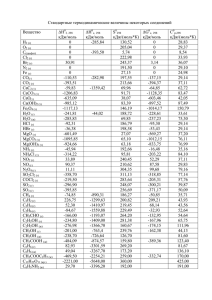
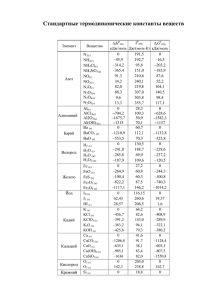
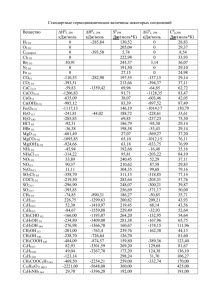
элементы химической термодинамики и кинетики
advertisement

ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ Государственное образовательное учреждение высшего профессионального образования Ульяновский государственный технический университет В.Т. ПИСЬМЕНКО ЭЛЕМЕНТЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ И КИНЕТИКИ Ульяновск 2008 УДК 541.1 (075) ББК 24.5я7 П 35 Рецензенты: доцент, к.х.н. О.А. Давыдова (ГОУ ВПО Ульяновский государственный университет) доцент, кан. биол. наук О.А. Индирякова (ГОУ ВПО Ульяновский государственный университет) Утверждено редакционно-издательским советом университета в качестве учебного пособия Письменко В.Т. П 35 Элементы химической термодинамики и кинетики: учебное пособие / В.Т. Письменко. – Ульяновск: УлГТУ, 2008. – 118с. ISBN 5-89146-001-7 Учебное пособие предназначено для студентов первых курсов нехимических специальностей вузов. Пособие написано в соответствии с программой курса химии для инженерной подготовки студентов. Может быть использовано для факультативной подготовки старшеклассников, учителями средней школы. Рассматриваются основные понятия, определения химической термодинамики и кинетики без применения высшей математики. Разделы сопровождаются многочисленными примерами, упражнениями, задачами и иллюстрациями, позволяющими самостоятельно усвоить прочитанный материал и приобрести навыки практических расчетов. УДК 541.1(075) ББК 24.5я7 © В.Т. Письменко, 2008 © Оформление. УлГТУ, 2008 ISBN 5-89146-001-7 2 ОГЛАВЛЕНИЕ ОГЛАВЛЕНИЕ ...................................................................................................... 3 Предисловие ........................................................................................................... 5 Глава I. ЭЛЕМЕНТЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ ................. 6 Введение ............................................................................................................... 6 1. НЕКОТОРЫЕ ОСНОВНЫЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ............................................................ 7 1.1. Система ......................................................................................................... 7 I.2. Параметры состояния.................................................................................... 8 1.3. Внутренняя энергия.................................................................................... 10 1.4. Теплота и работа ......................................................................................... 13 2. ИЗМЕНЕНИЕ ВНУТРЕННЙ ЭНЕРГИИ СИСТЕМ................................ 15 2.1 Изменение внутренней энергии системы при проведении процесса при постоянном объеме ............................................................................................ 15 2.2. Изменение внутренней энергии системы при проведении процесса при постоянном давлении ........................................................................................ 17 2.3. Энтальпия и ее изменение ......................................................................... 21 2.4. Термохимические уравнения..................................................................... 25 2.5. Энтальпия образования вещества ............................................................. 26 2.5. Стандартные условия ................................................................................. 26 2.7. Влияние температуры на тепловой эффект реакции. ............................. 29 3. ТЕРМОХИМИЧЕСКИЕ ЗАКОНЫ ............................................................. 30 3.1. Закон Лавуазье-Лапласа............................................................................. 30 3.2. Закон Гесса. ................................................................................................. 32 3.3. Термохимические расчеты......................................................................... 34 Глава П. НАПРАВЛЕННОСТЬ ХИМИЧЕСКИХ ПРОЦЕССОВ............ 37 Введение ............................................................................................................. 37 1. САМОПРОИЗВОЛЬНЫЕ И ОБРАТИМЫЕ ПРОЦЕССЫ ................. 38 2. ПРИНЦИП БЕРТЛО-ТОМСЕНА............................................................... 40 3. ЭНТРОПИЯ И ЕЁ ИЗМЕНЕНИЕ............................................................ 43 3.1. Энтропия...................................................................................................... 43 3.2. Изменение энтропии и направленность процессов................................. 46 3 3.3. Энтропия как мера беспорядка.................................................................. 50 3.4. Стандартные энтропии............................................................................... 56 4. СВОБОДНАЯ ЭНЕРГИЯ .............................................................................. 58 4.1. Свободная энергия – критерий направленности процессов................... 58 4.2. Примеры расчета свободной энергии....................................................... 62 4.3. Стандартный изобарно-изотермический потенциал и его изменение .. 66 4.4. Изохорно- изотермический потенциал..................................................... 67 3. ОСОБЕННОСТИ ТЕРМОДИНАМИЧЕСКОГО МЕТОДА ИССЛЕДОВАНИЯ.............................................................................................. 69 Глава III. ХИМИЧЕСКАЯ КИНЕТИКА И РАВНОВЕСИЕ ..................... 70 Введение ............................................................................................................. 70 1. ОСНОВНЫЕ ПОНЯТИЯ ХИМИЧЕСКОЙ КИНЕТИКИ...................... 70 1.1. Механизм химической реакции ................................................................ 70 1.2. Гомогенные и гетерогенные реакции ....................................................... 71 1.3. Скорость реакции........................................................................................ 72 2. ВЛИЯНИЕ НЕКОТОРЫХ ФАКТОРОВ НА СКОРОСТЬ РЕАКЦИИ 75 2.1. Зависимость скорости от концентрации реагирующих веществ........... 75 2.2. Зависимость скорости реакции от температуры ..................................... 82 2.3. Энтропия активации ................................................................................... 87 3. ХИМИЧЕСКОЕ РАВНОВЕСИЕ ................................................................. 89 3.1. Химическое равновесие и константа равновесия.................................... 89 3.2. Смещение химического равновесия. Принцип Ле Шателье .................. 96 3.3. Константа равновесия и свободная энергия Гиббса ............................... 99 4. ЯВЛЕНИЕ КАТАЛИЗА ............................................................................... 104 БИБЛИОГРАФИЧЕСКИЙ СПИСОК ......................................................... 107 ПРИЛОЖЕНИЕ ................................................................................................... 108 ОТВЕТЫ НА ВОПРОСЫ И ЗАДАЧИ .......................................................... 113 4 Предисловие Первое издание этого пособия было опубликовано в 1996 году. При подготовке настоящего издания текст его был тщательно просмотрен, исправлены опечатки, расширены некоторые разделы, внесены поправки. Пособие написано для студентов первых курсов нехимических специальностей технических вузов в соответствии с программой по химии. Оно также может быть использовано для факультативной подготовки старшеклассников учителями средней школы. Классическое изложение термодинамики и кинетики в курсе общей химии для студентов нехимических специальностей затруднено из-за явно недостаточного количества часов, отводимых в учебных планах на изучение курса химии, и трудностей, связанных с использованием аппарата высшей математики, с которым первокурсники не успели еще достаточно полно познакомиться. К тому же, в последнее время студенты первого курса демонстрируют, как правило, в большей степени алгоритмическое мышление (что, видно, связано с методологическим креном школьной педагогики): они относительно легко могут оперировать понятиями, не представляя их сущности, при решении школьных задач. Так, например, зная уравнение Менделеева-Клапейрона, они не могут им воспользоваться при решении, если один из параметров состояния газа задан в неявном виде. Студенты могут рассчитать тепловой эффект, свободную энергию реакции, используя табличные данные, не представляя, что скрывается за понятиями: энтальпия, энтропия, изобарно-изотермический потенциал. Такое положение обусловлено недостаточным развитием творческого мышления; поверхностным, формальным представлением о сущности тех или иных понятий, определений и отсутствием связи между ними. Главная задача данного пособия – помочь читателю усвоить основные понятия химической термодинамики и кинетики, установить взаимосвязь между ними и развить практические навыки решения конкретных задач, примеров. Почти каждый раздел сопровождается упражнениями по решению тех или иных задач. Кроме того, приведены вопросы и задачи для самостоятельной проработки прочитанного материала, ответы на которые даны в конце пособия. Поэтому рекомендуется читателю работать с карандашом в руке, т.к. прочные, глубокие знания приходят через труд, а не созерцание. Пособие написано по результатам многолетней работы на кафедре химии Ульяновского государственного технического университета. Автор благодарен коллективу кафедры за обсуждение рукописи, пожелания и замечания, а также преподавателям факультетов повышения квалификации МГУ и МХТИ, слушателем которых он был в 1970-1985 годах. 5 Глава I. ЭЛЕМЕНТЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ Введение Как известно, вещества состоят из отдельных частиц (молекул, атомов, ионов, электронов…), находящихся во взаимодействии. При химических превращениях происходит изменение состава, строения веществ, что связано с разрушением старых связей между частицами исходного вещества (для чего, заметим, требуются затраты энергии) и образованием новых связей между частицами вновь образующихся веществ, которое сопровождается выделением энергии. Так или иначе, химические превращения сопровождаются энергетическими изменениями. Выделение (поглощение) энергии в химическом процессе, обмен ею между окружающей средой и реагирующей системой и в пределах самой системы происходит эквивалентно в различных видах и формах. Например, взаимодействие цинка с сульфатом меди в гальваническом элементе протекает с превращением химической энергии в электрическую, а при электролизе протекает обратный процесс превращения электрической энергии в химическую; энергия сгорания топлива в ракетных двигателях преобразуется в механическую энергию поступательного перемещения ракеты и выделения части её в окружающую среду в виде тепла и света; энергия окисления бензина в двигателе внутреннего сгорания автомобиля сопровождается передачей ее окружающей среде в форме тепла и работы по перемещению автомобиля; поглощение квантов лучистой энергии фотопленкой приводит к протеканию химических (фотографических процессов) и так далее. В связи с тем, что в первых изучаемых реакционных процессах обмен энергией чаще всего протекал через тепловые эффекты, это нашло отражение в исторически сложившимся названии данного раздела химии – химическая термодинамика (от греческого thermo – теплота, dynamic - сила). Естественно, современная химическая термодинамика не ограничивается этим одним способом передачи энергии. Превращения веществ, химические реакции протекают при определенных условиях (температуре, давлении, концентрации и других). При изменении таковых процессы могут прекращаться и более того – в системе смогут протекать процессы в обратном направлении, сопровождаясь энергетическими изменениями. Поэтому химическая термодинамика изучает и условия, при которых процессы становятся возможными, а также равновесное состояние веществ и реакционных систем. Таким образом, можно сказать, что химическая термодинамика является разделом химии, который изучает главным образом превращение энергии одних видов в другие, энергетические эффекты, сопровождающие различные 6 физико-химические процессы, а также условия самопроизвольного протекания процессов и их равновесия. Термодинамика опирается только на опыт. Все ее понятия и законы являются результатом человеческого опыта и имеют общий характер. Прежде чем перейти к рассмотрению энергетических закономерностей химических процессов, познакомимся кратко с некоторыми основными определениями химической термодинамики. 1. НЕКОТОРЫЕ ОСНОВНЫЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ. 1.1. Система Объектом изучения в термодинамике является система. Под системой принято понимать произвольно выбранную совокупность тел, веществ, находящихся во взаимодействии, которая (совокупность) мысленно или фактически выделяется из окружающей среды (рис. 1.I.I.) Под понятием «тело» в химической термодинамике подразумевается вещество (вещества), заполняющее определенный объем, внешний вид которого Окру(цвет, форма) представляется несущественРеакционная жающая система среда ным. Под словом «тело» может, например, подразумеваться вода, воздух, железо, соль, раствор или какое-либо другое вещество, взятое в определенном объеме и характеризующееся какими либо свойствами: плотностью, температурой, давлением, электропроводностью и другими признаками. В зависимости от характера взаимоВСЕЛЕННАЯ действия системы с внешней средой различают изолированные, замкнутые и открытые системы. Изолированная система не обмениваетРис. 1.1.1. Схематическое ся с окружающей средой ни веществом, ни изображение системы энергией и находится при постоянном объеме (V = const.). В природе не существует изолированных систем. Любая реальная система взаимодействует с окружающей средой. И понятие изолированная система это воображаемое теоретическое построение. Например, представим себе реагирующую смесь их водорода и кислорода в запаянном сосуде со стенками из теплоизоляционного материала. Такая система не обменивается с окружающей средой ни энергией, ни веществом, хотя мы понимаем, что идеальной теплоизоляции нет и, рано или поздно, данная система обменяется энергией с окружающей средой. 7 Кстати, экспериментальное исследование изолированной системы невозможно, т. к. для получения информации о ее состоянии нужно вводить в нее сигналы и получать их обратно, что противоречит определению понятия изолированная система. Но мы будем пользоваться этим понятием, помня о его идеализированном характере. Замкнутая система не обменивается с другими системами, окружающей средой, веществом, но может получать и отдавать энергию. Например, запаянный сосуд с реагирующим веществом – это замкнутая система, поскольку у нее имеется возможность обмениваться энергией с окружающей средой (например, способом теплопередачи, излучения, изменением объема системы), но не веществом. Ок р у ж а ю щ а я с р е д а Э Н Е Р Г И Я V = const Вещество Теплоизоляция а б в Рис.1. I.2. Схематическое изображение трех типов систем (в виде реакционной колбы): а- изолированная; б – замкнутая; в – открытая. Открытая система обменивается и энергией и веществом с другими системами и с окружающей средой. Например, горящая спичка, работающий двигатель автомашины, растущее дерево и т.д. На рисунке 1.I.2. представлены схематически все три типа систем. I.2. Параметры состояния Состояние системы характеризуют параметрами состояния: объемом, давлением, температурой, количеством вещества, концентрацией, энергией и т.д. Термодинамическим параметром может быть любое свойство системы, если оно рассматривается как одна из независимых переменных, определяющих состояние системы. Если все параметры состояния не изменяются во времени, то система, говорят, находится в равновесном состоянии, или просто – в равновесии. В химической термодинамике свойства системы рассматриваются в ее равновесных состояниях: начальном (исходном) и конечном. 8 Предположим, например, что некая система из равновесного состояния «1» с параметрами T1, P1, V1 переходит в новое (конечное) состояние «2», характеризующееся параметрами T2, P2, V2. Естественно, что в процессе перехода параметры состояния системы непрерывно изменяются, но химическая термодинамика изучает не промежуточные их значения, а параметры состояния системы лишь в начальном и конечном равновесных состояниях и на основании этого делает выводы о возможности процессов в указанном направлении, энергетических изменениях. Применительно к рассматриваемой нами системе, о ее свойствах приходится судить по изменению параметров: ∆ T = T2 -T1; ∆ P = P2 - P1 и ∆ V = V2 - V1 в результате перехода из состояния «1» в состояние «2». Кстати, заметим, что в термодинамике принято из параметров конечного состояния вычитать параметры начального состояния. Термодинамические параметры состояния системы характеризуют лишь данное состояние, никак не свидетельствуя о предшествующих или промежуточных состояниях системы. Другими словами, - при переходе системы из одного состояния в другое изменение ее свойств не зависит от пути перехода (процесса), а определяется лишь начальным и конечным состоянием системы (можно сказать: система не «помнит» своей истории). Можно представить себе, в частности, что система, выйдя из некоторого начального состояния и претерпев ряд изменений, вновь возвращается в него. Следовательно, она вновь приобрела те же свойства, которые имела в исходном состоянии. Мы же не имеем никакой информации о природе процесса (и был ли он?), но можем характеризовать систему, рассматривая ее начальное и конечное состояние. Вопросы и задачи для самостоятельной работы 1.1.1. Рассмотрите, какой тип системы реализуется в ниже перечисленных примерах: а) пробирка с реагирующими веществами; б) колба с реагирующими веществами, закрытая пробкой; в) космонавт в космическом корабле; г) планета Земля; д) вся Вселенная в целом. 1.1.2. На практике иногда оказывается, что система, приведенная вновь в начальное состояние, обладает различными свойствами по сравнению с исходными. Например, нагрев сплава до определенной температуры и затем быстрое его охлаждение (закалка) до обычных температур дает сплав того же состава, но с более высокой твердостью, чем в исходном состоянии. Чем это можно объяснить? 1.1.3. Перечислите признаки систем, свойства которых изучает химическая термодинамика. 1.1.4. Почему в одних случаях энергия выделяется, а в других поглощается системой? 1.1.5. Почему химические и фазовые превращения сопровождаются энергетическими изменениями? 1.1.6. Состояние любой химической системы, в которой идет реакция, определяется ее параметрами (например, для газообразной системы: давлением, температурой, объе9 мом, концентрацией). Этих параметров достаточно для описания, если реакцию сопровождают энергетические эффекты? 1.3. Внутренняя энергия Поскольку химические превращения сопровождаются выделением (поглощением) энергии, спрашивается, откуда же она берется (или куда девается)? По-видимому, взаимодействующие вещества и каждое вещество в отдельности содержат и содержали определенный запас энергии, но только в скрытой форме. А проявляется она при их взаимодействии. Этот запас энергии тела или системы тел называется внутренней энергией, которую обычно обозначают латинской буквой U. Она включает в себя кинетическую и потенциальную энергию частиц, составляющих данные вещества системы. Внутренняя кинетическая энергия обусловлена тепловым хаотическим движением частиц и непосредственно связана с температурой. Внутренняя потенциальная энергия – это энергия взаимодействия ядер с ядрами, электронов с ядрами, электронов с электронами и т.п. В понятие внутренней энергии входит и ядерная энергия и много других составляющих, которые далеко не все нам известны в настоящее время, а поэтому абсолютное значение ее экспериментально определить или рассчитать не представляется возможным. Но важно другое, что каждое тело, вещество, система имеет определенное значение этого запаса внутренней энергии U. Заметим, в понятие внутренней энергии системы не входит потенциальная энергия положения системы в пространстве и кинетическая энергия движения всей этой системы как целого. То есть внутренняя энергия какой-либо системы, (например, колбы с реагирующими веществами) и на поверхности Земли и в лаборатории на восьмом этаже, а так же в летящем самолете, одна и та же. Внутренняя энергия системы является ее свойством и зависит только от ее сиюминутного состояния и не зависит от предыстории системы в том смысле, что, говоря о ее значении в данном состоянии, мы не можем, например, обнаружить, за счет чего система изменила свою энергию: или за счет тепла, или за счет работы. Внутренняя энергия, говорят, является функцией состояния. Функция состояния обладает двумя особенностями: – при переходе системы из одного состояния в другое изменение параметра не зависит от пути перехода; – если система совершает круговой процесс, возвращаясь в исходное состояние, то изменение параметра системы равно нулю. На рисунке 1.1.3А схематично представлено изменение внутренней энергии системы в процессе перехода ее из состояния «1» в состояние «2» по пути а и б. Независимо от пути перехода изменение внутренней энер10 гии ∆U = U2 – U1 оказывается одним и тем же, поскольку оно определяется лишь начальным и конечным состоянием системы. В случае же совершения какого-то процесса и возвращения системы в исходное состояние (рис.1.1.3.Б) изменение внутренней энергии оказывается равным нулю ∆U = 0 . В этом случае мы даже не можем сказать, а был ли процесс, если будем судить только по начальным и конечным параметрам состояния. Внутренняя энергия U определяется параметрами состояния системы: температурой T, давлением P, объемом V: U = f (P, T, V), которые в свою очередь характеризуют кинетическую и потенциальную энергию частиц системы. U 1 ○ U U1 = U2 а 1 • ∆U = 0 ∆U U1 б 2 • 2 U2 ○ Ход Ход процесса А процесса Б Рис. 1.1.3. Изменение внутренней энергии системы: А – при переходе из состояния 1 в состояние 2 по пути а и б; Б – при переходе через различные состояния в исходное состояние 1 Энергия системы складывается из энергии ее составных частей, т.е. она обладает свойством аддитивности1: Uсистемы = U1 + U2 + U3 + U4 + …..+ Un Так как запас внутренней энергии U зависит и от количества вещества, то для определенности и простоты при расчетах в химической термодинамике условились относить его к 1 молю вещества. Единицей измерения внутренней энергии служит Джоуль (Дж) или в старых литературных изданиях - калория (кал), которые связаны между собой простым соотношением: 1 кал = 4.184 Дж. Вычислить или экспериментально определить абсолютное значение внутренней энергии системы в начальном U1 и в конечном состоянии U2 мы не можем (ведь, нам даже не известны все виды энергии). Но при изменении состояния системы: переход ее из начального состояния с запасом внутренней энергии U1 в конечное с запасом внутренней энергии U2, 1 От английского «addition» - сложение. 11 которое сопровождается энергетическими эффектами, мы можем найти изменение запаса внутренней энергии ∆U системы (рис.1.1.4): ∆U = U 2 - U 1 . (1.1.1) На первый взгляд это парадокс: не зная величин U 2 и U 1 .можно найти их разницу U 2 - U 1. Но этот случай подобен той бытовой ситуации, когда вы не знаете, сколько денег в вашем кошельке до какой-то покупки в магазине и сколько их осталось после покупки, но, зная стоимость покупки, вы можете оценить, насколько изменилось содержимое кошелька. Кстати, говоря, о переходах системы в различные состояния, представим себе, что переход изолированной системы из состояния «1» в состояние «2» связан с изменением внутренней энергии ∆U1 , а при обратном переходе (из состояния «2» в состояние «1») в исходное состояние с изменением внутренней энергии ∆U2. Если ∆U1 и ∆U2 не равны ∆U1 ≠ ∆U2, то мы получаем при этих переходах или выигрыш, или потерю внутренней энергии системой, которая вследствие изолированности системы ничем не компенсируется. Однако это невозможно, т.к. в противном случае нарушается закон сохранения энергии: энергия возникает из ничего или теряется неизвестно куда. Поэтому одной из формулировок первого закона Начальное состояние 1 термодинамики является утверждение: ∆U = U2 – U1 в изолированной системе сумма Конечное состояние 2 всех видов энергии постоянна. U1 Внутренняя энергия системы зависит от ее состояния, определяемого U2 параметрами T, P, V, и, соответственно, изменение внутренней энергии ∆U также будет зависеть от них. Рис. 1.1.4. Изменение внутДля многих практических целей ренней энергии системы при важно знать энергетические изменепереходе из одного состояния в ния в системах при постоянном объедругое ме и постоянном давлении, так как большинство химических процессов протекает при постоянном давлении (чаще всего - атмосферном), и при постоянном объеме (объеме раствора, сосуда). Вопросы и задачи для самостоятельной работы. 1.1.7. Перечислите Вам известные виды энергии, входящие в понятие внутренней энергии, например, 1 моля воды. 1.1.8. Каковы свойства внутренней энергии? Можно ли измерить внутреннюю энергию, и в каких единицах ее выражают? 1.1.9. Можно ли определить изменение внутренней энергии изолированной системы? 12 11.10. Увеличится ли внутренняя энергия колбы с реагирующими веществами, если перенести ее с первого этажа на девятый? 1.1.11. Вы поднялись пешком с 1 этажа на восьмой кафедры химии для выполнения лабораторной работы. Какие из перечисленных величин являются функциями состояния: а- пройденное расстояние; б- количество совершенной работы; в- количество времени, затраченного на подъем; г- количество остановок; д- изменение высоты над уровнем Куйбышевского водохранилища; е- количество теплоты выделенное вашим организмом? 1.4. Теплота и работа Прежде чем перейти к рассмотрению изменения внутренней энергии U в зависимости от параметров состояния системы, вспомним и уясним понятия теплота и работа. Энергия, теплота и работа измеряются в одних и тех же единицах Джоулях. На основании этого можно ли сказать, что эти понятия равнозначны? Когда мы говорим об энергии1, то представляем себе запас возможной, но еще неосуществленной работы. Можно ли сказать, что теплота и работа есть «запас теплоты» или «запас работы»? Когда проявляется теплота, то имеется, по меньшей мере, два тела: одно, которое отдает энергию (с более высокой температурой), и другое (с меньшей температурой), которое получает её за счет разности температур. Точно так же, когда выполняется работа, имеется тоже, по меньшей мере, два тела: одно, которое развивает силы, совершающие работу, и другое, к которому эти силы приложены. Первое тело, производящее работу, отдает энергию, второе тело – получает ее. То есть понятия «теплота» и «работа» связаны с процессом передачи энергии, и не относятся к состоянию системы. Теплота и работа представляют два возможных способа передачи энергии от одного тела к другому. Теплота представляет собой совокупность микрофизических актов процесса передачи энергии (обмен энергией при соударении молекул, атомов, излучения квантов света и т.д.), т.е. неупорядоченного способа обмена энергии между системами вследствие хаотичности перемещения частиц, что более вероятно, чем направленное движение всех частиц. Возникновение теплоты в процессе всегда свидетельствует о малоэффективном способе передачи энергии, так как вероятность того, что хаотическое движение получит определенную направленность для совершения работы маловероятно. Поэтому полный переход энергии посредством теплоты в работу невозможен, хотя работа может перейти в теплоту целиком. Работа - это любая макрофизическая, упорядоченная, направленная форма передачи энергии за счет передвижения масс под действием какихлибо направленных сил. Работа может быть использована на пополнение запасов различных видов энергии: электрической, магнитной, кинетической и других. Теплота же, 1 Энергия – слово греческого происхождения. Приставка «эн»-означает «в» (или «содержащаяся»), «эрг»-корень слова – «работа». 13 как способ передачи энергии, без преобразования её в работу может пополнять только запас внутренней энергии системы. Количество поглощенной (выделенной) теплоты в процессе, как и совершенной работы, зависят от способа проведения процесса. Например, проводится ли процесс при постоянном объеме или постоянном давлении. Кстати, системе энергия может быть сообщена двумя единственно возможными способами: в виде работы А и тепла Q. Соответственно, и изменение внутренней энергии должно складываться из двух этих величин: количества энергии переданной в виде тепла Q, и энергии переданной посредством работы – А ∆U = Q + A (1.1.2) Энергию тела, системы можно изменять, подводя её в виде тепла и работы. Но эти вклады обезличиваются, превращаясь в единую величину – энергию U. Не существует величины, которую можно было бы назвать теплотой тела Q, как нет внутри тела величины, которую можно назвать работой А. Следовательно, нельзя говорить о теплоте, как об отдельной (независимой) «форме энергии», «запасе теплоты», «приращении теплоты» системы (тела) и тому подобное, что мы порой делаем. Это историческое наследие от вещественной теории теплоты (теплороде), развитой во второй половине XVIII века. Этой теории мы также обязаны введением понятий «теплотворная способность», «теплоемкость», «скрытая теплота» и некоторых других. В дальнейшем, употребляя термин «теплота», «тепловой эффект реакции», мы будем однозначно представлять себе передачу какой-то определенной части энергии за счет разности температур между телами. А количество энергии, переданной системой (системе) в процессе работы, будем называть количеством работы, или работой. Таким образом, принятая для теплоты и работы размерность энергии, не доказывает тождества их последней. Вопросы и задачи для самостоятельной работы 1.1.11. Найдите ошибки в следующем рассуждении: если энергия есть функция состояния, а теплота есть энергия, то и теплота есть функция состояния. 1.1.12. При каких условиях изменение внутренней энергии равно теплоте, получаемой системой из окружающей среды? 1.1.13. Точен ли автор, утверждая в учебнике (Фролов В. В. Химия. М. Высшая школа, 1975 стр. 116 и 121): «Чаще всего в результате химических реакций выделяется или поглощается тепловая энергия»; «Q – энергия, сообщенная системе, может быть тепловой или другой формой энергии….»? 14 2. ИЗМЕНЕНИЕ ВНУТРЕННЙ ЭНЕРГИИ СИСТЕМ 2.1 Изменение внутренней энергии системы при проведении процесса при постоянном объеме Для начала рассмотрим изменение внутренней энергии на конкретном процессе системы, состоящей из одного моля цинка и одного моля раствора серной кислоты. Поместим эти вещества в специальное устройство - калориметр, схема которого представлена на рисунке 1.2.1. Устройство представляет собой цилиндр 1, закрытый поршнем 2, который фиксируется стопорным штифтом 3, чтобы поршень не перемещался, а объем реагирующей системы оставался постоянным. Цилиндр опущен в сосуд 4 с водой, температуру которой можно контролировать с помощью термометра 5. Стенки сосуда 4 выполнены из теплоизоляционного материала. Реакционная система, которая состоит из металлического цинка 6 и раствора серной кислоты в сосуде 7, в начальном состоянии (и затем про3 2 1 дуктов реакции в конечном состоянии) будет находиться при постоянном объеме, а так как она не об4 менивается с окружающей средой веществом, будем считать, и энергией, то система является изолиро5 ванной. В начальном состоянии (до взаимодействия цинка с раствором 6 серной кислоты) система обладает запасом внутренней энергии U1, который можно рассматривать как сумму внутренней энергии частей 7 системы: раствора серной кислоты Рис. 1.2.1. Схема установки и цинка. С помощью какого-либо для определения энергетических приспособления приведем в эффектов при постоянном объеме соприкосновение цинк с раствором кислоты. Начнется реакция. Она будет продолжаться некоторое время. Это состояние системы назовем переходным, т.к. при этом происходит образование новых веществ (продуктов), исчезновение исходных. Процесс сопровождается выделением энергии в виде теплоты Qv. По окончании реакции будем иметь систему в конечном состоянии, состоящую их раствора сульфата цинка и водорода согласно уравнению реакции: Znкр + H2SO4 p-p = ZnSO4 p-p +H2 газ1 1 Буквенными индексами при химических формулах указываются агрегатные состояния: кр – кристаллическое; p-p – раствор; г – газ; ж – жидкое. 15 и обладающую внутренней энергией U2. Изменение внутренней энергии системы в результате взаимодействия веществ (отдельных частей системы) должно соответствовать количеству энергии, переданной в виде тепла воде установки Qv (индекс «v» указывает на то, что теплота определена при постоянном объеме реагирующей системы) (1.2.1.) ∆U = Qv . Выделившееся количество теплоты Qv можно экспериментально определить, зная температуру воды до опыта и после реакции, ее массу m и теплоемкость c согласно уравнению: Qv = m c ∆t , а, следовательно, и изменение внутренней энергии системы. Однако, идеальной теплоизоляции не существует и выделившееся количество теплоты Qv, рано или поздно, рассеется через теплоизоляционные стенки сосуда в окружающую среду. Следовательно, наша система уменьшит свою внутреннюю энергию на величины выделившегося (положительного) эффекта реакции: - ∆U = Qv (1.2. 2.) Знак минус в этом выражении подчеркивает тот факт, что в результате процесса внутренняя энергия системы уменьшилась, а внешняя среда получила количество энергии в форме тепла Qv (экзотермическая реакция). Действительно, если U2 < U1 , то и разность величина отрицательная U2 - U1 = - ∆U. В противном случае, когда энергия подводится из внешней среды к реагирующей системе (отрицательной, эндотермический тепловой эффект -Qv), она пойдет на увеличение внутренней энергии системы ∆U = - Qv U U1 С учетом теплового эффекта термохимическое уравнение можно записать так: Znкр., H2SO4 р- Znкр + H2SO4 p-p = ZnSOp-p + H2 газ + 165,7 кДж. -∆U= Qv ZnSO4 р-р, Н2 газ U2 Ход реакции Рис. 1.2.2. Изменение внутренней энергии в процессе Тепловой эффект реакции при постоянном объеме равен Qv = + 165,7 кДж. А энергосодержание системы уменьшилось на эту величину положительного теплового эф- фекта реакции: ∆U = - 165,7 кДж 16 Обратим внимание на то, что тепловым эффектом реакции мы назвали то количество энергии, которое отведено от продуктов реагирующей системы, но таким образом, чтобы они имели ту же температуру, что и исходные вещества до реакции. Энергетические изменения в процессе можно изобразить графически, если по оси ординат отложить внутреннюю энергию системы U а по оси абсцисс ход реакции (рис. 1.2.2). Из рисунка 1.2.2. наглядно видно, что изменение внутренней энергии системы количественно соответствует тепловому эффекту реакции. Таким образам, если система находится при постоянном объеме, то изменение внутренней энергии системы связано с потерей или приобретением энергии в виде теплового эффекта реакции. Так, соотношение - ∆U = + Qv соответствует потере системой энергии, а соотношение + ∆U = - Qv отвечает увеличению (приобретению) системой энергии. Отметим, что здесь применена определенная система знаков. Тепловой эффект экзотермической реакции, идущей с выделением энергии, обозначают со знаком «+», а эндотермической (с поглощением тепла) – со знаком «-». То есть тепловые эффекты рассматриваются, как бы с точки зрения окружающей среды: если она (среда) получает энергию от реакционной системы, значит тепловой эффект положителен, а если внешняя среда отдает энергию реакционной системе – отрицателен. Вопросы и задачи для самостоятельной работы 1.2.1. Если в изолированной системе происходит горение магния в кислороде, как изменится ее энергия? (внутренняя?) Куда тратится выделившееся количество энергии в форме тепла, излучения? 1.2.2. Как изменилась внутренняя энергия реакционной замкнутой системы, если в результате процесса, прошедшего в ней при постоянном объеме, тепловой эффект реакции оказался равным Qv= -41 кДж/моль? 1.2.3. Что называют тепловыми эффектами реакции? 1.2.4. В чем различие энергетических изменений с точки зрения системы и внешней среды процессов обмена энергией? 2.2. Изменение внутренней энергии системы при проведении процесса при постоянном давлении Воспользуемся приведенной ранее установкой и проведем тот же процесс взаимодействия цинка с раствором серной кислоты, но при постоянном давлении. Для этого нужно, чтобы по мере выделения водорода в реакции система занимала все больший и больший объем. Этого можно достигнуть, если убрать стопорный штифт 3, который ограничивал движения поршня 2 в 17 цилиндре 1, содержащем реагирующие вещества (рис.1.2.3.). Будем считать, что поршень в цилиндре перемещается свободно, без трения. Тогда реакционная система будет автоматически изменять свой объем, и давление в системе будит оставаться постоянным, а сама система окажется замкнутой. Начальному состоянию системы (вещества не реагируют) будет соответствовать внутренняя энергия системы U1, объем системы V1 (поршень расположен на отметке h1 ) и давление в системе Р. Приведем в соприкосновение цинк и раствор серной кислоты какимлибо способом. Начнется реакция, в результате которой будет происходить образование сульфата цинка, водорода, и выделяться какое-то количество 2 1 3 энергии в форме теплоты Qp, о чем можно судить по изменению темпера4 туры воды. Одновременно крышка системы в виде поршня 2 начнет подниматься по мере выделения водоро5 да, увеличивая объем системы. По истечении какого-то времени, когда, наh конец, последний атом цинка прореаh 6 гирует с последней молекулой кислоты и образуется последняя молекула сульфата цинка и водорода, система перейдет конечное состояние с внут7 ренней энергией U2, объемом V2 Рис. 1.2.3. Схема установки для (поршень расположен на отметке h2) и определения энергетических эффектов при том же самом давлении P. при постоянном давлении Изменение внутренней энергии системы и объема выразится через разности этих величин в начальном и конечном состояниях: ∆U = U2 – U1 ∆V = V2 - V1. и Из чего будет складываться изменение внутренней энергии в этом процессе? Конечно, из теплового эффекта Qp, который можно подсчитать, замеряя температуру воды в сосуде 4 до реакции и после нее, и зная ее теплоемкость с и массу m, а также из работы А, которую система совершила, расширяясь. Действительно, система увеличивает свой объем, расширяется, воздействуя на окружающую среду, поршень поднимается против внешней силы тяжести, на все это требуются затраты энергии виде работы по преодолению внешних сил. Численное значение работы А можно рассчитать, как произведение силы F на путь (h2 – h1): A = ∆h · F = ( h2 - h1 ) ·F (1.2.3) Причем, эта часть изменения внутренней энергии в форме работы А, заметим, равносильна отдаче (потере) энергии реагирующей системой непосредственно внешней среде, в противоположность тепловому эффекту, который остается в системе благодаря теплоизоляционным стенкам сосуда. 2 1 18 Таким образом, изменение внутренней энергии системы складывается из передачи энергии двумя способами: теплового эффекта реакции Qр и работы А, которую совершает система против сил внешней среды: ∆U = Qр + ( - A) (1.2.4) Знаки, заметим, в этом выражении перед Qр и A разные. Данное уравнение устанавливает тот факт, что изменение внутренней энергии системы равноценно выделившемуся количеству энергии за счет теплового эффекта Qр (который остается присущим самой системе, по крайней мере, в первый момент времени из-за её термоизоляции) и той её части, которая в форме работы А отдана непосредственно расширяющейся системой внешней среде. То есть изменение внутренней энергии ∆U системы в результате реакции при постоянном давлении сопровождалось уменьшением её на величину работы А: ∆U = Qр - A Поэтому работе и тепловому эффекту приписываются разные знаки. Соотношения между Qр и А в системах может быть различным. В случае, если бы системой не производилась работа (А = 0), то изменение внутренней энергии соответствовало бы выделившейся энергии в форме теплового эффекта ∆U = U2 – U1 = Qр А если бы в процессе отсутствовали бы тепловые изменения (Qр = 0), то работа производилась бы за счет убыли внутренней энергии системы ∆U = U2 – U1 = - A . В рассмотренном процессе при постоянном давлении в самом начале мы допустили, что внутренняя энергия системы уменьшилась (∆U < 0), реакция сопровождается увеличением объема системы (∆V > 0). Если же рассмотреть противоположный случай с увеличением внутренней энергии (∆U > 0), за счет подвода энергии извне (эндотермическая реакция), с уменьшением объема системы при постоянном давлении, то изменение внутренней энергии будет складываться из теплового эффекта реакции и работы, т.е. ∆U = Qр + A. В каком же соотношении находятся тепловые эффекты реакции Qр и Qv? Изменение внутренней энергии при постоянном объеме равно тепловому эффекту Qv как мы выяснили ранее: ∆U = Qv, а при постоянном давлении равно: ∆U = Qр + A. Сопоставляя два последних выражения (приравнивая правые части уравнений), получим: Qv = Qр + A (1.2.5). 19 Мы видим, что тепловые эффекты Qv и Qр разные, то есть тепловой эффект зависит от того, в каких условиях протекает процесс, от способа его организации. Отличаются они на величину работы А совершаемой системой (или над системой). Действительно, если экспериментально определить тепловой эффект при постоянном давлении Qр, то термохимическое уравнение этого процесса взаимодействия цинка с раствором серной кислоты запишется так: Znкр + H2SO4 p-p = ZnSOp-p + H2 газ + 163,2 кДж. Сравнивая его с тепловым эффектом при постоянном объеме Qv = + 165,7 кДж, мы видим, что они разные. И разница между ними составляет величину работы А совершенной системой при расширении A = Qv - Qp = 165,7 - 163,2 = 2,5 кДж. Данная работа была совершена за счет изменения (в нашем случае увеличения) объема системы А = Р·∆V, что было связано с выделением одного моля газообразного водорода. Величина ∆V = V2 – V1 представляет собой изменение объема системы, вследствие того, что число молей газообразных продуктов реакции изменяется на величину ∆n = n2 - n1, где n1 и n2 - число молей газообразных веществ исходных и конечных продуктов. В соответствии с уравнением Менделеева - Клапейрона Р ·V = n· RT имеем Р· ∆V = ∆n·RT. Поэтому выражение 1.2.5. можно записать так: Qv = Qр + ∆n·RT, т.е. тепловой эффект изобарного и изохорного процесса отличаются на величину работы расширения, совершаемой в результате изменения числа молей газообразных исходных веществ и продуктов реакции. Если в реакции не происходит изменение числа молей газообразных веществ (∆n=0), то Qv = Qр. Например, в реакциях: а) СОгаз + Н2Огаз = СО2 газ + Н2 газ, б) Н2 газ + Сl2 газ = 2 НСlгаз изменение числа молей газообразных продуктов равно нулю и Qv = Qр. Если при химической реакции происходит увеличение числа молей газообразных продуктов, как, например, в случае взаимодействия цинка с раствором серной кислоты, то Qv > Qр. Это значит, что часть внутренней энергии системы расходуется на работу расширения (при постоянном давлении). В противном случае, если при химической реакции происходит уменьшение числа молей газообразных продуктов, то Qv < Qр. 20 Например, в реакции Н2 газ + ½ О2 газ = Н2О жид происходит уменьшение числа молей газообразных продуктов (∆n= -1.5), поэтому Qv < Qр и тепловой эффект при постоянном давлении равен Qр = Qv + ∆n·RT = Qv + 1,5 RT Это означает, что работа внешних сил, сжимающих систему, превращается в тепловой эффект реакции. Вопросы и задачи для самостоятельной работы 1.2.5. Предположим, что в результате процесса при постоянном давлении изменение внутренней энергии системы оказалось равным нулю (∆U = 0). Что можно сказать о таком процессе? 1.2.6. Может ли изменение внутренней энергии полностью превращаться в работу? 1.2.7. В каком процессе (какова его особенность) окажется, что Qv = Qp ? 1.2.8. Рассматривая выражение изменения внутренней энергии системы при постоянном давлении ∆U = Qp + A, можно ли сказать, что это первый закон термодинамики? 1.2.9. Опишите физический смысл следующего выражения: Qр = ∆U + A. 1.2.10. Изменение внутренней энергии ∆U, как известно, является функцией состояния, то есть не зависит изменение энергии ∆U от пути перехода, а работа и теплота зависят. Как это понимать, если ∆U = Qp + A? 2.3. Энтальпия и ее изменение В рассмотренном процессе при постоянном давлении взаимодействие цинка с раствором серной кислоты изменение внутренней энергии системы описывается уравнением ∆U = Qp - A. В этом выражении работу А выразим через произведение силы на путь уравнение (2.2.1): ∆U = Qp - A = Qр - (h2 - h1 ) F (1.2.6) Силу F можно представить через произведение давления на площадь поршня S ∆U = Qр - (h2 - h1 ) P· S ∆U = Qр - (h2S - h1S ) P или (1.2.7). Произведение высоты цилиндра на площадь дна цилиндра (дна поршня) есть объем цилиндра, поэтому выражение Sh2 – Sh1 есть не что иное, как изменение объема системы в результате процесса: Sh2 – Sh1 = V2 - V1 = ∆V. (1.2.8) Учитывая все это, уравнение (2.3.2) примет вид: ∆U = Qр - (h2S - h1S ) P = Qр - P·∆V 21 (1.2.9) или U2 – U1 = Qр - P (V2 - V1). (1.2.10) Раскроем скобки в выражении (2.3.5) и перегруппируем члены уравнения, выразив в явном виде тепловой эффект реакции при постоянном давлении Qр Qр = (U2 + P V2) – (U1 + P V1). (1.2.11) Упростим последнее выражение, обозначив суммы в скобках (U + PV) через Н (1.2.12) (U1 + P V1) = Н1 (1.2.13) (U2 + P V2) = H2 Тогда получим: Qр = H2 - H1, или ∆Qр = ∆H (1.2.14). Величину «Н» в термодинамике называют энтальпией (или теплосодержанием, хотя правильнее было бы называть энергосодержанием), а ∆H изменение энтальпии. Чтобы уяснить себе смысл этих понятий, представим себе нашу реагирующую систему, которая находится не при комнатной температуре, при которой мы проводили эксперимент, а при нулевой температуре (0 К). Следовательно, чтобы перевести вещества в состояние с повышенной температурой (к комнатной), их нужно нагреть, т.е. передать им какую-то часть энергии от какого-то другого внешнего источника окружающей среды за счет разности температур. При нагревании с веществами происходят изменения: они плавятся, испаряются, меняют тип кристаллической решетки, изменяют объем, и, в конце концов, мы имеем с ними дело в том виде, в котором они находятся при комнатной температуре. Естественно, что вещества в этом процессе нагревания изменили и свое энергосодержание. Изменилась и внутренняя энергия системы при этих превращениях, и её объём. Весь этот запас энергии системы при комнатной температуре и включает в себя понятие энтальпии. Кстати, слово энтальпия греческого происхождения и в переводе обозначает «нагреваться». Нжид. ∆Нплавления= ∆Нж – ∆Нкрис. 200 жидкость 100 газ ∆Нпарообраз. кр. кДж моль ∆Нплавлен. ∆Ниспарения =∆Нгаз - ∆Нж Нкристалл Н, газ ∆Нсублимации = ∆Нгаз - ∆Нкрис. Нгаз 0 0 1000 2000 ТК Рис. 1.2.5. Зависимость энтальпии свинца от температуры. Рис. 1.2.4. Изменение энергосодержания вещества при нагревании 22 На рисунке 1.2.4 представлено схематическое изображение изменения энергосодержания вещества при постоянном давлении и нагревании, а на рис. 1.2.5 зависимость энтальпии свинца от температуры.1 Так как при нагревании веществ системы при постоянном давлении происходит увеличение их объема, то понятие энтальпии часто связывают с энергосодержанием расширенной системы. Принимая во внимание сказанное, можно считать, что моль атомов цинка и моль молекул серной кислоты при постоянном давлении имеют энтальпию HZn кр. и H H 2 SO 4 раствор соответственно, а моль молекул сульфата цинка и водорода соответственно H ZnSO 4 раствор и H H 2 газ . Если в процессе исчезли исходные вещества с соответствующим энергосодержанием, равным их сумме H H 2 SO 4 раствор + HZn кр , и образовались вещества продуктов реакции с суммарным энергосодержанием H ZnSO 4 раствор + H H 2 газ , то изменение энтальпии процесса составило некоторую величину ∆H реакции реакции, равную их разности: ∆H реакции = (H ZnSO 4 раствор + H H 2 газ ) – (H H 2 SO 4 раствор + HZn кр ). А так как Q = - ∆Hреакции , то другими словами: тепловой эффект реакции при постоянном давлении есть не что иное, как разность сумм энтальпий продуктов и исходных веществ2. Или это то же самое, что изменение энтальпии системы в процессе (реакции). На практике часто для краткости говорят просто – «энтальпия процесса (реакции)», опуская слово «изменение». Изменение энтальпии процесса связано с изменением внутренней энергии системы простой зависимостью. Если сопоставлять уравнения (1.2.4) и (1.2.14), то получим: ∆H = ∆U + P· ∆V Реакционная Реакционная система система Окружающая среда 100 кДж ∆Н= +100 кДж а 100 кДж ∆Н= - 100 кДж б Рис. 1.2.6. Изменение энергосодержания системы: а – энтальпия системы увеличивается на 100 кДж; б – энтальпия системы уменьшается на 100 кДж 1 (1.2.15) Следовательно, изменение энтальпии включает в себя изменение внутренней энергии системы с учетом той работы А = P·∆V, которую она совершила, расширяясь. Энтальпия H и ее изменение ∆Н, как и внутренняя энергия, являются функциями состояния. Все системы, с которыми мы имеем дело на практике, обмениваются График представлен в книге: Карапетьянц М. Х. Введение в теорию химических процессов. М. Высшая школа, 1981 на стр. 10. 2 Это соотношение известно, как следствие из закона Г.И. Гесса, который рассмотрим несколько позже. 23 энергией с окружающей средой. Так и рассматриваемая система с образовавшимися продуктами (сульфатом цинка и водородом) рано или поздно передаст выделившееся количество энергии в форме теплоты Qр окружающей среде. Соответственно, система уменьшит свое энергосодержание, а окружающая среда приобретет энергию в виде положительного теплового эффекта реакции Qр при постоянном давлении, что можно отразить в виде различных знаков для изменения энтальпии и теплового эффекта реакции: - ∆H = Qр . Знак «-» при ∆H подчеркивает тот факт, что энергозапас системы уменьшился (рис. 1.2.6). Наглядно энергетические процессы при постоянном давлении часН то изображают графически. Для этого по оси ординат отклаНачальное дывают энтальпию, а по абсциссе состояние – ход реакции (рис.1.2.7). Из диаН1 граммы приведенной на рис.1.2.7. Конечное -∆Н=Q состояние видно, что изменение энергосоН2 держания системы в ходе реакции соответствует положительному Ход реакции тепловому эффекту реакции Qр . Разность же энтальпий H2 – H1 – Рис. 1.2.7. Графическое изображение изменевеличина отрицательная, так как ния энергосодержания системы в ходе реакH2 < H1 и ∆H < 0. ции р Вопросы и задачи для самостоятельной работы 1.2.11. В результате химического процесса, оказалось, что ∆H > 0. Энергосодержание системы увеличилось? Уменьшилось? Каков знак теплового эффекта реакции? 1.2.12. Процесс диссоциации молекул водорода H2 →2 H протекает с изменением энтальпии ∆H = 436 кДж/моль. Энергосодержание системы при этом процессе уменьшилось? Увеличилось? 1.2.13. Процесс горения водорода в кислороде сопровождается изменением энтальпии ∆H = -241 кДж/моль. Что произошло с энергосодержанием системы? 1.2.14. При растворении 1 моля атомов цинка в разбавленной серной кислоте при 250 С выделяется 143,1 кДж теплоты. Одновременно выделяется 1 моль водорода, причем против внешнего давления совершается работа. Определите изменение внутренней энергии системы. 1.2.15. Отличается ли, а если да, то на сколько, тепловой эффект реакции при постоянном давлении, от такового этой же реакции в приведенном предыдущем примере при постоянном объеме? 24 2.4. Термохимические уравнения Термохимические уравнения записываются как обычные химические уравнения реакций, но с указанием величины, и знака теплового эффекта реакции. Поскольку технологический процессы и химические эксперименты проводятся а большинстве случаев при постоянном давлении, поэтoму в дальнейшем тепловые эффекты реакции будут выражаться через изменение энтальпии ∆H, которые приведены в большинстве термодинамических таблиц и справочниках. Пользуясь известными соотношениями - ∆U = Qv, -∆H = Qр, можно рассчитать и остальные величины. Например, термохимическое уравнение реакции восстановления оксида свинца оксидом углерода запишется так: PbOкр. + COгаз = Pbкр. + CO2 газ ; ∆ H = -64 кДж. Возможен несколько иной вариант записи этого же уравнения: PbOкр. + COгаз = Pbкр. + CO2 газ + 64 кДж. Изменение энтальпии в реакции составляет ∆ H = -64 кДж, а тепловой эффект реакции Qр равен +64 кДж. Следовательно, энергосодержание реагирующей системы уменьшилось (знак минус перед величиной изменения энтальпии) на 64 кДж, а в окружающую среду рассеялось 64 кДж энергии (положительный эффект реакции). Обратите внимание на то, что в термохимических уравнениях указываются агрегатные состояния веществ: кристаллическое (кр.), газообразное (газ), жидкое (ж), т.к. тепловой эффект реакции зависит от состояния реагирующих веществ и продуктов реакции (сравните, например, вода в жидком или парообразном состоянии будет обладать большим энергосодержанием). Приведем еще несколько примеров термохимических уравнений. Экзотермические реакции протекают с выделением энергии. Например, H2 газ + ½ O2 газ = H2 Oпар + 241,8 кДж. Тепловой эффект данной реакции Qр = 241,8 кДж, а изменение энтальпии ∆ H = -241,8 кДж. Эндотермические реакции протекают с поглощением энергии. Например, диссоциация воды H2Oж = H+ водный раствор + OH-водный раствор - 57,5 кДж. Реакция диссоциации воды на ионы протекает с поглощением тепла Qр = 57,5 кДж, а энтальпия этого процесса – величина положительная ∆ H = + 57,5 кДж. В термохимических уравнениях допускаются дробные коэффициенты, поскольку приводимые тепловые эффекты или изменения энтальпии относят на 1 моль исходного вещества или продукта реакции. Например, в термохимическом уравнении N2 газ +½ O2 газ = N2 Oгаз - 82,0 кДж Приведенный тепловой эффект Qр = -82,0 кДж относится к 1 молю азота или к 1 молю продукта реакции N2O. 25 2.5. Энтальпия образования вещества В реакции, описываемой термохимическим уравнением H2 газ + ½ O2 газ = H2 Oжид. + 285,85 кДж, из простых веществ водорода и кислорода образуется 1 моль сложного вещества воды в жидком состоянии, и при этом выделяется 285, 85 кДж энергии. Следовательно, эту величину теплового эффекта можно назвать теплотой (энтальпией) образования жидкой воды, если считать, что теплоты образования простых веществ при тех же самых условиях равны нулю ∆Hн2газ = ∆HО2 газ = 0 Количество теплоты, которое выделяется или поглощается при образовании 1 моля химического соединения из простых веществ, носит название теплоты образования химического соединения. Или, используя понятие энтальпии, можно сказать, что энтальпией образования вещества называется изменение энтальпии в реакции образования 1 моля химического соединения из простых веществ. Например, образование оксида алюминия протекает согласно термохимическому уравнению: 2 Alкр. + 1,5 O2 газ = Al2 O3 кр.; ∆ H = - 1675 кДж. Так как оксид алюминия образовался из простых веществ, то изменение энтальпии этого процесса связано с его образованием, и поэтому величину ∆H = - 1675 кДж можно назвать энтальпией образования оксида алюминия (1 моля). Энтальпии (теплоты) образования веществ являются индивидуальными характеристиками соединений, точно так же, как и плотность, электропроводность, температура плавления и другие. И точно также энтальпии образования зависят от условий, при которых происходит их определение. 2.5. Стандартные условия Тепловые эффекты реакций зависят от условий, при которых они протекают. Поэтому, для того, чтобы можно было сравнивать полученные значения тепловых эффектов реакций, энтальпии образования веществ, условились определять или сводить их к определенным, одинаковым, так называемым, стандартным условиям, коими принять считать состояние 1 моля чистого вещества при давлении 101325 Па (1 атм или 760 мм рт. ст.) и температуре 250 С1 (298 К). Для веществ, находящихся в растворе, принимают их стандартную концентрацию, равную один моль в литре (С =1моль/литр). Причем предполагается, что раствор ведет себя при этой концентрации, точно так же, как и при бесконечном разбавлении, т.е. является идеальным. Это же предположение относится к веществам, которые находятся газообразном состоянии: газ как Опыты проще проводить при атмосферном давлении 101325 Па и комнатной температуре 250 С, поэтому и приняты эти параметры за стандартные. Внимание - не путать стандартные условия и нормальные, которые отличаются температурой (298 К и 273 К). 26 1 бы является идеальным и при давлении 1 атмосфера, и при давлении также значительно более низком. Стало быть, изменение энтальпии реакционной системы при переходе из одного состояния в другое при стандартных условиях также будет носить стандартный характер. И энтальпия образования этого моля сложного вещества из простых веществ при стандартных условиях также будет называться стандартной энтальпией (теплотой) образования. Стандартные изменения энтальпии образования обозначают ∆H0f,2981. В дальнейшем будем их называть просто стандартными энтальпиями образования веществ или энтальпиями реакции (опуская слово изменение). Например, стандартная энтальпия образования воды в жидком состоянии обозначается так: ∆H0f н2о ж.,298 = - 285,85 кДж/моль. Эта запись означает, что в стандартных условиях образование одного моля воды в жидком состоянии из простых веществ сопровождается потерей реагирующей системой 285,85 кДж. Для термохимического уравнения реакции такая запись выглядит, например, так: 2 NH3 газ + 1,5 O2 газ = N2 газ + 3 H2Oпар ; ∆ H0298 = - 618 кДж. Для большинства известных веществ стандартные энтальпии образования определены опытным путем или рассчитаны и сведены в справочные таблицы термохимических свойств веществ (частично они представлены в таблице 1 Приложения и для наглядности в таблице 2.6.1). Таблица 2.6.1. Стандартные энтальпии образования ∆H f,298 некоторых веществ 0 Элемент Литий Натрий Калий Рубидий Цезий Бериллий Магний Кальций Стронций Барий 1 Энтальпия образования ∆ H0f,298 в кДж/моль для: Фторидов Хлоридов Бромидов Иодидов -616,7 -408,8 -351,0 -272,7 -570,3 -410,9 -359,8 -287,9 -568,1 -435,9 -392,0 -327,6 -552,3 -430,9 -393,2 -328,4 -531,4 -432,9 -410,0 -336,7 1033,4 -334,7 -330,5 -63,2 -1123,4 -641,8 -516,0 -359,8 -1219,6 -785,8 -682,8 -535,5 -1216,2 -815,9 -740,0 -556,5 -1207,1 -859,8 -753,1 -602,5 Индексы обозначают: «0» - стандартные условия, f –образования (от английского formation), 298 – стандартная температура в градусах Кельвина. В дальнейшем стандартные эн0 тальпии будем обозначать с одним индексом ∆H . 27 Стандартные значения энтальпий образования простых веществ (например, H2 газ, O2 газ, Cu кр) для тех агрегатных состояний, в которых эти вещества устойчивы, принимаются равными нулю 1, т.е. 0 0 ∆H f, H2 газ 298 = 0,0 кДж/моль; ∆H f,О2 газ 298 = 0,0 кДж/моль и т.д. Стандартная энтальпия образования соединения является мерой его термодинамической устойчивости, прочности, и носит периодический характер для одного класса, группы однотипных веществ (смотрите, например, таблицу 2.6.1.). Иногда в выборе стандартного состояния бывают исключения, например, когда мы говорим о стандартной теплоте образования парообразной воды, мы подразумеваем, что образуется водяной пар, давление которого1 атм, а температура 250С. Но при 250С водяной пар имеет значительно более низкое равновесное давление. Значит, теплота образования воды в парообразном состоянии ∆H0f н2о пар.,298 это чисто условное состояние. Вопросы и задачи для самостоятельной работы 1.2.16. Что называется тепловым эффектом реакции? Чему равен тепловой эффект реакции при постоянном объеме? При постоянном давлении? 1.2.17. Какова связь между энтальпией и внутренней энергии системы? 1.2.18.. Какие условия являются стандартными? 1.2.19. Что называют стандартной теплотой (энтальпией) образования вещества? 1.2.20.. Воспользовавшись таблицей Приложения 1, найдите значение энтальпии образования следующих веществ: Fe2O3 кр., CH4 газ , O2 газ , CO2 газ , Feкр. . Чему равна теплота образования соответствующих веществ? 1.2.21. Перечислите, какие особенности у термохимических уравнений? 1.2.22. Запас внутренней энергии больше у 1 моля воды в жидком или кристаллическом виде? 1.2.23. Какие из следующих реакций являются эндотермическими: 1. 0,5 N2 газ + O2 газ + 33,5 кДж = NO2 газ , 2. 0,5 N 2 газ +1,5 H2 газ = NH3 газ + 46 кДж, 3. 0,5 N2 газ + 0,5 O2 газ = NOгаз ; ∆H0 = 90 кДж, 4. 0,5 O2 газ + H2 газ = H2 O пар ; ∆ H0 = - 242 кДж? 1.2.24. Сколько энергии надо затратить для разложения 9 г воды на водород и кислород в стандартных условиях? 1.2.25. Напишите термохимическое уравнение реакции образования оксида кальция, если ∆H0СаО кр. = - 635,б1 кДж/моль. 1.2.26. Определите изменение внутренней энергии при испарении 250 г воды при 25 С, допуская, что пары воды подчиняются законам идеальных газов. Объемом жидкости по сравнению с объемом газа можно пренебречь. Удельная теплота парообразования воды ∆Hпарообразования = 2451 Дж/г. 1.2.27. При взаимодействии водорода и кислорода с образованием воды в жидком состоянии при 250С выделяется 285,84 кДж на моль водорода. Вычислите изменение внутренней энергии системы. 1 Если вещество при стандартных условиях может существовать в нескольких модифика0 циях, то к нулю приравнивается ∆H f,298 наиболее устойчивой модификации. Например, 0 кислород, при стандартных условиях может быть и О2 и О3 , но ∆H f о2газ, 298 = 0, как более устойчивой модификации. 28 1.2.28. В чем различие между понятиями внутренняя энергия системы U и энтальпия системы H? При каких условиях тепловой эффект реакции равен изменению внутренней энергии? При каких условиях тепловой эффект реакции совпадает с изменением энтальпии? 1.2.29. Почему при вычислении теплового эффекта реакции необходимо указывать состояние веществ? 2.7. Влияние температуры на тепловой эффект реакции. Изменится ли тепловой эффект реакции, если провести его не при стандартной температуре? Известно, что энтальпия H = U + PV является мерой энергии, накопленной веществом, системой, и она зависит от температуры, и от давления. При постоянном давлении тепловой эффект реакции есть изменение энтальпии системы: 0 0 0 ∆H реакции = Σ∆H f,298 продуктов - Σ∆H f,298 исходных веществ А что произойдет с энтальпиями тех веществ (и исходных веществ и продуктов реакции), если процесс проводить не при стандартных условиях? Например, не при Т1 = 298 К, а, скажем при более высокой температуре T2 ? Это означает, что исходные вещества, и, соответственно, продукты реакции нужно нагреть до этой температуры Т2, т.е. сообщить им определенный запас энергии, который будет зависеть и от количества вещества, участвующего в реакции, и от его теплоемкости. В Н общем случае теплоемкости продукΣ Н исходных тов реакции их исходных веществ невеществ одинаковы. Следовательно, и количе∆Н Т2 ство затраченной энергии на нагрев исходных веществ и продуктов будут ∆Н разными, а соответственно и изменение энтальпии при температуре Т2 реакции и при стандартной Т1=298 К Σ Н продуктов тоже будут различными, т.е. 0 0 ∆Н 298 ≠ ∆Н Т2 . Т 298 Т= 298К Т2 На рис. 1.2.8 представлена эта зависимость графически. Как видно в приведенном на рис.1.2.8, суммарная энтальпия исходных веществ 0 Σ∆H f,298 исходных веществ с температурой растет значительно быстрее, чем сумма энтальпий продуктов реакции Σ∆H0f,298 продуктов, и, как результат: измене0 ∆H Т2 ние энтальпии при стандартных условиях ∆H0298 не равно таковой при температуре Т2 реакции и, согласно рисунку, заметно меньше, т.е. ∆Н0298 < ∆НТ2. разница между ними Рис. 1.2.8. Изменение энтальпии исходных веществ и продуктов реакции при нагревании. ∆ НТ2 - ∆Н0298 будет определяться разностью изобарных теплоемкостей продуктов реакции и исходных веществ и зависимостью последних от температуры 29 ∆Н Т2 - ∆Н или 0 = ∆Ср ∆Т 298 ∆НТ2 = ∆Н0298 + ∆Ср (Т2-Т1), (1.2.16) (1.2.17) где ∆Н0298 – изменение энтальпии реакции при стандартной температуре Т1=298 К, ∆НТ2- изменение энтальпии реакции при температуре Т2, ∆Ср- разность молярных изобарных теплоемкостей всех продуктов и исходных веществ реакции, ∆Т – разность температур. Если учесть зависимость теплоемкости веществ от температуры, то выражение 2.7.2 можно представить в интегральной форме: T 0 (1.2.18) ∆H T = ∆H 298 + ∫ C p dT 298 2 Эта закономерность известна как уравнение Кирхгофа1 (1858 г), которую можно сформулировать так: изменение энтальпии в реакции при температуре Т2 равно изменение ее при температуре Т1 (в нашем случае при стандартных условиях, т.е. ∆Н0298) плюс разность мольных теплоемкостей продуктов реакции и исходных веществ, умноженная на изменение температуры (выражение 1.2.16 и 1.2.17). Теплоемкости в уравнениях 1.2.16 и 1.2.17 принимаются как бы не изменяющиеся с температурой. Для широкого интервала температур и с учетом зависимости теплоемкости от температуры следует воспользоваться уравнением 1.2.18. Расчеты по уравнению Кирхгофа и практическое определение энтальпии при различных температурах, показывают небольшую чувствительность ∆Н к изменению температуры. Например, для реакции Сграфит+СО2 газ= 2 СОгаз изменение энтальпии при температуре 500 К составляет величину ∆Н0500= 173,47 кДж, а при температуре 1500 К изменение энтальпии равно ∆Н01500 = 165,11 кДж, т.е. изменение температуры на 10000 изменяет тепловой эффект всего на 8,36 кДж. Поэтому при выполнении термохимических расчетов, допуская незначительную ошибку, можно пользоваться стандартными значениями энтальпий образования для условий протекания процессов, отличных от стандартных. 3. ТЕРМОХИМИЧЕСКИЕ ЗАКОНЫ 3.1. Закон Лавуазье-Лапласа Термохимических законов всего два, и оба являются частными случаями общего закона сохранения энергии. Один из них установлен в период с 1774 по 1764 года французскими учеными Лавуазье2 и Лапласом1, в честь которых и назван. Закон читается так: 1 Кирхгоф Густав Р. (1824-1887). Немецкий физик, профессор в Бреслау,Гейдельберге и Берлине; разработал вместе с Бунзеном метод спектрального анализа. 2 Антуан Лоран Лавуазье (1743- 1794) – великий французский химик, президент Парижской академии, один из основоположников современной химии. Выяснил роль кислорода 30 теплоты образования и разложения вещества равны по абсолютной величине, но противоположны по знаку. Или в несколько более общей формулировке: тепловой эффект прямой реакции равен по абсолютной величине тепловому эффекту обратной реакции, но с противоположным знаком. Иначе говоря, осуществив какой- либо процесс, а затем ему противоположный (обратный), мы возвращаем систему в исходное состояние с той же внутренней энергией, какую она имела. Например, образование 1 моля воды сопровождается выделением 285,84 кДж Н2газ + 0,5 О2газ = Н2Ож + 285,84 кДж. В результате этой реакции энергосодержание системы уменьшилось на 0 ∆Н 298= - 285,84 кДж (рис.1.3.1). Разложение же 1 моля жидкой воды на Н2 газ + 0,5 О2 газ исходные составляющие: водород и кислород потребует подвода извне того же количества энергии т.е. ∆Н0298= + 285,84 кДж в соответствии с уравнением: Н2Ож = Н2газ+0,5 О2газ. – 285,85 кДж - 285,85 +285,85 кДж Таким образом, энтальпия образования кДж вещества равна по абсолютной величине энтальпии разложения, но имеет противоположный знак: . ∆Нf Н2О жобраз.= - ∆Н Н2Ожразлож . Н2О жидкая Следствием из этого закона является положение: чем больше теплота образования Рис.1.3.1. Иллюстрация закона Лавуазье-Лапласа вещества, тем оно более устойчиво, прочнее. Например, если известно, что теплота образования оксида скандия Sc2O3крис. ∆Н0 = 1907,4 кДж/моль, а оксида ртути HgOкрис. ∆Н0 = - 90,4 кДж/ моль, то ясно, что оксид скандия очень прочное соединение, и для разложения его на простые вещества потребуются очень высокие температуры. В то же время оксид ртути HgOкрис. разлагается при температуре около 1000 С на металлическую ртуть и кислород. в процессах горения, чем опроверг теорию флогистона. Один из основателей термохимии. Автор первого классического курса «Начальный учебник химии». При монархии был одним из откупщиков (сборщиков налогов), имел огромные доходы, значительную часть которых тратил на научные исследования. В числе других откупщиков гильотирован по суду ревтрибунала. В 1796 г. посмертно реабилитирован. 1 Пьер Симон Лаплас (1749-1829)- великий французский ученый-астроном, математик, физик. Автор классических трудов по теории вероятности, небесной механики, по дифференциальным уравнениям, теория капиллярности, теплоте, акустике, геодезии и др. 31 Знание стандартных энтальпий образования соединений и применение закона Лавуазье-Лапласа позволяет в какой-то степени оценить реакционную способность веществ. Непрочные соединения обычно более реакционноспособные. А в отношении простых веществ можно сказать, что чем более экзотермичнее реакция (больше выделяется энергии) с их участием, тем они химически более активны, поскольку вероятность протекания выше той реакции, в которой больше выделяется энергии. Вопросы и задачи для самостоятельной работы. 1.3.1. Какие из галидов наиболее устойчивы : KF, KCl, KBr или KJ? Ответ дайте, анализируя таблицу 2.6.1. 1.3.2. Каковы теплоты разложения хлоридов бериллия, магния, кальция, стронция, бария? Какое соединение из перечисленных более прочное? 1.3.3. Хлор не соединяется непосредственно с кислородом. Оксиды хлора Cl2О? ClO2 и Cl2O7 получают косвенным путем. Можно ли определить на опыте теплоту образования этих оксидов или рассчитать её? 3.2. Закон Гесса. Другой закон термохимии открыт русским ученым Г.И. Гессом1 в 1836 г. и читается он так: тепловой эффект химической реакции не зависит от способа ее проведения, а зависит лишь от начального и конечного состояния реагирующих веществ. Или в другом распространенном варианте: если химический процесс протекает в несколько стадий, то общий тепловой эффект процесса равен алгебраической сумме тепловых эффектов отдельных, промежуточных стадий. Представим себе, что имеется реакционная система, в которой вещества А и В превращаются в продукты D и Е, согласно термохимическому уравнению: А + В = D + Е; ∆Н0реакции. Изменение энтальпии этой реакции ∆Н0реакции. Продукты реакции D и Е можно получить прямо и непосредственно из исходных веществ А и В, как это схематически представлено на рис. 1.3.2а по пути 1-2, минуя какие-либо промежуточные стадии. Тепловой эффект при этом способе превращения будет равен Q0реакции= - ∆Н0реакции. (рис. 1.3.2б). Получить те же самые продукты D и Е можно, осуществив процесс через образование каких-либо промежуточных веществ по пути, например, 1-3-4-52 или 1-6-7-2 (рис.1.3.2а). 1 Герман Иванович Гесс (1802-1850) выдающийся русский химик, профессор, член Петербургской академии наук. Основоположник термохимических исследований в России. Открыл и исследовал состав нескольких минералов, изучал катализ, доменный процесс, нефть. Написал учебник «Основания чистой химии». 32 Причем каждая стадия образования промежуточных веществ будет характеризоваться своим тепловым эффектом или изменением энтальпии: ∆Н1, ∆Н2, ∆Н3 , ∆Н4 и ∆Н5, ∆Н6, ∆Н7, соответственно, для каждого пути процесса (рис.1.3.2б). Если же рассмотреть конечный итог энергетических изменений процесса через промежуточные стадии, то окажется, что он равен алгебраической сумме изменения энтальпий промежуточных стадий: ∆Н0реакции= ∆Н01 + ∆Н02+ ∆ Н03+ ∆ Н04 ( для пути 1-3-4-5-2) или ∆Н0реакции= ∆ Н05 + ∆Н06+ ∆Н07 . ∆Н01 А+В 3 ∆Н02 1 А+В А +В ∆Н01 4 ∆Н05 6 А+В (для пути 1-6-7-2). ∆Н03 ∆Н05 ∆Н02 ∆Н0реак. ∆Н06 ∆Н06 ∆Н0реак. ∆Н03 5 7 0 ∆Н ∆Н04 7 0 ∆Н 2 ∆Н07 4 D+E а D +E Путь 1 -2 D + E путь 1,3,4,5,2 б D + E путь 1,6,7,2 Рис.1.3.2. Схематическая иллюстрация закона Гесса: а- возможные пути проведения процесса, б - схемы изменения энтальпий промежуточных стадий в зависимости от пути реакции. То есть тепловой эффект реакции не зависит от способа проведения процесса, а зависит лишь только от начального состояния исходных веществ и конечного состояния продуктов реакции (рис.1.3.2б). На конкретной реакции, например, окисления железа кислородом, проверим выполнимость закона Гесса. Термохимическое уравнение этого процесса: 2Feкр+ 1,5 О2 газ+Fe2O3 кр+821,3 кДж. (1.3.1). Проведем этот процесс по стадиям. Вначале окислим железо до оксида (II) согласно уравнению: I стадия: 2 Feкр+ O2 газ = 2 FeOкр+ 2· 263,7 кДж 33 (1.3.2) с тепловым эффектом 2 · 263,7 кДж, а затем доокислим оксид железа (II) по второй стадии до оксида (III) согласно уравнения: II стадия: 2 FeOкр + 0,5 О2газ = Fe2O3кр+ 293,9 кДж, (1.3.3) в которой выделится 293,9 кДж. Складывая уравнения первой и второй стадии реакций, получим: 2 Feкр+ 1,5 О2газ = Fe2O3кр+ 821,3 кДж. Суммарный тепловой эффект этих стадий также равен 821,3 кДж, как если бы проводили процесс без промежуточных стадий согласно уравнению 1.3.1. То есть закон Гесса выполняется. Графически процесс представлен на рис. 1.3.3. Начальное состояние Обратим внимание на то, что термохимические уравнение можно складывать и вычитать, как обычные алгебраические уравнения. 2 Feкрис. +1,5 - 527,4 кДж - 821.3 кДж 2 FeOкрис. 3.3. Термохимические расчеты. Термохимические расчеты, связанные с определением тепловых эффектов реакций, теплот Конечное состояние Fe2O3 крис. образования соединений, дают Рис.1.3.3. Общее изменение энтальпии возможность в какой-то степени реакции окисления железа складываетпредсказать и вероятное направся из энтальпий отдельных стадий. ление процесса и приближенно охарактеризовать прочность соединения. Все расчеты основываются на двух законах термохимии и ее основных понятиях, и определениях. Рассмотрим несколько конкретных примеров термохимических расчетов. - 293,9 кДж Пример 1. Рассчитать, какое количество теплоты выделиться при сгорании 1 моля метана. Решение. Запишем термохимическое уравнение горения метана СН4газ+ 2 О2га з= СО2газ+ 2Н2Ож + Qр. Из справочной литературы по термодинамическим свойствам веществ (например, Рябин В.А., Остроумов М.А., Свит Т.Ф. «Термодинамические свойства веществ. Справочник». Л., Химия, 1977) выпишем стандартные значения энтальпий образования (теплот образования) исходных веществ и продуктов реакций: ∆Н0СН4газ = -74,86 кДж/моль, ∆Н0О2 газ = 0,00 кДж/моль, или Q0СН4газ = 74,86 кДж/моль; или Q0О2га = 0,00 кДж/моль; 34 ∆Н0СО2газ = 0,00 кДж/моль, или Q0О2га ∆Н0Н2Ож = - 286,0 кДж/моль, или Q0Н2Ож = 286,0 кДж/моль. = 393,8 кДж/моль; Поскольку в процессе горения метана образуются диоксид углерода (1 моль) и 2 моля воды в жидком состоянии, составим термохимические уравнения их образования из простых веществ: Сграф.+О2 газ=СО2 газ + 393,8 кДж 2 Н2 газ+ О2 газ= 2 Н2Ож + 2 · 286,0 кДж, а так как при горении метана разлагается метан СН4 газ, превращаясь в воду в жидком состоянии и диоксид углерода, запишем термохимическое уравнение разложения метана на простые вещества: СН4 газ= Сграф. + 2 Н2газ – 74,86 кДж. Сложив эти три последних уравнения, получим термохимическое уравнение реакции горения метана: СН4 газ + 2 О2 газ= СО2 газ + 2 Н2Ож + 890,94 кДж. Таким образом, тепловой эффект этой реакции при стандартных условиях равен Q0р=890,94 кДж/моль или изменение энтальпии реакции составляет ∆Н0реакции= - 890,94 кДж/моль. Если внимательно посмотреть на то, каким образом получилось это значение, то окажется, что из суммы теплот образования продуктов реакции вычиталась сумма теплот образования исходных веществ. Это следствие из закона Гесса, которое читается так: тепловой эффект реакции равен разности между суммой теплот образования продуктов реакции и суммой теплот образования исходных веществ, что можно записать так Qреакции= Σ Qобразов.продукты - ΣQобразов.исход. в-ва Или применительно к понятию изменения энтальпии реакции ∆Н0ракции= Σ∆ Н0продукты- Σ∆Н0исход в.ва. Применительно к нашей задаче тепловой эффект реакции можно рассчитать, не составляя уравнений образования и разложения веществ: Q0реакции= (Q0СО2газ+ 2 Q0Н2Ож) - (Q0СН4газ+2 Q0О2газ) Или, подставляя численные данные, получим : Q0реакции= (393,8 + 2· 286,0) - (74,86 +2· 0)= 890,94 кДж. Аналогичный расчет можно провести, используя не теплоты образования, а энтальпии: ∆Н0реакции= (∆Н0СО2 газ + 2· ∆Н0Н2Ож) – ( ∆ Н0СН4 газ + 2 · ∆Н0О2газ) ∆Н0реакции= (-393,8 +2·(-286,0)) - (74,86 +2·0) = - 890,94 кДж Пример 2. Рассчитать тепловой эффект реакции превращения графита в алмаз при стандартных условиях, зная теплоты сгорания алмаза и графита, которые соответственно равны 393 кДж и 395 кДж. Решение. Напишем термохимические уравнение реакции горения графита и алмаза: 1. Сграф. +О2 газ = СО2 газ; ∆Н0СО 2 газ= - 393 кДж/моль, 35 2. Салмаз +О2 газ = СО2 газ; ∆Н 0СО2 газ = - 395 кДж/моль. Вычтем из первого уравнения второе и после упрощения получим: Сграф.= Салмаз; ∆ Н0 = 2,0 кДж/моль. Таким способом можно легко вычислить изменения энтальпии процессов, которые не поддаются прямому экспериментальному определению. Пример 3. Вычислить теплоту превращения жидкой воды в пар. Решение. Напишем термохимические уравнения реакций образования воды из простых веществ для жидкого и парообразного состояния воды: Н2 газ + ½ ·О2 газ= Н2О пар; ∆ Н0Н2Опар= - 241,84 кДж, Н2 газ + ½· О2 газ= Н2Ож ; ∆Н0Н2Ож= - 285,84 кДж, используя справочные данные энтальпий образования воды. Вычтем из первого уравнения второе и получим Н2Ож = Н2О пар; ∆Н0 = 44,0 кДж. Таким образом, теплота испарения одного моля воды составляет 44,0 кДж. Пример 4. Теплота сгорания этана 1560 кДж/моль. Вычислить теплоту образования этана, если известно, что теплоты образования воды и углекислого газа соответственно равны 285,8 кДж/моль и 393,5 кДж/моль. Решение. Запишем термохимическое уравнение реакции горения этана С2Н6 газ+ 3,5 ·О2 газ = 2 СО2 газ + 3· Н2Ож + 1560 кДж. Согласно закону Гесса тепловой эффект реакции равен Q0реак.= ΣQ0продук. - ΣQ0исход. в-ва. В соответствии с термохимическим уравнением реакции горения 1560 кДж = (2· Q0СО2 газ + 3· Q0H2Oж)- (Q0С2Н6 газ + 3,5·Q0О2 газ). Подставляя значения теплот образования, получим: 1560 = (2·393,5 + 3·285,8) - (Q0С2Н6 газ +3,5· 0) или 1560 = 1644,4 - Q0C2Н6 газ Q0С2Н6 газ= 84,4 кДж/моль. и Пример 5. Вычислить энергию связи в молекуле НС1, если известны энтальпия образования НС1 ∆Н0нсlгаз = - 92,3 кДж/моль и энергии разложения молекул водорода Н2 на атомы ∆НН газ = - 218,0 кДж/моль и молекул хлора Cl2 на атомы ∆НCl2 газ= 121,кДж/моль. Решение. Энергия связи между атомами водорода и хлора в молекуле НСl это та энергия, которую нужно затратить, чтобы молекула распалась на свободные атом водорода и хлора, т.е. нужно найти изменение энтальпии процесса: 2 НС1газ= Нгаз + С1газ; ∆Н0 разл. НС1 газ. Энтальпия этого процесса неизвестна. Но известны другие энергетические эффекты для следующих процессов: 1. Образование НС1 из молекул: ½ Н2 газ + ½ С12 газ = НС1газ; ∆ Н0НС1 газ= - 92,3 кДж, 2. разложение Н2 на атомы: ½ Н2 газ=Нгаз; ∆Н0разл. Н2 газ = 218,0 кДж 3. Разложение С1газ на атомы: ½ С12 газ= С1газ; ∆Н0разл.С12газ = 121,1 кДж. Исходя из уравнения: 36 НС1газ= Нгаз + С1газ ; ∆Н0 разл. НС1 газ , и в соответствии с законом Гесса изменение энтальпии процесса разложения НС1 на свободные атомы можно представить в виде: ∆Н0разл. НС1газ=( ∆Н0образ.Н газ + ∆Н0образ. С1газ)- ∆Н0образ.НС1газ Подставим численные значения известных энтальпий образования: ∆Н0разлНС1газ= (18,0+121,1) - (-92,3) = 431,4 кДж. Полученное значение представляет собой энергию связи между атомами водорода и хлора в одном моле молекул НС1, или тепловой эффект реакции Нгаз+С1газ= НС1газ; ∆ Н0образ.НС1 газ= - 431,4 кдж/моль. Приведенная величина относится к 1 молю НС1, т.е. к образованию 6.02 23 10 молекул НС1. А для одной молекулы она будет в это же количество раз меньше: 431,4·103 Дж : 6.02·1023 = 7,66·10-20 Дж/молекула. Задачи для самостоятельной работы 1.3.4. Определите теплоту сгорания жидкого бензола при стандартных условиях. 1.3.5. Реакция горения этилового спирта выражается термохимическим уравнени∆Н0 =? ем С2Н 5ОН ж + 3·02газ = 2·СО2газ+ 3· Н2Ож; Вычислите тепловой эффект реакции. Какое количество тепла выделится при сгорании 92 г этилового спирта? 1.3.6. Пользуясь данными таблицы Приложения 1, вычислите тепловой эффект реакции: 2Mgкр+ СО2газ = 2MgОкр + Сграф. 1.3.7. Исходя из энтальпии образования газообразного СО2 и термохимического уравнения Сграф.+2·N2Огаз = СО2газ + 2· N2 газ +557,5 кДж, вычислите теплоту образования N2Огаз , если ∆Н0СО2газ= -393,5 кДж/моль. 1.3.8. Вычислите изменение энтальпии реакции SiО2 кр.+ 2·Mg кр.= Siкр.+ MgОкр. 1.3.9. Исходя из уравнения реакции MnО2кр + 2·Сграфит = Mnкр + 2·СО2 газ –293 кДж, вычислите энтальпию образования диоксида марганца. Глава П. НАПРАВЛЕННОСТЬ ХИМИЧЕСКИХ ПРОЦЕССОВ Введение Знание энергетики химических процессов, в частности, энтальпий образования, тепловых эффектов реакций между веществами позволяет рассчитывать энергетический баланс в реагирующей системе, устанавливать устойчивость соединений, подбирать условия (материалы, температуру, давление и т.д.) для проведения процессов. Но важно не только это. Часто необходимо заранее знать, возможен ли такой процесс (реакция) взаимодействия данных веществ вообще при данных условиях с образованием именно таких, а не других веществ. А если процесс не возможен, то при каких условиях он будет протекать? То есть нужны ко37 личественные критерии, на основании которых мы могли бы оценить принципиальную осуществимость процесса, его полноту (степень превращения). С помощью этих критериев (критерия) определить, как влияют на течение процесса температура, давление, концентрация, состав; можно ли заставить исследуемую реакцию протекать в обратном направлении и т.д. Другими словами: необходима возможность прогноза химических превращений. Прежде чем перейдем к рассмотрению основных факторов, обуславливающих направления протекания химических реакций, выясним некоторые понятия связанные с особенностями реагирующих систем. А именно: самопроизвольные, обратимые и необратимые процессы (реакции) и равновесие. 1. САМОПРОИЗВОЛЬНЫЕ И ОБРАТИМЫЕ ПРОЦЕССЫ При обычных, стандартных условиях, как показывает опыт, имеется множество процессов, которые протекают самопроизвольно. Например, достаточно смешать водород и кислород, как начнется их взаимодействие с образованием воды и выделением энергии. Если бросить металлический цинк в раствор серной кислоты, сразу же происходит образование продуктов реакции: сульфата цинка и водорода. И многие другие химические процессы: коррозия металлов и сплавов, горение топлива в двигателях внутреннего сгорания, хемосорбция протекают самопроизвольно, т.е. без внешнего воздействия. Это все примеры одностороннего течения процессов. К этому же типу взаимодействия относятся и физические процессы: передача энергии от более нагретого тела к холодному, расширение газа в вакууме, взаимная диффузия газов или жидкостей, сток воды с возвышенности в виде ручья или реки, падение метеоритов и т.д. Для всех этих процессов характерно, во первых, то, что их можно использовать для получения работы. Падающая с высоты вода может вращать турбины; химическое взаимодействие цинка с серной кислотой можно организовать в гальваническом элементе и получить электрическую энергию и т.д. Но по мере окончания самопроизвольного процесса система теряет способность производить работу. Второй особенностью самопроизвольных процессов является то, что при их протекании происходит выравнивание в системе каких-то параметров по всему объему: температуры, массы, энергии, концентрации, давления, количества движения и других, т.е. они всегда направлены в сторону приближения к равновесному состоянию и прекращаются по мере его достижения. Еще одним существенным общим признаком самопроизвольных процессов является превращение различных видов энергии, и, в конечном итоге, равномерное ее распределении в форме теплоты между всеми составными частями системы. Самопроизвольный процесс обратно не может протекать при данных условиях так же самопроизвольно, т.е. нельзя из продуктов, приведя их в контакт, получить исходные вещества. Например, из продуктов сгорания топли38 ва CO2, СО и Н2О снова получить бензин. Эти процессы получили название несамопроизвольных или необратимых. В принципе заметим, что косвенными путями можно вернуть систему в первоначальное состояние, однако при этом неизбежно придется провести какие-то энергетические изменения в окружающей среде. Так, из СО2, СО и Н2О можно синтезировать бензин, из воды снова получить водород и кислород, но самопроизвольно эти процессы протекать не будут и потребуют и затрат энергии, и, чаще всего, придется проводить их обходными путями. Таковы характерные черты самопроизвольных и необратимых процессов. Тем не менее, существуют реакции, в которых образовавшиеся продукты взаимодействуют друг с другом, образуя исходные вещества. Например, при температуре 8100 С взаимодействие диоксида углерода с водородом протекает обратимо: СО2газ + Н2газ ↔ СОгаз + Н2Огаз. В этом состоянии концентрации исходных веществ и продуктов не меняются, тем не менее, обе реакции (прямая и обратная) продолжают протекать. Просто у них одинаковы скорости образования монооксида углерода с водой и диоксида углерода с водородом (количество прореагировавших исходных веществ и образовавшихся исходных же веществ по обратной реакции за одно и то же время одинаково). Это состояние подобно тому, которое осуществляется в бассейне с водой, в который по одной трубе вода поступает, а по другой выливается. Если поступление воды в бассейн равно ее расходу, то количество воды (уровень) в нем перестает изменяться. Установившееся равновесие может сохраняться как угодно долго, но достаточно извне изменить какой-либо параметр состояния (температуру, состав, например), как автоматически нарушится равновесие и изменятся концентрации всех веществ. Со временем система перейдет в новое равновесное состояние, которому будут соответствовать новые равновесные концентрации всех веществ. Явление перехода системы из одного равновесного состояния в другое называют смещением или сдвигом равновесия. В нашем (вышеприведенном) примере, если увеличить концентрацию монооксида углерода или воды (или понизить температуру), то увеличатся концентрации исходных веществ. Чтобы тот или иной процесс был обратим, необходимо чтобы он был равновесным, т.е. в системе одновременно протекали бы оба процесса: прямой и обратный при данных условиях. Характерной особенностью обратимых реакций является то, что они не доходят до конца: до превращения последней частицы исходного вещества в продукты реакции, если продукты реакции не удаляются из системы. Тем не менее, существуют ложные или кажущиеся равновесия, которые не являются таковыми в термодинамическом смысле. Например, если при обычных условиях (250С) смешать газообразные водород или кислород и 39 пары воды, то они будут существовать достаточно долго без особо видимых изменений. Нам же может казаться, что идут процессы образования воды (из водорода кислорода) и разложения её на составляющие: водород и кислород. В действительности просто скорость взаимодействия водорода с кислородом настолько ничтожно мала при этой температуре, что мы даже за длительный промежуток времени не замечаем уменьшения концентрации кислорода и водорода. Но достаточно нагреть систему или внести в нее катализатор, ускоряющий взаимодействие, как начнется энергичное взаимодействие водорода с кислородом. И выясняется, что никакого равновесия и не было. А, просто-напросто, были трудности взаимодействия веществ кинетического характера, которые мы рассмотрим в следующей главе. В термодинамическом отношении же система явно была неустойчивой, и мы имели дело с самопроизвольным процессом, но с очень малой скоростью. Самопроизвольные процессы всегда направлены в сторону приближения к равновесному состоянию и прекращаются, когда это состояние достигнуто. При химическом взаимодействии равновесие определяется равенством скоростей прямой и обратной реакции; при теплопередаче – равенством температур; при расширении газа – равенством давлений; при диффузии – равенство концентраций. Если в какой-то системе совершается равновесный процесс, то в окружающей среде никаких изменений не происходит. Это тоже один из характерных признаков равновесного состояния системы: говорят, система находится в равновесии с окружающей средой. Вопросы и задачи для самостоятельной работы 2.1.1. В открытой системе происходит взаимодействие цинка с соляной кислотой. Является ли данная реакция обратимой. 2.1.2. При электролизе водного раствора сульфата натрия графитовыми электродами получили газообразные водород и кислород. Практически это означает, что разложили воду на составляющие: Н2Ож = Н2газ + ½ О2газ. Этот процесс самопроизвольный? Обратимый ? 2.1.3. Является ли самопроизвольным процессом: а) окисление бензина кислородом воздуха в двигателе внутреннего сгорания, б) растворение сахара в воде? Обратимые ли эти процессы? 2.1.4. В колбу налили воду и закрыли пробкой. Процесс испарения воды в колбе будет самопроизвольным? Обратимым? Система будет изолированной, открытой, замкнутой? 2.1.5. Самопроизвольный процесс это тот, который: а) обладает способностью к осуществлению без воздействия со стороны, б) который протекает быстро, в) в котором всегда выделяется теплота? 2. ПРИНЦИП БЕРТЛО-ТОМСЕНА Что же является определяющим при установлении возможности того или иного процесса при данных условиях? В шестидесятых годах прошлого столетия выдающиеся ученые своего времени – французский химик Пьер Бертло (1827-1907) и датский химик Ханс Томсен (1826 - 1909) предложили 40 принцип, определяющий направление течения реакций (впоследствии получивший название по имени их авторов). Согласно принципу Бертло-Томсена самопроизвольно протекают те процессы (с образованием тех продуктов), которые сопровождаются выделением наибольшего количества энергии, т.е. все самопроизвольные процессы являются экзотермическими. Кстати, термины «экзотермические», «эндотермические» реакции были введены П. Бертло. Примером, иллюстрирующим этот принцип, может служить реакция горения водорода в кислороде. Она протекает самопроизвольно, и при этом выделяется 285,84 кДж/моль: Н2газ+ ½ О2газ = Н2Ож + 285,84 кДж. Свой принцип П. Бертло и Х. Томсен сформулировали по аналогии с механической формой движеu1 ния материи. Так любое тело, имеющее большую потенциальную энергию (например, ∆u шар на наклонной плоскости) стремится ее самопроизвольно уменьшить (рис. 2.2.1.). u2 Выделение энергии в химических, механических процессах отражает общий физический принцип стремления Рис.2.2.1. Изменение потенциальной энерсистем к минимуму потенцигии шара. альной энергии, поэтому П. Бертло и Х. Томсен и предполагали, что при химических реакциях будут протекать только те процессы, которые сопровождаются выделением энергии. Во многих случаях опыт подтверждает данный принцип. Но известно много процессов, реакций, протекающих самопроизвольно с поглощением энергии. Самопроизвольные процессы растворения некоторых веществ происходят с поглощением энергии. Например, растворение хлорида калия и хлорида натрия протекает с поглощением тепла (∆Н>О): КС1кр + 400 · Н2Ож = КС1раствор - 17,57 кДж, NаС1кр + 400 · Н2Ож = NаС1раствор – 4,27 кДж. Взаимодействие хлора с водой протекает также с поглощением тепла: 2 С12газ+ 2 · Н2Огаз =4·НС1газ+ О2газ – 114,5 кДж. А с повышением температуры еще большее число реакций начинает самопроизвольно протекать с поглощением тепла. Например, реакция разложения карбоната кальция, диоксида углерода, восстановления оксида кремния углеродом: СаСО3кр = СаОкр + СО2газ – 157 кДж, 41 СО2газ = Сграф. + О2газ - 94 кДж, SiО2кр+ 3 Сграф.+ 526,7 кДж = 2 СОгаз+ SiСкр. и многие другие. Следовательно, принцип Бертло-Томсена нельзя распространять на все реакции и сведений только о тепловом эффекте процесса явно не достаточно для предсказания возможности и направления реакции. К тому же существуют атермические процессы (без энергетических изменений). Например, рассмотрим сосуд, содержащий при одном и том же давлении и температуре два газа: гелий и аргон, разделенных перегородкой (рис. 2.2.2а). Осторожно, чтобы не внести в систему Начальное состояние 1 Конечное состояние 2 энергетических изменений (мысленно это можно сдеHe, Ar Не Ar смесь лать легко), удалим перегородку между газами. Через некоторое время спустя мы а б обнаружим, что газы в сосуРис.2.2.2. Процесс смешения газов без энергеде перемешались тических изменений: (рис.2.2.2б). Произошел проа) газы находятся в разных половинках сосуцесс смешения газов без да, б) газы смешались. энергетических изменений (данные газы химически не взаимодействуют). А вот обратный процесс – разделение газов потребует уже затрат энергии для их разделения. Если быть наблюдательным, то можно заметить, что и принцип стремления тел, систем к минимуму потенциальной энергии и в физических процессах в природе выполняется как-то избирательно. Метеориты, снаряды, пули падают на поверхность Земли, вода течет с гор, но почему молекулы воздуха, облаков не упали на ее поверхность? Ведь, это отвечало бы минимуму их потенциальной энергии. Более того, если существует только один принцип направленности процессов к состоянию с минимальной энергией, то исключается возможность существования обратимых реакций! Однако, такие реакции существуют. Видимо, существует еще какой-то фактор, критерий, определяющий возможность протекания процессов, реакций. У систем (физических и химических), которые не подчиняются принципу Бертло-Томсена, есть одна общая особенность: они состоят из огромного количества отдельных, самостоятельных частиц (молекул, атомов). Которые практически не связаны между собой связями, как, например, это происходит в конденсированном состоянии вещества. В этих системах присутствуют вещества в газообразном состоянии1. В приведенных примерах дейстКстати, отметим, что «газ» в переводе с греческого означает «хаос» - беспорядок, что является полной противоположностью конденсированному состоянию вещества. 42 1 вительно все процессы связаны с газообразным состоянием веществ, частицы которых находятся в состоянии непрерывного, независимого, теплового движения, за счет которого изолированная система самопроизвольно стремится к максимальной однородности и неупорядоченности. 3. ЭНТРОПИЯ И ЕЁ ИЗМЕНЕНИЕ 3.1. Энтропия В рассмотренном ранее примере (в главе 1) взаимодействия цинка с раствором серной кислоты при постоянном давлении: Znкрис. + H2SO4раст. → ZnSO4раствор + Н2газ + Q1 мы отметили, что изменение внутренней энергии системы ∆U складывалось из теплового эффекта (обозначим его Q1), отданного внешней среде, и работы А, совершенной системой над окружающей средой: ∆U = Q1 + А (2.3.1.) Попытаемся вернуть систему в исходное состояние, т.е. получить вновь металлический цинк и раствор серной кислоты. Одновременно необходимо восполнить энергозапас системы, восстановив его до значений исходного состояния. Для чего системе нужно передать необходимое количество энергии Q2 от внешней среды с помощью теплообмена. Одна часть подведенной энергии Q2, переданной за счет разности температур, пойдет на компенсацию рассеянной энергии системой в окружающую среду из-за теплообмена Q1 при ее прямом переходе в конечное состояние, а другая её часть Q2 - Q1 на восполнение энергии системы, потерянной ею при совершении работы А против внешних сил, т.е. Q2 = Q1 + А, или (2.3.2) Q2 = ∆U Подведенное из внешней среды количество энергии Q2 за счет разности температур (теплоты) расходуется на увеличение энергосодержания системы. Уместно заметить, что в прямом процессе не всё значение изменения внутренней энергии передавалось в форме работы А (часть её рассеивалось в виде теплового эффекта реакции Q1). Соответственно и в обратном процессе не все подводимое количество энергии в форме тепла Q2 можно использовать для получения работы1. Часть ее, равная Q1, также рассеется в системе, не совершая никакой работы. Эту часть энергии Q1, которая получена системой в форме тепла в обратном процессе и рассеяна в системе без совершения работы, называют рассеянной энергией, и ее доля от подведенной общей энергии Q2 составляет Хотя, отметим, что работу вполне возможно полностью превратить в теплоту (например, в процессе трения). 43 1 Q1 Q2 (2.3.3.) Количество отведенного тепла Q1 от системы в прямом процессе при переходе ее в конечное состояние пропорционально температуре системы Т1: Q1 ~ Т1, так же как и количество подведенного тепла Q2 из окружающей среды (при переводе нашей системы в начальное состояние) должно быть пропорционально ее температуре Т2. Q2 ~ Т2. Поэтому долю рассеянного тепла Q1/Q2 можно оценить и по соотношению температур системы и окружающей среды Т1 Т2 (2.3.4) А поскольку это одна и та же доля, то выражения 2.3.3 и 2.3.4 можно приравнять Q1 T1 = Q2 T2 (2.3.5) В самом деле, если имеется два тела - нагретое и холодное, то тепло будет передаваться от первого второму до тех пор, пока температуры тел не сравняются. Соответственно, и доли переданной теплоты должны находиться в том же соотношении, что и температуры, поскольку температура является мерой энергии. Таким образом, в обратимом процессе, измеряя отношение количества теплоты, взятой от окружающей среды и отданной системе, мы сможем определить соотношение температур системы и окружающей среды. Полученное соотношение 2.3.5 перепишем в таком виде: Q1 Q2 = T1 T2 (2.3.6). Отношение количества теплоты, полученное системой в изотермическом процессе к температуре этого процесса Q T называют приведенной теплотой. Последнее выражение устанавливает тот факт, что приведенная теплота Q1/ Т1 при переходе системы из начального состояния в конечное (теплота Q1 передана внешней среде системой) равна полученной приведенной теплоте Q2/Т2 от внешней среды. Но количества тепла Q1 и Q2 имеют противоположные знаки, потому что одна энергия реагирующей системой теряется, а другая - приобретается внешней средой. Поэтому можно было бы записать так: Q1 Q =− 2 , T1 T2 44 или Q1 Q2 + =0 T1 T2 (2.3.7). Это означает, что сумма приведенных теплот в обратимом процессе равна нулю. Не сумма теплот, что отражало бы просто первый закон термодинамики – сохранения энергии, а сумма приведенных теплот! Это похоже на закон сохранения, но некоторой новой функции состояния, свойства системы. Действительно, реакционная система, совершив какой-то процесс, вернулась в исходное состояние, и изменение этого свойства приведенной теплоты Q T в обратимом процессе оказалось равным нулю, что присуще функции состояния. Это физическое свойство системы (подобно энергии, массе, давлению, температуре) характеризует состояние системы и сохраняется в обратимом процессе. Его называют энтропией1 и обозначают буквой S S= Q (2.3.8) T Размерность энтропии легко определить из уравнения 2.3.8. Она представляет собой единицы энергии, деленные на градус. Поскольку энтропия зависит от количества вещества, то её также принято относить обычно к 1 молю вещества. Таким образом, энтропия имеет следующую размерность: Дж . моль × градК Размерность энтропии совпадает с размерностью теплоемкости. Но теплоемкость характеризует количество тепла, необходимого для нагревания тела на 10, включая и работу, которую совершает вещество (тело) при нагревании. Энтропия же характеризует количество рассеянной энергии, отнесенное к одному градусу данной температуры. Следовательно, ее можно рассматривать, некоторым образом, как удельную энергию. Так как функция состояния обладает свойством аддитивности, то и энтропия всей системы складывается из энтропий ее отдельных частей: Sсистемы = S1 + S2 + S3 + S4 + ……+ Sn На этом же основании формально к изменению энтропии ∆S применим закон Гесса, т.е. изменение энтропии системы ∆S не зависит от способа перехода системы из начального в конечное состояние, а определяется только начальным и конечным состоянием составных частей системы: ∆Sреакции = 1 ΣSпродуктов - Энтропия – от греческого корня слова τροπη – превращение. 45 ΣSисход. в-в. (2.3.9) Все виды энергии могут целиком превращаться в теплоту. Но обратный полный переход теплоты в другие виды энергии невозможен. Такой процесс будет всегда сопровождаться накоплением теплового запаса энергии. Физический смысл понятия энтропии и в том состоит, что в процессе передачи системе (системой) тепловой энергии часть её переходит за счет разности температур системы и среды (от более нагретых к более холодным), а в работу преобразуется только часть переданной тепловой энергии. Энтропия это мера рассеяния, рассредоточивания (деградации) энергии. Особенность этого термина «энтропия» состоит в том, что это непревращаемая энергия (буквальный перевод с греческого) в работу. Энтропия характеризует молекулярный беспорядок системы, который связан с её температурой и уменьшается с понижением температуры. М. Планк1 (1912) обратил внимание на такую связь энтропии с температурой и выдвинул постулат, именуемый третьим законом термодинамики: энтропия любой системы, тела при температуре абсолютного нуля равна нулю. Энтропия является мерой «бесполезного» тепла, мерой обесцененной, «связанной» энергии. Если мы хотим, чтобы какая-то система (или машина) совершала работу, то за это необходимо заплатить не только расходом внутренней энергии, но и возрастанием энтропии, теплового запаса системы. Энергию можно вернуть, отдав обратно полученную работу, но возросшую энтропию системы уменьшить (без дополнительной работы) нельзя. В нашем мире любой выигрыш в работе приведет обязательно к увеличению энтропии. А какова связь между тем или иным состоянием системы, возможностью самопроизвольного протекания в ней процессов и изменением энтропии, которое сопровождает эти процессы? 3.2. Изменение энтропии и направленность процессов Пусть в начальном состоянии система имеет значение энтропии S1= Q1/T1, а в конечном состоянии: S2 = Q2/T2, При переходе системы из начального состояния в конечное изменение энтропии ∆S составит величину, равное разности этих значений: ∆S = S2 – S1 (2.3.10) Или подчеркивая, что речь идет о порции тепла Q (полученного или отданного системой в изотермическом процессе), изменение энтропии определится выражением Планк Макс (1858 – 1947), немецкий физик, основоположник квантовой теории, ин. ч.- к. Петербургской АН (1913) и поч. ч. (1926) АН СССР. Труды по термодинамике, теории относительности, философии естествознания. Нобел. премия (1918). 46 1 (2.3.11) Применительно к рассмотренной системе взаимодействия цинка с серной кислотой изменение энтропии ∆S будет равно ∆S= Q/Т ∆Sреакции = Sконеч.сост. – Sисход.сост., (2.3.12) или ∆Sреакции = S2 - S1. Если то ∆S= Q/T, T ∆ S· = Q, (2.3.13) а это означает, что чем больше изменение энтропии ∆S, тем большая часть тепла рассеивается системой, оставаясь непревращенной в полезную работу и тем более процесс необратим. То есть энтропия является и мерой необратимости процесса. Кстати, термин «энтропия» был введен великим немецким ученым Р. Клаузиусом1 в 1865 году по аналогии с термином энергия, отражая их близкое физическое сходство. В обратимом круговом процессе, в котором энтропии прямого и обратного процессов (реакции) равны: Q1/T1 = Q2/T2 , и, соответственно, изменение энтропии равно нулю (2.3.14) А это означает, что система находится в состоянии равновесия с окружающей средой. То есть изменение энтропии системы ∆ S = 0 в данном случае выступает критерием равновесного состояния системы. Рассмотрим изменение энтропии в необратимых процессах. Энтропия есть функция состояния. Поэтому, если система переходит из какого-либо состояния «1» в состояние «2», то изменение энтропии ∆ S будет тем же самым независимо от того каким был процесс: обратимым или необратимым. На частном примере теплообмена между двумя различно нагретыми телами с температурами Т1 и Т2 (пусть Т1 > Т2) проанализируем изменение энтропии системы. В процессе теплообмена одно тело (более нагретое) теряет количество теплоты – Q, другое (менее нагретое) приобретает это же количество теплоты + Q. Изменение энтропии системы двух тел ∆S составит алгебраическую сумму их энтропий , равную: ∆ S = 0. ∆ S = S2 - S1 = 1 Q T2 + (− Q T1 ). Рудольф Ю.Э. Клаузиус (1822-1888) один из основателей термодинамики, член Петербургской АН. Дал одновременно с У. Томсоном первую формулировку второго начала термодинамики (1850 г), доказал теорему вириала (1870 г), разработал теорию поляризации диэлектриков. 47 Поскольку энтропия системы S2 в конечном состоянии больше таковой S1 в начале процесса, то изменение энтропии ∆ S составит положительную величину ∆ S = S2 - S1 > 0 (2.3.15) А в отношении процесса можно сказать, что протекает процесс перехода тепла от более нагретого тела к более холодному. Следовательно, в изолированной системе при протекании самопроизвольных процессов энтропия растет ∆S > 0, и критерием возможности протекания этих процессов является это условие 2.3.15. Энтропия изолированной системы растет до тех пор, пока система не придет в состояние термодинамического равновесия, когда в ней прекратятся все самопроизвольные процессы, т.е. система самопроизвольно стремится к состоянию максимальной однородности, молекулярной неупорядоченности! Принцип возрастания энтропии изолированной системы при протекании самопроизвольных процессов и есть второй закон термодинамики. Существует несколько формулировок второго закона термодинамики. Например, еще две из них: теплота не может самопроизвольно переходить от холодного тела к теплому; невозможен процесс полного превращения теплоты в работу Естественно, если изменение энтропии в процессе величина отрицательная ∆S < 0, то это может служить критерием неосуществимости самопроизвольного прямого процесса в изолированной системе. В такой системе протекает самопроизвольно обратный процесс. Или такой процесс протекает в неизолированной системе и он не является самопроизвольным. Однако подчеркнем, что условие ∆S > 0, охватывающее все возможные случаи, относится только к изолированной системе и может только возрастать или, в крайнем случае, остается неизменной. Если система неизолированная, то ее энтропия может убывать. Тем не менее, суммарное изменение энтропии системы и внешней среды всегда либо положительно, либо равно нулю. Все реальные процессы теплообмена самопроизвольны, не обратимы, сопровождаются увеличением энтропии. Возрастание энтропии уменьшает техническую ценность внутренней энергии, ее превращаемость. Так как изменение энтропии равно ∆S = Q T , а согласно первому закону термодинамики Q = ∆U + A, то получим ∆S = 48 ∆U + A , T или (2.3.16) Это основное уравнение термодинамики. Оно объединяет первое и второе начала. Смысл его становится понятным, если представить уравнение в виде T ∆ S = ∆U + A . T ∆ S - ∆U = + A. Из которого следует, что не все изменение запаса внутренней энергии при постоянной температуре превращается в работу, а только его часть. Член T ∆ S отображает ту часть внутренней энергии, которая не переходит в работу, её называют связанной энергией. Энтропия показывает величину связанной энергии, приходящейся на один градус температуры системы. Для изобарного процесса, в котором тепловой эффект реакции равен изменению энтальпии, Qp= ∆H, Изменение энтропии можно связать с изменением энтальпий. Так как то и ∆S= Qp /T ∆S= ∆H / T, или ∆Н= T·∆S. (2.3.17) Увеличение энергосодержания системы за счет тепла, связанного с ростом энтропии ( Qр = Т ·∆S или ∆Н = Т ·∆S) для изобарного процесса, приводит к ослаблению, а в конечном итоге, и разрыву связей между частицами вещества системы, если оно находилось в конденсированном состоянии, и, соответственно, увеличивается вероятН2Опар Н2Окрис. Н2Ож S, ность существования отдельДж/моль К ных частиц, что и характерно для газообразного состояния 200 вещества. Поэтому мы наблюдаем плавление и испарение ∆Sиспар кристаллических и жидких веществ, разложение газообраз100 ных веществ на составные час∆Sплав ти. Заметим, что в газообразном состоянии расположение частиц более беспорядочное, хао200 400 Т,К тичное, но система имеет более 273 373 равномерное распределение внутренней энергии по всему Рис.2.3.1. Изменение энтропии 1 моля воды в зависимости от температуры. объему системы, что и отвечает равновесному состоянию. На рис.2.3.1 представлен график изменения энтропии одного моля воды в зависимости от температуры при давлении 101 кПа. Как видно из графика, с повышением температуры, а, следовательно, и количества связанной 49 теплоты ∆Н = T ∆S, происходит рост энтропии. Особенно резко изменяется значение энтропии в точке фазовых переходов: температуры плавления 273 К и температуры кипения 373 К, при которых происходит явная деструкция воды на составляющие ее молекулы, и как следствие - более не упорядоченное положение частиц (беспорядок). Априори мы можем всегда предсказать, что энтропия кристаллического состояния вещества Sкр меньше таковой жидкого состояния Sж, а последняя меньше энтропии газообразного состояния этого же вещества Sгаз, т.е. Sкр < Sж < Sгаз. 3.3. Энтропия как мера беспорядка При описании состояния систем мы использовали такие параметры, как температура, давление, объем. Они являются среднестатистическими величинами состояния огромного множества частиц составляющих систему и характеризуют макросостояние системы. В то время как каждая частица системы имеет свое собственное значение и кинетической энергии, и занимаемого удельного объема, и координат, и т.д., которые отличаются от усредненных параметров макросостояния. Другими словами, каждая частица системы индивидуальна по своим характеристикам. Возможно ли, зная микросостояние каждой структурной частицы системы, описать всю систему в целом? Что при этом мы будем вкладывать в понятие обратимости процесса при переходе системы из конечного состояния в исходное, и какие понятия и определения будут основными? Попытаемся ответить на эти вопросы на нескольких примерах. Рассмотрим, например, один моль любого газа. Если определим координаты каждой частицы в этом объеме, направление и скорость ее перемещения в каждый данный момент времени, мы тем самым охарактеризуем микросостояние каждой частицы. Результатом взаимодействия микросостояний всех частиц будет являться данное макросостояние системы. Но частицы перемещаются, меняют свои координаты, параметры, и в следующий момент времени каждой частице соответствует новое микросостояние, но все вместе они в этой новой комбинации, тем не менее, отвечают макросостоянию системы, характеризуемому теми же среднестатистическим параметрами Р,Т,V. Подсчитаем, сколько таких микрораспределений будет соответствовать макросостоянию системы. Для этого мысленно разобьем объем V0, занимаемый газом, на такое количество микрообъемов (ячеек) n, чтобы в каждом могла поместиться лишь одна молекула. Видимо, число таких микрообъемов будет явно больше числа молекул N, т.е. n > N, так как между молекулами газа есть свободный объем (собственный объем молекул газа много меньше объема занимаемого газом). В результате хаотического перемещения молекул по ячейкам в каждый данный момент времени свободное число (незанятых) ячеек будет 50 n-N, а число занятых ячеек равно числу молекул N. Тогда число всех возможных микрораспределений молекул по ячейкам W есть число сочетаний из n микрообъемов по N молекул W = n! . N (n − N )! Это число микросостояний W, посредством которых данное макросостояние реализуется, описываемое среднестатистическими параметрами Р,Т,V, называется термодинамической вероятностью. То есть величина W есть число способов реализации данного состояния системы из N частиц, составляющих ее. Это очень большое число в отличии от математической вероятности, которая выражается обычно числами меньше единицы. Рассчитаем, например, число комбинаций распределения для 10 молекул по 30 ячейкам: W = 30! 21 ⋅ 22 ⋅ 23 ⋅ 24 ⋅ 25 ⋅ 26 ⋅ 27 ⋅ 28 ⋅ 29 ⋅ 30 = = 30 ⋅ 10 6 10!(30 − 10)! 1 ⋅ 2 ⋅ 3 ⋅ 4 ⋅ 5 ⋅ 6 ⋅ 7 ⋅ 8 ⋅ 9 ⋅ 10 С увеличением объема системы V (а соответственно и числа микрообъемов n) и числа частиц N, содержащихся в нем, термодинамическая вероятность возрастает. Следовательно, расширение газа можно представлять, как переход системы из состояния с меньшей вероятностью в состояние с большей вероятностью. Рассматривая приведенный пример, мы молчаливо предполагали, что молекулы одинаковы и распределяются равномерно по всему объему системы. Но возможен и другой случай, когда молекулы газа распределяются неравномерно по всему объему системы V, а собираются одновременно в какой-то его части, занимая объем V0 , который много меньше первоначального V (рис.2.3.2). Правда, такое микрораспределение молекул не будет соответствовать макросостоянию системы, хотя бы потому, что ее объем V0 будет меньше первоначального V, соответственно, и плотность вещества будет больше, и т.д. Но попытаемся оценить вероятность такого микрораспределения частиц и, соответственно, возможность реализации такого макросостояния системы. V Естественно предположить, что каждая молекула этого газа, блуждая по всему объему V, какую-то часть времени находитV0 ся в выделенном объеме V0. Если мысленно разделить объем V пополам, то можно сказать, что половину времени «жизни» одна из Рис.2.3.2. Соотношение объемов начального V состояния гамолекул находится в одной половине объеза и конечного V0 ма, другую- во второй. Если же объем разделить на четыре части, то в каждой из четвертинок объема V молекула находится четверть времени «жизни». Продол51 жая делить объем занимаемый газом до объема V0, можно тогда сказать, что доля времени, проводимая в нем молекулой, будет равна V0/V. То есть вероятность найти молекулу в объеме произвольной формы V0 прямо пропорционально их соотношению W= V0/V. Поскольку мы молекулу выбрали произвольно из всего количества имеющихся в объеме, то и для второй и для третьей, и для любой другой, если они независимы в своем поведении, вероятность нахождения их в этом объеме V0 будет также определяться этим же соотношением W =V0/V. А это значит, что, если взять некоторый небольшой промежуток времени t0, первая молекула будет находиться в объеме V0 в течение времени t1 равном t1 = V0 t0 . . V За это же время t1 вторая молекула будет находиться в том же объеме V0 за время t2: t2 = V0 V ⋅V t1 = 0 0 t 0 V V ⋅V Соответственно, для третьей молекулы время нахождения в объеме будет равно t3 = V0 V ⋅V ⋅V t2 = 0 0 0 t0 V V ⋅V ⋅V И так далее для последующих молекул. Рассуждая, таким образом, найдем, что вероятность W нахождения N молекул в объеме V0 одновременно равна: V W = 0 V N Но, если в объеме V0 собрались все молекулы одновременно, а объем V0 меньше первоначального объема V, тогда это соответствует самопроизвольному сжатию газа. Насколько вероятен этот процесс? Оценим это событие, например, для одного моля газа, занимающего объем 22,4 л, в котором содержится 6,02·1023 молекул, и самопроизвольно сжимающегося до объема в 1 см3. В соответствии с последним выражением вероятность такого события равна N V 0,001 W = 0 = 22400 V 6 , 02⋅10 23 = 1 ( 22400 ⋅ 10 3 )6,02⋅10 23 Вероятность такого события ничтожна. Напротив, вероятность заполнения всего объема V=22,4 л молекулами, когда V=V0 равна: 23 22400 W = 22400 6 , 02⋅10 23 =1 Состояние, при котором частицы занимают весь предоставленный объем, более вероятно, чем состояние, при котором только часть объема занята 52 частицами системы. Следовательно, можно оценить наиболее устойчивое, равновесное состояние системы через вероятное распределение микросостояний частиц, образующих систему. Кстати, расширение газа представляет самопроизвольный процесс перехода системы из менее вероятного, более упорядоченного в более вероятное состояние и более разупорядоченное. Самопроизвольное же уменьшение объема газа, вплоть до образования кристалла с упорядоченной структурой, невероятно и не возможно. То есть необратимый процесс есть переход системы из менее вероятного состояния в более вероятное. Неупорядоченность систем растет не только при расширении газа. Если имеется идеальный кристалл, например, поваренной соли NаС1 при 0 К, то мы можем точно указать координаты каждого иона Nа+ и С1- – в нем. Но достаточно его нагреть выше нуля 00 К, как отдельные ионы начинают колебаться относительно своих равновесных положений в кристаллической решетке, и возникает несколько способов построения кристалла из колеблющихся ионов, но отвечающих макросостоянию кристалла. С еще большим повышением температуры в кристалле появляются вакантные узлы, межузельные ионы, а при плавлении и тем более нарушается порядок расположения частиц, растет неупорядоченность (беспорядок). Описывать такую систему придется уже значительным числом способов, комбинаций микросостояний частиц, эквивалентных конкретному микросостоянию систем. Неупорядоченность растет и при кипении жидкостей, и при сублимации твердых веществ, растворении кристаллов. Соответственно, возрастает и вероятность состояния системы при этих переходах. Ее изменение ∆W будет положительной величиной, т.е. ∆W = W2 –W1 > 0, где W1 - термодинамическая вероятность состояние веществ системы в начальном состоянии; W2 – термодинамическая вероятность состояния веществ системы в конечном состоянии. Но такой же характер зависимости носит и изменение энтропии в самопроизвольных процессах. Действительно, в изолированной системе все процессы протекают в направлении возрастания энтропии. Впервые о существовании связи между энтропией и вероятностью состояния системы догадался и установил ее в 70-х годах XIX века Л.Больцман1. Связь можно выразить математически, полагая, что S = f(W), где W – термодинамическая вероятность; f- некоторая функция. Вид этой функции можно установить, если учесть, что энтропия системы равна сумме энтропий составляющих ее частей, а вероятность некоторого состояния сисБольцман Людвиг (1844-1906), австрийский физик, один из основателей статистической физики, иностр. Член Петербургской академии наук. Дал статистическое обоснование второго начала термодинамики, вывел закон излучения (закон Стефана-Больцмана). 53 1 темы равна произведению вероятностей состояний составляющих частей системы. Например, для двух компонентной системы энтропия равна S = S1+ S2, а вероятность состояния системы оценивается произведением вероятностей W = W1·W2, где S1 и S2- энтропия первого и второго соответственно компонента системы; W1 и W2 первого и второго соответственно компонента системы. Так как энтропия каждого из компонентов является функцией вероятности их состояния: S1=f(W1) и S2= f(W2), то энтропия двухкомпонентной системы в целом определяется выражением S = f (W1·W2). Но энтропия обладает свойством аддитивности. Она для системы должна складываться из энтропий отдельных компонентов. Этому условию удовлетворяет логарифмическая функция. Поскольку энтропия системы S должна быть равна S = S1 + S2., то логарифмическая функция энтропии системы, выраженная через произведение вероятностей S= k ln (W1·W2) , где k - коэффициент пропорциональности, названный в честь ученого постоянной Больцмана, также будет равна сумме логарифмов вероятностей: S= k ln W1+ klnW2. А это значит, что энтропия системы равна сумме энтропий её составных частей S = S1 + S2.. Таким образом, с помощью логарифмической функции достигается свойство аддитивности энтропии системы. Использование постоянной Больцмана предполагает расчет значения энтропии на одну частицу. Чтобы получить значение энтропии 1 моля вещества, умножим правую и левую части уравнения на число Авогадро NA: NA·S = NA·R·lnW. Произведение NA·k есть универсальная постоянная R=NAk, а произведение NAS заменим большой буквой S – символом энтропии одного моля вещества. В результате имеем: S = R lnW Формула Л. Больцмана показывает, что энтропия вещества, системы пропорциональна логарифму вероятности их состояния. Или (применительно к химической термодинамике) энтропия является логарифмическим выражением вероятности существования веществ или их различных форм в том или ином состоянии. 54 Зная энтропии исходных веществ S1 (их сумму) и продуктов реакции S2, можно определить ее изменение ∆S при переходе из начального состояния системы в конечное ∆S = S2 - S1 = R lnW2 - R lnW1 или ∆S = R ln W2 W1 Изменение энтропии – это соотношение вероятностей существования продуктов реакции и исходных веществ. Если ∆S > 0, то и W2 > W1, т.е. вероятность существования продуктов больше, чем таковая для исходных веществ. Следовательно, в отсутствии энергетических изменений (∆Н = 0), о направленности процесса можно судить по знаку ∆S. Порядок величины ∆S часто можно ориентировочно (качественно) оценить по знаку и величине изменения объема системы ∆V в процессе, не прибегая к расчетам. Например, если в реакции участвуют только кристаллические реагенты и продукты, то изменение энтропии в этом случае незначительно ∆S ≈ 0. Энтропия веществ в жидком состоянии несколько больше таковой этих же веществ в кристаллическом состоянии, поэтому, если в реакции из кристаллических исходных веществ образуются жидкие продукты, то изменение объема системы будет также несколько больше нуля (∆V > 0) и соответственно ∆S > 0. Но если в реакции участвуют газообразные продукты, то изменение объема системы будет заметным, особенно, если разное количество молей газообразных исходных веществ и продуктов реакции. Например, для реакции приводимой ранее СаСОз кр (исходное состояние) = СаОкр + СО2газ (конечное состояние), априори можно сказать, что изменение энтропии в этом процессе – величина положительная ∆S > 0, т.к. в конечном состоянии одно из веществ находится в газообразном состоянии, а стало быть ΣSпродуктов> ΣSисходных веществ и ∆Sреакции = ΣSпродуктов - ΣSисходных в-в > 0 Кстати, заметим, что с повышением температуры объем кристаллических и жидких веществ возрастают не так значительно, как объем газообразных веществ, а следовательно, должны симбатно изменяться и их энтропии: для первых - незначительно, других – заметно. 55 Таким образом, вместо термодинамической формулировки второго начала термодинамики «энтропия изолированной системы всегда возрастает» мы можем сказать: в изолированной системе возрастание энтропии наиболее вероятно; или в изолированной системе процессы протекают самопроизвольно в сторону увеличения беспорядка. 3.4. Стандартные энтропии Для того чтобы иметь возможность сопоставить энтропии различных веществ и определить их изменения в процессах, принято относить энтропии к определенным стандартным условиям. А именно: температура 250 С или 298 К, давление 101 325 Па или 1 атм и количество вещества 1 моль. Для растворов принята концентрация растворенного вещества 1 моль/л. Энтропия одного моля вещества при этих условиях обозначается S0298 и называется стандартной энтропией. Численные значения стандартных энтропий приводятся в термодинамических справочниках. Значения стандартных энтропий веществ определяют экспериментально по зависимости теплоемкости вещества от температуры. Принято считать, Cl что при абсолютном нуле (0 К) энS тропия вещества равна нулю, что Дж/моль К составляет суть третьего закона 100 термодинамики. На рис 2.3.3. представлена зависимость стандартной S0298 энтропии различных простых Na веществ третьего периода Периоди50 ческой системы Д.И. Менделеева от S Mg Al порядкового номера элемента Z. В Si P пределах одного периода Периодической системы элементов зависи12 14 16 18 Z мость имеет сложных характер. Тем не менее, переход от мягкого натрия Рис.2.3.3. Изменение энтропии простых с меньшей плотностью к твердому веществ третьего периода в зависимости от кремнию с большей плотностью сопорядкового номера элемента. провождается уменьшением энтропии. Затем она несколько возрастает и в конце периода достигает максимального значения у газообразного хлора и аргона. В большинстве случаев выполняется правило; чем более компактна структура вещества, тем ниже энтропия, тем тверже вещество. Сверху вниз в подгруппе Периодической системы Д.И. Менделеева для простых веществ с одинаковой структурой или для однотипных соединений энтропия возрастает. Энтропия, как правило, тем больше, чем сложнее химический состав. Например, 0 56 Вещество NaCl MgCl2 AlCl3 Энтропия, Дж/моль К 72,4 89,5 167 Задачи и вопросы для самостоятельной работы 2.3.1. Не производя вычислений, качественно оцените знак изменения энтропии ∆ S в следующих процессах: а) 2 NH3газ = N2газ + 3 Н2 газ; б) СО2кр= СО2газ; в) 2 NOгаз+О2газ =2 NO2газ, г) 2 Н2Sгаз + 3 О2газ= Н2Ож+ 2 SO2газ,; д) 2 СН3ОНгаз + 3 О2газ = 4 Н2О газ + 2 СО . 2газ 2.3.2. Укажите знаки изменения энтальпии и энтропии (∆Н и ∆S) для процессов: испарения воды с поверхности водоема, для процесса адсорбции водяных паров на холодном предмете, внесенном в теплое помещение с улицы. 2.3.3. Почему процесс растворения происходит самопроизвольно? 2.3.4.Почему при низких температурах критерием, определяющим направление реакции, может служить знак ∆Н, а при достаточно высоких- таким критерием является знак ∆S? 2.3.5. Почему энтропия кристалла меньше, чем энтропия того же вещества в газообразном состоянии? 2.3.6. Почему растворение твердого или жидкого вещества в воде приводит к возрастанию энтропии, тогда как растворение газа вызывает уменьшение энтропии? 2.3.7. В каком случае энтропия системы больше: смесь диоксида углерода, воды, соединений азота и минеральных веществ и дерево, выросшее из них? 2.3.8. Вычислите изменение энтропии для каждой из следующих реакций, протекающих при стандартных условиях: а) СаСО3кр= СаОкр+ СО2газ,; б) Сакр+ 0,5 О2газ= СаОкр,; в) J2 кр= 2Jгаз. 2.3.9. Что можно сказать о прочности связей в простых веществах между атомами, если энтропия возрастает в ряду: Вещество Салмаз Ве крис. SiO2 крис. Pbкрис. Hg жид. Hg газ 0 S , кДж/(моль К) 2,44 9,54 41,8 64,9 77,4 174,9 2.3.10. Если бы принцип Бертло-Томсена был бы единственным и справедливым для газов и жидкостей, то что бы с ними произошло? 2.3.11. Расположите представленные вещества в порядке возрастания молярной энтропии: N2O4, Naкрис., NaClкрис., Br2 жид., Br2 газ. 2.3.12. Каким образом, согласно представлениям классической термодинамики, энтропия связана с температурой? 2.3.13. Как изменяется энтропия водного раствора ионов меди Сu2+ в результате процесса гидратации? 2.3.14. Взяли маленький кристаллик воды и поместили его в изолированную большую емкость, которую затем выдерживали длительное время при низкой температуре (ниже 00 С). Что произошло с кристаллом льда в емкости? 2.3.15. Как Вы можете объяснить то, что чем сложнее состав молекул, тем больше их энтропия? 57 4. СВОБОДНАЯ ЭНЕРГИЯ 4.1. Свободная энергия – критерий направленности процессов В предыдущих разделах, пытаясь разобраться с критериями направленности процессов, мы пришли к выводу, что в нашем мире, в котором мы живем, проявляются две тенденции, обуславливающие протекание процессов. Одна из них связана со взаимодействием частиц между собой и стремящаяся к объединению их в нечто единое целое структурно. Следствием этой тенденции является уменьшение энергосодержания системы, что и отражает принцип Бертло-Томсена: протекают самопроизвольно те процессы, реакции, в которых система понижает свою энергию. Критерием этой категории реакций при постоянном давлении служит изменение энтальпии реакции ∆Н. Если изменение энтальпии в реакции ∆Н < 0 (экзотермические реакции), то процесс протекать будет самопроизвольно, если изменение энтальпии реакции величина положительная ∆Н > 0 (эндотермические реакции), то самопроизвольно их течение исключено. Другая, противоположная тенденция, связана с разделением веществ на составляющие его частицы, беспорядочное расположение их с более равномерным распределением по всему объему системы, что является следствием тепловых свойств. Критерием направленности этих процессов является изменение энтропии ∆S. Если изменение энтропии величина положительная ∆S > 0, то процессы самопроизвольно протекают в сторону более вероятного состояния системы, характеризуемого бóльшим беспорядком. Если же изменение энтропии величина отрицательная ∆S < 0, то самопроизвольно такие процессы в изолированной системе не идут. В большинстве же процессов одновременно происходят изменения и энтальпии, и энтропии. Следовательно, должна быть функция, которая объединяла бы оба эти критерия. Приведенное ранее уравнение (2.3.17) ∆Н = T ·∆ S устанавливает тот факт, что обе тенденции одинаковы, т.е. энтальпийный ∆Н и энтропийный T·∆S. факторы компенсируют друг друга. Уравнение является условием равновесия прямой и обратной реакции (процесса), т.е. количество энергии, выделившееся в результате процесса при постоянном давлении, полностью расходуется (утилизируется) обратимо на тепловое движение частиц системы. В этом случае говорят, система находится в равновесии. Например, при испарении кипящей воды происходит увеличение энергосодержания пара (энтропийный фактор), которое возвращается при его конденсации (энтальпийный фактор). При их равенстве мы имеем состояние равновесия между кипящей водой при 1000 С и насыщенным водяным паром при давлении водяных паров равных 101325 Па. 58 Уравнение 2.3.17. является условием равновесия обратимого процесса, характеризуя такое состояние системы, когда скорости протекающих в ней противоположных процессов становятся равными. Кстати, с помощью последнего равенства можно рассчитать изменение энтропии, энтальпии и температуру в данном равновесном состоянии. Применительно к физическим фазовым переходам можно записать, что, например, температура плавления Tплав. равна Tплав. = ∆Нплав. /∆ Sплав.. А в случае химических процессов равновесная температура Трав. равна Травн.= ∆Нреак. / ∆ Sреак. Мы рассмотрели случай равновесия. Но большинство протекающих процессов сопровождаются одновременным изменением и энтальпийного и энтропийного фактора далеко в неравной степени. Что брать в таком случае за критерий направленности процесса? Предположим, что только часть выделившейся (или подведенной) энергии ∆Н в результате перехода реагирующей системы из начального состояния в конечное оказалась связанной и равной T·∆S. Она израсходовалась на увеличение теплового движения частиц системы. Оставшаяся, другая часть энергии ∆Н - T ·∆ S может быть использована для получения максимальной полезной работы в результате этой реакции. Эту часть энергии назовем свободной энергией (свободной энтальпией) и обозначим её ∆G. Тогда получим: ∆G = ∆H – T ∆S. (2.4.1.) ∆G – это фактическая полезная работа А, которую может совершить сис- тема при переходе из данного состояния к равновесному. Проведем формальный анализ полученного выражения 2.4.1. 1. Предположим, что в системе не происходит ни энергетических изменений ∆Н = 0, ни изменений энтропии ∆S = 0. Тогда ∆G = 0. Если в системе не происходит никаких изменений, значит, она находится в состоянии равновесия. 2. Пусть изменение энтропии в системе в результате процесса равно нулю (∆ S = 0). Следовательно, ∆G = ∆H. • Если в результате процесса системой выделяется энергия в форме тепла Qp > 0, а ее энергосодержание ∆H, соответственно, уменьшается на эту величину, то ∆G < 0. Такой процесс самопроизвольно может протекать, что соответствует принципу Бертло- Томсена. • Если же реакция эндотермическая Qp< 0, а энергосодержание системы увеличивается на эту же величину ∆H > 0, то самопроизвольно система не может увеличить свою энергию, и такой процесс не протекает. 59 Рассмотренный вариант является не чем иным, как принципом БертлоТомсена, когда возможность и направленность процесса определяется энергетическими изменениями. 3. Представим себе, что в системе не происходят энергетические изменения, т.е. изменение энтальпии ∆Н = 0. В этом случае ∆G = - Т∆S. • И если ∆S > 0, то ∆G оказывается отрицательной величиной (∆G < 0). Так как изменение энтропии положительная величина, то следовательно система из менее вероятного переходит в более вероятное, т.е процесс будет протекать самопроизвольно. • Если же окажется, что изменение энтропии будет отрицательной величиной ∆S < 0, то ∆G > 0 и система из более вероятного состояния в менее вероятное не перейдет. Стало быть, в этом варианте направленность процесса определяется изменением энтропии. 4. В случае, когда энтальпийный и энтропийный факторы не равны нулю, возможность процесса можно определить по знаку ∆G. А именно: • если ∆G < 0, то прямой процесс возможен (он будет протекать самопроизвольно); • при ∆G > 0 прямой процесс самопроизвольно протекать не будет (идет самопроизвольно обратный процесс); • и при ∆G = 0 система будет находится в равновесии. Итак, мы получили критерий принципиальной осуществимости процесса. Возможность протекания реакции, процесса в прямом направлении без затраты энергии (работы), является неравенство: ∆G р,Т < 0. (2.4.2) При тех же условиях (Р-давление, Т-температура) протекание обратного процесса невозможно, так как: ∆G p,T > 0 (2.4.3) Любая реакция может протекать самопроизвольно только по направлению к состоянию равновесия. Если в системе наступило химическое равновесие, то ∆G p,T = 0 (2.4.4) В выражении 2.4.1. ∆G = ∆H – T ∆ S, свободная энергия системы представляет движущую силу процесса, которая зависит лишь от ее значения (потенциала) G1 системы в начальном и конечном состоянии G2 , т.е. ∆G = G2 –G1, 60 (2.4.5) где G1- потенциал свободной энергии системы в начальном состоянии, равный G1 = Н1 - Т S1; G2- потенциал свободной энергии системы в конечном состоянии, равный G2 = H2 - T S2 . Таким образом, изменение свободной энергии в процессе представляет собой изменение и энтальпии ∆H, и энтропийного фактора ∆S: ∆G = G2 – G1= (H2 - H1) - T (S2 – S1) G Р,Т = const ∆h Исходные вещества Направление самопроизвольного про- равновесие Направление самопроизвольного про- б Продукты реакции а Рис.2.4.1. Изменение потенциала системы при достижении равновесия: а) изменение свободной энергии при постоянном давлении и температуре в случае прямой и обратной реакции; б) изменение потенциальной энергии шара в гравитационном поле. Для реакций, протекающих при постоянном давлении, G - называют изобарным потенциалом, свободной энергией, или свободной энтальпией, или энергией Гиббса в честь американского ученого Дж.У. Гиббса1 На рис.2.4.1а представлено графическое объяснение приведенной функции: любая реакция (прямая или обратная), самопроизвольно протекающая при постоянном давлении и температуре, должна обязательно сопровождатьДжозайя Уиллард Гиббс (1839-1903). Американский физико-химик, теоретик. Один из создателей термодинамики и статистической механики. Разработал теорию термодинамических потенциалов, правило фаз, распределение Гиббса. 61 1 ся уменьшением свободной энергии G. Когда реакционная система достигает положения равновесия, изобарно-изотермический потенциал G принимает минимальное значение, а изменение его становится равным нулю. В таком виде изобарный потенциал аналогичен в механике гравитационному потенциалу (рис.2.4.1б), когда тенденция перемещения тела сверху вниз определяется разностью уровней в его начальном и конечном состоянии независимо от траектории падения и независимо от того, с какой стороны шарик скатывается: слева или справа, переходя в равновесное состояние. Обратим внимание на то, что, решая принципиально вопрос с возможности реакции по знаку изменения энергии Гиббса, мы совершенно ничего не говорили о том, как, по какому пути будет протекать данный процесс. Знак изменения энергии Гиббса не дает ни какой информации о самом процессе, механизме реакции. Такое положение обусловлено свойством изобарно- изотермического потенциала - он является функцией состояния. Величина изменения энергии Гиббса ∆G, как и изменение энтальпии ∆H, и изменение энтропии ∆S, не зависит от пути процесса, а определяется лишь начальным и конечным состоянием реагирующей системы. Соответственно, и изменение изобарно-изотермического потенциала можно рассчитывать в общем виде по закону Гесса: ∆Gреакции = ∑∆Gпродуктов - ∑∆Gисходных веществ, или, используя стандартные значения: ∆G0 реакции = ∑∆G 0продуктов - ∑∆G0 исходных веществ . 4.2. Примеры расчета свободной энергии На примере двух конкретных процессов рассмотрим, как рассчитывается свободная энергия (энергия Гиббса) и на основании полученных результатов сделаем выводы о возможности и условиях протекания процессов. Пример 1. Возможен ли при обычных (стандартных) условиях процесс восстановления оксида железа (Ш) водородом? Процесс протекает в соответствии с уравнением Fe2O3 кристал.+ 3 H2 газ = 2Feкристалл. + 3 H2Oпар . Решение. Для ответа на вопрос задачи необходимо знать изменение изобарно-изотермического потенциала при стандартных условиях для вышеприведенной реакции, т.е. ∆G0 = ∆H0 – T ∆S0. Для этого, как видно, нужно знать стандартное изменение энтальпии и энтропии в процессе. Из термодинамического справочника выписываем энтальпии образования и энтропии всех веществ, входящих в уравнение реакции (смотрите Приложение), и сводим данные в таблицу 2.5.1. 62 Таблица 2.5.1. Термодинамические характеристики исходных веществ и продуктов реакции Вещество Fe2O3 кристал H2 газ Feкристалл. H2Oпар ∆H0 , кДж/моль - 821,3 0 0 - 241,84 S0, Дж/моль К 89,96 130,65 27,15 188,74 Рассчитаем изменение энтальпии и энтропии реакции при стандартных условиях в соответствии с законом Гесса. ∆H0реакции= Σ ∆H0продуктов - Σ ∆H0исходных веществ = ∆H0реакции= (2 ∆H0Fe крис.+ 3 ∆H0Н2О пар) – (∆H0Fe2O3 крис + 3 ∆H0Н2 газ) = (2 ·0 + 3·( - 241,84) ) - ( -821,3 + 3 · 0) = 95,78 кДж/ моль Таким образом, изменение энтальпии в реакции при стандартных условиях составляет величину ∆H0реакции = 95,74 кДж/моль. Изменение энтропии ∆ S0реакции также рассчитаем в соответствии с законом Гесса: ∆S0реакции = ΣS0продуктов – ΣS0исходных веществ = ∆S0реакции = (2 ·S0Feкрис. + 3·S0H2Oпар ) - ( S0Fe2O3 крис. + 3 ·S0H2 газ ) = (2 ·27,15 + 3 · 188,74) - (89,96 + 3 · 130,65) = 138,61 Дж/моль К. Изменение энтропии в реакции при стандартных условиях оказалось равным ∆S0реакции = 138,61 Дж/моль К. При температуре 298 К изменение энергии Гиббса составит величину ∆G0реакции = ∆H0реакции – T ∆S0реакции = 95,78 - 298 · 0,13861 = + 54,48 кДж/ моль. Большая положительная величина ∆G0реакции== + 54,48 кДж/ моль указывает на невозможность восстановления Fe2O3 кристал водородом до металлического железа при стандартных условиях. Наоборот, противоположный процесс 2Feкристалл. + 3 H2Oпар = Fe2O3 кристал.+ 3 H2 газ Характеризуется отрицательной величиной изменения энергии Гиббса ∆G0 обратной реакции= - 54,48 кДж/ моль. Из чего следует, что такая реакция возможна. Действительно, этот процесс самопроизвольно протекает, и результатом его является окисление (коррозия) железа, что мы и наблюдаем повседневно. А при каких условиях (при какой температуре) будет наблюдаться равновесие в данной системе? В равновесном состоянии изменение энергии Гиббса системы равно нулю, т.е. Обратите внимание на то, что изменение энтропии представлено в кДж/(моль К), потому что величина изменения энтальпии представлена в кДж. 63 1 ∆G0реакции = 0 и Откуда 1 ∆G0равновес. = ∆H0равновес. – Tравновес.· ∆ S0равновес. = 0 T равновес. = ∆H 0 равновес. 95780 = = 691,0 К ∆S 0 равновес. 138,61 При этой температуре обе реакции: восстановление и окисление железа равновероятны, их скорости одинаковы. При температуре ниже 691 К железо самопроизвольно окисляется водяными парами до оксида железа с выделением водорода, а при температуре выше 691 К, наоборот, водород восстанавливает оксид железа до металлического. Таким образом, используя основные понятия и термодинамические закономерности, мы оценили принципиальную возможность протекания как прямой, так и обратной реакции, а также и температуру равновесного состояния реакционной системы. Пример 2. Возможен ли процесс испарения воды при стандартных условиях? Каково влияние температуры на этот процесс? Решение. Для решения вопроса о возможности данного процесса нужно знать изменение энергии Гиббса при его протекании. Поэтому вначале составим термохимическое уравнение требуемого перехода: Н2Ож = Н2Опар; ∆Н0испар.. Для расчета изменения энергии Гиббса этого перехода, равного ∆G0 испар = ∆H0испар – T ∆S0 испар., Нужно рассчитать изменение энтальпии ∆H0испар и энтропии ∆S0испар этого процесса. Рассчитаем изменение энтальпии этого перехода при стандартных условиях: ∆H0 испар = ∆Н0Н2Опар – ∆Н0Н2Ож И изменение энтропии в этом процессе при этих же условиях: ∆S0 испар .= S0Н2Опар- S0Н2Ож. Подставляя табличные данные (см. приложение), получаем: ∆H0 испар .= -241, 84 – (-285, 84) = 44 кДж/моль, ∆S0 испар =188,74 – 69,96 = 118, 78 Дж/ (моль К) Зная эти величины, рассчитываем изменение энергии Гиббса при этом переходе для стандартных условий: ∆G0 испар = ∆H0 испар – T ∆S0 испар. = 44000 – 298·118,78 = +8603,6 Дж Полученная величина энергии Гиббса ∆G0 испар .= +8,6 кДж явно величина положительная (∆G0 испар> 0), и, следовательно, процесс испарения воды при 250С невозможен. Но наш повседневный опыт говорит об обратном: вода испаряется при комнатных условиях. В чем же дело? При расчете температуры равновесия предполагаем, что изменение энтальпии и энтропии не зависит от температуры, и используем стандартные их значении для 298 К. 64 1 Расчет нами проведен для стандартных условий, когда водяные пары имеют парциальное давление 101 325 Па при температуре 250С. В реальных же условиях парциальное давление водяных паров много меньше (всего 3 647 Па) при этой температуре, что отвечает равновесному состоянию системы: «вода жидкая – пар». Если бы в реальных условиях парциальное давление водяных паров оказалось бы равным 101 325 Па, то, естественно, никакого самопроизвольного испарения жидкой воды в этих условиях не происходило бы (поэтому ∆G испар > 0), а вот обратный процесс- конденсации водяного пара наблюдался бы. Приведенный пример, показывает, что нужно аккуратно обращаться с понятием стандартные условия и результатами термодинамических расчетов. Рассчитаем, при какой температуре наступит равновесие между скоростью испарения и конденсации водяных паров, если их парциальное давление будет 101325 Па. В состоянии равновесия изменение энергии Гиббса равно нулю ∆G испар= 0 и ∆G испар = 0 = ∆H испар – T· ∆Sиспар., Стало быть, температура равновесного состояния Трав. Будет определяться выражением: Травн = ∆H испар/ ∆Sиспар. = 44000/118,78 ≈ 370,43 К, или t = 97,40С. Значение 97,40С близко температуре кипения воды, равной 1000С. Различие в 2,60 обусловлено тем, что мы воспользовались стандартными значениями энтальпии и энтропии для 250С и не учитывали их зависимость от температуры. Превышение температуры системы над ее равновесным значением приведет к преобладанию скорости испарения воды по сравнению со скоростью конденсации, и тогда изменение энергии Гиббса окажется отрицательной величиной. А при понижении температуры системы относительно ее равновесного значения возобладает скорость конденсации водяных паров, и поэтому ∆G испар будет положительной величиной. Вопросы и задачи для самостоятельной работы 2.4.1. Энтальпийным или энтропийным фактором определяется направление химических реакций при очень низких температурах? 2.4.2. При каких по знаку изменениях энтальпий ∆Н и энтропии ∆S в системе возможны только экзотермические процессы? 2.4.3. Чем определяется направленность процесса в данной системе? 2.4.4. Что больше: энтальпия моля водяных паров или энтальпия моля жидкой воды? Как это понять, если самопроизвольно происходит испарение воды? 2.4.5. Почему неверно утверждение, что во всех самопроизвольных процессах системы стремятся к состоянию с минимальной энергией? 2.4.6. Почему неверно утверждение, что во всех самопроизвольных процессах происходит повышение энтропии? Когда оно справедливо? 65 2.4.7. В каком из следующих случаев реакция возможна при любых температурах: а) ∆Н<0, ∆S >0; б) ∆H>0; ∆S<0; в) ∆ H<0, ∆S<0. 2.4.8. Вычислите, при какой температуре начнется диссоциация пентахлорида фосфора, протекающая по уравнению: РС15газ= РС13газ+С12газ; ∆Н0= + 92,6 кДж. 2.4.9. При какой температуре наступит равновесие в реагирующей системе: СОгаз+ 2 Н2газ=СН3ОНж; ∆Н0= -128, 05 кДж? 2.4.10. Восстановление оксида железа оксидом углерода (П) отражает уравнение: Fe3O4kp+ COгаз = 3 FeOкр + СО2газ. Вычислите изменение энергии Гиббса и сделайте вывод о возможности самопроизвольного протекания этой реакции при стандартных условиях. При какой температуре возможен этот процесс? 4.3. Стандартный изобарно-изотермический потенциал и его изменение Изобарно-изотермический потенциал G является функцией состояния, зависит только от начального и конечного состояния системы, вещества. А поэтому эта функция также проявляет аддитивные свойства, т.е. общий изобарно-изотермический потенциал системы равен сумме потенциалов отдельных ее частей (отдельных веществ): Gсистемы = G1 + G2 + G3 + …….+ Gn, и, как следствие, его изменение ∆G определяется также законом Гесса: ∆Gреакции = Σ ∆Gпродуктов - Σ ∆Gисход. в-ва. (2.4.6) Чтобы можно было сравнивать изобарно- изотермические потенциалы, производить с ними алгебраические действия, необходимо их относить к одним и тем же условиям. По аналогии со стандартными значениями изменения энтальпии ( ∆Н0298 и ∆Н0Т) и энтропии (S0298 и S0T) пользуются системой стандартных энергий Гиббса ∆G0298 и ∆G0Т образования данного вещества. Для простых веществ (О2газ , F2газ , N2газ , Feкр , Cuкр и т.д.) устойчивых в стандартном состоянии принимается значение изобарно-изотермического потенциала ∆G0298 = 0. Обычно в термодинамических справочниках значения функций состояний приведены для Т = 298 К. Значение ∆G зависят от концентрации. Принято для характеристики химических процессов пользоваться значениями ∆G0298, отвечающими условиям, когда парциальное давление (концентрации) всех реагентов в течении всего процесса остаются неизменными и равными единице. То есть это условие предполагает состав реакционной смеси неизменным, и молчаливо допускается, что количество всех веществ в реакционной зоне несоизмеримо больше, чем количество прореагировавших и образовавшихся веществ по уравнению реакции. Так, например, для реакции: Н2газ+ 0,5 О2 газ= Н2Огаз изменение изобарного потенциала ∆G0298 = - 228,8 кДж/моль предполагает следующее: если бы в достаточно большом количестве смеси (водорода, кислорода, водяных паров), взятой при 250С, в которой парциальные давления всех компонентов равны 66 РН2 = РО2 = РН2О = 101 325 Па, т.е. общее давление 3·101 325 Па и все газы идеальны, прореагировало бы по одному молю Н2 и 0,5 моля О2, то в результате энергия Гиббса уменьшилась бы на 228,8 кДж/ моль. Применим закон Гесса к указанной реакции и рассчитаем изменение свободной энергии ∆G0, без отдельных расчетов изменения энтальпии и энтропии процесса, используя табличные данные энергии Гиббса образования исходных веществ и продуктов реакции: ∆Gреак.= Σ∆Gпродук. – Σ∆G0исход.в-ва Или ∆G0реак. = (∆G0Н2Огаз) – (∆G0Н2 газ + 0,5 · ∆G0 О2газ) = -226,8 кДж/ моль. Рассчитанные ∆G0298 для стандартных условий и соответствующие выводы о характере процессов, нужно осторожно переносить на другие условия протекания процессов. Значения ∆S и ∆G сильно зависят и от концентрации реагирующих веществ и температуры. Конечно, если для стандартных условий ∆G0298 значительно меньше нуля, то можно считать, что и при любых реальных условиях прямой процесс возможен (и наоборот, если ∆G0298 > 0, то осуществима обратная реакция). Другими словами, нельзя подменять величину ∆G0Т при любой другой температуре величиной ∆G0298. Для получения сугубо ориентировочных данных можно считать, что: ∆G0T ≈ ∆Н0298 – Т · ∆S0298, Но здесь не учтена зависимость ∆Н и ∆S от температуры. Воспользоваться приведенным выражением можно, но с одним еще условием, что при переходе системы от стандартных условий (температуры) к изучаемым при температуре Т, не происходят фазовые превращения, т.к. это связано с резкими изменениями энтропии системы. 4.4. Изохорно- изотермический потенциал Для характеристики процессов, идущих при постоянном объеме, используется изохорно-изотермический потенциал F (эту функцию еще называют свободной энергией Гельмгольца, или потенциалом Гельмгольца1. Вспомним, что при проведении процессов при постоянном объеме и температуре изменение внутренней энергии системы связано с тепловым эффектом простым соотношением: ∆U = Qv, следовательно, если изменение изобарно-изотермического потенциала рано: ∆G = ∆ H - T ·∆ S, nо изменение изохорно– изотермического потенциала будет равно: Герман Людвиг Гельмгольц (1821-1894) немецкий ученый, разработавшего термодинамическую теорию химических процессов. Ввел понятия свободной и связанной энергии, обосновал математически закон сохранения энергии и многое другое.) и его изменение ∆F. 67 1 ∆F = ∆U – T·∆S, (2.4.7) А так как изменение изохорно- изотермического потенциала ∆F есть разность потенциалов системы в начальном и конечном состоянии ∆F = F2 – F1, где F1 и F2 – изохорно-изотермический потенциал системы в начальном и конечном состоянии соответственно, которые отвечают следующим выражениям: F1 = U1 + T· S1 F2 = U + T· S2. Таким образом, изменение изохорно-изотермического потенциала будет складываться из изменения внутренней энергии и энтропии системы: ∆F= F2 – F1 = (U2 - U1) - T (S2-S1), ∆F= ∆U - T· ∆S. Критерием самопроизвольной реакции при протекании ее в изохорноизотермических условиях является ∆Н также условие: отрицательная величина изменения изохорно-изотермического ∆U P∆V потенциала ∆F < 0, T∆S ∆F P∆V как и ∆G < 0 для изобарно- изотермических условий. T∆S ∆G Численно ∆G и ∆F отличаются друг от друга на работу расширения системы Р∆V. В справочниРис.2.4.2. Диаграмма соотношения терках обычно приводятся только измодинамических функций менения изобарного потенциала ∆G (как и ∆H). Но из значений ∆G и ∆H легко получить и все остальные, используя ранее выведенные соотношения: ∆Н = ∆ U + P∆V, ∆G = ∆ H – T∆S, ∆F = ∆U - T ∆S. Так, например, ∆G= ∆H - T∆ S = ∆ U+ P∆V – T∆S = ∆ F+ P∆V = ∆ F + ∆n RT. Для более удобного запоминания соотношения термодинамических функций пользуются диаграммой представленной на рисунке 2.4.2. 68 3. ОСОБЕННОСТИ ТЕРМОДИНАМИЧЕСКОГО МЕТОДА ИССЛЕДОВАНИЯ Итак, мы рассмотрели энергетические изменения, сопровождающие химические превращения, основные закономерности, условия превращения веществ, ввели новые термодинамические функции и проанализировали их. Установили, что самопроизвольно происходят лишь взаимодействия, которые характеризуются уменьшением изобарно-изотермического потенциала (∆G<0); условию равновесия отвечает равенство нулю изменения изобарноизотермического потенциала (∆G = 0), в котором отражается противоборство двух тенденций в физико-химических системах: одна из них уменьшение энергосодержания системы за счет взаимодействия частиц с образованием упорядоченной структуры, другая – возрастание энтропии, увеличение беспорядка в системе; переход к независимому, самостоятельному существованию частиц вещества, равномерному распределению тепловой энергии по всему объему системы. Термодинамический подход позволяет не вдаваться в подробное изучение химических связей в рассматриваемых соединениях, их природы, строения и т.д. Он (метод) исходит только из факта, что при взаимодействии таких-то веществ с образованием определенных продуктов выделилось (поглотилось) столько-то энергии и в дальнейших рассуждениях опирается только на этот факт. Зная энергетические характеристики отдельных веществ (опятьтаки, не спрашивая, почему они таковы), мы можем понять причину, почему одни реакции протекают, другие – нет; одни вещества устойчивы, другие – нет. Это достоинство метода, когда требуется лишь только принципиальное заключение о возможности того или иного процесса. Но термодинамический метод, позволяя сделать принципиальный вывод о возможности процесса, не говорит однозначно – будет ли в действительности протекать процесс с практически измеримой скоростью и как. Например, известная реакция взаимодействия водорода с кислородом: Н2газ+ 0,5 О2газ= Н2Ож при стандартных условиях имеет значение изменения изобарноизотермического потенциала равное ∆G0298 = - 237,5 кДж/моль (явно ∆G0298<0). Следовательно, реакция должна протекать. Но если смешать кислород и водород, то даже по истечении длительного времени, мы не сможем обнаружить и следов воды в этой системе. Тем не менее, этот процесс протекает, но с ничтожно малой скоростью, которая определяется какими-то другими факторами, о которых термодинамический метод ничего не говорит. Термодинамический метод, фиксируя начальное и конечное состояние системы, совершенно игнорирует переходный процесс, временной фактор. Этот пробел устраняется в другом разделе химии – химической кинетике. 69 В химической кинетике вводятся новые понятия, и устанавливается их связь с термодинамическими величинами. Все это и является предметом рассмотрения следующего раздела. Глава III. ХИМИЧЕСКАЯ КИНЕТИКА И РАВНОВЕСИЕ Введение Любой процесс, любая реакция протекает во времени. Одни процессы заканчиваются относительно быстро; система переходит в конечное, равновесное состояние за доли секунды, минуты, часы; другие требуют для своего завершения десятки, сотни, миллионы лет. От каких факторов зависит скорость процессов, как она связана со строением, свойствами веществ, их природой? И если химическая термодинамика дает принципиальный ответ о возможности процессов в данных условиях, то химическая кинетика изучает закономерности протекания химических процессов во времени, их механизмы; зависимости скорости процесса от состава, строения, природы реагирующих веществ, температуры и присутствия катализаторов. Знание закономерностей и механизма, в свою очередь, позволяет сознательно управлять химическими процессами. Но прежде чем начнем рассматривать кинетические закономерности, вначале познакомимся с некоторыми основными понятиями химической кинетики. 1. ОСНОВНЫЕ ПОНЯТИЯ ХИМИЧЕСКОЙ КИНЕТИКИ. 1.1. Механизм химической реакции Большинство химических реакций являются сложными в том смысле, что они состоят из нескольких простых, одновременно протекающих процессов, суммарный результат которых отражает обычная запись уравнений реакции. Как правило, химические реакции протекают через образование промежуточных частиц, взаимодействия их между собой, с исходными веществами и опять с образованием новых частиц, которые только в конечной или промежуточной стадии процесса превращаются в продукты реакции. Очень редко встречаются процессы, в которых исходные частицы (молекулы, атомы) столкнувшись, образуют сразу же частицы продуктов реакции. Например, казалось бы, на первый взгляд простая реакция в записи уравнения: Н2 + С12 = 2 НС1, должна протекать путем непосредственного столкновения молекулы хлора с молекулой водорода и с образованием сразу же двух молекул соляной кислоты (в соответствии с уравнением реакции). Но эта реакция протекает совершенно не так. Вначале происходит распад молекулы хлора на возбужденные (активные) атомы хлора под действием кванта энергии, полученного или при 70 нагревании, облучении или при столкновении с более активными частицами системы, стенками сосуда: (первая стадия) С12 + hν = 2 Cl* Затем один из возбужденных атомов хлора с молекулой водорода Cl*, в свою очередь, сталкивается (вторая стадия) С1*+ Н2 = НС1 + Н* с образованием первой молекулы соляной кислоты НС1 и возбужденного атома водорода Н*, который далее взаимодействует с молекулой хлора (третья стадия) Н* + С12 = НС1+ С1* Возбужденные атомы водорода и хлора, образовавшиеся во второй и третьей стадии, или продолжают реакции взаимодействия с выделением молекул соляной кислоты и новых активных частиц по цепному механизму или инактивируются (отдают свою энергию) на стенках сосуда. Этот сложный путь реакции оказывается, тем не менее, более выгодным, чем непосредственное столкновение молекул водорода и хлора с образованием молекул соляной кислоты. Совокупность стадий, из которых складывается химическая реакция, носит название механизма реакции. Каждая стадия процесса описывается уравнением простой (элементарной) реакции. 1.2. Гомогенные и гетерогенные реакции По фазовому признаку различают два типа реакций: гомогенные и гетерогенные. Фаза представляет собой часть системы, отделенная от других ее частей поверхностью раздела, при переходе через которую свойства системы изменяются скачкообразно, резко. Примером однофазной, однородной (гомогенной) системы может служить любая газовая смесь – все газы при стандартных условиях неограниченно растворимы друг в друге. Во всех участках объема данной системы параметры ее состояния (плотность, температура, состав, упругость и т.д.) одинаковы (рис.3.1.1а). Аналогичным примером гомогенной системы может служить раствор нескольких веществ в одном растворителе, например, раствор хлорида, натрия, нитрата магния, кислорода и азота в воде. Как в том, так и в другом случае все свойства системы в любой ее части тождественны. Соответственно, и реакции (например, между хлором и водородом, находящимся в газообразном состоянии), протекающие в такой системе будут являться гомогенными. Химические реакции, протекающие в пределах одной фазы, называют гомогенными. 71 Особенностью протекания гомогенной реакции является то, что она протекает во всем объеме системы. То есть в гомогенной системе (однородной системе) нет каких-то особых, приоритетных участков, микрообъемов, где реакция между веществами протекает, а в других не происходит. О2 газ Н2Опар Н2 газ + Сl2 газ лед уголь а - гомогенная Н2Ож б – гетерогенные системы Рис.3.1.1. Иллюстрация гомогенных и гетерогенных систем и соответствующих реакций: а – гомогенная (однородная) система из водорода и хлора, в которой реакция протекает в любой части занимаемого объема; б – примеры гетерогенных систем: двухфазной в случае взаимодействия угля с кислородом и трехфазной для состояния воды (вода в кристаллическом виде, вода в жидком состоянии и вода в газообразном состоянии). В гетерогенном состоянии имеется граница раздела между частями системы, через которую и осуществляется взаимодействие В качестве примера гетерогенной системы может служить, например, уголь с кислородом; вода в жидком, кристаллическом и парообразном состоянии (рис.3.1.1б). в том и другом случае имеется поверхность раздела фаз, обладающих различными свойствами. А реакции (например, горение угля и кислорода), протекающие на границе раздела фаз называют гетерогенными. Особенностью таких взаимодействий является то, что они протекают на границе раздела фаз в данной системе. Так, уголь окисляется кислородом только на поверхности раздела фаз, железо коррозирует тоже с поверхности, вода испаряется или с поверхности жидкости, или кристалла – это все примеры гетерогенных взаимодействий. Поэтому, когда говорят о скорости того или иного процесса (реакции), всегда имеют в виду особенность его протекания в гомогенной и гетерогенной системе. 1.3. Скорость реакции При взаимодействии реагентов (исходных веществ) происходит изменение количества реагирующих веществ: уменьшение количества исходных веществ и увеличение количества продуктов реакции. 72 Количество реагирующих веществ и образовавшихся продуктов реакции взаимосвязаны стехиометрическим уравнением реакции, поэтому о скорости реакции можно судить по изменению количества любого из веществ, участвующих в реакции. Используя стехиометрические коэффициенты, можно всегда скорость реакции по одному веществу пересчитать на скорость реакции по другому веществу. Однако, чтобы изменение количества вещества не было безотносительным (действительно, в реакции можно использовать разные количества вещества) его относят к единице объема в случае гомогенной реакции или к единице поверхности раздела фаз – в случае гетерогенной реакции. В самом деле, происходит ли изменение массы реагирующего вещества (например, того же хлора при взаимодействии с водородом) в объеме пробирки или цистерны; между куском угля или его крошкой с кислородом – разница есть и довольно существенная. В первом случае скорость расходования вещества будет обратно пропорциональна объему, во втором – прямо пропорциональна величине поверхности. Поэтому в определении скорости реакции должно фигурировать понятие концентрации вещества, а в случае гетерогенных реакций скорость превращения следует относить и к единице поверхности раздела фаз. А поскольку изменение количества вещества определяют во времени, его нужно относить к единице времени. Таким образом, общее определение скорости реакции можно сформулировать так: это число элементарных актов превращения вещества, происходящих в единице объема (для гомогенных реакций) или на единице поверхности раздела фаз (для гетерогенных реакций) за единицу времени. Принимая во внимание, что количество вещества в единице объема – не что иное, как концентрация вещества, можно дать определение С скорости реакции и так: С0 А скорость реакции это измеСD нение концентрации реагирующеD го вещества за единицу времени. С2 Предположим, что имеется реC1D акция, описываемая следующим уравнением : C1А mA + nB = pD + gF, A А С С2 где А и В исходные вещества; D и О F – продукты реакции; m, n, р и g t1 t2 время, t – стехиометрические коэффициенты. Рис. 3.1.2. Изменение концентрации реаО скорости этой реакции гирующих веществ и продуктов во времожно судить по скорости мени уменьшения концентрации исход73 ного вещества А и В; или же по скорости увеличения (накопления) продуктов D и F. Графически изменение концентрации веществ А и D во времени отображаются зависимостями, представленными на рис. 3.1.2. (кривые CA и CD соответственно). Скорость химической реакции изменяются с течением времени из-за различных факторов: изменения концентрации исходных веществ и их свойств и многих других, поэтому различают среднюю и истинную скорость реакции. Средней скоростью химической реакции называют изменение концентрации ∆С реагирующего вещества, отнесенное к конечному интервалу времени ∆t, в течение которого это изменение произошло. Применительно к рассматриваемой реакции, согласно рис. 3.1.2, можно написать выражение для средней скорости реакции за интервал времени ∆t по веществу А: V А сред. С 2А − С1А ∆C A , =− =− t 2 − t1 ∆t или по компоненту (веществу) D: D Vсред . = С 2D − С1D ∆C D . =+ ∆t t 2 − t1 Скорость реакции всегда считается положительной величиной. Отношение же ∆C/ ∆t может быть как положительным, так и отрицательным, в зависимости от соотношения концентраций в начальном и конечном моменте времени. В соответствии с этим перед выражением скорости химической реакции в общем случае принято ставить знаки «±»: Vсред. = ± ∆C ∆t Истинной скоростью реакции V в данный момент времени называется изменение концентрации какого-либо компонента, отнесенное к бесконечно малому промежутку времени V = lim ∆t →0 dC ∆C , =± ∆t dt т.е. производная концентрации по времени. Размерность скорости выражается в единицах концентрации, отнесенной к единице времени. Концентрацию в химической кинетике принято выражать либо числом молекул в единице объема (например, число молекул/ см3, или просто см -3 ), либо числом молей в одном литре (моль/л). Соответственно и скорость будет измеряться в соответствующих единицах: молекул / см3 · с; молекул / м3 · час и т.п., либо: моль / л· час , : моль / л· с и т.д. Скорость реакции зависит от ряда факторов: природы реагирующих веществ, концентрации, температуры и присутствия катализаторов, что мы и рассмотрим в следующих разделах. 74 Вопросы и задачи для самостоятельной работы 3.1.1. Какие из ниже перечисленных систем являются однофазными а) лед и вода жидкая; б- водород и хлор при стандартных условиях; в- водород и оксид железа; г- воздух и раствор сахара в воде; д- водный раствор солей NaCl, NaNO3, CaCl2? 3.1.2. В чем отличие гомогенных и гетерогенных реакций? 3.1.3. Какие из перечисленных реакций являются гетерогенными : а- взаимодействие водорода и кислорода при стандартных условиях; б- окисление железа кислородом воздуха; в – горение природного газа в газовой кухонной плите; г- взаимодействие металлического натрия с водой; д– разложение известняка на оксид кальция и диоксид углерода; евзаимодействие известняка с раствором соляной кислоты? 3.1.4. На сколько уменьшилась концентрация вещества В, если концентрация вещества А уменьшилась на 0,7 моль/л в реакции: 2А+В = С? 3.1.5. На сколько увеличилась концентрация вещества Д, если концентрация вещества В уменьшилась на 0,3 моль/л в реакции: А+3В = С + 2Д? 3.1.6. Сгорает 1 кг кускового угля и 1 кг угольного порошка, приготовленного из того же куска, при одном и том же давлении (концентрации) кислорода. С какой скоростью протекали эти процессы: с одинаковой, равной? 3.1.7. При постоянной температуре, в одинаковых по форме и равных по объему сосудах образовалось 36 г Н2О, 18 г Н2, 136 г Н2S за одно и то же время. Скорость какой реакции была наибольшая? Почему? Какие скорости сравниваются: истинные, средние? 2. ВЛИЯНИЕ НЕКОТОРЫХ ФАКТОРОВ НА СКОРОСТЬ РЕАКЦИИ 2.1. Зависимость скорости от концентрации реагирующих веществ Рассмотрим простейшую реакцию между двумя молекулами вещества А и В, протекающую согласно уравнения А + В = АВ в объеме системы V (рис.3.2.3а). Продукт реакции АВ образуется лишь только после столкновения исходных молекул А и В, что определяется вероятностью столкновения этих молекул за определенный промежуток времени. Следовательно, скорость реакции будет пропорциональна вероятности столкновения молекул А и В. Если же в этом объеме увеличить количество молекул, например, компонента А в два раза, т.е. увеличить концентрацию молекул А в два раза (рис.3.2.3б), то, соответственно, увеличится и частота столкновений молекул А с молекулой В во столько же раз. Значит, и скорость реакции возрастет в два раза. Если же теперь еще увеличить и концентрацию (число молекул в том же самом объеме) вещества В в два раза (рис.3.2.3.в), то вероятность столкновения молекул А и В увеличиться в 4 раза. Соответственно, возрастет и скорость реакции тоже в 4 раза. Следовательно, скорость образования вещества АВ будет пропорциональна концентрациям веществ А и В: 75 V ~ CА · CВ . А В а б в Рис. 3.2.3. Реакционный объем для: а – двух молекул, б – трех молекул, в – четырех молекул А если концентрацию компонента А изменить в m раз, концентрацию компонента В в n раз, то число столкновений (вероятность) измениться в m·n раз. Во столько же раз измениться и скорость реакции: V ~ CmА · CnВ . Чтобы заменить символ пропорциональности «~» в этих выражениях знаком равенства для составления уравнений скорости реакции, введем коэффициент пропорциональности «k». Тогда для простой реакции, когда участвуют только молекулы двух веществ А и В, уравнение запишется в виде следующего выражения: V = k · СА · СВ, а для реакций более общего вида типа mA + n B = Am Bn уравнение отобразится таким образом: V = k · CmА · CnВ . (3.2.1) Это и есть аналитическое выражение зависимости скорости реакции от концентрации реагирующих веществ. Закон установлен в 1867 году норвежскими учеными Като Максимилианом Гульдбергом (1836-1902) и Петером Вааге (1833-1900). Оно известно под названием закона действующих масс1, который читается так: скорость химической реакции прямо пропорциональна произведению концентрации реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции при условиях постоянства всех остальных факторов (температуры, состава). Под действующими массами во времена К.М. Гульдберга и П. Вааге (профессора математики, химии и технологии университета в Осло) понимали то, что мы сейчас называем концентрациями. 76 1 Для сложных процессов, протекающих в несколько стадий, этот закон применим только к отдельным, элементарным (простым) стадиям процесса. В качестве примера приложения закона действующих масс приведем зависимость скорости простой реакции восстановления оксида азота водородом, протекающей согласно уравнению 2NOгаз + H2газ = N2Oгаз + H2Oпар , в виде выражения: V = k C2NO · CH2. Для гетерогенных реакций, у которых принято относить скорость реакции к единице поверхности контакта фаз, закон действующих масс будет записываться с учетом концентрации только того из реагирующих средств (газообразного для системы: «газ – твердое», «газ- жидкость», и жидкого для системы «твердое- жидкость»), которым определяется скорость реакции. Например, для реакции горения угля: Сграфит + О2 газ = СО2 газ скорость реакции при заданных условиях (температура, состав) будет зависеть лишь от концентрации кислорода, и не будет зависеть от объемной концентрации угля, т.к. скорость взаимодействия относят к единице поверхности раздела фаз (рис.3.2.4а). О2 СО2 Zn Н2Ож уголь а б в Рис. 3.2.4. К понятию скорости гетерогенной реакции и применимости закона действующих масс: а – взаимодействие угля с кислородом, б – взаимодействие СО2 с водой, в - взаимодействие раствора НСl с цинком Поэтому выражение закона действующих масс для этой реакции записывается так: V1 = k1Co2 без учета концентрации угля. Гетерогенная реакция взаимодействия диоксида углерода с водой СО2 газ + Н2Ож = Н2СО3 ж определяется лишь концентрацией диоксида углерода, что и отражается в выражении закон действующих масс для этой реакции: V2 = k2 CCO2 . В выражении закона действующих масс для реакции взаимодействия цинка с раствором соляной кислоты 77 Znкрис. + HClж = ZnCl2 раствор + Н2 газ будет фигурировать лишь концентрация соляной кислоты: V3 = k3 C2 HCl . Физический смысл коэффициента пропорциональности «k» в выражении закона действующих масс становится ясным, если принять концентрации реагирующих веществ (или их произведение) равным единице. Так, для той же самой формальной реакции, описываемой уравнением скорость реакции равна mA + n B = Am Bn , V = k CmA · CnB. Если использовать концентрации реагирующих веществ А и В равными СА = СВ = 1 моль/л , то скорость реакции окажется равной коэффициенту пропорциональности k: V =k Коэффициент пропорциональности k в выражении закона действующих масс называют константой скорости. Численное значение константы скорости реакции равно количеству вещества, прореагировавшего за единицу времени при концентрации исходных веществ, равных единице (например, 1моль/л). Следовательно, константа скорости это удельная скорость химической реакции, отнесенная к единичным концентрациям реагирующих веществ, а для гетерогенных реакций и к единичной поверхности раздела фаз (к 1см2 или 1 м2). Константа скорости химической реакции показывает, какая доля из общего числа соударений молекул веществ А и В приводит к химическому взаимодействию. Если реакция осуществляется в результате столкновения молекулы А с молекулой В, то число столкновений, а следовательно, и скорость реакции будет пропорциональна концентрациям веществ А и В. Если для химического превращения необходимо, чтобы одновременно сталкивались по две или по три одинаковы[ молекул, то скорость реакции будет пропорциональна квадрату или соответственно кубу концентраций этого вещества. Константа скорости реакции зависит от температуры, присутствия катализатора и строго индивидуальна для каждой реакции, т.е. определяется и природой взаимодействующих веществ. Порядок реакции. В зависимости от вида кинетического уравнения, связывающего скорость реакции с концентрацией реагирующих веществ, различают реакции нулевого, первого, второго и третьего порядка. Если скорость реакции не зависит концентрации, то реакция имеет нулевой порядок; если скорость реакции зависит от концентрации в первой степени, то это реакция первого порядка; если – во второй, то второго порядка и так далее. В общем случае, например, для формальной реакции αА + βB + δD + ….. = fF + lL + …. 78 зависимость скорости от концентрации выражается кинетическим уравнением: υ = k C αA ⋅ C Bβ ⋅ C Dδ ...., где показатели степени α, β и δ могут быть любыми небольшими числами, чаще всего равными 1 или 2, редко 3. Показатели степени концентрации определяются на основании экспериментальных данных. При некоторых условиях показатель может быть равным и нулю. Кроме того, известно немало реакций с дробными показателями. Показатель степени концентрации реагирующего вещества в кинетическом уравнении реакции (α, β, δ) называется порядком реакции по данному веществу ( А, В и D соответственно). Общим порядком химической реакции, или просто порядком реакции, называется величина, равная сумме показателей степени концентраций реагентов в кинетическом уравнении реакции. Общий порядок реакции = α + β + δ + …. Показатель степени концентрации данного вещества в кинетическом уравнении, как правило, не совпадает с его стехиометрическим коэффициентом в уравнении реакции. Для элементарных, простых реакций, т.е. реакций протекающих в одну стадию, показатели степени в кинетическом уравнении совпадают со стехиометрическими коэффициентами реагентов в уравнении реакции. Единицы измерения константы скорости k зависят от суммы показателей степеней при концентрациях (порядка реакции) в выражении закона действующих масс и типа реакции (гомогенная или гетерогенная реакция). В таблице 3.2.1 приведены примеры размерности константы скорости реакции в зависимости от порядка реакции, которые легко получить, помня, что k= V . C A ⋅ CB Таблица 3.2.1 Размерность константы скорости в зависимости от порядка реакции Порядок реакции 0 1 2 Размерность k для гомогенной реакции Размерность k для гетерогенной реакции моль/л·с 1/с л/ моль·с моль/л·с·см2 1/с·см2 л/ моль·с·см2 Например, для гетерогенной реакции второго порядка, описываемой уравнением Акрис. + 2 Вгаз = АВ2 крис. , выражение закона действующих масс запишется в виде зависимости: 79 V = k C2B. Из такой записи видно, что это реакция второго порядка (показатель степени концентрации реагирующего вещества В равен 2), следовательно, размерность константы скорости этой реакции равна Единицы измерения k = единицы скорости реакции / ( единицы концентрации)2. После подстановки получаем: Единицы измерения k = моль л = 2 л ⋅ см ⋅ с ⋅ ( моль / л) моль ⋅ см 2 ⋅ с 2 По числу частиц, фактически участвующих в элементарном акте химического превращения, реакции делят на мономолекулярные, в которых осуществляются реакции при участии одной молекулы, бимолекулярные – для осуществления взаимодействия требуется участие двух частиц, тримолекулярные – для осуществления реакции необходимо уже три молекулы. Вполне понятно, что вероятность одновременного столкновения сразу трех и большего числа молекул меньше, чем вероятность столкновения двух молекул. Поэтому тримолекулярные реакции менее вероятны, чем бимолекулярные. Если в реакцию и вступает формально (в соответствии с уравнением реакции) более трех молекул, то в действительности оказывается, что реакция проходит через ряд промежуточных стадий, в каждой из которых участвуют одна, две молекулы. Молекулярность и порядок реакции не всегда совпадают. Практически никогда не встречаются реакции выше третьего порядка, так как для осуществления тримолекулярной реакции требуется столкновение трех частиц одновременно. Вероятность такого столкновения быстро убывает с увеличением числа частиц. Подавляющее большинство реакций протекает в несколько стадий, т.е. они являются сложными. При этом каждая стадия имеет свой порядок реакции. Скорость же всей реакции в целом определяется скоростью наиболее медленной стадии, которая, следовательно, и определяет кажущийся порядок всей реакции. Например, в реакции, выражаемой молекулярным уравнением 6 FeCl2раств. + KClO3раствор + 6 HClраствор = 6 FeCl3раств. + KClраствор + 3 H2Oж , общий порядок должен быть равен 13, а в действительности реакция протекает как реакция третьего порядка. Рассмотрим несколько примеров использования закона действующих масс при решении практических задач. Пример 1. Реакция между кислородом и железом протекает по уравнению 2 Fekp + 3·О2газ = 2 Fe2O3 кр. Как измениться скорость реакции при увеличении давления кислорода в 2 раза? Решение. Данная реакция гетерогенная, и закон действующих масс для нее запишется в виде выражения 80 V = k C3О2 без учета концентрации железа. Предположим, что концентрация кислорода при исходном давлении равна 1 моль/л, тогда скорость реакции в этих условиях будет равна V1 = k 13 моль/л. После увеличения давления в два раза, концентрация кислорода также увеличивается в 2 раза, т.е. CО2 = 2 моль/л, соответственно и скорость реакции будет определяться следующим выражением: V2 = k 23 моль/л. Разделив скорость реакции полученную после увеличения давления на таковую в начальном состоянии V2/V1, узнаем во сколько раз изменилась скорость реакции V2 k ⋅ 2 3 = = 8 раз V1 k ⋅ 13 Пример 2. Во сколько раз следует увеличить давление, чтобы скорость образования диоксида азота по реакции 2 NOгаз + O2газ = 2 NO2 газ Возросла в 1000 раз? Решение. Это гомогенная реакция. И выражение закона действующих масс запишется для исходных условий таким образом: V1 = k C2NO · CO2 . После увеличения давления в n раз возросла и концентрация реагирующих веществ в n раз. Выражение закона действующих масс для возросших концентраций в реагирующей системе выразится уравнением: V2 = k (nCNO)2 ·(n CO2) Согласно условию задачи отношение скоростей должно различаться в 1000 раз, т.е. V2 k ⋅ (nC NO ) 2 ⋅ (nCo 2 ) = = 1000 . 2 V1 k ⋅ C NO ⋅ Co 2 Упростив выражение, получим n3 = 1000. Откуда n = 10. Так как давление и концентрация газа пропорциональны, то изменение давления тоже должно быть увеличено в 10 раз. Вопросы и задачи для самостоятельной работы 3.2.1. Напишите выражение закона действующих масс для реакции, протекающей между водородом и кислородом; между оксидом азота (П) и кислородом до получения N2O5; между азотом и водородом с образованием аммиака; между оксидом железа (Ш) и водородом с получением железа и воды; раствором медного купороса и железом. 3.2.2. Напишите выражение закона действующих масс для химической реакции, протекающей в гомогенной системе по уравнению: А + 2В = АВ2 и определите, во сколько раз увеличится скорость этой реакции, если: а – концентрацию вещества А увеличить в два раза; б – концентрацию вещества В увеличить в два раза; в – концентрацию и вещества А и В увеличить в два раза? 3.2.3. Во сколько раз следует увеличить концентрацию водорода в системе: N2 газ + 3 Н2 газ = 2 NH3 газ , 81 чтобы скорость реакции возросла в 64 раза? 3.2.4. Во сколько раз изменится скорость простой гомогенной реакции: а) Н + Cl = HCl б) 2 NO + Cl2 = 2 NOCl в) N2O4 = 2 NO2 при увеличении давления в системе в два раза? 2.2. Зависимость скорости реакции от температуры Для подавляющего числа реакций повышение температуры приводит к увеличению скорости реакции. Вант-Гоффом1 опытным путем было установлено, что: при увеличении температуры на 10 градусов скорость гомогенной химической реакции возрастает в γ = 2—4 раза, т.е. скорость реакции при температуре t2 = t1 ± 10, будет отличаться от таковой при температуре t1 в γ раз. Если же температуры различаются в несколько раз, то и скорости будут отличаться соответственно в γ Vt 2 = Vt1 ⋅ γ t 2 −t1 10 раз: t 2 −t1 10 (3.2.2) Число γ, которое показывает, во сколько раз возрастает скорость реакции при увеличении температуры на 10 град, называют температурным коэффициентом скорости реакции. Вычисляют температурный коэффициент скорости реакции по уравнению: γ10 = k t +10 . kt (3.2.3) Нижний индекс при γ показывает температуру, которой соответствует константа. Зависимость позволяет рассчитать скорость реакции при другой температуре, если известна она для данной температуры, или оценивать, во сколько раз измениться скорость реакции при изменении температуры реагирующей системы. Например, во сколько раз изменится скорость реакции при повышении температуры от 200 до 700 С, если температурный коэффициент равен двум? Воспользуемся правилом Вант –Гоффа, согласно которому скорость реакции при температуре равной t2 = 700 C, будет равна t 2 −t1 10 Vt 2 = Vt1 ⋅ γ Разделив левую и правую часть уравнения на значение скорости реакции Vt1 при температуре 200 C, получим: 70 − 20 Vt 2 = 2 10 = 2 5 = 32 . Vt1 Таким образом, скорость реакции при температуре 700 С больше таковой при 200 С в 32 раза. 1 Якоб Генрих Вант – Гофф (1852-1911) знаменитый нидерландский физико-химик, первый лауреат Нобелевской премии по химии (1901), один из основателей учения о растворах, химической кинетики. 82 Самое простое объяснение факта возрастания скорости реакции с увеличением температуры, которое первым приходит в голову, связано с тепловым движением частиц (молекул, атомов). С увеличением температуры повышается скорость движения частиц, возрастает вероятность их столкновения, и как следствие, увеличивается скорость реакций. Следовательно, повышение температуры на 10 градусов должно привести и к повышению скорости движения молекул 2 – 4 раза. Но анализ, если воспользоваться кинетической теорией газа, показывает, что повышение температуры на 100, приводит к незначительному увеличению скорости движения частиц. Например, соотношение скоростей перемещения частиц при температуре Т1 = 293 К и Т2 = 3030 К, равной 293 + 100, составит величину: u2 T2 = = u1 T1 303 ≈ 1,017 раза , 293 где u1 и u2 – скорости движения молекул газа при температуре Т1 и Т2, соответственно. И, тем не менее, скорость реакции изменяется, примерно, в 2 – 4 раза! Оказывается, что не любое столкновение частиц приводит к химическим превращениям (на это указывают экспериментальные данные). Объясняя опытные данные, С. Арn рениус1 предположил, что столкновения молекул, частиц будут эффективны (приn1сред Т1 ведут к химическим превращениям) при условии, что Т2 n2сред сталкивающиеся молекулы обладают некоторым избытn2ак. ком (запасом) энергии (необходимым для разрыва старых n1ак. связей в молекулах и образоЕак Е`сред. Е``сред. Е вания новых) по сравнению Рис. 3.2.5. Распределение доли молекул по со средней энергией молекул энергиям для температур Т1 и Т2 = Т1 + 100 в рассматриваемой системе при данной температуре. Молекулы, несущие эту избыточную энергию, называют активными, а сам избыток энергии - энергией активации, которую обозначают Еак. Химическое взаимодействие наиболее вероятно при столкновении молекул, имеющих большой запаса энергии. Чем меньше энергия активации Еак, тем большее число столкновений частиц оказываются эффективными при той же температуре, и соответственно, выше скорость реакции. 1 Сванте Август Аррениус (1859-1927) – выдающийся шведский физико-химик, автор теории электролитической диссоциации, один из основателей учения о химической кинетики, лауреат Нобелевской премии (1902). 83 Кинетическая энергия молекул определяется температурой Т лишь в среднем. В каждый момент времени в газе, если взять, например, один моль газа, имеются молекулы, у которых энергия больше энергии активации Еак., так и те , у которых она меньше. В среднем количество молекул, обладающих той или иной энергией, в состоянии теплового равновесия не меняется. Распределение молекул по энергиям при температуре Т1 и Т2 отображаются зависимостями представленными на рис.3.2.5 в координатах: доля молекул n энергия. Наибольшее число молекул обладают энергией близкой к средней (наивероятнейшей) Е`сред. и Е``сред.. соответственно для температур Т1 и Т2. Причем, чем больше энергия молекул отличается от среднего значения, тем их меньше. При любой температуре имеется некоторое количество молекул, обладающих запасом энергии Е ≥ Еак., необходимым для осуществления реакции (взаимодействия). Но доля таких молекул, как видно из рисунка 3.2.5 при температуре Т1 и Т2 разная. При температуре Т2 она явно больше раза в два, как представлено на рисунке, по сравнению с температурой Т2 (сравните значения n1ак. n2ак.). То есть нагревание газа приводит к увеличению доли активных молекул. Доля активных молекул α есть отношение числа активных молекул nак к общему числу молекул: α = nак. / N , а её связь с температурой и энергией активацией дается распределением Максвелла-Больцмана α = (3.2.4) Принимая во внимание, что взаимодействие между молекулами осуществимо, лишь при условии наличия у них энергии Еак достаточной для разрушения старых связей и образования новых, следовательно, и скорость реакции будет пропорциональна доле активных молекул в реагирующей системе: V ~ α. Применительно к удельной скорости реакции, т.е. к константе скорости реакции k можно также записать: k ~ α, или k~ Освободимся от знака пропорциональности в последнем выражении, для чего введем коэффициент пропорциональности k0. Тогда получим уравнение для зависимости скорости реакции от температуры и энергии активации: (3.2.5) Это и есть уравнение Аррениуса, которое им найдено при обработке экспериментальных данных и изначально было представлено в логарифмической форме: ln k = ln k 0 − 84 E aк. . RT (3.2.6) Энергию активации выражают в джоулях на моль (Дж/моль). После преобразования уравнения Аррениуса, если известно значение константы скорости k1 и k2 при температуре Т1 и Т2 соответственно, получают довольно простое выражение для аналитического расчета энергии активации процесса: kt 2,3 ⋅ R ⋅ lg 2 ⋅ Т 1 ⋅ Т k t1 Ea.к. = , (3.2.7) Т 2 − Т1 где kt2 — константа скорости реакции при температуре Т2; kt1 — константа скорости реакции при температуре T1; R — газовая постоянная. Следовательно, найдя из lnk опытных данных константы скорости реакции при двух температурах, аналитически можно рассчитать энергию активации данной реакции. Чаще всего прибегают к α lnk0 графическому способу определения энергии активации. Для чего экспериментальные данные представляют в координатах: lnk 1/Т - 1/T. На графике получается прямая линия, тангенс угла наРис. 3.2.6. Зависимость логарифма константы клона которой равен: скорости реакции от обратной температуры tga = Еак. / R . Тангенс угла наклона рассчитывают по отношению катетов треугольника. В выражении константы скорости реакции − Еак . RT k = k0e остается не совсем ясным физический смысл коэффициента пропорциональности «k0» и соответственно всего выражения в целом. Постоянная k0 является произведением двух величин: числа столкновений частиц Z и стерического множителя «р»: k0 = Z ·p. (3.2.8) Подставляя в выражение константы скорости произведение этих двух величин, р − Еак . RT (3.2.9) Множитель e является долей молекул, обладающих энергией активации. Произведение числа столкновений Z на долю активных столкновений, есть не что иное, как число активных (эффективных) соударений: 85 − Еак . RT Z ⋅e . Теперь становится понятнее смысл выражения 3.2.9: константа скорости реакции – это есть число активных соударений, умноженных на некий поправочный коэффициент «р», называемый стерическим множителем. Суть стерического множителя выясним несколько позже, а пока рассмотрим несколько примеров решения задач с использованием уравнения Аррениуса. Пример 1. Известно, что энергия активации некоторого процесса равна Еак. = 100 кДж/моль. Во сколько раз увеличится скорость реакции, если увеличить температуру с 200 С до 300 С? Решение. При температуре t1 = 200 C (или 293 К) константа скорости реакции k1, согласно уравнению Аррениуса, будет равна: k1 = k 0 e − Еак . RT1 , 0 а при 30 С (303 К) она равна: − Еак . RT2 k2 = k0e . Разделим второе выражение на первое и подставим численные значения известных величин. − Eak RT2 Eak ⋅(T2 −T1 ) 100000⋅( 303− 293) k2 k0 ⋅ e = = e R⋅T1 ⋅T2 = e 8,31⋅303⋅293 = 3,88 раза − Eak k1 k 0 ⋅ e RT2 При увеличении температуры на 100 скорость реакции вырастет в 3,88 раза, что хорошо совпадает с эмпирическим правилом Вант-Гоффа. Пример 2. Скорость реакции возросла в два раза при повышении температуры на 100. Рассчитайте энергию активации этой реакции вблизи 300 К. Решение. Согласно условию задачи отношение констант скоростей реакции при температуре, скажем, Т1 = 295 К и Т2 = 305 К должно быть равно двум: k2 / k1 = 2. Воспользуемся уравнением 3.2.7. и подставим наши численные данные: 2,3 ⋅ R ⋅ lg Еак. = k t2 k t1 ⋅Т1 ⋅Т Т 2 − Т1 = 2,3 ⋅ R ⋅ lg 2 ⋅ 295 ⋅ 305 = 51,815кДж / моль 305 − 295 Отсюда получаем значение энергии активации данного процесса, равное 51,815 кДж/моль. Вопросы и задачи для самостоятельной работы 3.2.5. Константа скорости некоторой реакции при 200 С равна 3·0-2, а при 500С она составляет величину 4·10-1 (условных единиц). Определите энергию активации и рассчитайте константу скорости при 300С. 3.2.6. Вычислите, во сколько раз возрастет скорость реакции при уменьшении энергии активации на 100 кДж/моль, если энергия активации равна 150 кДж/ моль (при 300К). 86 3.2.7. Пользуясь уравнением С Аррениуса изобразите графически зависимость скорости реакции от температуры ( температура меняется от 0 до ∞) и от энергии активации (Еак меняется от 0 до ∞). Укажите пределы применимости этого уравнения. Каков физический смысл экстремальных значений Т и Еак и каковы значения константы скорости k в этих условиях? 2.3. Энтропия активации Большинство изученных реакций не подчиняются закономерности, учитывающей лишь только число активных соударений. Частицы, имеющие достаточный запас энергии для химического взаимодействия, при столкновении могут и не прореагировать с образованием нового соединения. Необходимо, чтобы энергия активации была сосредоточена на определенной химической связи и чтобы частицы сталкивались бы именно этими связями. Большое значение имеет конфигурация электронных облаков, их размеры; месторасположение атомов, групп атоZ Z Z Z мов в молекулах и их ориентации в пространстве относительно друг X X друга в момент столкX X новения. Поэтому далеY Y Y Y ко не всякое столкновеH H H H ние частиц с энергией, большей или равной Рис. 3.2.7. Схема взаимодействия двух атомов водоэнергии активации, морода жет привести к превращению. Например, рассмотрим конфигурационный фактор в случае взаимодействия двух атомов водорода между собой и атома водорода со фтором. При взаимодействии атомов водорода (рис.3.2.7) в силу сферической симметрии электронных орбиталей Is1 как одного, и так другого атома, перекрывание их произойдет в любом месте соприкосновения их, независимо от ориентации атомов относительно друг друга. Но взаимодействие атомов водорода и фтора будет зависеть от их относительной ориентации валентных орбиталей атомов (рис.3.2.8). Внешняя электронная конфигурация Z Z Z Z атома фтора…..2 s22р5. Х X на одной из орбиталей, а X X именно 2рх (более светлая орбиталь), согласно Y Y Y Y нашему рисунку3.2.8, Рис. 3.2.8. Схема взаимодействия атомов водорода и находится одиночный фтора валентный электрон. Остальные орбитали 2s2, α β β α α α β β α β β α α β α β 2 2 1 2 1 1 2 2 1 2 1 87 2р2у, 2р2z фтора содержат по паре электронов и в образовании связи с атомом водорода участвовать не будут. Перекрытие валентных орбиталей (2р,х атома фтора и Is атома водорода) с образованием ковалентной связи возможно только в направлении локализации орбитали 2рх. Столкновение атомов фтора и водорода по всем остальным направлениям (в любой точке соприкосновения) не приведет к образованию связи, т.е. к химическому превращению. Поэтому, говоря о скорости взаимодействия, нужно учитывать из всего числа активных соударений только те, при которых сталкивающиеся частицы ориентированы относительно друг друга определенным образом, а именно, – валентными орбиталями. Еще сложнее требование надлежащей ориентации в случае сложных атомов, молекул (например, особенно органических молекул с длинными цепями). Действительно, одна ситуация, когда сталкиваются два атома водорода, и совершенно другая, когда сталкивается этот же атом водорода с большой молекулой, например, 1- гептилена СН2=СН-СН2-СН2-СН2-СН2-СН3, у которой реакционноспособными являются только первые два атома углерода. И чем сложнее сталкивающиеся частицы, тем менее вероятно их соударение определенными валентными орбиталями (надлежащей взаимной ориентации), тем меньше скорость реакции. Поэтому, например, реакции между органическими веществами протекают медленнее, т.к. в них участвуют сложные молекулы, где общее число возможных ориентаций очень велико, а благоприятных для протекания реакции – весьма ограничено. Вероятность того или иного события «р» (в нашем случае: необходимой ориентации сталкивающихся частиц) будет определяться отношением числа способов ориентации, приводящих к взаимодействию, к общему числу возможных способов ориентации: . Пользоваться этой величиной в таком виде неудобно, т.к. численное значение её очень велико. Действительно, число возможных способов ориентации даже одной молекулы достаточно велико, а для одного моля частиц и подавно. Поэтому пользуются логарифмической зависимостью стерического множителя р. Если взять натуральный логарифм величины р и умножить его на постоянную Больцмана, то получим новую величину s = к· ln р, (3.2.10) называемую энтропией активации S одной частицы. Для одного моля вещества можно получить её значение, если умножить на число Авогадро NA левую и правую части выражения: s NA = NA · к ·ln р, Обозначив энтропию одного моля вещества Sак = s NA. , получим 88 Saк. = R· ln p. После потенцирования выражения 3.2.11 представим стерический в явном виде: 1 (3.2.11) фактор Чем больше вероятность надлежащей ориентации частиц для взаимодействия, т.е., чем больше стерический фактор, тем выше скорость реакции. Полученное значение стерического фактора подставим в уравнение константы скорости реакции 3.2.9: Таким образом, скорость реакции при одной и той же концентрации реагирующих веществ будет определяться той долей активных соударений, в которых выполняется требование надлежащей ориентации, определяемой стерическим фактором 3. ХИМИЧЕСКОЕ РАВНОВЕСИЕ 3.1. Химическое равновесие и константа равновесия Все химические реакции условно подразделяются на необратимые и обратимые. Необратимые протекают в данных условиях только в направлении образования конечных продуктов реакции. Так, взаимодействие между водородом и кислородом идет в сторону образования воды 2Н2 + О2 → 2Н2О. В обратимых реакциях (тоже при определенных условиях), наряду с образованием конечных продуктов реакции, происходит взаимодействие их между собой с выделением исходных веществ. О таких реакциях говорят, что они протекают как в прямом, так и в обратном направлении. В качестве классического примера приведем реакцию взаимодействия водорода с иодом: прямая реакция обратная реакция: Н 2 + I2 → 2 НI Н 2 + I2 ← 2 НI, Н 2 + I2 ↔ или уравнения этих реакций можно записать в виде одного выражения: 2 НI. Реакция является обратимой, т.е. в ней одновременно протекает как прямая реакция образования НI, так и обратная - с выделением вновь исходных веществ Н2 и I2.. В принципе можно считать, что все реакции обратимые. Понятие необратимости условно. Оно определяется параметрами состояния системы, 1 Стерический – этот термин греческого происхождения: «стерео» - объемный, пространственный. 89 свойствами веществ, ее составляющих, организацией осуществления процесса. Обратимые реакции носят более общий характер, поэтому они представляют и больший интерес. Ведь, зная закономерность протекания обратимых процессов, можно избирательно осуществлять как прямой, так и обратный процесс, что требуется в практической деятельности. Рассмотрим обратимую формальную реакцию, описываемую уравнением: а А + b В ↔ d D + f F, где А и В – молекулы исходных веществ; D и F – молекулы продуктов реакции; а, b, d и f – стехиометрические коэффициенты. В начальный момент времени, когда исходные вещества еще не взаимодействовали, в реакционной системе продуктов не было. И, естественно, обратная реакция не протекает. Но вот началась прямая реакция взаимодействия исходных веществ А и В. Их концентрации со временем начнут уменьшаться (соответственно и скорость прямой реакции будет снижаться), а концентрации продуктов D и F начнут увеличиваться и скорость обратной реакции начнет расти. Скорость реакции, как известно, в каждый данный момент времени пропорциональна произведению концентраций реагирующих веществ при постоянстве всех остальных условий реакции (закон действующих масс). Поэтому скорость прямой реакции Vпрям. образования продуктов реакции в любой момент времени равна: Vпрям = kпрям.· C Aa ⋅ C Bb , где Vпрям. – скорость прямой реакции, протекающей слева направо; kпрям.- константа скорости прямой реакции; C Aa ⋅ и ⋅ C Bb - концентрации исходных веществ; а и b – стехиометрические коэффициенты. По мере накопления веществ продуктов реакции D и F происходит увеличение скорости обратной реакции равной: Vобрат. = kобрат.· C Dd ⋅ C Ff , где Vобрат. – скорость обратной реакции, протекающей справа налево; kобрат.константа скорости обратной реакции; C Dd ⋅ и ⋅ C Ff - концентрации продуктов реакции в каждый данный момент времени; d и f – стехиометрические коэффициенты. В какой-то определенный момент времени по мере протекания реакции скорости прямой и обратной окажутся одинаковыми (равными): Vпрям = Vобрат (3.2.12) или kпрям.· C Aa ⋅ C Bb = kобрат.· C Dd ⋅ C Ff . (3.2.13) Практически это означает, что число молекул исходных веществ А и В израсходованных в прямой реакции равно числу молекул А и В, образовавшихся в обратной реакции, за единицу времени. Такое состояние реагирующей системы называется равновесным. В равновесном состоянии процессы в 90 системе не прекращаются: одновременно протекает и прямой, и обратный процессы. Преобразуем последнее равенство 3.2.13, выделяя постоянные величины в левую часть, а переменные – в правую часть уравнения: k прям. k обрат. = C Dd ⋅ C Ff C Aa ⋅ C Bb (3.2.14) В левой части уравнения стоит отношение двух постоянных величин – констант скоростей прямой и обратной реакции: k прям. k обрат. . Отношение двух постоянных величин есть тоже величина постоянная. Обозначим ее Крав. Крав. = k прям. k обрат. и назовем константой равновесия. Перепишем уравнение константы равновесия с учетом равновесных концентраций Крав. = k прям. k обрат. C Dd ⋅ C Ff = a b C A ⋅ CB (3.2.15) Уравнение константы равновесия 3.2.15. показывает, что в условиях равновесия концентрации всех веществ (исходных и продуктов), участвующих в реакции, связаны между собой. Изменение концентрации любого из них автоматически влечет за собой изменения концентрации всех остальных веществ, но со временем устанавливаются новые равновесные концентрации, а соотношения между ними вновь отвечают константе равновесия. Концентрации веществ при установившемся равновесии называются равновесными концентрациями. Поскольку константа равновесия Крав. является отношением констант скоростей (прямой и обратной реакции), то она зависит от тех же самых факторов, что и константа скорости реакции: природы реагирующих веществ и температуры. Численное значение константы равновесия Крав. в первом приближении характеризуют выход реакции: соотношение между получаемым количеством продукта и тем количеством, которое получилось бы при протекании реакции до конца. Например, если Крав.> > 1, то выход реакции велик, т.к. произведение концентраций продуктов реакции (числитель) значительно больше такового для исходных веществ (знаменатель): C Dd ⋅ C Ff >> 1. C Aa ⋅ C Bb То есть в системе в основном присутствуют продукты реакции и совсем незначительное количество исходных веществ. При обратном соотношении, когда Крав. << 1 , или 91 C Dd ⋅ C Ff << 1, C Aa ⋅ C Bb концентрация продуктов незначительна. Больше концентрация исходных веществ, а, следовательно, реакция практически не идет в прямом направлении. Равновесные величины в выражении 3.2.15 можно представлять в различных единицах, а потому и константа равновесия Крав. будет выражаться разными значениями. Например, константу равновесия можно выразить через молярные концентрации веществ, участвующих в реакции. В этом случае она обозначается Кс и выражаться будет вышеприведенным уравнением Кс. = C Dd ⋅ C Ff . C Aa ⋅ C Bb Если же выразить её через парциальные давления газообразных веществ, участвующих в реакции, то её обозначают Кр и она будет равна для приведенной формальной реакции: Кр = PDd ⋅ PFf PAa ⋅ PBb Иногда константу равновесия выражают через мольные доли Кγ и число молей участвующих веществ Кn. Между всеми способами выражения константы равновесия существует определенная связь. Поскольку чаще используется Кс. и Кр, то установим связь между ними. Для чего воспользуемся уравнением состояния идеальных газов РV = n RT, где Р – давление, V – объем, n – число молей газа, R – газовая постоянная, Т – абсолютная температура. Представим уравнение газового состояния в таком виде: P= n RT = CRT , V где С – концентрация какого либо вещества (компонента), выраженная в моль/л. Из этого выражения видно, что C= P . RT Тогда константа равновесия для рассмотренной ранее реакции после подстановки значений давления соответствующих компонентов запишется так: Кр = PDd ⋅ PFf C Dd ⋅ C Ff d+f -a–b = a b (RT) , a b PA ⋅ PB C A ⋅ CB или Кр = Кс (RT ) ∆n , где ∆n= (d + f) - (a + b) есть изменение числа молей реагирующих веществ и продуктов в результате реакции. Если ∆n= 0, то есть реакция не сопровождается изменением числа молей компонентов, то Кр = Кс . 92 Приведем несколько примеров записи выражения константы равновесия для гомогенных и гетерогенных реакций. Например, для гомогенной реакции 2 СОгаз + О2газ ↔ 2 СО2газ Константа равновесия запишется так: Кс. = 2 C CO 2 Кр = или 2 C CO ⋅ C O2 2 PCO 2 2 PCO ⋅ PO2 . В случае гетерогенной реакции Fe2O3крис. + 3 H2газ ↔ 2 Feкрис. + 3 H2Oгаз в выражении константы равновесия Kc = C H3 2O C H3 2 или Кр = PH32O PH32 фигурируют концентрации (парциальные давления) тех веществ, от которых зависит скорость прямой и обратной реакции, и отсутствуют концентрации (парциальные давления) твердых веществ, так как скорость гетерогенных реакций принято относить к единице поверхности раздела фаз. Примером гетерогенного равновесия является состояние кристаллов малорастворимого вещества (например, AgCl) в насыщенном растворе этой соли. Кристаллы хлорида серебра, диссоциирующие на ионы серебра и хлора, находятся в равновесии с ионами раствора AgClкрис. ↔ Ag+раств. + Cl-раств. и константа равновесия запишется в виде выражения: Kc = CAg + раств.·CCl раств.. Применительно к равновесным процессам растворимости веществ константа равновесия, равная произведению концентраций ионов, называется также произведением растворимости (сокращенно ПР), т.е. Kc = ПР Смысл понятия произведение растворимости аналогичен понятию константы равновесия. Например, при уменьшении концентрации ионов серебра и хлора в растворе вместе или порознь, в силу каких–то причин, для сохранения постоянства значения произведения растворимости (Kc = ПР) в раствор должны перейти новые порции ионов из кристалла (кристалл будет растворяться). При повышении содержания ионов в растворе, по сравнению с равновесным значением, будет происходить обратный процесс: осаждение ионов из раствора на поверхности кристалла (образование осадка, кристаллизация). Все сказанное о константе равновесия относится к равновесию простых, элементарных реакций, протекающих в одну стадию. Для сложных реакций, протекающих через несколько последовательных стадий, константа 93 равновесия равна произведению констант равновесия отдельных стадий. Действительно, предположим, что имеется сложная реакция: A ↔ F, которая характеризуется константой равновесия Кс. = CF , CA и протекает через ряд последовательных обратимых реакций: A ↔ B ↔ D ↔ F. Константы равновесия каждой промежуточной стадии этой сложной реакции соответственно равны: ` Кс = CB , CA `` Кс = CD , CB ``` Кс = CF . CD Перемножим константы равновесия промежуточных стадий ` `` ``` Кс Кс Кс .= CB CD C A CB CF C = F. CD CA Полученное выражение и есть константа равновесия сложной реакции, т.е. константа равновесия сложной реакции Кс. = CF , CA равна произведению констант равновесия отдельных простых обратимых реакций Кс = CF = Кс` Кс`` Кс```, CA из которых она складывается. На практике часто возникают вопросы, связанные с расчетами начальных, конечных и равновесных концентраций в реакционных системах. На двух примерах покажем, как можно использовать понятие константы равновесия для решения тех или иных задач. Пример 1. В обратимой реакции Н2 газ + I2газ ↔ 2 HIгаз наступило равновесие, когда равновесная концентрация HI оказалась равной 0,03 моль/л. Начальные концентрации водорода и иода составляли 0,02 моль/л. Каковы равновесные концентрации водорода и иода в реакционной системе и чему равна константа равновесия этой реакции при данной температуре? Решение. В соответствие с уравнением обратимой реакции видно, что на образование двух молей HI нужно затратить 1 моль Н2 и 1 моль I2. Если в реакционной системе образовалось 0,03 моля HI, то водорода и иода было израсходовано по 0,015 моля. Следовательно, равновесные концентрации водорода и иода составят разницу между исходными концентрациями и израсходованными на образование HI в концентрации 0,03 моль/л, т.е. 0,02 моль/л - 0,015 моль/л = 0,005 моль/л. 94 Константа равновесия Кс для данной реакции запишется в виде выражения: Кс = 2 C HI , C H2 ⋅ C I2 и, подставляя равновесные концентрации, получим значение константы равновесия: 0,03 2 Кс = = 36 . 0,005 ⋅ 0,005 Константа равновесия больше единицы, значит в системе концентрация продуктов значительно больше, чем концентрация исходных веществ. Пример 2. При температуре 8500 С в реакционной системе, описываемой уравнением реакции СОгаз + Н2Огаз ↔ СО2 газ + Н2 газ константа равновесия Кс = 1. Нужно рассчитать равновесные концентрации всех веществ, если исходные концентрации реагентов были: Ссо начальн.= 3 моль/л, Сн2о начальн. = 2 моль/л. Решение. Из уравнения реакции следует, что из 1 моля СО и 1 моля Н2О образуется по 1 молю СО2 и Н2. Если предположить, что прореагировало х моль/л СО2, то столько же прореагировало и Н2О и образовалось по х моль/л СО2 и Н2. Тогда оставшиеся, равновесные концентрации исходных веществ СО и Н2О будут равны: и Сн2о равновес.= 2 – х. Ссо равновес.= 3 – х Запишем выражение константы равновесия для данной гомогенной реакции: Кс = C СO2 равн. ⋅ С H 2 равн. C CO. равн ⋅ C H 2O. равн. . Подставим равновесные значения концентраций веществ в выражение константы равновесия: Кс = x⋅x . (3 − x) ⋅ (2 − x) Согласно условию задачи Кс = 1. Отсюда следует (3 - х) (2 - х) = х2. Решая уравнение, получим: х = 1,2 моль/л. Таким образом, искомые равновесные концентрации равны: ССО2 равновес.= 1,2 моль/л; Сн2 равновес.= 1,2 моль/л; Ссоравновес.= 3 -1,2 = 1,8 моль/л; Сн2о равновес.= 2 – 1,2 = 0,8 моль/л. Вопросы и задачи для самостоятельной работы 3.3.1. Напишите выражение константы равновесия для следующих реакций: 1. Fe2O3 кр. + 3 Н2 газ ↔ 2 Feкр. + 3 Н2О пар 2. 2 СОгаз + О2 газ ↔ 2 СО2 газ, 3. Znкр. + Н2SO4 раствор ↔ ZnSO4раствор + Н2 газ, 95 4. 2 SO2 газ + О2 газ ↔ 2 SO3 газ, 5. AgClкр. ↔ Ag+раствор + Cl-раствор, 6. 3 Feкр. + 4 Н2Ожид. ↔ Fe3O4 крис. + 4 Н2 газ. 3.3.2. В гомогенной системе А + 2 В ↔ D равновесные концентрации реагирующих веществ СА = 0,06 моль/л; СВ = 0,12 моль/л; СD =0,216 моль/л. Вычислите константу равновесия и исходные концентрации веществ А и В. 3.3.3. В гомогенной системе А + В ↔ D + F равновесие установилось при концентрациях: СВ = 0,05 моль/л; СD = 0, 02 моль/л. Константа равновесия равна Кс = 0.4. Вычислите исходные концентрации веществ А и В. 3.2. Смещение химического равновесия. Принцип Ле Шателье Равновесное состояние системы определяется постоянством внешних условий, воздействий. Истинное равновесное состояние системы характеризуется тем, что при отсутствии внешних воздействий состояние системы остается неизменным во времени. Если же во внешней среде произошли какиелибо изменения, то система следует за ними. При внешнем воздействии, вызывающем изменения в системе (температуры, давления, объема, концентрации, состава), происходит изменение и в соотношении между скоростью прямой и обратной реакции, т.к. эти факторы могут в разной степени влиять на них. Следовательно, в системе нарушается равновесное состояние. Тем не менее, по истечении некоторого времени в системе опять устанавливается равновесие. Переход системы из одного равновесного состояния в другое называется смещением или сдвигом равновесия. Новому равновесному состоянию будут отвечать и новые равновесные концентрации исходных веществ и продуктов реакции. В связи с этим часто необходимо качественно оценить: в какую сторону (вправо или влево) смещается равновесие в системе при воздействии того или иного фактора. Когда равновесие сместилось вправо, то подразумевают увеличение скорости прямой реакции и, соответственно, возрастание концентрации продуктов реакции. Если равновесие смещается влево, то имеют в виду, что увеличивается скорость обратной реакции и, как следствие, возрастает концентрация исходных веществ. Для предсказания влияния условий на положение равновесия пользуются правилом, которое получило название принципа Ле Шателье1, сформулированного в 1884 году: если на систему, находящуюся в равновесии, воздействовать извне, изменяя какое-либо условие, определяющее положение равновесия, то в системе усилится то из направлений процесса, которое ослабляет эффект этого воздействия, и положение равновесия сместится в том же направлении. Анри Луи Ле Шателье (1850-1936) – выдающийся французский ученый, действительный член Парижской Академии наук (1907), почетный член АН СССР (1927). Исследовал процессы при высоких температурах, металлические сплавы, свойства материалов и керамики. 96 1 Принцип Ле Шателье справедлив и для равновесных систем, в которых не протекают химические превращения: растворение, кипение, кристаллизация и т.д. Рассмотрим на конкретном примере влияние некоторых факторов на смещение равновесия. Возьмем в качестве примера реакцию взаимодействия сернистого газа с кислородом (широко используемую в промышленности), и протекающую согласно уравнению: 2 SO2 газ + О2 газ ↔ 2 SO3 газ + 198 кДж. Оценим, как повлияет на состояние равновесия этой системы, увеличение, например, концентрации кислорода, увеличение давления в системе (уменьшение объема), увеличение температуры? Влияние концентрации. Повышение концентрации кислорода должно привести к увеличению скорости прямой реакции согласно закону действующих масс: 2 ⋅ CO1 , Vпрям = kпрям.· C SO 2 2 К этому же выводу можно прийти из анализа выражения константы равновесия данной реакции: Кс. = 2 C SO 3 2 ⋅ C O2 C SO 2 Увеличение концентрации кислорода (знаменатель этого выражения) должно привести к увеличению и концентрации серного ангидрида SO3 (числитель выражения), т.к. константа равновесия – величина постоянная. То есть увеличение концентрации кислорода сдвинет равновесие вправо: увеличится концентрация продуктов реакции. К этому же выводу можно прийти, применяя принцип Ле Шателье. Увеличение концентрации кислорода по сравнению с равновесным значением в системе должно усилить тот процесс (реакцию), который свел бы это увеличение к минимуму. Таким процессом является прямая реакция, в ходе которой потребляется кислород, и концентрация его понижается. Вообще избыток исходного вещества (исходных веществ) вызывает смещение равновесия вправо, как и уменьшение (удаление) концентрации продуктов реакции. Добавление продукта (продуктов) реакции вызывает смещение равновесия влево. Добавляя или удаляя исходные вещества, продукты реакции можно смещать равновесие в ту или иную сторону, что и делается на практике. Влияние давления на равновесие. В соответствии с принципом Ле Шателье, повышение давления в системе должно сместить равновесие в ту сторону, в направлении того процесса, в котором происходит противодействие оказанному повышению, т.е. усиливается реакция, идущая с меньшим объемом образующихся продуктов. Прямая реакция идет с выделением двух молей газообразного триоксида серы, обратная – с образованием трех молей газообразных веществ: двух молей диоксида серы и одного моля кислорода. 97 Поэтому повышение давления приведет к увеличению скорости прямой реакции, т.е. равновесие сместится вправо. На рисунке 3.3.1 представлена схема влияния давления на реакционное равновесие: повышение давления (уменьшение объема) в системе приводит к уменьшению числа частиц в газовой фазе за счет их объединения и, наоборот: понижение давления (увеличение объема) сопровождается увеличением их числа в результате распада крупных частиц. Р а б Рис. 3.3.1. Схема влияния давления на реакционное равновесие. Увеличение давления в равновесной системе (состояние “а”) приводит к уменьшению числа частиц в газовой фазе (состояние “б”). Понижение давления (переход из состояния “б” в ”а”) в системе будет сопровождаться увеличением числа частиц в системе. Влияние температуры на равновесие. Повышение температуры, в соответствии с принципом Ле Шателье, должно привести к усилению роли той реакции, которая ослабляет производимое увеличение температуры. Прямая реакция протекает с выделением Q = +198 кДж/моль, т.е. она является экзотермической, и температура в системе будет повышаться по мере ее протекания. Обратная реакция – разложение триоксида серы является эндотермической, т.е. она протекает с поглощением тепла Q = -198 кДж/моль, и температура системы должна, соответственно, понижаться. Поэтому повышение температуры системы, произведенное извне, усилит роль обратной реакции. Равновесие сместится влево. На рис.3.3.2 представлено соотношение энергий активации прямого и обратного процессов в случае эндотермической и экзотермической реакции. Из которого видно, что в случае эндотермической реакции, увеличение температуры приведет к значительному увеличению константы скорости обратной реакции, т.к. энергия активации обратной реакции много меньше энергии активации прямого процесса (рис.3.3.2а.) 98 Таким образом, направление смещения равновесия в результате изменения температуры определяется знаком теплового эффекта реакции: Н Н Еак.об Еак.прям. Н2 Еак .прям. Еак. обр. Н1 ∆Н >0 Н1 ∆Н < 0 Н2 Ход реакции Ход реакции а б Рис. 3.3.2. Соотношение энергий активации прямого и обратного процессов для: а - эндотермической и б - экзотермической реакции. нагревание вызывает смещение равновесия в сторону того из двух процессов, протекание которого сопровождается поглощением теплоты; понижение температуры приводит к смещению равновесия в сторону той реакции, протекание которой сопровождается выделением теплоты, иначе говоря, охлаждение способствует экзотермическому процессу и вызывает увеличение константы скорости этого процесса. Вопросы и задачи для самостоятельной работы 3.3.4. В каком направлении произойдет смещение равновесия в системе: N2 газ+3H2 газ ↔ 2 NH3 газ; H0 = -92 кДж/моль при понижении температуры, повышении давления? 3.3.5. В каком направлении произойдет смещение равновесия при повышении давления в реакционной системе: 1. Fe3O4 крис. + 4 Н2 газ↔ 3 Feкрис. + 4 Н2О пар. 2. 2 NOгаз + О2 газ ↔ 2 NO2 газ ? 3.3.6. В каком направлении произойдет смещение равновесия в системе: 4 НСl газ + О2 газ ↔ 2 Н2Опар + 2 Cl2 газ при увеличении концентрации кислорода? Хлора? При понижении концентрации HCl? 3.3. Константа равновесия и свободная энергия Гиббса Рассмотрим простую, обратимую, формальную реакцию А + В ↔ АВ с энергетической точки зрения. Со времен С. Аррениуса в химии широко принято символическое изображение пути реакции в виде энергетической диаграммы, изображенной на рис.3.3.3. Где по оси ординат откладывается энергия реагирующей системы H (энтальпия), а по оси абсцисс ход реакции. 99 В начальном состоянии до взаимодействия веществ между собой, если рассматривать изобарно-изотермические условия протекания реакции, система обладает энтальпией H1, а в конечном состоянии после окончания реакции в условиях равновесия - энтальпией H2. В ходе реакции выделяется тепловой эффект Qp, равный изменению энтальпии: Qр = - ∆Н = H2 - H1. Активированный комплекс А ·················В Н Еак. прям. Н1 Еак А, В .обрат -∆Н = Qp Н2 Исходное состояние Промежуточное состояние Ход АВ Конечное состояние реакции Рис.3.3.3. Энергетическая схема реакции По оси абсцисс откладывается ход реакции – в первом приближении это изменение расстояния между реагирующими молекулами. Условно ход реакции можно разделить на три этапа, три участка. Участок начального, или исходного состояния, когда в системе отсутствует превращение частиц, и они расположены на относительно больших расстояниях друг от друга. Переходный этап связан с уменьшением расстояния между реагирующими молекулами А и В, а следовательно, и с увеличением энергии системы, которая необходима для преодоления взаимного отталкивания электронных оболочек атомов, молекул. Уменьшение расстояния между молекулами при сближении приводит к “расшатыванию” старых связей в них, а в конечном итоге и к разрыву, если энергия реагирующих молекул, по крайней мере, равна энергии активации или больше ее. Энергия активации – это тот минимальный избыток энергии, который необходим системе, чтобы в ней происходило превращение молекул. При наличии избытка энергии, равного энергии активации прямой реакции Eак..пр., происходит в течение какого-то промежутка времени разрыв старых связей в реагирующих молекулах, и образуются новые связи между ними. Возникает так называемый переходный активный комплекс, когда нельзя во взаимодействующих частицах, комплексе различить исходные молекулы А и В и выделить молекулы продуктов АВ. Но в следующий момент времени активный 100 комплекс распадается на конечные продукты, и выделяется энергия, затраченная на его образование. Система переходит в конечное состояние с энергосодержанием системы H2. Образовавшиеся конечные продукты реакции АВ могут реагировать между собой через образование опять того же переходного активного комплекса и распада его на исходные продукты А и В. Но для этого молекулы продуктов должны уже преодолеть потенциальный барьер энергии активации обратной реакции Eак.об., который является минимальным избытком энергии относительно состояния системы с энергосодержанием H2. Разница между энергиями активации обратного и прямого процессов есть изменение энтальпии ∆H, или тепловой эффект реакции (рис.3.3.3): Eак.об. – Eак.пр. = - ∆H (3.3.1.) Константа скорости прямой реакции согласно уравнению С. Аррениуса равна: а обратной: Для состояния равновесия обратимой реакции, согласно уравнению 3.2.15, константу равновесия можем представить как отношение констант прямой реакции и обратной: Крав. = k прям. k обрат. Разделив одно выражение на другое, получим константу равновесия через приведенные выражения: . Крав. = k прям. k обрат. = Z прям. ⋅ e Z обрат. ⋅ e − Eak .прям RT − Eak .обрат RT ⋅e Sak .прям R ⋅e Sak .обрат R . (3.3.2) Упростим данное выражение. Отношение числа столкновений при прямой реакции Zпрям. и обратной Zобрат. в условиях равновесия есть величина постоянная. Сгруппируем её с величиной константы равновесия Крав. Крав. Z обрат. Z прям. =K, и будем данное соотношение считать новой константой равновесия К. После преобразования правой части выражения 3.3.2 получим: . К = Принимая во внимание, что разность Е ак.обрат. – Е ак.прям. = - ∆ Н (смотрите рисунок 3.3.3), а разность энтропий обратной и прямой реакции 101 S ак.обрат. – S ак.прям. = - ∆S - изменение энтропии ∆S, то после подстановки этих величин в последнее выражение получим: − ∆H RT ⋅e K =e После перемножения экспонент, получим: K= e ∆S . R − ∆H +T∆S RT . . Прологарифмируем полученное выражение: ln K = − ∆H + T ⋅ ∆S , RT или RT lnK = -∆H + T∆S. Если полученное выражение умножить на -1, то получим уравнение - RT lnK = ∆H - T∆S. (3.3.3) Правая часть этого уравнения ∆H - T∆S = ∆G, есть не что иное, как изменение изобарно- изотермического потенциала ∆G реакции, следовательно, - RT lnK = ∆G, (3.3.4) или в экспоненциальной форме K= e − ∆G RT . Данное соотношение представляет связь между изменением энергии Гиббса процесса и его константой равновесия. Зная значение константы равновесия, можно рассчитать изменение энергии Гиббса ∆G, и наоборот. Если реакция проводится в стандартных условиях, когда концентрации всех участников реакции равны 1 моль/л или их парциальное давление равно 101 325 Па, то выражение 3.3.4 будет связано со стандартным изменением энергии Гиббса ∆G0 = - RT lnK (3.3.5) последнее уравнение используют также для экспериментального определения ∆G0 реакций, если возможно определить концентрации веществ в состоянии равновесия. Проанализируем уравнение 3.3.4, для чего представим его в таком виде: - RT ln = ∆G, заменив константу равновесия отношением констант скоростей прямой и обратной реакций k прям. k обрат. . Если константы скорости прямой и обратной реак- ции равны kпрямой = kобрат, то их соотношение равно 1, а ∆G = 0. Значит, система находится в состоянии равновесия. 102 Если константа скорости прямой реакции kпрямой реакции больше константы скорости обратной реакции kобрат., т.е. kпрямой > kобрат., то ∆G < 0 и, следовательно, в реакционной системе преобладает протекание прямой реакции. В противоположном случае, когда kпрямой < kобрат, изменение изобарно-изотермического потенциала ∆G будет больше нуля ∆G > 0 , то преобладающей реакцией в системе будет обратная. Проиллюстрируем полученную зависимость на трех примерах. Пример 1. В реагирующей обратимой системе H2 газ + Cl2 газ↔ 2 НСl газ При 298 К установилось равновесие. Нужно определить константу равновесия этой реакции, если известно, что изменение энтальпии реакции ∆Н0= 92,34 кДж/моль, а изменение энтропии при стандартных условиях составляет 20,05 Дж/(моль К). Решение. Поскольку ∆G0 = ∆Н0 – Т∆S0 , то ∆Н0 – Т∆S0 = - 8,31·298·2,303·lgK.= 5,69 lgK, если значение ∆G0 использовать (и соответственно изменение энтальпии и энтропии) в кДж/ моль. Подставляя численные значения известных величин, и решая относительно К, получим: lgK = 92,34 + 298 ⋅ 20,05 ⋅ 10 −3 = 17,24 . 5,69 И от сюда получаем, что К = 1,7·1017. Следовательно, в данной системе равновесие сдвинуто практически полностью в сторону образования НСl, об этом говорит огромная величина константы равновесия. Пример 2. Гидроксид алюминия Al(OH)3 может диссоциировать в водном растворе в соответствии с уравнением: Al(OH)3 ↔Al3+ + 3 OH . Насколько возможен такой процесс, если произведение растворимости ПР при 250 С равно 1,9·10-33? Решение. Возможность этого процесса оценим по значению изобарноизотермического потенциала ∆G0, который связан с константой равновесия этого процесса, равной Кс = C Al 3 COH C Al (OH )3 Система эта гетерогенная. Следовательно, Отсюда . Кс = ПР = 1,9·10-33. 103 ∆G0 = - 5,69 lgKс = -5,69 lg1,9·10-33 = + 0, 186 кДж/моль. Положительная величина изменения изобарно-изотермического потенциала говорит о невозможности самопроизвольного распада гидроксида алюминия – на ионы Al3+ и 3 OH при данных условиях. Пример 3. Вычислите ∆G0 процесса распада аммиачного комплекса иона серебра: [Ag (NH3)2]+ ↔ Ag+ + 2 NH3 и сделайте вывод о возможности этого процесса в прямом и обратном направлениях, если константа нестойкости иона [Ag (NH3)2]+ при 200 С равна 6,8 · 10-8. Решение. Выражение константы равновесия для процесса распада аммиачного иона серебра, исходя из уравнения его распада, имеет вид: Кс = и является также и выражением константы нестойкости К.. В этом случае изменение изобарно-изотермического потенциала этого процесса равно: 0 -8 ∆G = - 5,69 lgKн = - 5.69 lg 6,8·10 = + 40,78 кДж/моль. 0 Положительная величина ∆G > 0 указывает на то, что процесс распада ком- плексного иона на составные частицы практически невозможен при данных условиях. То есть данный ион – довольно устойчивое образование. Задачи и упражнения для самостоятельной работы 3.3.7. Вычислите константу равновесия для системы 2 СН4 газ ↔ С2Н2 газ + 3 Н2 газ 0 При стандартных условиях, если ∆G = 310 кДж/моль. 3.3.8. Рассчитайте константу равновесия системы Fe3O4 крис. + 4 Н2 газ ↔ 3 Feкрис. + 4 Н2О пар При стандартных условиях. О чем говорит полученный результат? 0 3СООН при 25 С равСН 3.3.9. Константы диссоциации воды и уксусной кислоты -16 -5 0 ны, соответственно, Кдис. воды = 1,86·10 , Кдис. сн3соон = 1,8·10 . Рассчитайте ∆G для этих процессов и сравните их. 4. ЯВЛЕНИЕ КАТАЛИЗА Под катализом понимают процесс изменения скорости реакции, происходящий в присутствии различных химических веществ- катализаторов. Катализаторами называют вещества, оказывающие влияние на скорость реакции и участвующих в ней таким образом, что их химический состав после реакции оказывается практически неизменным. При одной и той же температуре, если формально рассмотреть факторы, влияющие на скорость реакции, в соответствии с выражением константы скорости реакции 104 ими окажутся энергия активации Еак. и энтропия активации Sак, незначительные изменения которых приводят к существенному изменению константы скорости., так как они входят в степень данного выражения.. Число столкновений z, можно считать, не оказывает влияния, поскольку для данной реакции (природы веществ), температуры, концентрации число столкновений постоянно. Механизм катализа может быть представлен следующим образом. Например, для символической реакции А + В = АВ, (3.4.1) протекающей без катализатора, скорость равна V1. Этот процесс связан с определенными значениями энергии активации Еак. и энтропия активации Sак., которые для данного пути (механизма) реакции- величины постоянные (рис.3.4.1 сплошная кривая). Но этот же процесс получения вещества АВ из веществ А и В можно провести по иному пути через промежуточные стадии (предположим через две стадии, пунктирная кривая рис. 3.4.1). Представим, что один из исходных реагентов, пусть это будет вещество А, взаимодействует с катализатором kat с образованием промежуточного соединения Аkat по первой стадии реакции А + kat = Аkat (3.4.2.) Этот этап взаимодействия характеризуется своими значениями энергии активации Е`ак. и энтропии активации S`ак (рис.3.4.1 пунктирная кривая). Образовавшееся промежуточное соединение Аkat, в дальнейшем реагирует с компонентом В в соответствии с уравнением реакции: Akat + В = АВ + kat. (3.4.3.) Это взаимодействие протекает также с присущими для него энергией активации E``ак. и энтропией активации S``ак. По окончании второй стадии реакции выделяется конечный продукт АВ и вещество катализатора kat почти в неизменном виде. Если сложить уравнения первой стадии реакции и второй с участием катализатора, то получим: А + kat = Аkat Аkat + В = АВ + kat А + В + kat = АВ + kat Тот же результат по составу продуктов, что и в простой реакции 3.4.1, которая проводилась без катализатора. Однако скорости протекания этих процессов (с катализатором и без него) будут разными. Причем для приведенного примера скорость реакции в присутствии катализатора больше таковой для реакции без катализатора, потому что энергии активации реакций промежуточных стадий E`ак и E``ак (каждая в отдельности) меньше энергии активации прямой реакции без катализатора Еак. 105 Активированный комплекс А ·················В Н Еак. прям. А ·····kаt Е` ак Н1 А, В Akat·····B Akat -∆Н = Qp Е``ак АВ kat Н2 Ход реакции Рис.3.4.1 Энергетическая схема реакции: без катализатора – сплошная кривая; с катализатором – пунктирная кривая. А это означает, что число активных молекул, соответственно и соударений, больше в единице объема и за единицу времени при протекании реакции по пути в присутствии катализатора. Энергетически такой более сложный путь реакции оказывается, тем не менее, выгоднее. В ряде случаев с помощью катализаторов изменяется не только механизм, энергия активации, но и взаимное расположение реагирующих молекул, т.е энтропия активации. Возможно как увеличение энергии активации с помощью катализаторов, так и уменьшение. Характер влияния зависит от природы реагирующих веществ и свойств катализатора. Различают положительный катализ, если скорость реакции увеличивается, и отрицательный, если - уменьшается. В первом случае катализаторы носят название промоторы, во втором – ингибиторы. По фазовому признаку катализ делят на гомогенный, если катализатор находится в одной фазе с реагирующими веществами, и гетерогенный, если катализатор представляет самостоятельную фазу в реакционной системе. Еще раз подчеркнем, что катализаторы изменяют скорость реакции, которая протекает в данных условиях, но не вызывают её, если она термодинамически невозможна. Катализаторы не влияют на константу равновесия, т..к. они в одинаковое число раз изменяют скорость и прямой и обратной реакции, понижая или увеличивая на одну и ту же величину энергию активации прямой и обратной реакции. 106 БИБЛИОГРАФИЧЕСКИЙ СПИСОК 1. Путилов К.А. Термодинамика/ К.А. Путилов, - М.: Наука,1971.375с.ил. 2. Шамбадаль П. Развитие и приложения понятия энтропии / П. Шамбадаль,-М.:Наука,1976.-280с.ил. 3. Крестовников А.Н. Химическая термодинамика / Крестовников А.Н., Вигдорович В.Н.,Гос.науч.-техн.Изд-во лит. по черной и цветной металлургии.-М.,1961.-280с.ил. 4. Карапетьянц М.Х. Примеры и задачи по химической термодинамике: Учебник. -4-е изд.,перераб. и доп. / М.Х.Карапетьянц. –М.: Химия,1974.-310с.ил. 5. Карапетьянц М.Х. Химическая термодинамика / М.Х. Карапетьянц, - М.: Химия,1975.-583с.ил. 6. Карапетьянц М.Х. Введение в теорию химических процессов / М.Х. Карапетьянц, - М.: Высшая школа,1970. 7. Карапетьянц М.Х. Общая и неорганическая химия М.Х./ Карапетьянц, С.И. Дракин. –М.: Химия,1981. 8. Романцева Л.В. Сборник задач и упражнений по общей химии / Л.В. Романцева, З.Л. Лещинская, В.А. Суханова. – М.: Высшая школа,1991. 9. Зайцев О.С. Общая химия. Направление и скорость химических процессов. Строение вещества / О.С. Зайцев. – М.: Высшая школа, 1983. 10. Зайцев О.С. Задачи и вопросы по химии / О.С.Зайцев. – М.: Химия, 1985. 11. Зайцев О.С. Состояние вещества и химические реакции / О.С.Зайцев. – М.: Химия, 1990. -352с.ил. 12. Киреев В.А. Краткий курс физической химии / В.А Киреев. – М.: Химия, 1978. 13. Ипполитов Е.Г. Физическая химия /Е,Г. Ипполитов, А.В. Артемов, В.В. Батраков; под ред. Е.Г. Ипполитова. –М. : Академия, 2005. -448 с., ил. 14. Панченков Г.М. Химическая кинетика и катализ /Г.М. Панченков, В.П. Лебедев.– М.: Изд-во МГУ,1961. -560с.ил. 15. Эмануэль Н.М. Курс химической кинетики / Н.М. Эмануэль, Д.Г. Кнорре. –М.: Высшая школа,1962.415с.ил. 16. Коровин Н.В. Общая химия / Н.В. Коровин. –М. : Высш. шк., 1998. -559с., ил. 107 ПРИЛОЖЕНИЕ Таблица 1 Термодинамические константы некоторых веществ Вещество Ag AgBr AgCl AgI AgF Ag2O Ag2CO3 Al Al2O3 Al(OH)3 AlCl3 Al2 (SO4)3 B B2O3 B2H6 Ba BaCO3 Be BeO BeCO3 Br2 HBr C C CO CO2 COCl2 CS2 CS2 C2H2 C2H4 CH4 C2H6 C6H6 CH3OH С2H5OH CH3COOH Ca CaO CaF2 CaCl2 CaC2 Ca(OH)2 CaSO4 Состояние Крис. -“-“-“-“-“-“-“-“-“-“-“-“-“газ Крис. -“-“-“-“газ -“алмаз графит газ -“-“-“Жид. газ -“-“газ жид. жид. -“-“крис. -“-“-“-“-“-“- - ∆Н0f 298 , - ∆G0f 298 , кДж/моль кДж/моль 0 99,16 126,8 64,2 202,9 30,56 506,1 0 1675,0 1275,7 697,4 3434,0 0 1264,0 31,4 0 1202,0 0 598,7 981,57 -30,92 36,23 -1,897 0 110,5 393,51 223,0 -115,3 -87,8 -226,75 -52,28 74,85 84,67 -49,04 238,7 227,6 484,9 0 635,1 1214,1 785,8 62,7 986,2 1424,0 0 95,94 109,7 66,3 184,9 10,82 437,1 0 1576,4 1139,72 636,8 3091,9 0 1184,0 82,8 0 1138,8 0 581,6 944,75 -3,14 53,22 -2,866 0 137,27 394,38 210,5 -65,1 -63,6 -209,2 -68,12 50,79 32,89 -124,50 166,31 174,77 392,46 0 604,2 1161,0 750,2 67,8 896,76 1320,3 108 S0298 , Дж/(моль К) 42,69 107,1 96,07 114,2 83,7 121,7 167,4 28,31 50,94 71,1 167,0 239,2 5,87 53,85 232,9 64,9 112,1 9,54 14,10 199,4 245,35 198,48 2,38 5,74 197,4 213,6 289,2 237,8 151,0 200,8 219,4 186,19 229,5 173,2 126,7 160,7 159,8 41,62 39,7 68,87 113,8 70,3 83,4 106,7 Продолжение табл. 1 Вещество CaSiO3 Ca3(PO4)2 CaCO3 Cl2 HCl HCl HClO Cu Cu2O CuO Cu(OH)2 CuF2 CuCl2 CuBr2 CuI2 CuS2 CuS CuSO4 CuCO3 Fe FeO Fe2O3 Fe3O4 FeCl2 FeCl3 Fe(OH)3 FeSO4 FeCO3 H2 H2O H2O H2O2 HF Hg HgCl2 I2 I2 HI HIO K K2O KOH KH Mg MgO Mg(OH)2 Состояние -“-“-“газ -“жид. -“крис. -“-“-“-“-“-“-“-“-“- -“-“-“-“-“-“-“-“-“-“-“газ -“жид. -“газ крис. -“-“газ -“жид. крис. -“-“-“-“-“-“- - ∆Н f 298 , 0 кДж/моль 1579,0 4125,0 1206,0 0 92,30 167,5 116,4 0 167,36 165,3 443,9 530,9 205,9 141,42 21,34 82,01 48,5 771,1 594,96 0 263,68 821,32 1117,9 341,0 405,0 824,25 922,57 744,75 0 241,84 285,84 187,36 270,9 0 230,12 0 62,24 25,94 158,9 0 361,5 425,93 56,9 0 601,24 924,66 - ∆G f 298 , 0 кДж/моль 1495,4 3899,5 1128,8 0 95,27 131,2 80,0 0 146,36 127,19 356,90 489,3 166,1 126,78 23,85 86,19 48,95 661,91 517,98 0 244,35 740,99 1014,84 302,08 336,39 694,54 829,69 637,88 0 228,8 237,5 117,57 272,992 0 185,77 0 19,4 1,30 98,7 0 193,3 374,47 38,49 0 569,6 833,7 109 S0298 , Дж/(моль К) 87,45 240,9 92,9 223,0 186,7 55,2 129,7 33,8 93,93 42,64 79,50 84,5 113,0 142,34 159,0 119,24 66,5 113,3 87,9 27,5 58,79 89,96 146,29 119,66 130,1 96,23 107,51 92,9 130,6 188,74 69,96 105,86 173,794 76,1 144,35 116,73 260,58 206,33 24,32 64,35 87,0 59,41 67,95 32,55 26,94 63,14 Окончание табл. 1 Вещество MgCO3 N2 N2O NO NO2 N2O4 NH3 NH4Cl NH4OH Na Na2O NaOH NaCl Na2CO3 O2 O3 P PCl3 PCl5 Pb PbO PbO2 PbSO4 PbS S SO2 SO3 H2S H2S H2SO4 SiO2 SnO SnO2 SrO SrCO3 Ti TiO2 TiCCl4 WO3 Zn ZnO ZnS ZnSO4 Состояние -“газ -“-“-“-“-“крис. жид. крис. -“-“-“-“газ -“красный газ -“крис. -“-“-“-“ромбич. газ -“-“жид. жид. крис. -“-“-“-“-“-“жид. крис. -“-“-“-“- - ∆Н f 298 , - ∆G f 298 , кДж/моль 1096,21 0 -81,55 -90,37 -33,89 -9,37 46,19 315,39 366,69 0 430,6 426,6 410,9 1129,0 0 142,24 18,41 277,0 369,45 0 217,86 276,6 918,1 94,28 0 296,9 395,2 20,15 39,33 811,3 859,3 286,0 580,8 590,4 1221,3 0 943,9 804,2 842,7 0 349,0 201,0 978,2 кДж/моль 1029,3 0 -103,6 -86,69 -51,84 -98,29 16,64 343,64 263,8 0 376,6 377,0 384,0 1047,7 0 162,86 13,81 286,27 324,55 0 188,49 218,99 811,24 92,68 0 300,37 370,37 33,02 27,36 742,0 803,75 257,32 519,65 559,8 1137,6 0 888,6 737,4 763,9 0 318,19 198,32 871,57 0 110 0 S0298 , Дж/(моль К) 65,69 191,5 220,0 210,62 240,45 304,3 192,5 94,56 179,9 51,42 71,1 64,18 72,36 136,0 205,03 238,98 22,8 311,7 362,9 64,9 67,4 76,44 147,28 91,20 31,88 248,1 256,23 205,64 122,2 156,9 42,09 56,74 52,34 54,4 97,1 30,6 50,3 252,4 75,9 41,59 43,5 57,7 124,6 Таблица 2 Произведение растворимости (ПР) некоторых малорастворимых электролитов при 250 С Электролит AgBr AgCl Ag2CrO4 AgI Ag2S Ag2SO4 Al(OH)3 BaCO3 BaCrO4 BaSO4 CaCO3 CaC2O4 CaF2 CaSO4 Ca3(PO4)2 Cd(OH)2 CdS ПР 6·10-13 1,8 ·10-10 4 ·10-12 1,1 ·10-16 6 ·10-50 2 ·10-5 1,9 10-33 5 ·10-9 1,6 ·10-10 1,1· 10-10 5 ·10-9 2 ·10-9 4 ·10-11 1,3 ·10-4 1 ·10-29 2 ·10-14 7,9 ·10-27 Электролит Cu(OH)2 CuS Fe(OH)2 Fe(OH)3 FeS HgS MnCO3 MnS PbBr2 PbCl2 PbCrO4 PbI2 PbS PbSO4 SrSO4 Zn(OH)2 ZnS ПР 2,2·10-20 6 ·10-36 1 ·10-15 3,8 ·10-38 5 ·10-18 1,6 ·10-52 5,05 10-10 2,5 ·10-10 9,1 ·10-6 2 ·10-5 1,8 ·10-14 8,0 ·10-9 1 ·10-27 1,6 ·10-8 3,2 ·10-7 1 ·10-17 1,6 ·10-24 Таблица 3 Константы нестойкости некоторых комплексных ионов в водных растворах при 250 С Схема диссоциации комплексного иона Константа нестойкости [Ag(NH3)2 ]+ ↔ Ag+ + 2 NH3 [Ag(NO2)2 ]- ↔ Ag+ + 2 NO2[Ag(S2 O3)2 ]3- ↔ Ag+ + 2 S2O32[Ag(CN)2 ]- ↔ Ag+ + 2 CN[HgCl4]2↔ Hg2+ + 4Cl [HgBr4]2↔ Hg2+ + 4Br [HgI4]2↔ Hg2+ + 4I [Hg(CN)4]2↔ Hg2+ + 4CN [Cd(NH3)4]2+ ↔ Cd2+ + 4NH3 [Cd(CN)4 ] 2↔ Cd2+ + 4CN [Cu(NH3)4]2+ ↔ Cu2+ + 4NH3 [Cu(CN)4]2↔ Cu2+ + 4 CN[Ni(NH3)6]2+ ↔ Ni2+ + 6NH3 9,3 ·10-8 1,8 ·10-3 1,1 ·10-13 1,1 ·10-21 8,5 ·10-16 1,0 ·10-21 1,5 ·10-30 4,0 ·10-42 7,6 ·10-8 7,8 ·10-18 2,1 ·10-13 5,0 ·10-31 1,9 ·10 -9 111 Таблица 4 Константы диссоциации некоторых слабых электролитов Электролит Константа диссоциации 1,8 ·10-16· 5,1 ·10-4 K1 = 5,83 ·10-10 1,772·10-14 K1 = 1,3 ·10-2 K1 = 1,1 ·10-7 4,9 ·10-10 K1 = 4,45· 10-7 1,754 ·10-5 K1 = 7,11· 10-3 K1 = 5,36 ·10-2 1,77 ·10-5 Вода Н2О Азотистая кислота НNO2 Борная кислота H3BO3 Муравьинная кислота HCOOH Сернистая кислота H2SO3 Сероводородная кислота H2S Синильная кислота HCN Угольная кислота H2CO3 Уксусная кислота CH3COOH Фосфорная кислота H3PO4 Щавелевая кислота H2C2O4 Гидроксид аммония NH4OH Таблица 5 Соотношения между некоторыми внесистемными единицами и единицами СИ Величина Длина Объем Давление Энергия, работа, количество теплоты Единица Эквивалент СИ Ангстрем (Ǻ) Микрон или микрометр (мкм) Литр (л) Миллилитр (мл) Атмосфера физическая (атм) Миллиметр ртутного столба (мм рт. ст.) Электронвольт (эВ) Калория (кал) Килокалория (ккал) 1·10-10 м 1 ·10-6 м 10-3 м3 10-6 м3 1,01325 ·105 Па 133.322 Па 1,60219 ·10-19 Дж 4,1868 Дж 4186,8 Дж Таблица 6 Значения некоторых фундаментальных физических постоянных Постоянная Обозначение Скорость света в вакууме Постоянная Планка Элементарный электрический заряд Число Авогадро Число Фарадея Газовая постоянная c h e NA F R 112 Численное значение 2,9979246 ·108 м/с 6,62618 ·10-34 Дж/с 1,602189 ·10-19 Кл 6,022045 ·1023 моль-1 9,64846 ·104 Кл/моль 8,3144 Дж/моль ОТВЕТЫ НА ВОПРОСЫ И ЗАДАЧИ ГЛАВА I. 1.1.1. а,г – открытая; б,в – замкнутая; д – изолированная по определению. 1.1.2. Зависимость свойств системы от пути перехода в исходное состояние связана с различной скоростью изменения состояния системы и скоростью изменения внешних условий. В приведенном примере скорость структурных преобразований явно меньше таковой изменения внешних условий – температуры. Быстрое охлаждение сплава позволяет сохранить его структуру, соответствующую высокотемпературному режиму. При достаточно медленном охлаждении сплав возвратится в исходное состояние. 1.1.3. Согласно определению: выделению из окружающей среды веществ, взаимодействие веществ. 1.1.4. Зависит от условий состояния системы и окружающей среды. 1.1.5. В том и другом случае происходят структурные преобразования, связанные с взаимодействием частиц. 1.1.6. Нет. Недостаточно для полного описания системы. 1.1.7. Энергия поступательного и вращательного движения молекул и колебаний атомов; энергия межмолекулярного взаимодействия, внутримолекулярная энергия связей; внутриатомная энергия электронных уровней; внутриядерная энергия; спиновая энергия. 1.1.8. Свойства функции состояния. Абсолютное её значение измерить и рассчитать нельзя, а изменение внутренней энергии можно. Выражают в Дж/моль. 1.1.9. Нет. 1.1.10. Нет. 1.1.11. д – изменение высоты над уровнем водохранилища. 1.1.12. Во-первых, свойство есть свойство, а функция это - зависимость. Нельзя ставить знак равенства или тождества между ними. Энергия системы функционально зависит от её состояния (внешних условий). Не сама же функция является энергией? Это чисто стилистическая ошибка, которая дает возможность подменить понятия: в частности, материальное свойство системы на математическую зависимость. Во-вторых, энергия есть энергия, а теплота является способом передачи этой энергии. А поэтому правильнее нужно говорить о энергии, переданной за счет разности температур. А способ передачи не тождественен передаваемому. 1.1.13. При постоянном объеме. 1.1.14. Нет. Теплота и работа лишь способы передачи энергии, механизм передачи. 1.2.1. Внутренняя энергия не изменится. Она перераспределится внутри системы более равномерно. 1.2. 2. Увеличилась на 41 кДж/моль. 113 1.2.5. Поскольку ∆U = Qp - A при постоянном давлении, а ∆U = 0, то Qp =A, т.е. все подводимая энергия теплопередачей к системе полностью расходуется на производство работы. А работа, производимая над системой, полностью превращается в тепло и выделяется системой. Равновесие. 1.2.6. Если процесс протекает в отсутствии тепловых эффектов (Qp =0), то ∆U = - A. Следовательно, есть возможность использования внутренней энергии, т.е. при исключении теплового обмена системы с окружающей средой производство работы расширения системой возможно только за счет убыли запаса её внутренней энергии. 1.2.7. В этом процессе должно практически отсутствовать изменение объема системы, т.е. исходные вещества и продукты реакции должны быть кристаллическими или жидкими. Могут быть и газообразными, но количество молей исходных газообразных веществ и газообразных продуктов должны быть одинаковыми. 1.2.8. Да. При всех взаимных превращениях энергия не теряется и не создается. 1.2.9. Количество энергии, подведенное к системе теплопередачей, расходуется на увеличение внутренней энергии системы и совершение работы расширения системой. 1.2.10. Порознь тепловой эффект реакции и работа, совершаемая системой, зависит от условий, при которых происходит процесс. Но в сумме они Qp + A определяют полное изменение внутренней энергии системы при переходе из начального состояния в конечное. И в пределах этой суммы энергии значения теплового эффекта и работы “вольны“ зависеть от пути перехода, способа передачи, определяемых условиями. 1.2.11. Если ∆Н > 0, то из окружающей среды системой поглощается энергия. Следовательно, её (системы) энергосодержание увеличилось. Тепловой эффект реакции Qp < 0. 1.2.12. Увеличилось на 436 кДж/моль. 1.2.13. Уменьшилось на 241 кДж/моль. 1.2.14. Изменение внутренней энергии и энтальпии системы связано соотношением ∆U = ∆H - P∆V . По условию задачи Qp = +143,1кДж/моль, а изменение энтальпии равно -143,1кДж/моль. Работа произведенная системой над внешней средой А = Р∆V. Величина ∆V= V2 – V1 представляет собой увеличение объема, поскольку в результате реакции возрастает число молей газообразных продуктов на ∆n = n2 – n1 (изменением объема участников реакции в конденсированном состоянии пренебрежем), где n1 и n2 – число молей газообразных участников реакции исходных веществ и продуктов, соответственно. В нашем случае ∆n= 1 – 0 =1, а согласно уравнению газового состояния P∆V = ∆nRT. Откуда А = ∆nRT = 1·8,31·(273 + 20) = 2,43 кДж. Тогда изменение внутренней энергии системы равно ∆U = ∆H - P∆V = -143,1 – 2,43 = - 145,53 кДж. 114 1.2.15. Разница между Qp и Qv определяется работой, которую совершает система расширяясь (сжимаясь): А = Qp - Qv. Работа расширения А = 2,43 кДж. Следовательно, Qp < Qv на величину 2,43 кДж. 1.2.16. При постоянном объеме ∆U = Qv. При постоянном давлении Qp = ∆H. 1.2.17. ∆U = ∆H + P∆V. 1.2.22. В жидком состоянии больше на ∆Hплавления= 5,99 кДж/моль. 1.2.23. Первая и третья. 1.2.24. Энтальпия образования воды в жидком состоянии ∆Н0f Н2О ≈ -286 кДж/моль. 9 граммов воды составляет 0,5 моля. Значит, нужно затратить как минимум 286 : 2 = 143 кДж. 1.2.25. Cакрис. + О2 газ = СаОкрис.; ∆H0СаО крис. = - 635,6 кДж/моль. 1.2.26. ∆U = ∆H – А или ∆U = ∆H - ∆nRT. Число молей пара ∆n = 250/18 =13,87 молей. Тогда ∆U =2451·250 – 13,87 · 8,31 ·298 =579 кДж. 1.2.27. ∆U = -282,49 кДж. 1.2.28. ∆U = ∆H + А или ∆U - ∆H =А. При постоянном объеме системы ∆U = Qv, при постоянном давлении Qp = - ∆H. 1.3.3. Можно. Если разложить эти оксиды на простые вещества и замерить тепловые эффекты. Ведь теплоты разложения и образования равны по величине и противоположны по знаку. В частности: ∆Н0f 298 = + 75,7 кДж/моль для Cl2Oгаз; для ClO2 газ ∆Н0f 298 = +104 кДж/моль и для Cl2O7 газ ∆Н0f 298 =+271 кДж/моль. 1.3.4. ∆Н0 = - 3270 кДж/моль. 1.3.5. ∆Н0 = - 1366,9 кДж/моль. При сгорании двух молей спирта (92 г) выделится Qp = 2733,7 кДж. 1.3. 6. ∆Н0 = - 810,1 кДж/моль. 1.3. 7. ∆Н0Н2О пар = 82,0 кДж/моль. 1.3. 8. ∆Н0 = 258,1 кДж/моль. 1.3. 9. ∆Н0МnO2 = - 404 кДж/моль. ГЛАВА П. 2.1.1. Реакция необратимая. 2.1.2. Несамопроизвольный, обратимый. 2.1.3. Процессы а) и б) самопроизвольные. При стандартных условиях процессы необратимы. 2.1.4. Процесс самопроизвольный, обратимый. Система замкнутая. 2.1.5. Обладает способностью к осуществлению без воздействия извне. 2.3.1. а, б, д – изменение энтропии – величина положительная (∆S>0), в, г – изменение энтропии величина отрицательная ((∆S<0). 2.3.2. Процесс испарения воды протекает с ∆Н0испар.> 0 и ∆Sиспар. >0; адсорбция водяных паров протекает с уменьшением энтропии ∆Sадсорб.< 0 (в адсорбированном состоянии вода находится в более упорядоченном виде) и с уменьшением энтальпии (∆Н0испар.< 0). 115 2.3.3. Процесс растворения подобен процессу расширения газа в вакууме, который сопровождается увеличением энтропии системы. 2.3.4. При низких температурах значение энтропийного фактора незначительно, а вблизи 0 К стремится к нулю. Поэтому решающее значение приобретает энергетический фактор ∆Н. При высоких температурах наоборот. 2.3.5. В газообразном состоянии больше возможностей реализации данного состояния системы, чем в кристаллическом. 2.3.6. Переход газа из газообразного (более вероятного состояния) в водный раствор сопровождается взаимодействием молекул газа и воды, что приводит к некоторому упорядочиванию их в растворе, а, следовательно, и к уменьшению энтропии газа. При растворении кристаллов или жидких веществ в воде происходит более равномерное, беспорядоченное расположение в растворе частиц этих веществ, т.е. увеличивается степень реализации состояния системы. 2.3.7. Энтропия системы больше в случае смеси индивидуальных веществ. В выросшем дереве ои взаимосвязаны, упорядочены. 2.3.8. а) ∆S0 = 160,4 Дж/моль К; б) ∆S0 = - 104,28 Дж/моль К; в) ∆S0 = 244,7 Дж/моль К. 2.3.9. В этом ряду она уменьшается. 2.3.10. Они находились бы в конденсированном состоянии: кристаллическом или жидком. 2.3.11. Naкрис., NaClкрис., N2O4 крис., Br2 жид., Br2 газ. 2.3.12. При 0 К энтропия равна нулю. 2.3.13. В результате гидратации ионов меди происходит упорядочивание расположения молекул воды и, соответственно, энтропия водного раствора должна возрасти. 2.3.14. Испарился. Молекулы воды равномерно распределились по всему объему. 2.4.1. Энтальпийным, т.к. можно считать ∆G ≈ ∆H. 2.4.2. ∆H< 0; ∆S > 0. 2.4.3. ∆G = 0. 2.4.4. Энтальпия жидкой воды ∆Н0f Н2О ≈ -286 кДж/моль, а энтальпия воды в парообразном состоянии ∆Н0f Н2О пар ≈ -241 кДж/моль. Это значит, что энергосодержание моля воды в парообразном состоянии выше. Испарение связано с энтропийным фактором. 2.4.5. Не учитывается энтропийный фактор. 2.4.6. Возможность процесса определяется двумя факторами. Суммарным выражением которых является ∆G = ∆H – Т∆S . 2.4.7. а). 2.4.8. 509 К. 2.4.9. 385,5 К. 2.4.10. ∆G = - 21,3 кДж/моль; Т = 602,8 К. 116 ГЛАВА Ш. 3.1.1. б), д). 3.1.3. Гетерогенные реакции: б), г). д), е). 3.1.4. На 0,35 моль/л. 3.1.5. На 0,2 моль/л. 3.1.6. С одинаковой скоростью. Реакция гетерогенная. 3.1.7. За одинаковое время в единице объема образовалось водорода 9 молей, сероводорода 4 моля, воды 2 моля. Скорость образования водорода наибольшая. Сравниваются средние скорости. 3.2.1. 1) V = k·[H2]2·[O2] ; 2) V = k·[NO]4·[O2]3; 3) V = k·[N2]·[H2]3; V = k·[H2]3; 4) V = k·[CuSO4]. 3.2.2. V = k· [A]· [B]2. а) 2 раза; б) 4 раза; в) 8 раз. 3.2.3. В 4 раза. 3.2.4. а) 4раза; б) 8 раз; в) 2 раза. 3.2.5. Еак. = 68 кДж/моль; k = 7,9· 10-2. 3.2.6. k2/k1 = exp(150· 103 – 100· 103)/( 8,31· 300) = 5,13· 108 раз. 3.3.1. а) Кс = [H2O]3/[H2]3; б) Kc =[CO2]2/([CO]2 ·[O2]; в) Кс = [H2]/[H2SO4]; г) Kc =[SO3]2/([SO2]2 ·[O2]); д) Кс = [Ag+раствор.]·[Cl-раствор]; е) Кс = [H2 ]4 /[H2 О]4. 3.3.3. [A] = 0,04 моль/л; [B] = 0,07 моль/л. 3.3.4. При понижении температуры смещение произойдет вправо. При повышении давления равновесие сместится так же вправо. 3.3.5. а) практически не сместится; б) сместится влево. 3.3.6. При увеличении концентрации кислорода равновесие сместится вправо; при увеличении концентрации хлора и понижении концентрации HCl – влево. 3.3.7. Кс = 5,35 ·10-55. 3.3.8. lgКс = 1,78 ·105. 3.3.9. ∆G0Н2О жид. = 89,5 кДж/моль; ∆G0СН3СООН = 27 кДж/моль. 117 Учебное издание Письменко Валерий Терентьевич Элементы химической термодинамики и кинетики Учебное пособие Редактор Н.А. Евдокимова Подписано в печать . . 2008. Формат 60х84/16. Бумага писчая. Печать трафаретная. Усл. печ. л. 7,68. Уч.-изд.л. 7,40. Тираж 100 экз. Заказ Ульяновский государственный технический университет 432027, Ульяновск, Сев. Венец, 32. Типография УлГТУ, 432027, Ульяновск, Сев. Венец. 32. 118