике. 101 Если температура реакционной смеси не слишком отличается от стандартной или не требуется высокая точность, вычисления можно упростить, пренебрегая зависимостью теплоемкости веществ от температуры. Например, при расчете теплового эффекта приведенной выше реакции синтеза метанола при Т = 350 К принимают: ∆CP = CP,CH3OH − CP,CO − 2 ⋅ CP, H 2 = const, где CP,CH3OH , CP,CO , CP, H 2 – средние теплоемкости СН3ОН, Н2, СО в интервале 298 – 350 К. Можно пойти по пути упрощений еще дальше и считать ∆СP постоянной величиной равной разности стандартных теплоемкостей конечных и исходных веществ при комнатной температуре Т = 298 К. ∆CP = CP0,CH3OH ,298 − CP0,CO,298 − 2 ⋅ CP0, H 2 ,298 = const Тогда по закону Кирхгофа: ∆H°350 = ∆H°298 + ∆CP⋅(350 – 298) ∆H 350 = −90470 + (44,13 − 29,14 − 2 ⋅ 28,83) ⋅ (350 − 298) = −92689 Вычисление ∆S 298 Дж = моль Дж кДж = −92,69 моль моль 3. Рассчитываем ∆S 298 - изменение энтропии при протекании реакции (1) в стандартных условиях при T = 298 K. Согласно (3), получаем: ∆S298 = S 298 ,CH3OH − S298,CO − 2 ⋅ S 298, H 2 = = (239,76 – 197,55 – 2⋅130,52) Дж/(моль⋅К) = –218,83 Дж/(моль⋅К) Анализ результата Задание ∆S 298 - отрицательная величина, это означает, что при протекании в стандартных условиях данной реакции энтропия системы уменьшается, т.е. система становится более упорядоченной. ∗∗∗ на уравнение химической реакции. Подумайте, Посмотрите можно ли было предсказать, что изменение энтропии должно 102 быть отрицательным? Вычисление ∆ST 4. Рассчитываем ∆S 500 - изменение энтропии при T = 500 K. Разность абсолютных энтропий продуктов реакции исходных веществ при температуре T определяется выражением (4). 500 ∆CP Вычисляем ∫ ⋅ dT T 298 500 ∆CP ∆a + ∆в ⋅ T + ∆c'⋅T − 2 + ∆c ⋅ T 2 ⋅ dT = ∫ T ⋅ dT = ∫ T 298 298 500 ∆ a⋅ln(500/298) + ∆ в⋅(500 – 298) – (∆ c′/2 ⋅(1/5002 – 1/2982) + + (∆ c/2)⋅(5002 – 2982)= – 18,43 Дж/(моль⋅К) Тогда согласно (4) ∆S500 = ∆S 298 + 500 ∆CP ⋅ dT = (– 218,83 – 18,43) Дж/(моль⋅К) = T 298 ∫ = – 237,26 Дж/(моль⋅К) В нешироком температурном интервале можно пренебречь зависимостью теплоемкости веществ от T и считать ∆CP = const. Тогда ∆ST = ∆S298 + Вычисление ∆GT T ∆CP T T ⋅ dT = ∆S298 + ∆CP ln 298 = ∆S298 + ln T 298 298 ∫ 5. Рассчитываем ∆GT - стандартный изобарный потенциал химической реакции (1) при T = 500 K. = ∆H − 500 ⋅ ∆S = - 97,61 - 500·(- 237,26·10-3) = ∆G500 500 500 = 21015,4 Дж/моль = 21,015 кДж/моль Анализ результата ∆G500 исследуемой реакции оказался значительной положительной величиной, следовательно, константа равновесия окажется ... (большой, маленькой?) величиной, и значит равновесие в системе, состоящей из СО, Н2 и СН3ОН будет сдвинуто в сторону ... (начальных, конечных?) веществ. 103 Вычисление K0 6. Рассчитываем константу равновесия реакции (1) (K0). Чтобы вычислить по справочным данным константу равновесия реакции следует рассчитать ее стандартный изобарный потенциал – ∆GT ; а затем воспользоваться уравнением изотермы химической реакции: ∆GT0 = − RT ⋅ ln K 0 Так константа равновесия реакции 2 Н2 + СО → СН3ОН при T = 500 К определяется из соотношений: ∆GT 21,015 ln K 0 = − =− = −5,055 R ⋅T 8,31 ⋅10 − 3 ⋅ 500 K0 = e– 5,05 = 6,38⋅10– 3 Анализ результата Какие выводы можно сделать из численного значения константы равновесия? Выражение (P~CH OH )равн. = K0 (P~H )2равн. ⋅ (P~CO )равн. 3 (∗) 2 означает, что в условиях равновесия парциальные давления всех веществ, участвующих в реакции, связаны между собой. Если изменить парциальное давление одного из них, изменятся и парциальные давления остальных веществ, но соотношение (∗) между парциальными давлениями веществ, участвующих в реакции, останется строго определенным при данной температуре. Так как K0 = 6,38⋅10– 3 – маленькая величина, то в равновесной смеси продукта реакции (CH3OH) во много раз меньше, чем исходных веществ (CO и H2), в таком случае говорят, что равновесие сдвинуто в сторону исходных веществ. Константа равновесия зависит от температуры. Так, для реакции синтеза метанола константа равновесия при Т = 298 К (K0 298) значительно отличается от рассчитанной выше константы равновесия при Т = 500 K (K0 500); 104 при Т = 298 K имеем: 0 0 0 ∆G298 = ∆H 298 − 298 ⋅ ∆S 298 = – 90,47 – 298⋅(– 0,218) кДж/(моль⋅К) = = – 25,26 кДж/(моль⋅К) ln K 0 298 = − ∆G298 − 25,26 =− = 10,20 −3 R ⋅T 8,31 ⋅ 10 ⋅ 298 K0 298 = e10,20 = 2,69⋅104 ∗∗∗ Сравните численные значения констант равновесия реакции (1) при Т = 298 K и Т = 1000 K. Каких газов будет много, а каких мало в равновесной смеси при Т = 298 K и при Т = 1000 K? ∗∗∗ Как Если известен состав реакционной смеси, можно опредеопределить лить направление в котором должна протекать химическая реакнаправление ция, чтобы в системе наступило равновесие. химической Направление процесса определяют по изменению изобарреакции в но-изотермического потенциала системы (∆G), которое вычисреакционной ляют по уравнению изотермы химической реакции. смеси ∗∗∗ Пример Так, чтобы выяснить, может ли в смеси, состоящей из заданных количеств газообразных CO, H2 и CH3OH и находящейся при Т = 500 K протекать реакция Задание 2 Н2 + СО → СН3ОН, следует вычислить изменение избарно-изотермического потенциала системы (∆G), которое может вызвать данная реакция, если она произойдет в смеси заданного состава. Согласно уравнению изотермы химической реакции ~ P CH OH ∆GT = ∆GT + RT ⋅ ln ~ 3 ~ , или PH2 ⋅ PCO 2 ~ PCH3OH ∆GT = −RT ln K + RT ⋅ ln ~ ~ , PH2 ⋅ PCO 2 ~ ~ ~ где PCH 3OH , PH 2 , PCO – относительные парциальные давления газов в исходной смеси. 105 Допустим 1) в исходной газовой смеси парциальные давления газов имеют следующие значения: ~ PCH 3OH = 0,13атм ( PCH 3OH = 0,13 ), ~ PH 2 = 2,6атм ( PH 2 = 2,6 ), ~ PCO = 5,2атм ( PCO = 5,2 ). Тогда при Т = 500 K ∆GT = ∆G500 + R ⋅ 500⋅ ln 0,13 2 2,6 ⋅ 5,2 = 21015+ (−23268) = −2253 Дж / моль ∆G < 0, следовательно в этом случае возможно протекание процесса в прямом направлении: из Н2 и СО может образовываться СН3ОН; 2) если в исходной смеси ~ PCH 3OH = 10,3 атм., ( PCH 3OH = 10,3 ), ~ PH 2 = 0,7 атм., ( PH 2 = 0,7 ), ~ PCO = 0,3 атм., ( PCO = 0,3 ), то при Т = 500 K: ∆G500 = ∆G500 + R ⋅ 500 ⋅ ln = 38672 10,3 2 0,7 ⋅ 0,3 = 21015 + 17657 Дж кДж = 38,67 моль моль ∆G > 0, следовательно в такой смеси процесс в прямом направлении неосуществим, но возможен в обратном направлении: СН3ОН будет разлагаться на Н2 и СО; 3) если в исходной смеси ~ PCH 3OH = 0,033атм ( PCH 3OH = 0,033 ), ~ PH 2 = 1,49атм ( PH 2 = 1,49 ), 106 ~ PCO = 2,33атм ( PCO = 2,33 ) то при Т = 500 K: ∆G500 = ∆G500 + R ⋅ 500 ⋅ ln кДж =0 1,49 2 ⋅ 2,33 моль 0,033 следовательно, такая смесь находится в равновесии. 3.3.1. Задачи для самостоятельного решения В соответствии с выбранным вариантом вычислить тепловой эффект реакции № . . . (табл. 3.4) при температуре Т. Уравнение зависимости СP = f(T) взять из таблицы термоди намических величин [4]. Вычислите ∆G0 и K0 данной реакции при заданной температуре. Табл. 3.4 № 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 Уравнение реакции 2Н2 + СО = СН3ОН(Г) 4НС1 + О2 = 2Н2О(Г) + 2С12 2N2 + 6Н2О (Г) = 4NH3 + 3О2 4NO + 6Н2О (Г) = 4NH3 + 5О2 2NO2 = 2NO + О2 N2O4 = 2NO2 1/2S2 + 2H2O (Г) = SO2 + 2H2 1/2S2 + 2CO2 = SO2 + 2CO 2SO2 + O2 = 2SO3 (Г) SO2 + Cl2 = SO2C12 (Г) CO + 3H2 = CH4 + H2(Г) 4CO + 2SO2 = S2(Г) + 4CO2 CO2 + H2 = CO + H2O (Г) 2 CO + 6H2 = 2CH4 + 2H2O(Г) 2CO2 = 2CO + O2 CH4 + CO2 = 2CO + 2H2 C2 H 6 = С2 Н 4 + H 2 C2H5OH(Г) = C2H4 + H2O (Г) CH3COOH(Г) + H2 = C2H5OH + H2O T, К 700 500 500 650 600 800 900 950 1000 1000 1000 1000 1000 1000 1000 1000 1000 1000 1000 107 3.4. Расчет состава равновесных смесей. Алгоритм расчета Пример 1 Если известен исходный состав реакционной смеси и константа равновесия, протекающего в системе химического процесса, то можно вычислить концентрации (мольные доли или парциальные давления) веществ в смеси после установления равновесия, т.е. определить состав равновесной смеси. Сложность такого расчета зависит от нескольких факторов:1) вида химического уравнения и 2) от того какие величины уже известны. В общем случае для выполнения расчетов необходимо: 1) выбрать в качестве неизвестной величины x (моль) – изменение количества одного из реагентов или α – степень превращения исходного вещества в продукты к моменту равновесия; 2) выразить через x (или α) состав равновесной смеси, т.е. определить а) число молей компонентов смеси(ni), б) мольные доли компонентов (x i), в) парциальные давления газообразных компонентов смеси (Pi) в равновесной смеси. В таких задачах часто удобно составлять таблицу, иллюстрирующую материальный баланс исходной и равновесной смеси. 3) записать выражение для константы равновесия и получить уравнение с одним неизвестным x (или α). В зависимости от условий задачи пользоваться можно или стандартной константой равновесия K° или величинами KP ,KC ,Kx; 4) решить уравнение, вычислить неизвестную величину x (или α) и рассчитать состав равновесной смеси. ∗∗∗ Метиловый спирт получают по реакции: CO + 2 H2 → CH3 OH Решение Исходные вещества введены в реактор в эквивалентных количествах, т.е. CO и H2 находятся в исходной смеси в соотношении 1:2. Общее давление газовой смеси Pобщ. = 1атм. = 1,033 105 Па, температура 500 K. Стандартная константа равновесия при температуре 500 K составляет K° = 6,38·10-3. Определить равновесный выход продуктов. 1). Введем обозначения: пусть x моль – изменение количества CH3OH в смеси к моменту равновесия; тогда в системе, первоначально не содержащей метилового спирта, к моменту равновесия в результате реакции образовалось x моль CH3OH и исчезло x моль CO и 2x моль H2. 108 Если в исходной смеси содержались 1 моль CO и 2 моль H2 и к моменту равновесия прореагировало x моль CO и 2 x моль H2, то (1 – х) моль – количество CO, которое не прореагировало и находится в равновесной смеси; (2 – 2х) моль – количество H2 в равновесной смеси; 2). Учитывая, что согласно определению мольная доля компонента смеси равна (1) χi = ni , где ∑ ni ∑ ni – общее число молей компонентов смеси; а парциальное давление компонентов идеальной газовой смеси согласно закона Дальтона определяется выражением: (2) Pi = Pобщ.·χi, где Pобщ. – общее давление газовой смеси; легко выразить состав равновесной смеси в мольных долях (χi) и рассчитать равновесные парциальные давления (Pi). Результаты расчетов удобно заносить в таблицу (см. табл. 3.5). Таблица 3.5 уравнение реакции CO число молей компонентов в 1 + исходной смеси, ni изменение числа молей к мо- – х менту равновесия, ∆ni число молей компонентов в 1 – х равновесной смеси, ( ni ) равн → CH3OH 0 2H2 2 – 2х х 2 – 2х х (ni ) равн = ni + ∆ni всего молей в смеси, ∑ ni мольная доля компонентов в равновесной смеси, ( χ i ) равн ∑ ni = 1 − x + 2 − 2 x + x = 3 − 2 x 1− х 3 − 2х 2 − 2х 3 − 2х х 3 − 2х ( χ i ) равн = ( ni ) равн. / ∑ ni парциальное давление компонентов в равновесной смеси, ( Pi ) равн 1− х 3 − 2х Pобщ. 2 − 2х 3 − 2х Pобщ. х 3 − 2х ( Pi ) равн = ( χ i ) равн. ⋅ Pобщ. 3) Запишем выражение для константы равновесия KP Pобщ. 109 KP = (3) (P ) CH 3OH равн (PCO )равн ⋅ (PH ) 2 2 равн Подставим равновесные давления компонентов в (3). х (4) KP = 3 − 2х ⋅ Pобщ. 2 ( 2 − 2х) 2 ⋅ Pобщ. ⋅ ⋅ Pобщ. 2 3 − 2х (3 − 2 х ) 1− х = х ⋅ (3 − 2 x ) 2 4 ⋅ Pобщ. ⋅ (1 − х ) 2 3 Определить значение KP, если задана K0 нетрудно. Для данной реакции стандартная константа равновесия KP и K0 связаны соотношением: K P = K ⋅ p0∆ν , (5) где ∆ν = 1 – 3= – 2 P0 = 1атм. = 1,033·105 Па Из соотношения (5) видно, если давление газа в системе измерять в атм., то значение K° и KP численно совпадают. Для исследуемой реакции, следовательно, KP = 6,38·10–3 атм–2. Подставляя в (4), Pобщ. = 1атм., получаем 2 х(3 − 2 х ) = 6,38 ⋅10 − 3 3 4(1 − х ) (6) 4) Решаем уравнение (6) методом последовательных приближений, учитывая, что возможные значения x лежат в пределах 0 < x < 1. Получаем x = 1,5·10-3. 5) Итак, в равновесной смеси содержится 0,0015 моль CH3OH, если бы смесь 1 моль CO и 2 моль H2 превратилась полностью в конечный продукт, должно бы получиться 1 моль CH3OH. Следовательно, выход продукта в заданных условиях составляет (7) Анализ результата η= 0,0015моль 1моль ⋅ 100% ≈ 1,5% Выход продукта невелик, этого следовало ожидать, так как константа равновесия K0 << 1. Чтобы увеличить выход 110 Пример 2 продукта, можно попробовать провести синтез CH3OH при более низкой температуре, для которой значение K0 будет больше. ∗∗∗ 3 В сосуд объемом 1 дм поместили 0,50 моль HI и нагрели до 448 °C. Значение константы равновесия KC для реакции: H2(г) + I2(г) ↔ 2 HI(г) при указанной температуре равно 50,5. Определить концентрации H2, I2 и HI в сосуде при равновесии. Решение 1) Введем обозначения и составим таблицу, иллюстрирующую материальный баланс. уравнение реакции исходные концентрации, моль⁄дм3 изменение концентрации, моль⁄дм3 равновесные концентрации С, моль⁄дм3 H2(г) 0 + I2(г) → 2 HI(г) 0 0,50 х х – 2х 0+х 0+х 0,50 – 2x 2) Подставим равновесные концентрации в выражение для константы равновесия и вычислим с его помощью единственную неизвестную величину х: KC = (1) KC (2) (3) (0,50 − 2 х )2 х2 х CH2 ⋅ CI2 ; 2 ( 0,50 − 2 х ) = х2 = 50,5 = 50,5 ⇐ уравнение 2-го порядка относительно х. 0,50 − 2 х (4) 2 C HI = 50,5 ⇐ математические преобразования (5) 0,50 − 2 х = 7,11 ⋅ х 0,50 (6) х = 9,11 = 0,055 Таким образом, равновесные концентрации участников реакции 111 составляют: СH 2 = х = 0,055 моль ⁄дм3 3 СI 2 = х = 0,055 моль ⁄дм СHI = 0,50 – 2х = 0,50 – 2·0,055 = 0,39 моль ⁄дм3. Пример 3 ∗∗∗ В газовой фазе при давлении P = 1,5 атм. протекает реакция разложения вещества А: A(г) → 2B(г) + C(г) Решение 1) Вывести уравнения, связывающие константу равновесия (K°, KP, Kχ) и степень превращения вещества A (α). 2) Проанализировать полученные уравнения и ответить на вопрос: как зависит степень превращения (α) от давления газовой смеси? ∗∗∗ Введем обозначения и составим таблицу, иллюстрирующую материальный баланс химическое уравнение A число молей в исходной сме- n° → 2B 0 + C 0 си, ni – α· n° изменение числа молей, ∆ni число молей в равновесной n° – α· n° смеси, (ni ) равн. 2α· n° α· n° 2α· n° α· n° общее число молей в равновесной смеси, ∑ ni ∑ n = n ° − α ⋅ n ° + 2α ⋅ n ° + α ⋅ n ° = мольная доля компонентов в равновесной смеси, (χi ) равн. 2α ⋅ n/ ° n/ °(1 + 2α ) = n ° ⋅ (1 + 2α ) n/ °(1 − α ) n/ °(1 + 2α ) α ⋅ n/ ° n/ °(1 + 2α ) 1−α α 2α ⋅ Pобщ ⋅ Pобщ ⋅ Pобщ 1 + 2α 1 + 2α 1 + 2α P относительные парциальные 1 − α Pобщ α 2α Pобщ ⋅ общ ⋅ ⋅ ~ давления, Pi равн. 1 + 2α P0 1 + 2α P0 1 + 2α P0 парциальные давления в равновесной смеси, (Pi ) равн. ( ) (P~i )равн. = (Pi ) равн. / P0 , где P0 = 1атм. = 1,033·105 Па. Подставим равновесные величины в выражения для кон- 112 стант равновесия (Kχ, K°, KP): (1) Kχ ( χ B2 )равн. ⋅ (χ C ) равн. ; = (χ A ) равн. 2 (2) (3) 2α α ⋅ 4α 3 1 + 2α 1 + 2α Kχ = = 1−α (1 − α )(1 + 2α )2 1 + 2α K ~ ~ ( PB2 )равн. ⋅ (PC ) равн. = ; (P~A )равн. (4) 2 2 Pобщ α 2α Pобщ ⋅ ⋅ ⋅ 2 Pобщ P0 4α 3 1 + 2α P0 1 + 2α K = = ⋅ 2 1 − α Pобщ (1 − α )(1 + 2α ) P02 ⋅ 1 + 2α P0 K = (5) или (6) где 4α 3 (1 − α ) ⋅ (1 + 2α )2 ~2 ⋅ Pобщ ; P ~ Pобщ = общ , и P0 = 1атм. = 1.013·105 Па. P0 (7) KP ( PB2 )равн. ⋅ (PC ) равн. = , (PA ) равн. где (PA)равн., (PB)равн., (PC)равн. – парциальные давления газов в равновесной смеси. (8) K P = 4α 3 (1 − α )(1 + 2 α ) 2 2 ⋅ Pобщ 113 Обратите внимание Если давление измерять в атм., то численные значения K° и KP одинаковы. Уравнение (8) можно упростить, если степень превращения исходного вещества незначительны. Если α << 1, то (1 - α) ≈ 1 и (1 + 2 α) ≈ 1, тогда (9) 2 K P ≈ 4α 3 ⋅ Pобщ и α =3 Kp 2 4 ⋅ Pобщ Из (9) следует, что при повышении давления в реакционной смеси (Pобщ) степень превращения (α) уменьшается. Тот же вывод можно сделать, пользуясь правилом Ле-Шателье. 3.4.1. Задачи для самостоятельного решения Задача 1 (оценка 2*) Доказать, что для газовых реакций, приведенных в таблице (3.6), справедливы соответствующие уравнения, связывающие их константы равновесия (K°) и степени термической диссоциации вещества (α). Таблица 3.6 Уравнение K0 = f (α). Уравнения реакций K0 = NH3 = 0,5N2 + 1,5H2 CO2 = CO + 0,5O2 A = ν2 B + ν3 C K0 = 0,50,5 ⋅ 1,51,5 ⋅ α 2 ~ ⋅P (1 − α )[1 + α ] общ 0,50,5 ⋅ α 1,5 (1 − α )[1 + 0,5 ⋅ α ]0,5 ~ 0 ,5 ⋅ Pобщ ν ν2 2 ⋅ν ν3 3 ⋅ α ν ~ν −1 K = ⋅ P (1 − α )[1 + (ν − 1) ⋅ α ]ν −1 общ 0 где v = v2 + v3 Проанализировать уравнения K0 = f (α) для данных реакций. Как изменится степень диссоциации с повышением давления реакционной смеси? Соответствует ли Ваш вывод правилу Ле-Шателье? Задача 2 (оценка 2*) В вакуумированный сосуд объемом 1л поместили 80г Fe2O3 и ввели H2 с давлением 2,44 атм. при 25°С. Сосуд нагрели до 1000°С. В системе протекает реакция: 114 Fe2O3 кр. + 3H2 г → 2Feкр. + 3H2O(г) Рассчитать равновесный состав газовой фазы. Температурной зависимостью ∆H0 и ∆S0 пренебречь. Задача 3 (оценка 1*) Водяной газ получали при 900°С по реакции: CO2(г) + H2 ↔ CO(г) + H2O(г) Равновесные смеси анализировали и получили следующие результаты, приведенные в таблице 3.7. Таблица 3.7 номер опыта 1 2 3 CO 0,352 0,266 0,186 парциальные давления, атм. H2 O CO2 0,352 0,648 0,266 0,234 0,686 0,314 H2 0,148 0,234 0,315 Написать выражение для константы равновесия реакции K0 и показать, что ее величина постоянна. Задача 4 (оценка 1*) В замкнутой системе (рис.3.9) протекает реакция разложения карбоната кальция: CaCO3 (кр) ↔ CaO(кр) + CO2 (г) Как скажется введение дополнительного количества CaCO3 на величине давления CO2 над поверхностью твердой фазы, если температура системы постоянна. Рассчитать давление углекислого Рис. 3.9 Равновесная гетерогенная система состоящая из кристаллов газа в системе при T = 500 K. CaCO3, кристаллов CaO и углекислого газа. Задача 5 (оценка 2*) Процесс получения водорода идет по уравнению: H2O + CO = CO2 + H2 При 1000°С константа равновесия 1,54. Равновесная смесь содержит 25 % CO2 и 25 % H2 по объему. Найти исходный состав смеси. 115 Задача 6 (оценка 2*) Константа равновесия реакции: CO(г) + H2O(г) → CO2 (г) + H2 (г) равна 0,71 при 700°С. Оксид углерода (CO) смешивают с водяным паром в молярном соотношении 2:1, подают в реактор при 700°С и давлении 15 атм. Определить равновесный состав смеси. Задача 7 (оценка 1*) В системе протекает реакция: 2HI(г) ↔ H2 (г) + I2 (г) при 448°С парциальные давления газов в состоянии равновесия следующие: PHI = 4·10–3 атм. PH 2 = 7.5·10–3 атм. PI 2 = 4.3·10–5 атм. Рассчитать 1) стандартную константу равновесия реакции K°, 2) рассчитать K°, KC, Kχ. Задача 8 (оценка 2*) Метиловый спирт получают в соответствии с уравнением реакции: CO(г) + 2H2 (г) ↔ CH3OH (г) Какое влияние на равновесное давление CH3OH будет оказывать: а) повышение температуры; б) повышение давления; в) добавление некоторого количества CO. Как изменятся численные значения K°, KP, Kχ. Задача 9 (оценка 2*) В газовой смеси протекает экзотермическая реакция: 4HCl(г) + O2(г) ↔ 2H2O(г) + 2Cl2(г) Какое влияние на равновесную концентрацию Cl2 будут оказывать следующие изменения: а) повышение температуры в реакционном сосуде; б) уменьшение общего давления; в) увеличение концентрации O2; г) увеличение объема реакционного сосуда; д) введение катализатора. Как изменятся численные значения K0, KP, Kχ. 116 Задача 10 (оценка 2*) При взаимодействии NO и O2 с образованием NO2 выделится 56 кДж на 1 моль NO. Написать уравнение реакции и указать, как изменятся равновесные концентрации, численное значение стандартной константы равновесия и скорость образования NO2, если а) увеличить температуру; б) увеличить общее давление реакционной смеси при данной температуре 117 Заключение Химическая термодинамика является важнейшим инструментом, используемым физической химией для изучения закономерностей физических, химических и физико-химических процессов. Несмотря на большие достижения в развитии других методов физической химии (квантовомеханического и статистического), термодинамический метод продолжает оставаться главным. Термодинамика основана на нескольких фундаментальных законах, обобщающих накопленный человечеством опыт наблюдений над превращениями энергии. Опираясь на эти законы, построена строгая и логически связанная система выводов и следствий. Первый закон термодинамики – частный случай закона сохранения энергии. Если конкретно указан определенный процесс или превращение, первый закон позволяет, фигурально говоря, вести бухгалтерский учет выделяемого тепла, выполненной работы и т.п. Однако он ничего не говорит о том, может ли в действительности идти рассматриваемый процесс. Этот вопрос решается на основе второго закона термодинамики. Второй закон термодинамики выражает то наблюдение, что любая неравновесная система изменяет свое состояние в определенном, характерном для нее направлении. Для того чтобы состояние такой системы изменилось в противоположном направлении, необходимо подводить к ней энергию. Именно второй закон термодинамики позволяет определить возможность, направление и предел самопроизвольного течения различных процессов в тех или иных условиях, а также определить условия, при которых система будет находиться в равновесии. Термодинамический метод имеет свои особенности, основными из которых являются следующие: • термодинамика применяется только к макроскопическим системам; она не применима к отдельным атомам и молекулам и к процессам, в которых участвуют единичные атомы или молекулы; • термодинамика не изучает скорость процесса, для нее важны только начальное и конечное состояния системы, эта особенность позволяет применять термодинамический метод к сложным процессам, когда промежуточные стадии не известны. На основе законов термодинамики, можно вывести уравнения, позволяющие решить вопросы, связанные с расчетом химического равновесия и выяснением оптимальных условий режима в процессе химического превращения. Для студентов, приступающих к изучению химии, иногда оказывается сложным само понятие химического равновесия. Следует ясно представлять себе, что многие химические реакции не протекают до конца, другими словами, смесь исходных веществ (реагентов) не полностью превращается в продукты. По прошествии некоторого времени изменение концентраций реагентов прекращается. Реакционная система в таком состоя- 118 нии представляет собой смесь исходных веществ и продуктов реакции. Химическая система в таких условиях находится в состоянии химического равновесия. Представление о химическом равновесии играет важную роль в химии. Действительно, при изучении любой химической реакции возникают два важнейших вопроса: 1) насколько далеко в сторону завершения может протекать реакция, прежде чем она достигнет равновесия? 2) как быстро достигается состояние равновесия? Ответ на первый вопрос дает химическая термодинамика. Получить ответ на этот вопрос – означает узнать важные сведения о реакционной способности системы. Так, если предполагается провести реакцию, которая, как показали термодинамические расчеты, имеет благоприятную константу равновесия, т.е. протекающую с превращением значительной доли реагентов в продукты, то остается только позаботиться о том, чтобы она имела соответствующую скорость. Последнее может оказаться нелегким делом, но напряженная работа и упорные поиски, в конце концов, нередко позволяют найти катализатор, ускоряющий медленную реакцию, или какой-нибудь способ регулирования слишком быстрой реакции. Если же термодинамические расчеты показали, что химическое равновесие будет достигнуто при превращении совсем незначительного количества реагентов, то такая реакция не будет иметь практического значения, даже если бы имелся катализатор, ускоряющий ее. Овладение методикой расчетов термодинамических параметров химических процессов и умение делать выводы, анализируя их результаты – важная составляющая профессиональной подготовки специалиста. 119 СПИСОК ЛИТЕРАТУРЫ 1. 2. 3. 4. 5. 6. Физическая химия. Под ред. Краснова К.С. –М.: Высшая школа, 1982.. Стромберг А.Г., Семченко Д.П. Физическая химия. – М.: Высшая школа, 1999. – 527 с. Киреев А.В. Краткий курс физической химии. –М.: Высшая школа, 1978. Краткий справочник физико-химических величин. Под ред. Равделя А.А., Пономаревой А.М. –Л.: Химия, 1983. – 231 с. Кемпбелл Дж. Современная общая химия. т. 2. – М.: Мир, 1975. Практические задания и лабораторные работы по физической химии: Учебное пособие/ Под ред. М.И. Гельфмана, Н.В.Розаленок, Ю.В. Тарасовой. – Кемерово, 2003. – 144 с. 120 ОГЛАВЛЕНИЕ Введение…………………………………………………………………………….3 Глава 1. Основы химической термодинамики………………………………..4 1.1. Что такое химическая термодинамика……………...……………….4 1.2. Основные понятия и определения………………………….………..5 1.3. Первый закон термодинамики………………………………………12 1.4. Применение первого закона термодинамики к химическим процессам…………………………………….……...13 1.5. Второй закон термодинамики…………………………………….…27 1.6. Постулат Планка. Абсолютные энтропии………………..…………36 1.7. Уравнения Гиббса – Гельмгольца…………………………………...41 1.8. Парциальные величины. Химический потенциал………………….47 1.9. Зависимость химического потенциала компонента от состава смеси……………………………………………………..49 Глава 2. Химическое равновесие…………………………………………...….52 2.1. Динамический характер равновесия………………………………..52 2.2. Условие протекания химического процесса в закрытых системах при постоянных давлении и температуре…………………….……53 2.3. Условие химического равновесия в закрытых системах при постоянной температуре и давлении. Закон действующих масс...58 2.4. Химическое равновесие в гетерогенных системах………………...63 2.5. Влияние температуры на химическое равновесие. Уравнение изобары химической реакции………………………….67 Глава 3. Некоторые приложения законов термодинамики………………...72 3.1. Расчеты тепловых эффектов и других термодинамических характеристик химических процессов. Термохимические уравнения……………………………………….72 3.2. Применение законов термодинамики к идеальным газам………...80 3.3. Расчет термодинамических параметров химических реакций и определение направления процесса в системе…………………..96 3.4. Расчет состава равновесных смесей……………………………….105 Заключение………………………………………………………………………...115 Список литературы………………………………………………………………..117 121 УЧЕБНОЕ ИЗДАНИЕ Холохонова Лариса Ивановна Корокая Елена Валерьевна Законы термодинамики и химическое равновесие Учебное пособие Для студентов вузов Печатается в авторской редакции Художественный редактор Л.П. Токарева Подписано в печать_________. Формат 60x84/16. Отпечатано на ризографе с готовых печатных форм Уч. – изд. л. ________. Тираж _______. Заказ _______. Кемеровский технологический институт пищевой промышленности 650056, г. Кемерово, б–р Строителей, 47 Отпечатано в лаборатории множительной техники КемТИППа 650010, г. Кемерово, ул. Красноармейская, 52.