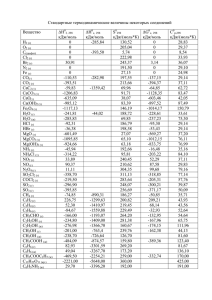
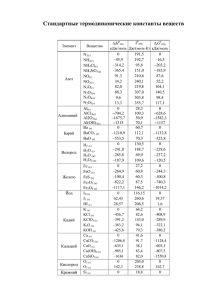
Энергии диссоциации связи С-NO 2 в ряду ароматических
advertisement

Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г УДК 544.144.7:544.18:547.546 ЭНЕРГИИ ДИССОЦИАЦИИ СВЯЗИ С-NO2 В РЯДУ АРОМАТИЧЕСКИХ НИТРОСОЕДИНЕНИЙ ПО ДАННЫМ КВАНТОВО-ХИМИЧЕСКОГО РАСЧЕТА Шарипов Д.Д., Егоров Д.Л.,Чачков Д.В., Шамов А.Г., Храпковский Г.М. Казанский государственный технологический университет 420015, Казань, ул. Карла Маркса, 68 E-mail: dmn@kstu.ru С использованием различных базисов (6-31G(d), 6-31G(d,p), 6-31G(df,p) и 6-311++ G(3df,3pd)) гибридного метода функционала плотности B3LYP определены геометрические параметры структур и энтальпии образования нитробензола, более 20 его монофункциональных производных и 1,3,5-тринитробензола, а также энтальпии образования радикалов, образующихся при гомолитическом разрыве связи C-NO2. На основе расчетных значений энтальпий образования изученных соединений и радикалов рассчитаны энергии диссоциации связи C-NO2 (D(C-N)) и энергии активации радикального газофазного распада. Установлено, что донорные заместители вызывают увеличение D(C-N), а акцепторные – ее снижение. Показано, что рассчитанные на основе D(C-N) значения энергий активации газофазного распада ароматических нитросоединений хорошо согласуются с экспериментом. Ключевые слова: ароматические нитросоединения, молекулярная структура, энтальпия образования, энергия диссоциации, квантово-химический расчет Введение Ароматические соединения широко используются в качестве бризантных взрывчатых веществ, промежуточных продуктов при синтезе красителей, лекарственных препаратов и в ряде других отраслей промышленности [1]. Экспериментальные данные по геометрии молекул в газообразном состоянии имеются для сравнительно небольшого числа молекул [2]. Приводимые погрешности в определении длин связей и валентных углов реакционного центра (C-NO2-группы) не позволяют проанализировать наблюдаемые изменения в ряду [3]. Не отличаются необходимой точностью и экспериментальные значения энтальпий образования свободных молекул нитроаренов: в ряде случаев результаты, полученные разными авторами, различаются на 15-20 кДж/моль [4]. Все основные характеристики молекул нитроаренов могут быть получены с использованием современных квантово-химических методов. В то же время, несмотря на достаточно большое число публикаций [5-10], сколько-нибудь надежные систематические данные о влиянии молекулярной структуры на изменение энтальпий образования в ряду ароматических соединений отсутствуют. В нашей работе представлены соответствующие результаты, полученные с использованием гибридного DFT-метода B3LYP с различными базисами. Результаты расчета достаточно хорошо согласуются с имеющимися экспериментальными данными по геометрии свободных молекул. 122 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г Результаты и их обсуждение Из-за ограничений по объему мы не приводим таблицу геометрических параметров рассчитанных нами с помощью различных базисов гибридного метода функционала плотности B3LYP, с частью из них можно ознакомиться в наших предыдущих работах [7,8,11]. Поэтому перейдем сразу к анализу полученных нами результатов. При проведении работы использовались пакеты квантово-химических программ Gaussian 98 [12] и Gaussian 2003 [13]. Использование различных базисов в рамках метода B3LYP приводит к достаточно близким результатам, вместе с тем можно выявить определенные тенденции в изменении значений геометрических параметров в зависимости друг от друга. Так, например, наиболее мощный из использованных в работе базисов 6-311++G(3df,3pd) предсказывает наименьшее значение длины для примыкающих к реакционному центру (группа C-NO2) связи C-C, а также r(N-O) и (в ряде случаев) углов CCNC и CCXC. Вместе с тем результаты расчета этим методом r(C-N) и угла ONO превышают оценки, полученные с использованием других базисов. Следует отметить, что различия в значениях геометрических параметров соединения, полученные с использованием различных базисов не велики и, как правило, не превышают 1 пм. Существенно также, что все использованные нами методы предсказывают хорошо согласующиеся между собой тенденции изменения геометрических параметров изученных соединений в ряду. Полученные нами данные показывают, что при замещении в молекуле нитробензола атомов водорода на донорные заместители F-, Cl-, Br-, H2N-, HO-, Н3С-, величина r(C-N) уменьшается, причем этот эффект наиболее сильно выраженный для о- и п-изомеров. Подобная тенденция изменения r(C-N) очевидно связана с проявлением прямого полярного сопряжения донорного заместителя с акцептором (NО2-группой). В молекулах о-галогеннитробензолов о-нитротолуола сопряжение нарушается в связи с проявлением стерических факторов, на что указывает выход NO2-группы из плоскости кольца, причем в случае фтора это нарушение проявляется в наименьшей степени, что связано с относительно небольшим размером атома фтора. В м-изомерах прямое полярное сопряжение отсутствует и величина r(C-N) для них мало отличается от нитробензола. Анализ геометрических параметров о-нитроанилина указывает на образования в этой молекуле внутримолекулярной водородной связи. В частности, значительно уменьшается r(C-N) по сравнению с м- и п-нитроанилинами, кроме того, в о-нитроанилине существенно увеличивается r(N-O). Подобная тенденция, но еще более сильно выраженная, наблюдается и для о-нитрофенола. Введение в молекулу акцепторных заместителей (COH, COOH и NO2групп) вызывает хотя и довольно незначительное, но увеличение r(C-N). В отличие от других ароматических нитросоединений, представленных в таблице, для динитробензолов наблюдается хоть и слабо выраженное, но вполне заметное уменьшение r(N-O). В отличие от соединений с донорными заместителями 123 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г длина связи С-Х для акцепторных групп в о-положении не уменьшается, а заметно увеличивается по сравнению с м- и п-изомерами. Тенденции изменения энтальпий образования изученных соединений в ряду наиболее удобно проследить по рис. 1. Сравнение с экспериментальными данными показывает, что в большинстве случаев расчет удовлетворительно передает как абсолютные значения энтальпий образования, так и в особенности тенденции их изменения в ряду. При этом следует отметить, что точность экспериментального определения энтальпий образования в ряду ароматических нитросоединений невысока. В ряде случаев различия в значениях энтальпий образования монофункциональных производных нитробензола, полученных в разных лабораториях, может достигать 15-25 кДж/моль [4]. Анализ результатов, представленных на рис. 1, показывает, что лучше всего экспериментальные значения передает базис 6-31G(d,p); средняя погрешность этого метода составляет 10.0 кДж/моль, что сопоставимо с различиями экспериментальных оценок разных авторов [4, 14]. Из других использованных в работе базисов близкие к результатам 6-31G(d,p) оценки дает базис 6-31G(df,p) (средняя погрешность его составляет 14.7 кДж/моль) и один из наиболее мощных базисов 6-311++G(3df,3pd). При этом интересно, что базис 6-311++G (3df,3pd), а также большинство других, использованных в работе базисов, завышают величину энтальпии образования по сравнению с экспериментальными значениями, а базис 6-31G(df,p) – занижает ее. 160.0 Эксперимент B3LYP/6-31G(d) B3LYP/6-31G(d,p) B3LYP/6-31G(df,p) B3LYP/6-311++G(3df,3pd) 120.0 80.0 кДж/моль 40.0 0.0 -40.0 -80.0 0 298, -120.0 ΔfH -160.0 -200.0 -240.0 -280.0 о-ф нит ро то бе рн нз ми ол тр фт о бе ор нз н ит п-ф ол ро то бе рн н з и о-х ол тр об ло ен рн зо ит мл ро хл бе ор п-х нитр нзол об ло ен рн зо ит о-б л р ро об мн ен мз и ол тр бр об ом ен ни зо п-б тр л ро об мн ен зо ит л ро бе о-н нз ит ол ро фе мни н о тр л п-н офе но ит л ро фе о-н но ит ро л ан мил ни и тр н п-н оани ли ит ро н ан о-н ил ит ин ро то млу ни о-н ол тр ит о ро т ол бе п-ни муо нз ни л ой трот тр п-н обен ный олуо ал зо л ит ьд йн ро еги ый бе нз о-н д а л ой ь ит д ны ро й а егид бе мнз ль ни ой де тр на гид п-н обен як зо и ит сл йн ро от ая бе а нз ки ой сл на от як а о-д ис ин ло ит т а ро мбе ди нз ни ол тр по 1,3 дини бенз ол ,5тр об тр ин ен ит ро зол бе нз ол -320.0 Рис. 1. Энтальпии образования некоторых производных нитробензола При обсуждении данных по геометрии о-нитротолуола, о-нитроанилина и о-нитрофенола мы обсуждали вопрос о возможности проявления внутримолекулярной водородной связи в этих соединениях. Этот вывод вполне согласуется и с расчетными и экспериментальными оценками энтальпий образования изо124 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г мерных нитрофенолов. Так, например, по данным различных используемых в нашей работе базисов энтальпия образования п-нитрофенола на 22.9-18.2 кДж/моль выше, чем у о-нитрофенола. Термохимическая оценка (14.1 кДж/моль) также согласуется с отмеченной тенденцией. Энтальпия образования о-нитроанилина на 2.5 кДж/моль ниже, чем у п-нитроанилина. Это указывает на существование внутримолекулярной связи существенно более слабой, чем в о-нитрофеноле. Этот вывод согласуется и с данными по геометрии и распределению электронной плотности в этих молекулах. Аналогичную тенденцию можно наблюдать и для нитроанилинов. Различия в экспериментальных значениях о-нитроанилина и п-нитроанилина составляет 4.2 кДж/моль [14]. В о-нитротолуоле энтальпия образования максимальная в ряду изомеров как по данным расчета с использованием различных базисов, так и по результатам эксперимента. Из этих данных можно сделать вывод об отсутствии в онитротолуоле внутримолекулярной водородной связи и о проявлении в этой молекуле стерических напряжений, о проявлении которых можно судить также и по повороту нитрогруппы и метильной группы относительно плоскости кольца. Занимающие большой объем функциональные группы в орто-положении могут вызывать увеличение энтальпии образования и в других нитроаренах, например, в полинитробензолах и галогенонитробензолах. Другим достаточно интересным результатом проявления влияния строения молекул на изменение значений энтальпий образования изученных нитроаренов является снижение энтальпии образования п-изомеров по сравнению с м-изомерами для производных нитробензола, имеющих донорные заместители. Это различие можно объяснить проявлением прямого полярного сопряжения донорного заместителя с акцепторной нитрогруппой. Этот эффект невозможен в мета-изомерах, а в орто-изомерах указанных соединений с ним могут конкурировать образование внутримолекулярных водородных связей и стерическое отталкивание. Мы не будем комментировать результаты рассчитанных энтальпий образования радикалов, поскольку надежные экспериментальные данные имеются только для фенильного радикала и NO2. Для этих соединений расчет удовлетворительно передает экспериментальные значения энтальпий образования, причем для фенильного радикала они систематически завышаются, а для NO2группы занижаются. Большой интерес представляет теоретическая оценка D(C-N) в ряду ароматических нитросоединений с использованием современных квантовохимических методов. Для этого наряду с расчетом энтальпий образования необходимо иметь расчетные значения продуктов реакции гомолитического разрыва связи C-NO2. Расчет D(C-N) проводится с использованием уравнения (1): o D(C − N ) = Δ f H Ro • + Δ f H NO − Δ f H Ro − NO2 (1) 2 На основе D(C-N) можно оценить также энергию активации радикального газофазного распада по уравнению (2): E = D(C − N ) + RT (2) 125 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г Интересно, что если мы с использованием уравнения (2) пересчитаем для температуры эксперимента (700К), то расчетные значения энергий активации газофазного распада хорошо согласуются с экспериментальными оценками энергий активации (Табл. 1). В определенной степени это происходит и потому, что при расчете энергий диссоциации по уравнению (1) происходит частичная компенсация погрешностей энтальпий нитросоединений и продуктов их распада. Таблица 1 Энергия активации радикального газофазного распада (Т=700К) Соединение нитробензол о-фторнитробензол м-фторнитробензол п-фторнитробензол о-хлорнитробензол м-хлорнитробензол п-хлорнитробензол о-бромнитробензол м-бромнитробензол п-бромнитробензол о-нитрофенол м-нитрофенол п-нитрофенол о-нитроанилин м-нитроанилин п-нитроанилин о-нитротолуол м-нитротолуол п-нитротолуол о-нитробензойный альдегид м-нитробензойный альдегид п-нитробензойный альдегид о-нитробензойная кислота м-нитробензойная кислота п-нитробензойная кислота о-динитробензол м-динитробензол п-динитробензол 1,3,5-тринитробензол 6-31G(d) 6-31G(d,p) 6-31G (df,p) 298.7 281.1 293.4 301.1 267.3 291.3 297.3 269.5 291.2 296.9 332.3 296.3 309.2 316.8 300.7 316.6 288.0 299.4 302.9 270.8 292.7 289.8 261.5 293.6 291.3 250.8 286.9 285.8 277.1 298.7 281.2 293.5 301.3 267.3 291.5 297.4 269.5 291.4 297.1 333.0 296.3 309.3 317.9 300.8 317.0 288.4 299.5 303.1 271.2 292.9 290.0 261.6 293.8 291.5 255.2 283.8 286.0 277.2 298.6 281.5 293.5 301.5 268.0 291.5 297.5 271.5 291.9 297.7 332.3 296.2 309.3 318.2 300.8 317.4 288.4 299.5 303.1 271.4 292.9 290.0 262.7 293.8 291.6 251.9 287.0 286.1 277.4 6-311++G (3df,3pd) 285.8 265.2 280.5 288.6 257.8 286.2 254.5 278.7 285.1 313.2 283.2 296.6 301.1 287.4 304.3 273.8 286.6 290.7 257.8 280.4 277.4 250.7 281.0 278.8 241.9 274.7 274.3 265.6 Эксперимент 291 281 293 300 298 301 308 284 275 284 286 281 Достаточно хорошее согласие эксперимента и расчета позволяет использовать квантово-химические оценки для корректировки в необходимых случаях экспериментальных данных. Так, например, давно уже вызывают сомнения приводимые в литературе значения энергий активации газофазного распада ми п-нитротолуолов, равные соответственно 284 и 275 кДж/моль, что ниже, чем для нитробензола. Объяснить эти данные с позиций радикального механизма первичного акта реакции очень сложно. В литературе уже были высказаны предположения, что экспериментальные значения энергии активации нитротолуолов не отвечают радикальному механизму. Мы с использованием методов B3LYP 6-31G(d,p) определили энергию активации радикального газофазного распада для м- и п-нитротолуолов. Полученные значения 299.4 кДж/моль и 126 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г 303.2 кДж/моль несколько выше, чем экспериментальные и расчетные значения энергии активации для нитробензола, что вполне согласуется с основными закономерностями влияния донорных заместителей на прочность связи C-NO2 в ряду ароматических нитросоединений. Приведенные выше значения можно использовать для рассмотрения конкуренции различных механизмов газофазного распада о-нитроанилина и онитрофенола, т.к. известно, что при повышении температуры должен возрастать распад по радикальному механизму, поскольку этому процессу отвечает существенно более высокое значение предэкспоненциального множителя. Выводы Впервые получены расчетные значения энтальпий образования для 29 ароматических соединений (нитробензола, его монофункциональных производных и тринитробензола). Определены энергии диссоциации связи С-NO2 и на их основе для Т=700К (средней температуры экспериментального изучения) рассчитаны энергии активации газофазного распада для 10 соединений, для которых имеются надежные экспериментальные данные. Метод B3LYP/6-31G(d,p) дает наилучшее согласие с экспериментом, его средняя погрешность составляет 4 кДж/моль, что не превышает погрешности экспериментального определения энергии активации газофазного распада нитроаренов. Литература 1. Орлова, Е.Ю. Химия и технология бризантных взрывчатых веществ./ Е.Ю. Орлова. – М. : Химия, 1973. – 650 с. 2. Храпковский, Г.М., Влияние строения молекул на кинетические параметры мономолекулярного распада С- и О-нитросоединений./ Г.М. Храпковский, Г.Н. Марченко, А.Г. Шамов – Казань : ФЭН, 1997. – 224 с. 3. Садова, Н.И., Геометрия молекул нитросоединений./ Н.И. Садова, Л.В Вилков. //Успехи химии. – 1982. – Т.51. – №1. – С.153-183. 4. Лебедев, Ю.А., Термохимия нитросоединений./ Ю.А. Лебедев, Е.А. Мирошниченко, Ю.К. Кнобель – М. : Наука, 1970. – 160с. 5. Николаева, Е.В. Особенности механизма первичного акта газофазного мономолекулярного распада С-нитросоединений по результатам квантово-химических расчетов.: дис.…канд. хим. наук./ Николаева Е.В. – Казань, 2005. – 212 с. 6. Glenewinkel-Meyer, T., The isomerization of nitrobenzene to phenylnitrite./ T. GlenewinkelMeyer, F.F. Crim// Journal of Molecular Structure (Theochem). – 1995. – Vol.337. – P. 209224. 7. Чачков, Д.В., Некоторые особенности влияния строения молекул на энергию активации радикального газофазного распада нитробензолов./ Д.В. Чачков, Е.В. Николаева, А.Г. Шамов, Г.М. Храпковский// VIII Всероссийская конференция "Структура и динамика молекулярных систем". Сборник статей. Йошкар-Ола. – 2001. – Часть 2. – С. 198-202. 8. Храпковский, Г.М., Теоретическое изучение механизмов молекулярного газофазного распада ароматических нитросоединений./ Г.М. Храпковский, Е.В. Николаева, Д.В. Чачков, Г.А. Шамов, А.Г. Шамов// VIII Всероссийская конференция "Структура и динамика молекулярных систем". Сборник статей. Йошкар-Ола. – 2001. Часть 2. – С. 202-206. 127 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г 9. Khrapkovskii, G.M., Some characteristic of the in-fluence of molecular structure on kinetic parameters of gas-phase monomolecular decomposition of aromatic nitrocompounds according to the results of quantum-chemical investigation./ G.M. Khrapkovskii, D.V. Chachkov, E.V. Nikolaeva, A.G. Shamov// 34th Inter-national Annual Conference of ICT. "Energetic Materials: Reactions of propellants, explosives and pyrotechnics". Collection of articles. Federal Republic of Germany, Karlsruhe. – 2003. – P. 137-1 – 137-22. 10. Храпковский, Г.М., Барьеры вращения функциональных групп и оценка предэкспоненциального множителя реакции радикального мономолекулярного распада нитробензола и его монофункциональных производных./ Г.М. Храпковский, Д.В. Чачков, А.Г. Шамов// IX Всероссийская конференция "Структура и динамика молекулярных систем". Сборник статей. Уфа-Казань-Йошкар-Ола-Москва. – 2002. – Том 2. – С. 234-237. 11. Шарипов, Д.Д., Молекулярная структура нитробензола и ряда его монофункциональных производных в газообразном состоянии./ Д.Д. Шарипов, Д.Л. Егоров, Д.В. Чачков// Бутлеровские сообщения. – 2008. – Том 14. – №5. – С. 46-51. 12. Frisch, M.J. Gaussian 98 / M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, V.G. Zakrzewski, J.A. Montgomery, R.E. Stratmann, J.C. Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K. Raghava-chari, J.B. Foresman, J. Cioslowski, J.V. Ortiz, B.B. Stefanov, G. Liu, A. Li-ashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challa-combe, P.M.W. Gill, B.G. Johnson, W. Chen, M.W. Wong, J.L. Andres, M. Head-Gordon, E.S. Replogle and J.A. Pople. – Pittsburgh PA: Gaussian Inc., – 1998. 13. Frisch, M.J. Gaussian 03 / M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox, H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y. Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S. Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K. Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, and J.A. Pople. – Pittsburgh PA: Gaussian Inc., – 2003. 14. Chen, P.C., Density functional calculations of the heats of formationfor various aromatic nitro compounds./ P.C. Chen, Y.C. Chieh, S.C. Tzeng // Journal of Molecular Structure (Theochem). – 2003. – Vol.634. – P. 215-224. 128 Электронный журнал «Структура и динамика молекулярных систем». №6,А, 2009 г C-NO2 BOND DISSOCIATION ENERGIES IN AROMATIC NITROCOMPOUNDS OBTAINED BY QUANTUM-CHEMICAL CALCULATIONS Sharipov D.D., Egorov D.L., Chachkov D.V., Shamov A.G., Khrapkovskii G.M. Kazan state technological university 68, Karl Marks street, 420015, Kazan, E-mail: dmn@kstu.ru Molecular structures and enthalpies of formation of nitrobenzene, more than it’s 20 monoderivatives, 1,3,5-trinitrobenzene and radical formed after C-NO2 bond homolytic cleavage were calculated at B3LYP level with different basis sets (6-31G(d), 6-31G(d,p), 6-31G(df,p) и 6-311++ G(3df,3pd)). Dissociation energies of C-NO2 (D(C-N)) bond and activation energies of gas phase radical decomposition were calculated based on enthalpies of formation of original molecules and formed radicals. It is found that donating substituents increase D(C-N)) and withdrawing decrease. Activation energies of gas phase radical decomposition calculated with D(C-N)) values are in good agreement with experimental data. Keywords: Aromatic compounds, molecular structure, enthalpy of formation, dissociation energy, quantum-chemical calculations. 129