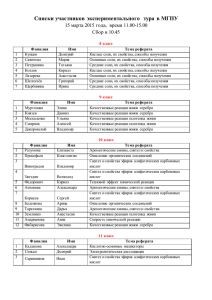
Молекулярно-термодинамическое моделирование ионных
advertisement

САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ На правах рукописи КОРОЛЕВА Софья Владимировна Молекулярно-термодинамическое моделирование ионных специфических эффектов в мицеллярных растворах ионных поверхностно-активных веществ Специальность 02.00.04 – физическая химия ДИССЕРТАЦИЯ на соискание ученой степени кандидата химических наук Научный руководитель д.х.н. профессор А.И. Викторов Санкт-Петербург 2015 2 ОГЛАВЛЕНИЕ ВВЕДЕНИЕ…………………………………………………………………………………………....4 ГЛАВА 1. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О ИОННЫХ СПЕЦИФИЧЕСКИХ ЭФФЕКТАХ В РАСТВОРАХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ…….……….…6 1.1 Последовательности Гофмейстера……………………………………………………………6 1.2 Экспериментальные данные о мицеллообразовании в водно-солевых растворах ионных поверхностно-активных веществ……………………………………………………………….…….9 1.3 Теоретические подходы к описанию влияния природы соли на термодинамические свойства флюидных систем……………………………………………………………………..……14 ГЛАВА 2. ИОННОГО МОЛЕКУЛЯРНО-ТЕРМОДИНАМИЧЕСКАЯ ПОВЕРХНОСТНО-АКТИВНОГО МОДЕЛЬ ВЕЩЕСТВА В АГРЕГАЦИИ ВОДНО-СОЛЕВОМ РАСТВОРЕ С УЧЕТОМ СПЕЦИФИКИ ИОНОВ……………………………………………....36 2.1 Модель свободной энергии агрегации…………………………………………..…………..36 2.2 Равновесное распределение подвижных ионов вокруг мицеллы……………………….....39 2.3 Критическая концентрация мицеллообразования, переход сфера-цилиндр, числа агрегации…………………………………………………………………………………..…….……40 2.4 Параметры модели…………………………………………………………………………....42 ГЛАВА 3. МЕТОДИКА РАСЧЕТА……………………………………………………………….45 3.1 Алгоритм расчета агрегативных характеристик мицеллярных растворов……………….45 3.2 Процедура подбора параметров модели……………………………………………….……46 ГЛАВА 4. РЕЗУЛЬТАТЫ МОДЕЛИРОВАНИЯ АГРЕГАЦИИ ИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ В РАСТВОРАХ НЕОРГАНИЧЕСКИХ 1:1 ЭЛЕКТРОЛИТОВ……………………………………………………………………………….….49 4.1 Влияние параметров модели на результаты расчетов характеристик мицеллярного раствора……………………………………………………………………………………………..…49 4.2 Влияние противоионов на образование и рост мицелл………………………………..…...61 4.3 Влияние коионов на образование и рост мицелл……………………………………….…..68 4.4 Сопоставление результатов настоящей работы с результатами предыдущих работ….…73 4.5 Значения параметров модели и структура агрегата……………………………………..….77 ЗАКЛЮЧЕНИЕ……………………………………………………………………………….….….85 СПИСОК ЛИТЕРАТУРЫ…………………………………..……………………………………...87 Приложение 1. Ссылки на экспериментальные данные о критической концентрации мицеллообразования, солевых порогах перехода сфера-цилиндр и числах агрегации………...101 Приложение 2. Дисперсионное взаимодействие иона со сферическим и цилиндрическим агрегатами……………………………………………………………………………………..……..101 3 Приложение 3. Свободная энергия, энтропия и химические потенциалы для смеси твердых сфер…………………………………………………………………………………………………...103 Приложение 4. Экспериментальное определение растворимости н-гексана и констант Сеченова в растворах солей NaCl, NaBr, NaNO 3 , KCl………………………………………………….….....104 4 ВВЕДЕНИЕ Химическая индивидуальность ионов оказывает существенное влияние на поведение широкого круга систем с надмолекулярной организацией: полимерных гелей, растворов блоксополимеров, белков, амфифильных синтетических и биологических мембран и т.п. Для растворов ионных поверхностно-активных веществ (ПАВ), замена противоиона или природы солевого фона может привести к радикальной перестройке структуры раствора, вызвать переходы сферических мицелл в червеобразные, образование пространственных сеток, жидкокристаллических структур, микроэмульсий и фазовое разделение. Указанная перестройка нередко сопровождается изменением макроскопических характеристик системы. Характер влияния природы ионов на агрегацию в растворах ионных ПАВ отражается рядами Гофмейстера [1], т.е. последовательностями, в которых влияние иона на критическую концентрацию мицеллообразования (ККМ), пороговые концентрации смены морфологий и рост образующихся агрегатов, а также макроскопические свойства (вязкость, поверхностное натяжение на границах раздела раствор – диэлектрик и пр.) усиливается или убывает. Коллинсом [2] была выдвинута концепция, в соответствии с которой положение иона в последовательности Гофмейстера зависит от компактности или рыхлости образующихся ионных пар. В последнее время достигнуты значительные успехи в теории ионных специфических эффектов для растворов электролитов [3], границ раздела раствор соли – диэлектрик (воздух, углеводород и др.) [4-10], растворов глобулярных протеинов [11-15] и для мембранных систем [16]. Эти теории показали, что наиболее важными факторами при проявлении специфики ионов являются их гидратация и поляризуемость, определяющая дисперсионные взаимодействия. Классические молекулярно-термодинамические модели [17-21] мицеллярных растворов ПАВ не учитывают указанные факторы. Различные модификации таких моделей [21-24] имеют лишь ограниченную применимость и не позволяют описать агрегацию в растворах различных катионных и анионных ПАВ в широком интервале концентрации добавляемой к раствору соли с учетом ее природы. Таким образом, разработка молекулярно-термодинамической модели для описания ионных специфических эффектов в растворах ионных ПАВ является актуальной задачей. Понимание закономерностей влияния природы соли на агрегацию ПАВ необходимо в разнообразных химико-технологических и биомедицинских приложениях: при оптимизации технологических схем процессов с участием водно-солевых растворов ПАВ (например, процессов мицеллярного разделения веществ, инкапсуляции лекарственных препаратов, снижения сопротивления потоку, повышения нефтеотдачи при гидроразрыве пластов др.), при 5 создании сенсоров, систем управляемой доставки лекарств, моющих средств и косметических препаратов, в фармакологии, нефтехимии. Выяснение механизмов влияния природы соли окажется полезным и при разработке новых подходов для описания поведения более сложных самоорганизующихся амфифильных молекул, в т.ч. белковых и фосфолипидных структур. Цель настоящей работы заключалась в учете природы ионов в молекулярнотермодинамической модели образования и роста мицелл ионных ПАВ. В связи с этим были поставлены следующие задачи: 1. Выяснение механизмов влияния природы ионов на агрегацию ПАВ в рамках предлагаемой модели; оценка роли различных факторов, отражающих химическую индивидуальность ионов (роль дисперсионных взаимодействий, эффектов гидратации, собственных размеров). 2. Разработка алгоритмов и программного обеспечения для проведения расчетов согласно предлагаемой модели. 3. Применение модели для описания критических концентраций мицеллообразования (ККМ), чисел агрегации и солевых порогов переходов сфера-цилиндр в водно-солевых растворах классических катионных и анионных ПАВ в широком интервале концентраций для различных солей; описание эффектов природы противоионов и коионов. 6 ГЛАВА 1. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О ИОННЫХ СПЕЦИФИЧЕСКИХ ЭФФЕКТАХ В РАСТВОРАХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ 1.1 Последовательности Гофмейстера Характер влияния природы добавленной соли на различные физико-химические характеристики флюидных систем (растворимость и теплоты растворения компонентов, осмотические коэффициенты, теплоемкости, pH, коэффициенты диффузии, проводимость и вязкость, межфазное натяжение на границах раздела, набухание высокомолекулярных веществ, стабильность коллоидов и др.) отражается с помощью ионных последовательностей Гофмейстера – первого ученого, сумевшего обобщить полученные данные о влиянии специфики ионов на стабильность коллоидных растворов (коллоидных оксидов железа, олеата натрия, коллагена и др.) [1]. В поисках объяснения положения ионов в этих последовательностях предлагались разнообразные концепции, рассматривающие структуру растворителя вокруг иона. Например, Воетом [1] была предложена концепция упорядочивания ионов в соответствии с их числами гидратации; кинетическая теория Самойлова [25] оказалась полезной при теоретическом обосновании высаливания компонента раствора в зависимости от гидратации ионов, количественно описываемой величинами поверхностной плотности молекул воды в первой координационной сфере. В соответствии с современной концепцией Коллинса [2] противоположно заряженные ионы образуют прочную контактную пару только тогда, когда оба иона либо космотропы (малые, слабо поляризуемые ионы с развитыми гидратными оболочками), либо хаотропы (большие, легко поляризуемые ионы с малыми гидратными оболочками). Данная концепция позволила объяснить специфические эффекты ионов в ряде флюидных систем; например, знак теплот растворения солей в воде в зависимости от природы ионов, составляющих соль [1]. Последовательность ионов в ряду Гофмейстера может изменяться как в зависимости от условий (pH раствора, концентрации соли, температуры), так и в зависимости от природы противоиона, составляющего пары с ионами последовательности, что может привести к частичному или полному обращению последовательности. В мицеллярных растворах ПАВ примером обращенной последовательности может служить последовательность связывания катионов щелочных металлов карбоксилатными полярными группами в сравнении с сульфатными [26]. Степень связывания катионов сульфатными агрегатами убывает в порядке Cs+>Rb+>K +>Na+>Li+, а карбоксилатными – в обратном. Проанализировав различные литературные данные и дополнив их квантово-механическими расчетами и расчетами по методу молекулярной динамики, Влачи и сотр. [26] подтвердили применимость концепции 7 Коллинса [2] для объяснения факта обращения ряда катионов щелочных металлов для карбоксилатных ПАВ в сравнении с сульфатными. По результатам моделирования [26] типичные анионные группы, входящие в состав полиэлектролитов, ПАВ и фосфолипидов, были упорядочены в ряд по уменьшению гидратации: карбоксилаты>фосфаты>сульфаты, аналогичный ряду Гофмейстера для анионов. Катионы щелочных металлов взаимодействуют с анионными группами ряда в соответствии с концепцией Коллинса: менее гидратированные катионы, например, Cs+ и Rb+, сильнее взаимодействуют со слабо гидратированными сульфатными группами и обнаруживают прямую последовательность Cs+>Rb+>K+>Na+>Li+, а более гидратированные катионы, например, Li+ и Na+, сильнее взаимодействуют с более гидратированными карбоксилатными группами и обнаруживают обратную последовательность Li+>Na+>K+>Rb+>Cs+ связывания ионов. Интересно рассмотреть пример обращения рядов катионов щелочных металлов для коэффициентов активности солей в водных растворах при замене галогенид-ионов ацетат-, формиат- или гидроксид-ионами. Робинсон и Харнед [1] предположили, что обращение ряда катионов в данном примере наступает благодаря процессу ―локализованного гидролиза‖, в котором могут участвовать ацетат-, формиат- и гидроксид-ионы. При ―локализованном гидролизе‖ одна из молекул воды, гидратирующих катион М +, смещает атом водорода к протон-акцепторному аниону A− : М+ - - - OH− — H+ - - - A−. При таком ‗гидролизе‘ положительный заряд на катионе М+ уменьшается, и он охотно теряет часть своей гидратной оболочки, уменьшая эффективный радиус (сильнее для лития, чем для цезия). Наблюдаемый эффект уменьшения эффективного радиуса катиона в присутствии протон-акцепторного аниона вызывает частичное или полное обращение ряда катионов щелочных металлов в растворах их солей для коэффициентов активности. В недавней работе Парсонс и сотр. [3] предположили, что отличие ацетат, формиат и гидроксид-ионов от галогенидов может быть связано с несферической симметрией первых, которая усиливает межчастичные неэлектростатические (дисперсионные) взаимодействия. Для линейно- и плоско-симметричных ионов были введены анизотропные поправки на пространственное распределение масс и поляризуемости атомов в ионе [3]. Было отмечено, что эти поправки должны сказаться на энергиях сольватации несимметричных ионов; еще существеннее они изменят межионные дисперсионные взаимодействия и дисперсионные взаимодействия ионов с границами раздела, поскольку несимметричные ионы могут быть ориентированы на определенные углы по отношению друг к другу и к границе раздела, упрочняя или ослабляя образующиеся ионные пары. При ориентационном взаимодействии ионов друг с другом и с границами раздела не исключается и изменение ионных гидратных оболочек, о котором упоминали Робинсон и Харнед [1]. Усиление или ослабление дисперсионного притяжения иона к границе раздела благодаря 8 асимметрии должно приводить к изменению адсорбции иона на отрицательно заряженной поверхности слюды [27], и, следовательно, сказываться на смачивании поверхности слюды раствором электролита. Так, растворы электролитов с ацетатом и другими органическими ионами создают конечный контактный угол на поверхности слюды, т.е. не смачивают, а растворы галогенидов смачивают слюду [27]. Частично обращенной последовательностью (колоколообразной) называется последовательность, в которой часть ионов сохраняет свой порядок расположения как в прямой последовательности, а другая часть, ввиду немонотонности изменения физико-химического свойства системы в зависимости от природы иона, переходит в обращенную. Примером колоколообразной последовательности может служить последовательность катионов щелочных металлов для роста додецилсульфатных мицелл [28]. Тенденция сферических мицелл расти с образованием длинных стержнеобразных структур увеличивается в последовательности Li+<<Na+<<K +<Cs+<Rb+, где обращение наступает не для всего ряда, а только для Cs + и Rb+. Миссель и сотр. [28] качественно объяснили такую последовательность ионов исходя из изменений размеров противоионов в гидратированном состоянии [28]. 1.2 Экспериментальные данные о мицеллообразовании в водно-солевых растворах ионных поверхностно-активных веществ Растворы ПАВ очень чувствительны к природе и концентрации добавленной соли и легко изменяют структуру и макроскопические свойства. Рассмотрим характерные особенности поведения растворов классических ионных ПАВ в присутствии различных электролитов. При измерении температур Крафта, степеней связывания ионов, ККМ, чисел агрегации, концентрационных порогов смены морфологий, фазовых переходов и других свойств ряды Гофмейстера прослеживаются достаточно четко. Как было показано [29], изменение физикохимических свойств агрегированных растворов ПАВ зависит от концентраций ионов вблизи агрегатов. Степени селективного связывания противоионов щелочных металлов с поверхностью пленок додецилсульфата натрия возрастают в ряду Na+<K+<Rb+<Cs+ [1], что совпадает с последовательностью Гофмейстера для понижения ККМ в растворах додецилсульфатов щелочных металлов [28]. Для экспериментального определения величин поверхностных концентраций ионов недавно предложен метод химического захвата (chemical trapping) [29-31]. Метод основан на захвате слабоосновного нуклеофила реактивной группой амфифильного арендиазониевого иона, добавляемого в систему и встраивающегося в агрегированную структуру (мицеллу или пленку ПАВ на границе раздела двух сред). Поверхностные концентрации нуклеофилов определяются по количеству продуктов реакции 9 захвата методом жидкостной хроматографии. Метод пригоден для определения концентраций различных противоионов, коионов и даже молекул воды. В качестве слабоосновных нуклеофилов могут выступать галогенид-ионы, OH-группы неионных и полярные группы ионных ПАВ (сульфатная, сульфонатная и карбоксилатная группы), спирты, вода и другие компоненты раствора. Исследования с помощью этого метода показывают, что вблизи мицеллярной поверхности концентрации полярных голов и противоионов высокие, порядка 14M; при структурных переходах (например, сфера-цилиндр) наблюдается существенное изменение поверхностных концентраций противоионов и воды. В недавней работе [30] было обнаружено, что с мицеллами алкилтриметиламмония бромид-противоион связывается в три раза сильнее, чем хлорид, а вблизи мицелл додецилсульфата натрия поверхностные концентрации коионов бромида и хлорида примерно одинаковы вплоть до 1М растворов, что согласуется с оценками, полученными при решении уравнения Пуассона-Больцмана [30]. В целом, самые сильные ионные специфические эффекты наблюдаются в случае гидротропных ионов – больших органических ионов типа салицилата и бензоата, – которые способны встраиваться в мицеллу [32]. Интересна статья Дженга и сотр. [31], в которой изучалось влияние 2,6- и 3,5-замещенных дихлоробензоатных (2,6ClBz¯ и 3,5ClBz¯, соответственно) противоионов на переходы сфера-стержень цетилтриметиламмониевых (С16 TA+) мицелл в бессолевых растворах. С помощью метода химического захвата было установлено, что увеличение содержания замещенного противоиона X¯ (X¯= 3,5ClBz¯ или 2,6ClBz ¯) в смеси C16 TACl+C16 TAX вызывает резкое увеличение концентрации X¯ и уменьшение концентрации воды вблизи мицеллярной поверхности при переходе сферических мицелл в стержнеобразные. При этом переход сфера-цилиндр в присутствии 3,5ClBz¯наступает при более низком его содержании в смеси, чем в присутствии 2,6ClBz¯. Полученные результаты показывают, что 3,5ClBz¯-ион охотнее образует контактную пару с полярной головой ПАВ, чем 2,6ClBz¯-ион, что увеличивает концентрацию противоиона на поверхности мицеллы, дегидратирует ее, вызывая более раннюю смену морфологии. Маджид и сотр. [33] исследовали влияние концентраций двух конкурирующих противоионов Cl¯ и 2,6ClBz¯ на морфологию образующихся мицелл в растворах цетилтриметиламмоний хлорида (C16 TACl) в соли. При низком содержании соли доминируют электростатические взаимодействия, и 2,6ClBz¯ сильно связывается с поверхностью мицеллы и проникает в мицеллярную корону, стимулируя одномерный рост мицелл, а непроникающий и сильно гидратированный хлорид вплоть до 1M растворов NaCl формирует глобулярные мицеллы. Если при общей концентрации соли ~1M начинают увеличивать содержание 2,6ClBz¯, то наблюдается резкий рост цилиндрических мицелл. В 0.12М растворе 2,6ClBz¯ числа агрегации цилиндров достигают своего максимума, а при последующем увеличении содержания 2,6ClBz¯ начинают 10 уменьшаться, так что в 1M растворе Na2,6ClBz мицеллы снова становятся сферическими. При общем увеличении концентрации соли максимум чисел агрегации смещается в область более низких содержаний 2,6ClBz¯. При низких концентрациях добавленной соли, когда в системе доминируют электростатические взаимодействия, при добавлении соли понижается поверхностный заряд мицеллы, и образование цилиндрической части становится выгоднее образования полусферических окончаний мицеллы, поэтому мицеллы растут. При этом противоион 2,6ClBz¯ понижает заряд мицеллы эффективнее, чем Cl¯. При высоких концентрациях соли, когда поверхность мицелл становится почти нейтральной, начинают действовать неэлектростатические факторы. Среди таких факторов Маджид и сотр. [33] отметили поверхностный вклад в свободную энергию образования агрегата, вклады стерического отталкивания полярных голов ПАВ и переноса углеводородного радикала молекулы ПАВ из раствора в мицеллу и специфического взаимодействия ароматического противоиона с триметиламмониевой полярной группой, приводящего к проникновению противоиона 2,6ClBz¯ в мицеллярную корону. По мнению авторов работы [33] указанные неэлектростатические факторы способствуют уменьшению тенденции роста цилиндрических мицелл при добавлении соли, при этом образование шапочек становится все более выгодным по сравнению с образованием цилиндров, мицеллы уменьшаются в размере и становятся сферическими. Для солей неорганических и небольших органических ионов специфические ионные эффекты также могут быть весьма значительными. Например, дидодецилдиметиламмоний сульфат (DDAS) почти нерастворим, но набухает в воде, образуя ламеллярную жидкокристаллическую фазу [1], а дидодецилдиметиламмоний ацетат (DDAAc) и гидроксид (DDAOH) чрезвычайно хорошо растворимы в воде и спонтанно образуют термодинамически стабильные везикулы, которые при добавлении ПАВ переходят в мицеллы [1, 34]. На качественном уровне особенности поведения этих систем объясняли образованием пар между полярными группами ПАВ и более или менее сильно гидратированными противоионами [1, 34], что в настоящее время может быть объяснено с помощью концепции Коллинса [2]. Специфические эффекты ионов в мицеллярных растворах наиболее изучены для классических ионных ПАВ в присутствии неорганических электролитов типа 1:1. Настоящая работа будет посвящена рассмотрению именно таких систем. Для катионных ПАВ эффект противоиона (аниона) сильнее, чем для анионных. Это связано с бóльшей поляризуемостью анионов [10]. Авторы работы [35] объясняют этот факт тем, что анионы – это частицы с избыточным количеством электронов, а значит, с более протяженными и рыхлыми электронными оболочками. 11 Среди катионных ПАВ наиболее систематически исследованы алкилпиридиниевые, алкилтриметиламмониевые и додецилдиметиламмониевые производные в присутствии солей с различными противоионами (см. Приложение 1). Для этих ПАВ влияние противоиона на понижение ККМ обычно следует последовательностям OH¯<F¯<Cl¯<Br¯<NO3 ¯<ClO3 ¯<I¯<Bz¯<Sal¯. Среди Гофмейстера [32, 36-38] анионных ПАВ наиболее изучены алкилсульфаты и алкилкарбоксилаты. Для алкилсульфатов ряд противоионов для понижения ККМ следующий Li+<Na+<K +<Cs+ [28]. Однако для алкилкарбоксилатов обнаружено обращение катионного ряда для ККМ Li+>Na+>K+>Cs+ [24]. При низких и средних содержаниях соли кривые ККМ-ионная сила раствора в логарифмических координатах (зависимость Коррина-Харкинса) линейны и почти параллельны для разных противоионов [39]. При средних и высоких концентрациях соли обнаруживается разброс экспериментальных данных для ККМ. Тем не менее, общие тенденции прослеживаются ясно. При средних и высоких концентрациях степень отклонения от линейности кривых ККМионная сила в логарифмических координатах сильно зависит от природы соли [39]. Для галогенидов додецилдиметиламмония Окуда и сотр. [39] получили кривые ККМ с отклонениями вниз, причем в случае с хлоридом отклонение получилось более сильным, чем с бромидом. Следует отметить, что не все используемые методы пригодны для измерения ККМ при умеренных и высоких концентрациях соли. В работах [40-41] даны ККМ в растворах додецилдиметиламмоний хлорида в присутствии хлорида натрия, измеренные разными методами (солюбилизации, рассеяния света, вискозиметрии и тензиометрии). В бессолевых системах и при низких концентрациях соли измеренные разными методами ККМ согласуются достаточно хорошо. При высоких концентрациях соли первые три метода (солюбилизации, рассеяния света и вискозиметрии) дают завышенные ККМ, кривые ККМ с изломами или неестественно резкими отклонениями вниз [41-43], что может быть связано с превращением сферических мицелл в цилиндрические [40]. В таком случае необходим детальный анализ структуры и размера образующихся агрегатов. Метод измерения поверхностного натяжения дает самые низкие значения ККМ, при которых ПАВ из мономерной формы переходит либо в сферические, либо в цилиндрические мицеллы. Влияние противоионов на рост чисел агрегации и понижение солевых порогов переходов сфера-цилиндр в общем усиливается в той же последовательности, что и для понижения ККМ [37]. В присутствии разных противоионов солевые пороги перехода сфера-цилиндр могут отличаться более чем на порядок. Примером такого сильного э ффекта служат хлориды и бромиды алкилпиридиния и алкилтриметиламмония. Более сильный рост наблюдается для бромидов этих ПАВ. Еще более сильной тенденцией к агрегации обладают иодиды 12 алкилпиридиния. Низкая ККМ и значительный рост цилиндров уже при низкой концентрации соли [44] в присутствии иодидов связаны с образованием особенно компактных пар пиридиний – иодид, при котором наблюдается перенос заряда от иодид-противоиона к пиридиниевой группе [45]. Концепция Коллинса [2] может быть применена для объяснения последовательностей Гофмейстера в мицеллярных системах [26]. В мицеллярных коронах хаотропные противоионы образуют более компактные пары с хаотропными головами, а космотропные противоионы – с космотропными полярными головами, теряя часть гидратных оболочек. Например, хаотропные сульфатные или триметиламмониевые головы образуют более компактные пары с хаотропными противоионами Cs+ и Br¯, соответственно [26]; космотропный Li+ образует более компактные пары с космотропной карбоксильной группой, приводя к обращению последовательностей Гофмейстера для алкилкарбоксилатов в сравнении с алкилсульфатами [26]. Более компактная упаковка пар противоион-голова уменьшает предпочтительную кривизну короны агрегата и, следовательно, благоприятствует мицеллообразованию и росту. При добавлении соли к раствору ионного ПАВ мицеллы испытывают рост, становятся длинными и переплетаются – раствор становится вязкоупругим. Природа коиона тоже оказывает влияние на поведение мицеллярных растворов ПАВ [46]. На ККМ и начало переходов сфера-цилиндр природа коиона оказывает слабое влияние [46-47]; поведение вязкоупругих свойств раствора при высокой концентрации соли может существенно зависеть от природы добавленного коиона. Например, добавление KCl [48] вместо NaCl [49] к водным растворам катионного ПАВ эруцилбис(гидроксиэтил)метиламмоний хлорида (EHAC) на полпорядка выше сдвигает значение концентрации соли, при которой наблюдается пик вязкости. 1.3 Теоретические подходы к описанию влияния природы соли на термодинамические свойства флюидных систем В классической молекулярно-термодинамической модели [17-21, 50] свободная энергия образования мицеллы из мономерных молекул ПАВ представляется в виде суммы вкладов: свободной энергии переноса углеводородного радикала молекулы ПАВ из раствора в ядро мицеллы, свободной энергии стерического отталкивания полярных голов, свободной энергии упругой деформации углеводородных радикалов молекул ПАВ, свободной энергии образования границы углеводород-раствор и свободной энергии взаимодействий заряженных голов с подвижными ионами. Влияние природы добавленной соли может отражаться на большинстве этих вкладов. Рассмотрим существующие подходы для учета влияния природы соли на 13 Рисунок 1. Зависимость логарифма растворимости S, моль/л, от числа углеродных атомов в цепи линейного углеводорода при T=298K, p=1бар. Незакрашенные кружки – эксперимент [51], треугольники – эксперимент Толлса и сотр. [52], пунктирная линия – расчеты групповым методом [53], сплошная линия – расчеты по методу молекулярной динамики [54]. указанные вклады в свободную энергию мицеллообразования. Гидрофобный вклад Вклад переноса углеводородных радикалов молекул ПАВ из воды в мицеллу (так называемый гидрофобный вклад) пропорционален логарифму растворимости углеводорода с той же длиной цепи [17]. Проблема экспериментального определения растворимости углеводородов связана с необходимостью измерения чрезвычайно малых величин. Считалось [55-56], что кривая зависимости логарифма растворимости коротких углеводородов с увеличением числа углеродных атомов в цепи спадает, претерпевая излом при более длинных углеводородов она почти постоянна [54], , после которого для рисунок 1. Завышенную растворимость длинных углеводородов по сравнению с короткими объясняли образованием метастабильных коллоидных суспензий алканов, приводящих к искусственному увеличению измеряемой растворимости, либо сворачиванием отдельных молекул в более компактные конформации [54]. В некоторых работах образующиеся коллоидные агрегаты частично удалялись с помощью микрофильтров [51], что позволяло получить более низкие значения растворимости, однако четкой зависимости логарифма растворимости от числа углеродных атомов в цепи не получалось, рисунок 1. Авторы работы [53], собрав большой массив экспериментальных данных о парциальных молярных объемах при бесконечном разбавлении, свободной энергии Гиббса, энтальпии и теплоемкости испарения и гидратации углеводородов и спиртов в воде и использовав групповой подход, рассчитали термодинамические функции отдельных групп атомов. Растворимости линейных углеводородов в воде, рассчитанные вплоть 14 до =22 при использовании группового подхода, показаны на рисунке 1. Авторам работы [53] удалось получить экспоненциально убывающую зависимость логарифма растворимости от числа углеродных атомов в цепи [54]. Позже Толлс и сотр. [52] экспериментально подтвердили эту зависимость для углеводородов с . С помощью метода молекулярной динамики Фергусон и сотр. [54] показали, что если сворачивание и характерно для молекул линейных углеводородов, то для углеводородов с длиной цепи . В расчетах по методу молекулярной динамики [54] была получена кривая зависимости логарифма растворимости углеводородов от числа углеродных атомов в цепи, которая совпала с экспериментальной кривой (с учетом данных Толлса и сотр. [52]) и кривой, рассчитанной групповым методом [53] для , рисунок 1. Для более длинных углеводородов ( ) рассчитанная с помощью метода молекулярной динамики [54] кривая отклоняется выше рассчитанной групповым методом [53], рисунок 1. Такое искривление линии растворимости авторы, однако, не связывают с гибкостью цепей и их конформационным свертыванием. При расчетах методом молекулярной динамики [54] было обнаружено, что зависимость избыточного химического потенциала углеводорода в воде, или свободной энергии переноса молекулы углеводорода из идеальной газовой смеси в раствор, от числа углеродных атомов немонотонна, и максимум ее приходится на . Немонотонность избыточного химического потенциала углеводорода индуцирует искривление линии растворимости при . В классических молекулярно-термодинамических моделях мицеллообразования [17-21] гидрофобный вклад оценивался на основе экстраполяции экспериментальной кривой растворимости, полученной при на более длинные цепи в расчете на СН2 группу [57]. Использование этой экстраполяции должно приводить к несколько заниженным оценкам ККМ для ПАВ с длиной цепи , а использование данных Фергусона и сотр. [54] должно улучшить согласие расчетов по молекулярно-термодинамическим моделям с экспериментом. Большинство неорганических солей обычно высаливает углеводороды, а значит, растворимость еще больше понижается [58-59]. Явления всаливания и высаливания углеводородов обычно характеризуются с помощью констант Сеченова [60-62]. Логарифм отношения растворимостей углеводорода в растворе данной соли и воде п ропорционален концентрации соли [63] (1) а – константа Сеченова, л/моль; – постоянная Больцмана, – концентрация соли, - температура. Константа Сеченова характеризует все виды взаимодействий в растворе, кроме взаимодействий углеводород- 15 углеводород, и является функцией температуры , давления и природы соли. Выражение 1 применимо при концентрациях растворов до 1-2M. Для расчета недостающих констант Сеченова используют правило аддитивности [24], в соответствии с которым эффект соли на данный углеводород складывается из эффектов отдельных ионов, составляющих раствор. Уравнение 1 используется как поправка (―вклад Мукерджи‖) к гидрофобному вкладу в свободную энергию мицеллообразования, учитывающая влияние соли [64-65]. Для качественного объяснения и количественного описания эффектов высаливания углеводородов и других неэлектролитов был разработан ряд теорий, которые можно условно разделить на несколько групп: теории, учитывающие гидратацию ионов; электростатические теории; теории, учитывающие ван дер ваальсовы взаимодействия; теории, учитывающие ионное давление растворов [63]. Первые гидратационные теории всаливания-высаливания неэлектролитов строились на основе утверждения о независимости чисел гидратации ионов от природы неэлектролита [63]. Однако вскоре эксперимент показал, что постоянство чисел гидратации ионов в разных объектах соблюдается не строго, и что порядок их изменения от иона к иону не всегда соответствует последовательностям ионов, наблюдаемых при исследовании всаливаниявысаливания неэлектролитов. Позже эффекты гидратации стали учитываться через эффективные размеры ионов и молекул неэлектролита в электростатических теориях при решении уравнения ПуассонаБольцмана (ПБ) для молекулы неэлектролита в растворе соли. Получаемое при решении уравнения ПБ распределение локального электрического потенциала вблизи молекулы неэлектролита используется при расчете коэффициента активности неэлектролита, который зависит от зарядов ионов, концентрации соли, температуры и диэлектрической проницаемости раствора неэлектролита. Для описания специфики ионов предлагались различные модификации уравнения ПБ и выражения диэлектрической проницаемости раствора [63], при этом учитывались собственные размеры ионов, поляризуемость неэлектролита и его дипольный момент (если неэлектролит полярный), зависимость диэлектрической проницаемости раствора от среднего расстояния между молекулой неэлектролита и ионом или от парциального молярного объема неэлектролита. Прогресс предложенных электростатических подходов заключался в возможности предсказывать верный порядок величин констант Сеченова. Для ряда электролитов (например, хлоридов натрия и калия), для которых специфические эффекты ионов незначительны, разработанные подходы позволили получить верные последовательности Гофмейстера. Однако существенных различий между константами Сеченова для некоторых солей с помощью этих 16 подходов получить не удалось. Фундаментальный недостаток предложенных теорий состоял в том, что помимо собственных размеров ионов они не учитывали роль неэлектростатических взаимодействий. В теориях, учитывающих ван дер ваальсовы взаимодействия, наряду с электростатическими стали учитываться дисперсионные взаимодействия ионов с молекулой неэлектролита. Линдерстрѐм и Ланг [63] обнаружили, что последовательности ионов при высаливании неэлектролита коррелируют с изменением молярных рефракций водно-солевых растворов, зависящих от поляризуемостей ионов, входящих в состав растворов. Кортум [63] предположил, что степень всаливания неэлектролита большим ионом может зависеть от силы ван дер ваальсова взаимодействия иона с неэлектролитом. МакДевит и Лонг [63] получили параметрическое выражение для коэффициента активности неэлектролита, содержащего электростатическое и дисперсионное слагаемые, параметры которых находились из эксперимента. Для слабо полярных и неполярных молекул неэлектролита (углеводорода) электростатическое слагаемое положительное и означает высаливание, а дисперсионное – отрицательное и означает всаливание. Как было отмечено МакДевитом и Лонгом [63], дисперсионное слагаемое должно быть пропорционально объему иона и, следовательно, его поляризуемости. Почти одновременно с МакДевитом и Лонгом Бокрис, Боулер-Рид и Китченер [63], взяв в качестве дисперсионного потенциала лондоновский, получили уравнение для коэффициента активности неэлектролита, в котором коэффициенты при электростатическом и дисперсионном слагаемых были выражены в явном виде. Интересным результатом применения предложенных подходов, учитывающих ван дер ваальсовы взаимодействия, явилась возможность предсказывать всаливание неэлектролита большими ионами, например, тетраалкиламмониевыми. Все же дисперсионное слагаемое получилось довольно слабо зависящим от природы иона, что не позволило описывать существенные различия между константами Сеченова для разных солей и наблюдаемое в некоторых случаях обращение последовательностей Гофмейстера для изменения растворимости неэлектролитов, например, последовательности катионов H+<<Cs+<Rb+~Li+<K+<Na+ при высаливании бензола [62-63] для Li+ и H+. Для получения более существенных различий между константами Сеченова и объяснения обращений последовательностей Гофмейстера необходим более строгий учет дисперсионных взаимодействий [63]. Несмотря на это, предположение о дисперсионных взаимодействиях как возможном источнике ионных специфических эффектов в растворах неэлектролитов [63] является важным результатом применения теорий, учитывающих ван дер ваальсовы взаимодействия. В основе теорий, учитывающих ионное давление, создаваемое раствором соли, лежит концепция Тамманна [63]. Внутреннее ионное давление определяется степенью, с которой 17 растворитель сжимается или расширяется в присутствии ионов. Поскольку для большинства неполярных неэлектролитов последовательности ионов получаются идентичными , МакДевит и Лонг [63] развили подход Тамманна, предположив, что специфические эффекты ионов в растворах неэлектролитов определяются свойствами индивидуальных растворов солей. В рамках теории МакДевита и Лонга [63] выражение для константы Сеченова для малых молекул неэлектролита имеет вид [63] (2) где – парциальный молярный объем неэлектролита при бесконечном разбавлении ; ―эффективное давление‖ соли в растворе; – - концентрация соли. Для линейных углеводородов, константы Сеченова должны быть пропорциональны числу углеродных атомов в цепи [53], поскольку парциальный молярный объем линейного углеводорода в растворе также примерно пропорционален числу углеродных атомов в цепи. Знак производной указывает на всаливание или высаливание неэлектролита. экспериментально получены линейные зависимости давлений Для ряда солей от концентрации были вплоть до высоких концентраций соли. Было показано, что хаотропные ионы, например, Cs+ и I-, разрушая структуру воды, уменьшают внутреннее давление раствора и могут вызвать всаливание, иногда значительное, как, например, большой катион (CH3 )4 N+. Космотропные ионы, в т.ч. высокозаряженные, встраиваясь в структуру воды и упрочняя ее, увеличивают внутреннее давление в растворе и обнаруживают значительный высаливающий эффект. Для больших молекул неэлектролита подходы, учитывающие давление ионов раствора, дают завышенные абсолютные значения констант Сеченова, а для малых неполярных молекул – вполне удовлетворительные результаты [63]. Рассчитанные для растворимостей относительные эффекты различных солей получились верными, в том числе и для ионов Li+ и H+ в ряду H+<<Cs+<Rb+~Li+<K+<Na+. Обобщим основные результаты существующих теоретических подходов для расчета влияния соли на растворимость неэлектролитов в водных растворах. Электростатические теории позволили описать экспериментально наблюдаемую линейность изменения растворимости неэлектролита в зависимости от концентрации соли, верный порядок величин констант Сеченова, эффекты размера и полярности молекул неэлектролита и эффекты гидратации ионов. Теории, учитывающие ван дер ваальсовы взаимодействия, и теории, учитывающие ионное давление раствора, позволили воспроизвести ионные последовательности Гофмейстера и их нарушение для коэффициентов активности слабо полярных неэлектролитов в растворах ряда солей, а также показали, что поведение неэлектролитов в растворах солей определяется ионной поляризуемостью и связанными с ней дисперсионными 18 взаимодействиями. Вследствие достаточно сильного разброса экспериментальных данных о растворимости длинных линейных углеводородов в растворах солей (из-за проблемы измерения низких значений растворимостей) предложенные теории могут быть использованы в классических молекулярно-термодинамических моделях агрегации при оценке солевого эффекта для переноса гидрофобного радикала молекулы ПАВ из раствора в ядро мицеллы. Вклад стерических отталкиваний полярных голов в короне мицеллы Полярные головы ПАВ располагаются на поверхности ядра мицеллы, где их можно рассматривать как монослой. В классических моделях мицеллообразования [17] полярные головы считаются жесткими, поэтому их стерические взаимодействия описывают, используя отталкивательную часть двумерного уравнения ван дер Ваальса. Свободная энергия стерического отталкивания между полярными группами ПАВ дается выражением [17] (3) где – эффективная площадь поперечного сечения полярной головы ПАВ; – площадь мицеллярного ядра, приходящаяся на молекулу ПАВ. Больший размер полярной головы усиливает стерическое отталкивание на поверхности мицеллы, дестабилизируя мицеллу и способствуя диспергированию. В некоторых вариантах молекулярно-термодинамических моделей мицеллообразования [21, 24] для описания ионных специфических эффектов в свободной энергии стерических взаимодействий учитывается взаимное отталкивание голов и проникающих в мицеллярную корону ионов. В таких теориях свободная энергия стерического отталкивания, , является функцией собственных размеров ионов и степеней их связывания с поверхностью мицеллы. Деформация углеводородного радикала молекулы ПАВ Вклад свободной энергии упругой деформации рассчитывается для сильно сегрегированных в мицеллярном ядре углеводородных радикалов молекул ПАВ в рамках аналитической теории самосогласованного поля, предложенной для растворов блоксополимеров [17, 66]. Один конец углеводородного радикала закреплен на поверхности ядра мицеллы, а другой свободно располагается в глубине агрегата. Углеводородный радикал моделируется гауссовой цепью, растяжение которой подчиняется закону Гука [17, 66]. В соответствии с законом Гука, самосогласованное поле, действующее на цепь, является квадратичной функцией ее растяжения. Для свободной энергии упругой деформации углеводородного радикала ПАВ в мицеллярном ядре имеем [17] (4) 19 где для сферических и для цилиндрических мицелл, сферического/цилиндрического ядра мицеллы, – радиус – длина сегмента Куна, – число сегментов Куна в углеводородном радикале [17]. В классических моделях [17] предполагается, что соль заметно не проникает в углеводородное ядро, поэтому свободная энергия деформации углеводородных радикалов ПАВ, , считается не зависящей от природы соли. Поверхностный вклад При образовании мицеллы необходимо учитывать свободную энергию формирования границы раздела вода – углеводородное ядро. В молекулярно-термодинамических моделях для этого пользуются выражением [17] (5) где – площадь ядра, экранированная от контакта с водой площадью поперечного сечения углеводородного радикала или полярной головы . Межфазное натяжение границы ядро-вода дается выражением (6) где – поверхностное натяжение алифатического углеводорода молекулярной массы , равной массе радикала ПАВ; – поверхностное натяжение воды. Все поверхностные натяжения выражены в единицах [мН/м]. Известно, что соли могут оказывать существенное и различное влияние на межфазное натяжение границы раздела водный раствор – диэлектрик (углеводород, воздух и др.) [1]. В работе Морейры и Фирузабади мицеллообразования, [24] поверхностный вклад в свободную энергию , зависит от природы и концентрации соли и оценивается эмпирически на основе экспериментальных данных об изменении поверхностного натяжения раствора соли на незаряженной границе раздела с углеводородом. Ионный вклад Для расчета ионных взаимодействий на заряженных и незаряженных, плоских и искривленных границах раздела применяют уравнение ПБ, решение которого позволяет рассчитать профили распределения электрического потенциала и ионов вблизи границы. Зная профили распределения ионов, рассчитывают поверхностное натяжение, свободные энергии ионных взаимодействий при образовании межфазных границ раздела и мицеллярных агрегатов и другие характеристики. Первым теоретическим подходом для описания ионных взаимодействий в растворе на границе с диэлектриком стала работа Онзагера и Самараса [67-68], в которой был предложен поверхностный аналог предельного закона Дебая-Хюккеля. Онзагер и Самарас [67-68] решали 20 уравнение ПБ для случая плоской незаряженной диэлектрической поверхности в разбавленном растворе соли, в котором помимо электростатически х межионных учитывались электростатические взаимодействия ионов с границей раздела, возникающие благодаря разнице диэлектрических проницаемостей воды и диэлектрика (взаимодействия ионов с мнимыми зарядами, индуцированными ими в воздушной среде). Выражение для потенциала средней силы взаимодействия иона с границей раздела является функцией длины Дебая и собственного размера иона (расстояния максимального приближения иона к границе раздела) – единственной специфической характеристики иона. Предложенный аналог закона Дебая-Хюккеля оказался применимым до взаимодействия концентраций электролита ионов с границей ~0.2M, раздела при становятся которых электростатические в значительной степени экранированными. Авторы рассматриваемой работы отмечают, что для предсказания зависимостей поверхностного натяжения при высоких концентрациях соли необходимо учесть дополнительные неэлектростатические силы, действующие на ионы вблизи границы раздела [27]. Как упоминалось ранее, предположение о том, что ионная поляризуемость и дисперсионные взаимодействия могут отражать природу иона, впервые было использовано при теоретическом описании коэффициентов активности неэлектролитов в растворах солей [63]. Однако, чтобы получить количественный подход, включения простой эмпирической зависимости потенциала дисперсионного взаимодействия иона с молекулой неэлектролита в электростатические уравнения оказалось недостаточно. Более строго задача учета ван дер ваальсовых взаимодействий в солевых растворах решалась Дэвисом, Нинэмом, Нэтсом и сотр. [69-71] на основе теории Лифшица [72] дисперсионных взаимодействий. Теория Лифшица дает общее выражение для потенциала дисперсионного взаимодействия между двумя полубесконечными плоскими диэлектрическими телами в вакууме [72] или в среде, в которую может быть погружена система [73-74], в зависимости от флуктуаций испускаемого телами электромагнитного поля [75]. Позже было показано, как на основании макроскопического уравнения Лифшица можно получить выражения для потенциалов дисперсионных взаимодействий атома с другим атомом [73] и с макроскопическим телом[74]. Вслед за макроскопической теорией Лифшица на основе микроскопических предположений был разработан ряд математически более простых подходов [76-77], позволивших для потенциалов дисперсионного взаимодействия получить выражения, идентичные уравнениям Лифшица [73, 75]. В рамках этих подходов было показано, как учесть кривизну и геометрию диэлектрического макротела [75, 78-83], неоднородности поверхностных слоев вблизи границ раздела сред [75], поправки на трехчастичные взаимодействия и взаимодействия более 21 высокого порядка [75, 84-86] (в т.ч. для систем достаточно высокой плотности [87-88]) и собственный размер атомов/молекул [84]. Для ионных систем Дэвис, Нинэм, Нэтс и сотр. [69-71] предложили применить выражения для потенциалов дисперсионных взаимодействий, полученные в рамках теории Лифшица и микроскопических подходов. При этом индивидуальность иона выразилась в силовой константе потенциала дисперсионного взаимодействия, которая зависит от поляризуемости иона, материала диэлектрика и окружающей среды [3, 13] (см. Приложение 2). Для разных ионов поляризуемость разная [3, 10, 12], следовательно, потенциалы дисперсионного взаимодействия двух ионов или иона с агрегатом специфичны [13, 69]. В силовой константе потенциала дисперсионного взаимодействия отражаются различные виды взаимодействий иона с окружением в широком спектре частот [75]: взаимодействия Кеезома (постоянных диполей), Дебая (постоянного диполя с индуцированным) и ван дер Ваальса (индуцированных диполей). Учесть все эти типы взаимодействий теоретически в настоящее время невозможно, поскольку необходимые частотные зависимости диэлектрических проницаемостей взаимодействующих сред и поляризуемостей ионов не могут быть измерены с достаточно большой точностью [75]. Следует отметить, что используемые в большинстве работ дисперсионные константы учитывают только высокочастотные вклады (УФ и видимого излучения) в поляризуемость и диэлектрические функции, т.е. собственно дисперсионные составляющие взаимодействия иона с границей раздела [27] или с другими ионами [3]. Низкочастотные вклады (ИК и микроволнового излучения), которые возникают при гидратации ионов полярными молекулами воды, не учитываются. Поэтому эффекты гидратации ионов приходится учитывать в моделях отдельно, вводя дополнительные параметры [27]. Предполагается, что строгий учет всего спектра взаимодействий в широкой области частот в рамках подхода Дэвиса, Нинэма, Нэтса и сотр. [69-71] позволит исключить введение дополнительных поправок на исключенный объем, гидратацию ионов и другие факторы в теориях, описывающих ионные специфические эффекты в разных системах [27]. В первое время подход Дэвиса, Нинэма, Нэтса и сотр. [69-71] применялся в основном для расчета профилей распределения ионов вокруг заряженных объектов, например, молекул глобулярных протеинов. Для глобулярного протеина в растворах различных солей были выполнены расчеты по методу Монте-Карло (МК), где вода рассматривалась как непрерывная среда, и интегральным уравнениям Орнштейна-Цернике с гиперцепным замыканием (ОЦ-ГЦ) [13]. Модель включала межионные дисперсионные взаимодействия [13] наряду с дисперсионными взаимодействиями между ионом и молекулой протеина и взаимодействиями исключенного объема ионов. Полученные распределения ионов сравнивались с результатами решения нелинеаризованного уравнения ПБ, модифицированного только дисперсионными 22 Рисунок 2. Зависимость рассчитанных [13] приведенных концентраций ионов от расстояния от поверхности молекулы протеина диаметром =3нм и зарядом +20e0 в растворе концентрации 1.0М для Na2SO4 . Незакрашенные кружки – профили противоионов (сульфат), серые кружки – профили коионов (натрий), рассчитанные методом Монте-Карло; сплошные линии – расчет по уравнению ПБ; пунктирные линии – расчет по уравнению ОЦ-ГЦ. потенциалами ион-протеин. Все используемые потенциалы дисперсионного взаимодействия рассчитывались в рамках подхода Дэвиса, Нинэма, Нэтса и сотр. [69-71] В общем, распределения ионов, полученные в рамках всех трех подходов, МК, ОЦ-ГЦ и ПБ, согласуются удовлетворительно [13]. Расхождение наблюдается, когда в системе присутствует сильнозаряженный противоион (например, двухзарядный), при этом профили распределения ионов, полученные при решении уравнения ПБ, качественно отличаются от результатов подходов ОЦ-ГЦ и МК, рисунок 2. В соответствии с расчетами по методам ОЦ-ГЦ и МК сильное (электростатическое и дисперсионное) связывание двухзарядного противоиона с молекулой протеина усиливает (электростатическое) связывание коионов, которые образуют второй плотный слой, следующий за слоем адсорбированных противоионов – максимум концентрации на профиле распределения коионов. Этот максимум не воспроизводится при решении уравнения ПБ. Причина состоит в том, что в случае двухвалентных противоионов более существенными становятся корреляции между ионами, возникающими благодаря наличию высокого заряда и собственным размерам ионов, которые уравнение ПБ не учитывает [13]. Однако концентрации ионов в непосредственной близости от молекулы протеина, полученные на основании решения уравнения ПБ и методами МК и ОЦ-ГЦ, различаются несущественно. Учитывая межионные дисперсионные взаимодействия в интегральных уравнениях ОЦ-ГЦ и расчетах по методу МК, авторы [13] нашли лишь небольшие изменения 23 профилей распределения ионов, из чего заключают, что роль этих взаимодействий по сравнению с дисперсионными взаимодействиями ион-протеин невелика. Подход Дэвиса, Нинэма, Нэтса и сотр. [69-71] применен Бострѐмом и сотр. [5] для расчета поверхностного натяжения на незаряженной границе воздух-раствор соли. При этом для усовершенствования подхода Онзагера-Самараса в потенциале средней силы в уравнении ПБ к электростатическому взаимодействию иона с границей раздела был добавлен потенциал дисперсионного взаимодействия иона с границей раздела, константа взаимодействия иона , где – силовая с диэлектриком. В работе [5] катионы имели отрицательные (притяжение), а анионы положительные (отталкивание) дисперсионные константы. Однако силовые константы дисперсионного потенциала для разных ионов не рассчитывались в рамках теории Лифшица [72], а подгонялись так, чтобы получить наилучшее согласование теории с экспериментом. Для всех ионов было выбрано одинаковое расстояние максимального приближения к границе раздела. Полученные при решении модифицированного уравнения Пуассона-Больцмана (мПБ) профили распределения ионов использовались в изотерме адсорбции Гиббса при расчете поверхностного натяжения на границе раздела раствора соли с воздухом. Рассчитанные в 1М растворах галогенидов и ацетатов щелочных металлов приращения значений поверхностного натяжения воды по сравнению с чистой водой удовлетворительно согласуются с экспериментальными. При высоких концентрациях соли благодаря экранированию электростатических взаимодействий дисперсионные взаимодействия доминируют. При этом специфическое влияние соли на поверхностное натяжение складывается из эффектов, производимых каждым из ионов соли в отдельности, что наблюдается и в эксперименте. Несмотря на то, что в работе Бострѐма и сотр. [5] удовлетворительный результат расчета был получен только при учете дисперсионных взаимодействий как причины специфического влияния ионов, авторы этой работы отмечают, что подобранные комбинации дисперсионных констант не уникальны, и что учет других эффектов тоже важен, например, эффектов гидратации и исключенного объема ионов. Анионы в сравнении с катионами являются более поляризуемыми [35]. ‗Мягкие‘, более поляризуемые ионы слабее гидратированы и должны располагаться ближе к границе раздела воздух-вода. ‗Жесткие‘, слабо поляризуемые ионы сильно гидратированы и должны располагаться дальше от поверхности. Подходя ближе к границе раздела, более ‗мягкие‘ анионы сильнее изменяют поверхностные натяжение и потенциал, чем более ‗жесткие‘ катионы [1]. Исключение составляет водород-ион, или ион гидроксония . Кислоты с ‗жесткими‘ протонами понижают поверхностное натяжение, что должно означать стремление протонов к поверхности. Основываясь на данных молекулярной динамики, Юнгвирс, Петерсен и сотр. [89- 24 90] нашли, что из-за геометрического строения иону удобнее подходить к поверхности, чем находиться в объеме. Бострѐм и сотр. [5] не учитывали способность разных ионов подходить к границе раздела на разные расстояния. В последующей работе [4] они попытались одновременно с дисперсионными взаимодействиями учесть гидратацию ионов. Потенциал, связанный с изменением гидратной оболочки иона при его переносе из объема раствора в точку вблизи границы раздела (потенциал гидратации), рассчитывался на основе зависимости энергии Борна иона от распределения молекул воды вблизи границы раздела, найденного в расчетах по методу молекулярной динамики [91]. Полученный потенциал гидратации зависит от заряда и радиуса иона. Радиус иона и силовая константа дисперсионного взаимодействия (рассчитывалась в рамках теории Лифшица) – специфические параметры иона в данной модели [4]. По результатам расчета в работе [4] были сделаны следующие выводы: – учет только дисперсионных взаимодействий (т.е. использование модели с отключенными потенциалами гидратации) не позволяет воспроизвести экспериментально наблюдаемую последовательность изменения величин поверхностного натяжения на границе раствор-воздух в 1М растворах галогенидов натрия; – учет взаимодействий, связанных с гидратацией ионов (т.е. расчеты с отключенными дисперсионными потенциалами), приводит к верной последовательности для галогенидов, но получаемые для разных солей значения поверхностного натяжения различаются меньше, чем в эксперименте [4]; – cовместный учет обоих видов взаимодействий позволяет получить достаточно хорошо согласующиеся с экспериментом величины поверхностных натяжений для галогенидов натрия в 1M растворах. Для описания эффектов гидратации ионов Манкью и Рукенштейн [68] использовали потенциалы жесткой стенки и потенциальной ямы для космотропных и хаотропных ионов, соответственно [68]. Взаимодействие хаотропных ионов с границей раздела описывалось также дисперсионным потенциалом, . В работе [68] показано, что при низких концентрациях соли доминируют электростатические взаимодействия. При умеренно высоких концентрациях, когда расстояния между ионами уменьшаются, и их гидратные оболочки взаимодействуют друг с другом, доминируют силы гидратации. При высоких концентрациях соли существенную роль начинают играть дисперсионные взаимодействия. С помощью молекулярной динамики с учетом поляризуемости ионов было показано, что локально в некоторых точках раствора вблизи границы раздела может происходить накопление ионов [9, 91-94], а глобально, т.е. суммарно по всему объему системы, может наблюдаться обеднение, при этом межфазное натяжение будет возрастать [9, 91-94]. Следовательно, 25 первоначальное убеждение, что ионы простых электролитов, вызывающие увеличение поверхностного натяжения по сравнению с чистой водой, всегда выталкиваются из приграничных слоев, оказывается неверным. В работах [9, 91-94] были получены немонотонные профили распределения ионов и молекул воды вблизи границы раздела, а также зависимость локальной диэлектрической проницаемости воды от расстояния до углеводородной фазы [94]. Полученные зависимости диэлектрической проницаемости и распределения молекул воды использовались в уравнениях мПБ и изотермы адсорбции Гиббса при расчетах поверхностного натяжения растворов солей на границе с воздухом [4] и при расчете расклинивающего давления между двумя пузырьками воздуха или двумя плоскими углеводородными слоями в растворе соли [94]. Несмотря на то, что подходы Бострѐма и сотр. и Манкью и Рукенштейна [4, 68] давали профили распределения ионов, не согласующиеся с профилями, полученными по методу молекулярной динамики, они позволили верно воспроизвести последовательности Гофмейстера для галогенид-ионов для поверхностного натяжения водно-солевого раствора на границе с воздухом [4] и объяснить наличие экстремума на экспериментальной кривой зависимости поверхностного натяжения от концентрации электролита (эффект Рэя-Джонса) при низких концентрациях соли [68]. При расчете поверхностного натяжения раствор соли – углеводород дос Сантос и Левин [8] проанализировали роль собственных размеров ионов. В рамках предложенной ими модели предполагается, что вблизи границы раздела на любой ион действуют силы Онзагера-Самараса, связанные с разницей диэлектрических функций двух граничащих сред, и электростатические силы, связанные с взаимодействием ионов раствора между собой. Предполагается, что на хаотропные ионы со стороны границы раздела действуют еще три составляющих: электростатическая энергия, учитывающая частичную дегидратацию иона в зависимости от его способности более или менее сильно проникать в углеводород (эта составляющая являлась аналогом энергии Борна, но зависела не только от заряда и собственного размера иона, но и от его поляризуемости и равновесной степени проникновения иона в углеводород [8]); дисперсионная составляющая (которая тоже зависела от поляризуемости иона [4-5, 68]) и гидрофобная составляющая, или энергия образования ионом полости в среде, как в воде, так и в углеводороде, если ион способен проникать в него хотя бы частично. Предложенная модель позволила количественно описать экспериментально наблюдаемые зависимости поверхностного натяжения галогенидов калия на границе раздела вода-углеводород при концентрациях до 1М. В общем, подход Дэвиса, Нинэма, Нэтса и сотр. [69-71] был вполне успешно применен для описания солевых эффектов на незаряженных границах раздела между раствором соли и диэлектриком (воздухом, углеводородом, слюдой, оксидами алюминия и кремния и др.) [4-9, 26 68, 94-95], для растворов индивидуальных электролитов [3, 10, 96], для систем с мембранами [16] и водно-солевых растворов глобулярных протеинов [11-15, 97]. Во всех упомянутых работах продемонстрирована важная роль дисперсионных взаимодействий [15-16]. Так в работе [16] на примере рассчитанных для ионов натрия и галогенов свободных энергий сольватации и переноса иона из фазы объемного раствора в фазу мембраны было показано, что дисперсионными взаимодействиями пренебречь нельзя, т.к. они могут вносить вклад от 10-30% всей энергии сольватации. Примером успешного описания ионных специфических эффектов является работа Парсонса и сотр. [3] Авторы этой работы [3] показали, что учет ион-ионных дисперсионных взаимодействий позволяет добиться количественного описания коэффициентов активности солей галогенидов и ацетатов щелочных металлов вплоть до высоких концентраций соли. Данная работа [3] замечательна тем, что впервые для весьма представительного ряда солей было достигнуто удовлетворительное описание ионных специфических эффектов без применения большого числа подгоночных параметров. Подгоночными параметрами в модели Парсонса и сотр. [3] служит только размер ионов. Силовые константы потенциалов межионного дисперсионного взаимодействия являются функциями поляризуемостей ионов, которые рассчитываются квантово-механически [3]. Подход Дэвиса, Нинэма, Нэтса и сотр. [69-71] применялся при расчете профилей распределения ионов, электрического потенциала и подвижного заряда вокруг заряженной коллоидной частицы фиксированного заряда, которую авторы работы [98] рассматривали в качестве модели мицеллярного агрегата. Получаемые профили распределения ионов согласуются с типичными последовательностями Гофмейстера, наблюдаемыми в мицеллярных растворах, однако агрегативные характеристики в работе [98] не рассматривались. Среди молекулярно-термодинамических моделей мицеллообразования существует лишь несколько, в рамках которых агрегативные характеристики зависят от природы иона [21-24]. В моделях Шринивасана и Бланкштейна (ШБ) [21-22] и Бауэра и сотр. (БВК) [23] в качестве параметра, отражающего природу иона, фигурируют степень связывания противоиона с мицеллой и константа диссоциации молекулы ПАВ в мицелле, соответственно. Величины этих параметров оптимизировались, чтобы достичь наилучшего согласия расчетов с экспериментом. Обе предложенные модели близки по физическому содержанию, поскольку степень связывания и константа диссоциации противоиона – два родственных параметра. Модель ШБ [21-22] опробована для одного катионного и двух анионных ПАВ в присутствии нескольких противоионов и их смесей: X-алкилтриметиламмония (Cn TAX, X=Cl-, Sal-), X-додецилсульфата (XDS, X=Li+, Na+, K+, Rb+, Cs+) и X-додецилдиоксоэтиленсульфата (XDE2 S, в смесях X=Na+/Ca2+, Na+/Al3+). Для анионных ПАВ результаты расчета степеней связывания противоионов, ККМ, чисел агрегации и значений параметров роста мицелл вблизи 27 солевого порога перехода сфера-цилиндр удовлетворительно согласуются с экспериментальными данными. Для катионного ПАВ влияние природы противоионов на ККМ рассматривалось лишь в бессолевой системе, а на рост агрегатов – только при изменении концентрации ПАВ в системе C16 TASal+0.2М NaCl, для которой модель ШБ недооценивает интенсивность роста цилиндрических мицелл. Модель БВК [23] применена для единственного ПАВ (C16 PX+KX, X=Cl-, Br-, I-, NO 3 -) в областях составов, расположенных вблизи переходов сфера-цилиндр, где цилиндрические агрегаты имеют относительно небольшие числа агрегации. Для описания ионных специфических эффектов для ККМ и чисел агрегации мицелл небольшого размера Морейра и Фирузабади (МФ) [24] предложили версию молекулярнотермодинамической модели [17], в которой эмпирически учли целый ряд факторов: эффективные размеры гидратированных полярных голов и проникающих в мицеллярную корону ионов, высаливание углеводородных радикалов молекул ПАВ, влияние природы и концентрации соли на натяжение границы раздела углеводородное ядро – раствор и на диэлектрическую постоянную раствора. В связи с учетом ряда факторов модель МФ включает внушительное число подгоночных параметров. В бессолевых растворах и в области низких концентраций соли рассчитанные зависимости ККМ и чисел агрегации сферических и малых глобулярных агрегатов удовлетворительно описывают экспериментальные данные для целого ряда катионных и анионных ПАВ в присутствии 1:1 электролитов. При умеренных и высоких концентрациях электролита, где образуются длинные цилиндрические мицеллы, данная модель применяться не может, т.к. не учитывает образование и рост цилиндрических агрегатов. В заключении работ как Шринивасан и Бланкштейн [21-22], так и Бауэр и сотр. [23] отмечают, что для расширения границ применимости предложенных ими моделей необходимо учитывать дисперсионные взаимодействия. Первоначальный вывод Морейры и Фирузабади [24] о том, что дисперсионные взаимодействия не способны описывать ионные специфические эффекты в мицеллярных растворах, был пересмотрен в более поздней работе [99]. В модели Иванова и сотр. [36] дисперсионные взаимодействия ионов с мицеллярными агрегатами учитываются при описании ККМ, однако область применимости этой модели ограничена низкими концентрациями соли. На основании проведенного обзора литературы можно заключить, что влияние природы иона определяется совокупностью факторов, наиболее существенные из которых – ионные дисперсионные взаимодействия и эффекты гидратации/дегидратации. Важную роль играет также собственный размер ионов. Теория ионных специфических эффектов успешно разрабатывалась в применении к растворам неорганических солей, границам раздела диэлектрик-раствор соли, водно-солевым растворам глобулярных протеинов и мембранам. Тем 28 не менее, до настоящего времени общая количественная теория специфических эффектов ионов в водно-солевых растворах ионных ПАВ отсутствует. Классические молекулярно- термодинамические модели агрегации неспособны отразить влияние природы соли, а предложенные в последние годы модификации этих моделей [21-24, 36] не достаточно полно учитывают указанные выше ключевые факторы, и имеют весьма ограниченную применимость для описания эффектов специфики ионов. В частности, указанные модели не позволяют с приемлемой точностью описывать мицеллообразование и интенсивный рост агрегатов для катионных и анионных ПАВ. Ни в одном из предложенных для мицеллярных растворов подходов не рассматривались эффекты коионов, хотя, как отмечалось ранее, роль коионов может быть существенна. Задача настоящей работы состоит в том, чтобы учесть перечисленные выше факторы при построении молекулярно-термодинамической модели агрегации ионных ПАВ в присутствии неорганических электролитов различной природы. 29 ГЛАВА 2. МОЛЕКУЛЯРНО-ТЕРМОДИНАМИЧЕСКАЯ МОДЕЛЬ АГРЕГАЦИИ ИОННОГО ПОВЕРХНОСТНО-АКТИВНОГО ВЕЩЕСТВА В ВОДНО-СОЛЕВОМ РАСТВОРЕ С УЧЕТОМ СПЕЦИФИКИ ИОНОВ Модель свободной энергии агрегации 2.1 Классические молекулярно-термодинамические модели мицеллярных систем описывают стандартную свободную энергию агрегации как сумму различных вкладов [17, 19]. Для агрегата любой формы ( ) можно записать [17] (7) где – вклад переноса углеводородного радикала молекулы ПАВ из раствора в мицеллу, – вклад упругой деформации углеводородных радикалов молекул ПАВ в ядре, поверхностный вклад, ПАВ, – – вклад стерического отталкивания между полярными головами – вклад ионных взаимодействий. Вклады и зависят от природы и концентрации соли и модифицировались в настоящей работе [100-101], а вклады , и не зависят от природы соли, были заимствованы из классической квазихимической модели мицеллообразования Нагаражана и Рукенштейна [17] и обсуждались в предыдущей главе. Гидрофобный вклад. Вклад переноса углеводородного радикала молекулы ПАВ из раствора в мицеллярное ядро определяется растворимостью углеводорода в растворе соли (8) где – растворимость углеводорода в водном растворе соли. Соль модулирует гидрофобный вклад в мицеллообразования, где классической молекулярно-термодинамической – стандартная свободная энергия переноса углеводородного радикала из воды в ядро мицеллы в отсутствии добавленной соли, углеводорода в воде; cлагаемое модели – растворимость – поправка к гидрофобному вкладу в присутствии соли, или ―вклад Мукерджи‖ [24, 64-65], где – константа Сеченова, – концентрация соли. Ионный вклад в свободную энергию агрегации описывает свободную энергию заряженного агрегата в облаке подвижных ионов. Все подвижные ионы свободно перемещаются в электрическом и дисперсионном полях вокруг мицеллы [100]. Ионы могут проникать в мицеллярную корону и подходить к ядру. Расстояние максимального приближения иона к ядру, , является параметром модели, специфичным для каждой пары голова – подвижный ион [100-101]. Дисперсионное притяжение иона к мицелле действует как ионная специфическая часть потенциала средней силы. Этот потенциал вводится в модифицированное 30 уравнение ПБ (мПБ) так же, как в предшествующих работах [13, 92]. Локальная энтропия подвижных ионов рассчитывается из модели смеси твердых сфер [102]. Влияние дисперсионных взаимодействий на свободную энергию ионных взаимодействий проявляется как неявно, так и явно [100]. Дисперсионные взаимодействия изменяют локальную концентрацию подвижных ионов вокруг мицеллы и, следовательно, локальную энтропию, электрический потенциал и электростатическую энергию. Энергия дисперсионных взаимодействий входит в свободную энергию агрегации явно как отдельное слагаемое [100] (9) где – энергия электростатических взаимодействий, взаимодействий; – энергия дисперсионных – вклад в свободную энергию от энтропии подвижных ионов; локальная плотность и химический потенциал иона , соответственно, интегрирование выполняется по всему объему и – - объемная плотность; мицеллярной ячейки. Электростатическая энергия дается выражением (10) где – локальный электростатический потенциал, создаваемый в точке (подвижными и фиксированными) вблизи мицеллярной поверхности, положительный заряд, – зарядовое число подвижного иона, всеми зарядами – элементарный – поверхностная плотность заряда агрегата, предполагаемая однородной; дельта- функция Дирака положение заряженной поверхности, описывает . Для энергии дисперсионных взаимодействий подвижных ионов с мицеллой записываем [94, 100] (11) где – локальный потенциал дисперсионного взаимодействия иона с агрегатом. Этот потенциал (см. Приложение 2) зависит от формы агрегата и содержит силовую константу материала агрегата и среды [3, 13]; кривизны , , которая зависит от поляризуемости иона, для цилиндра и сферы, соответственно, – радиус – расстояние от поверхности гидрофобного ядра агрегата. Ион может притягиваться или отталкиваться агрегатом [3] в зависимости от знака избыточной поляризуемости иона. Как знак, так и величина дисперсионного потенциала зависят также от соотношения диэлектрических функций, модельные выражения для которых выбираются в разных приближениях (одно- или мультимодальное приближения, квантово-механические расчеты) [6]. В настоящей работе мы учитываем дисперсионное взаимодействие между 31 подвижными ионами и массивным агрегатом, а дисперсионными взаимодействиями между подвижными ионами пренебрегаем. Вклад от энтропии подвижных ионов рассчитывается [100] с помощью уравнения состояния Бублика-Мансури-Карнахана-Старлинга-Леланда (БМКСЛ) для смеси твердых сфер [102-103] (12) здесь – плотность свободной энергии смеси твердых сфер для локальной ионной плотности вблизи мицеллы. Выражения для даны в Приложении 3. Конечный размер иона отражается диаметром твердой сферы [47] , индивидуальным параметром иона. В настоящей работе мы учитываем взаимодействия исключенного объема ионов, которые, вероятно, обеспечат более адекватные приповерхностные плотности подвижных ионов. Уравнение БМКСЛ вырождается в уравнение состояния идеального газа либо в пределе низкой концентрации соли (и при конечных размерах ионов), либо при исчезающе малых ионных диаметрах (и конечных концентрациях соли). 2.2 Равновесное распределение подвижных ионов вокруг мицеллы Минимизация свободной энергии агрегации, уравнение 7, относительно профилей , см. например работу [104], дает условие равновесного распределения ионов вокруг мицеллы [100] (13) Уравнение 13 показывает, что локальные химические потенциалы ионов в электрическом и дисперсионном полях вблизи мицеллы должны быть равны химическим потенциалам вдали от мицеллы, где оба эти поля исчезают. Равновесные профили и находим, решая уравнение 13 совместно с уравнением Пуассона. Уравнение Пуассона в сферических ( цилиндрических ( )и ) координатах выглядит следующим образом (14) где – диэлектрическая проницаемость вакуума; – относительная диэлектрическая проницаемость среды. Граничными условиями для уравнения Пуассона являются нулевой градиент электрического потенциала вдали от мицеллы и закон Гаусса на заряженной поверхности агрегата, соответственно: (15) (16) 32 где и – градиенты электрического потенциала соответственно с внешней и внутренней стороны от поверхности фиксированного заряда полярных голов; (17) – плотность заряда на поверхности фиксированного заряда полярных голов, цилиндра и для сферы, – заряд полярной головы ПАВ, для – расстояние от поверхности ядра агрегата до эффективного положения заряда полярной головы, (18) – площадь на молекулу ПАВ, – эффективный объем углеводородного радикала молекулы ПАВ, – число углеродных атомов в радикале. Параметр принимает индивидуальное значение в зависимости от строения заряженной головы ПАВ. Диэлектрическая проницаемость окружающего раствора и внутренней части агрегата предполагаются различными, но однородными. Для точечных ионов уравнение 13 значительно упрощается, давая профили явно (19) Из уравнений 9-12 и 19 после алгебраических преобразований получаем следующее выражение для равновесной свободной энергии для точечных ионов (20) Для ионов конечного размера свободная энергия ионных взаимодействий рассчитывается численно. 2.3 Критическая концентрация мицеллообразования, переход сфера-цилиндр, числа агрегации Как и в предшествующих моделях [17, 19], предполагается, что агрегаты имеют несжимаемые ядра. Радиус агрегата ( ) определяет площадь, , приходящуюся на молекулу ПАВ в мицелле, и поверхностную плотность заряда , уравнения 17-18. Сферические мицеллы предполагаются монодисперсными, имеющими наиболее вероятный размер Стержнеобразные мицеллы состоят из цилиндрического тела радиуса . различной длины, оканчивающегося двумя полусферическими шапочками. Число агрегации сферической мицеллы, , и число агрегации на единицу длины цилиндрической части агрегата, рассчитываются с помощью выражений (21) , 33 (22) Для ККМ ионного ПАВ, являющегося сильным 1:1 электролитом, имеем [105] (23) где – свободная энергия сферической мицеллы из расчета на одну молекулу ПАВ. Когда доля цилиндрических агрегатов существенна, уравнение 23 применяться не может. Для учета популяции цилиндрических агрегатов мы используем [100-101] уравнение материального баланса [106] (24) Здесь – общая мольная доля ПАВ в растворе; – мольная доля свободных мономеров; второе и третье слагаемые в правой части уравнения 24 учитывают сферические и стержнеобразные агрегаты, соответственно; – общее число агрегации цилиндрической мицеллы, имеющей две полусферические шапочки; свободная энергия молекулы ПАВ в цилиндрической мицелле, где – – параметр одномерного роста, связанный с разницей свободных энергий из расчета на молекулу ПАВ в шапочке и в цилиндрической части [106] (25) Большие избыточные энергии шапочек приводят к интенсивному росту цилиндрических мицелл [106]. Для заданной из уравнения 24 находят (условие ККМ ) и вычисляют соответственно среднечисловое, средневесовое и z-среднее числа агрегации цилиндрических мицелл: (26) (27) (28) Переход сфера-цилиндр обнаруживается по резкому возрастанию чисел агрегации при увеличении концентрации ПАВ или ионной силы. 2.4 Параметры модели Перечислим параметры предлагаемой модели [100], см. рисунок 3: - эффективная площадь поперечного сечения полярной головы, , уравнения 3 и 5; - положение эффективного заряда полярной головы относительно углеводородного ядра мицеллы, , уравнение 17; 34 - силовая константа дисперсионного взаимодействия иона с агрегатом, , связанная с ионной поляризуемостью, уравнение 11 и 2.9 из Приложения 2 [3, 13]; - ван дер ваальсов диаметр иона см. , Приложение 3 [47]; - константа Сеченова - расстояние максимального приближения иона к мицеллярному ядру , уравнение 8; , уравнения 9-12. Величины параметров модели, используемые в настоящей работе, приведены в таблицах 1-3. Рисунок 3. Мицелла катионного ПАВ в растворе соли (показана часть сечения агрегата). Параметры модели можно условно разделить на три группы. Параметры первой группы, и , Темным определяются строением ПАВ и не зависят от изображено углеводородное ядро. Как природы соли, таблица 1. Они такие же, как в противоионы, так и коионы способны классических проникать в мицеллярную корону. моделях [17, 20], не учитывающих специфику ионов. – Параметры второй группы, расстояния максимального приближения ионов к ядру, – ван дер ваальсовы диаметры ионов. молекулярно-термодинамических , и , определяются природой ионов и не зависят от строения ПАВ, – таблица 2. Они имеют фиксированные значения для расстояние от ядра до поверхности каждой соли в растворах любых ПАВ. Параметры эффективного заряда короны, третьей группы, расстояния поверхностная короны, плотность – заряда – площадь поперечного сечения головы ПАВ. параметрам подвижный и , относятся к ион-голова ПАВ и различаются для противоиона и коиона, таблица 3. Эти параметры отражают компактность образующихся пар полярная голова-ион, учитывая эффекты взаимной гидратации-дегидратации в мицеллярной короне. Так, потеря ионной гидратной оболочки, проникновение ионов в мицеллярную корону, подход ионов к мицеллярному ядру в рамках настоящей модели передается меньшими ; усиление гидратации мицеллярной короны и образование более ―рыхлых‖ пар голова-ион отражается в модели возрастанием . 35 Таблица 1. Рассматриваемые водные растворы ПАВ+соль и параметры модели, отражающие строение ПАВ. Система катионные ПАВ n-алкилтриметиламмоний-X (Cn TAX+NaX, X=Cl¯,Br¯,OH¯,NO3 ¯, n=12,14,16) n-алкилдиметиламмоний-X (Сn DAX+NaX, X=Cl¯,Br¯, n=12) n-алкилпиридиний-X (Cn PX+NaX, X=Cl¯,Br¯,I¯, n=10,12,14,16) эруцилбис(гидроксиэтил)метиламмоний хлорид (EHAC+XCl, X=Na+,K+,n=22) анионные ПАВ X-додецилсульфат (XDS+XCl, X=Li+, Na+, K+, Cs+, n=12) X-алкилкарбоксилат (Cn COOX+XCl, X=Na+, K+, n=9,11) ap, нм2 rh , нм 0.58 0.28 0.44 0.52 0.61 0.22 0.30 0.36 0.45 0.50 0.65 0.65 Таблица 2. Параметры модели, зависящие от природы соли. Ион di, нм -Bim,10-50 Дж м3 Ионные диаметры и константы дисперсионного взаимодействия F¯ Cl¯ Br¯ I¯ SCN¯ OH¯ NO3 ¯ Li+ Na+ K+ Cs+ 0.272 0.362 0.390 0.432 0.640 0.352 0.528 0.136 0.190 0.266 0.338 14.3 35.8 44.4 57.1 100.0 0 41.8 1.1 4.5 18.9 41.9 Константы высаливания на CH2 группу Соль NaF* NaCl NaBr NaI*) NaOH NaSCN*) NaNO3 LiCl KCl CsCl -kS, л/моль 0 0.05 0.03 0 0.10 0 0.03 0.11 0.04 0.03 *) из-за недостатка доступных экспериментальных данных о ККМ при высоких концентрациях ) соли для надежной оценки kS этот параметр принят равным нулю. Таблица 3. Расстояния максимального приближения ионов к мицеллярному ядру Тип полярной группы ПАВ Триметиламмоний TA+ Диметиламмоний DA+ Пиридиний P+ Бис(гидроксиэтил) метиламмоний HA+ Сульфат SO 4– Карбоксилат COO– . δi , нм 0.450 Cl¯ 0.375 Cl¯ 0.600 Cl¯ 0.325 Br¯ 0.330 Br¯ 0.390 Br¯ катионные ПАВ 0.750 0.295 0.600 0.240 OH¯ NO3 ¯ Na+ K+ 0.600 Na+ 0.267 0.360 0.350 I¯ NO3 ¯ Na+ 0.450 Cl¯ 0.600 Na+ 0.170 K+ 0.350 Na+ 0.500 K+ анионные ПАВ 0.255 0.300 0.375 0.600 K+ Cs+ F¯ Cl¯ 0.600 Cl¯ 0.675 Li+ 0.250 Na+ 0.680 Br¯ 0.800 I¯ 0.825 SCN¯ 36 ГЛАВА 3. МЕТОДИКА РАСЧЕТА Алгоритм расчета агрегативных характеристик мицеллярных растворов 3.1 Равновесные радиусы определялись минимизацией свободной энергии агрегации , уравнение 7, сферических и цилиндрических мицелл при условии, что радиус не может превосходить длину полностью растянутого углеводородного радикала . Полученные значения равновесных радиусов использовались [17] при расчете ККМ и чисел агрегации [100-101]. Оптимизация выполнялась методом дихотомии, причем необходимо было следить, чтобы радиус мицеллы и свободная энергия агрегации находились достаточно точно (радиус содержал бы не менее четырех значащих цифр, а свободная энергия – не менее пяти значащих цифр), в противном случае расчеты приводили к нестабильным зависимостям чисел агрегации от концентрации соли и ПАВ. На каждом шаге оптимизации свободной энергии агрегации равновесных профилей рассчитывался ионный вклад, и по радиусу из для сферического и цилиндрического агрегатов , уравнения 9-12. Равновесные профили определялись численным решением системы уравнений 13-14. Мицеллярная ячейка (радиусом от 10 до 20 Дебаевских длин при высоких и низких концентрациях соли, соответственно) разбивалась на неоднородную сетку, более частую вблизи мицеллы и более разреженную в объеме раствора [100]. Для более точных расчетов частоту сетки необходимо увеличивать либо в приповерхностной области раствора (при низких концентрациях соли), либо на всем протяжении мицеллярной ячейки (при высоких концентрациях соли). В зависимости от поверхностной плотности заряда агрегатов и объемной концентрации электролита общее число узлов сетки в конечно-разностной схеме изменялось в пределах от 2000 до 60000. Система уравнений 13-14 и граничные условия, уравнения 15-16, записывались в форме конечных разностей и решались методом Ньютона-Рафсона на узлах сгенерированной сетки. Уравнение Пуассона, уравнение 14, в форме конечных разностей принимает вид (29) где сетки, и – локальные значения электрического потенциала и плотности ионов , , в -ом узле – коэффициенты разложения в рамках метода конечных разностей левой части уравнения Пуассона. Уравнение 13 для противоиона и коиона записывалось в локальной форме (т.е. 37 зависело от локальных значений электрического и дисперсионного потенциалов и плотностей ионов). В соответствии с разработанным в настоящей работе алгоритмом расчета определение локальных значений электрического потенциала и плотностей ионов производится пошагово на узлах сгенерированной сетки в направлении из объема раствора к поверхности мицеллы. Значение электрического потенциала в объеме раствора выбиралось в соответствии с граничным условием, уравнение 15, методом дихотомии. На поверхности, на которой располагается эффективный заряд полярных голов в мицелле, для электрического потенциала должно выполняться другое граничное условие, уравнение 16. Получаемые профили электрического потенциала и распределения ионов также проверялись на соответствие условию электронейтральности мицеллярной ячейки – накопленный подвижный заряд должен быть равен поверхностному заряду мицеллы. В расчетах настоящей работы погрешность в накоплении заряда не превышала 0.001%. Интегрирование, необходимое для проверки условия электронейтральности, при расчетах составляющих свободной электростатической энергии и в других процедурах, производилось численно методом трапеции. Все численные процедуры расчета реализованы для Fortran Power Station 4.0. 3.2 Процедура подбора параметров модели Рассмотрим используемый в настоящей работе алгоритм подбора величин параметров модели для растворов заданного ПАВ в присутствии разных электролитов. На первом этапе подбирались параметры первой группы, параметра и , и начальная оценка для для противоиона. Величины этих параметров выбирались с учетом различий в молекулярном строении полярных голов разных ПАВ (геометрии и длин связей [47], положения заряда [107]) и различий в размерах полярной головы и ионов соли в гидратированном состоянии [26, 47]. Для того чтобы модель давала адекватные результаты для заданного ПАВ в присутствии разных противоионов, среди которых могут быть более или менее гидратированные, начальная оценка для должна немного превосходить сумму размеров полярной головы и большего из ионов в гидратированном состоянии. Подобранные величины , и должны быть таковы, чтобы при низких и умеренно высоких концентрациях электролита кривая ККМ-ионная сила в логарифмических координатах располагалась немного выше, переход сфера-цилиндр наблюдался позже, а наклон кривой чисел агрегации от ионной силы раствора в логарифмических координатах был немного 38 меньше, чем в эксперименте в растворах данного ПАВ в присутствии любого из рассматриваемых электролитов. На следующем этапе включается параметр его максимального приближения . Величины для противоиона и варьируется расстояние для ряда ионов даны в таблице 2 и могут быть использованы при расчетах с любыми ПАВ. Для других противоионов одновременным уменьшением расстояния максимального приближения подбираются с , при этом ККМ и солевые пороги переходов сфера-цилиндр понижаются, а числа агрегации растут. Подбор комбинаций и начинается с противоионов, поскольку, как упоминалось выше, эффект противоиона в общем намного сильнее эффекта коиона. В настоящей работе все ионы испытывают дисперсионное притяжение к мицелле ( ), которое уравновешивается отталкиванием от мицеллярного ядра, связанным с эффектами гидратации ( ). Кроме того, как будет показано позже, дисперсионное притяжение оказывает более существенный эффект на мицеллярные характеристики в сравнении с дисперсионным отталкиванием. Комбинация и для коионов не оказывает существенного влияния на ККМ и переходы сфера-цилиндр, но позволяет при высоких концентрациях соли корректировать наклоны кривых ККМ и чисел агрегации мицелл от ионной силы в логарифмических координатах. Так же, как и для противоионов, величины для коионов подбирались с учетом способности полярной головы и коиона образовывать контактные пары в связи с эффектами взаимной гидратации [26], а также с учетом молекулярного строения полярной головы [47, 107]. Например, для алкилпиридиниевого ПАВ, голова которого достаточно велика, слабо гидратирована, имеет плоское строение, располагается под углом к поверхности ядра мицеллы и обладает активными центрами взаимодействия, коионы и некоторые противоионы могут подходить особенно близко к поверхности мицеллы (малые также ван дер ваальсовы диаметры ). На данном этапе включаются как для противоиона, так и для коиона, таблица 2. При низких и умеренно высоких концентрациях соли, как будет показано позже, параметры почти не оказывают влияния, а при высоких концентрациях соли, хоть незначительно, но влияют на ККМ, солевые пороги переходов сфера-цилиндр и числа агрегации. Наконец подбираются константы Сеченова (из расчета на СH2 группу). Параметр вызывает отклонение кривой ККМ-ионная сила в логарифмических координатах вниз ( или вверх ( ) от линейности при высоких концентрациях соли. Величины ) подбираются с использованием надежных экспериментальных данных о ККМ при высоких концентрациях соли или рассчитываются по правилу аддитивности [24]. В настоящей работе при отсутствии надежных экспериментальных данных и при невозможности воспользоваться правилом аддитивности [24] недостающие величины приравнивались к нулю, таблица 2. 39 Для заданной системы ПАВ+электролит набор параметров и , , , , , , , , таблицы 1-3, позволяет одновременно воспроизвести ККМ и числа агрегации в широком интервале концентраций электролита [100-101]. 40 ГЛАВА 4. РЕЗУЛЬТАТЫ МОДЕЛИРОВАНИЯ АГРЕГАЦИИ ИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ В РАСТВОРАХ НЕОРГАНИЧЕСКИХ 1:1 ЭЛЕКТРОЛИТОВ Влияние параметров модели на результаты расчетов характеристик мицеллярного 4.1 раствора Рисунок 4. Независимые от природы соли вклады в свободную энергию агрегации (a) и соответствующие силы (б) как функции площади на голову в сферическом агрегате. Штрихпунктирная линия: поверхностный вклад, уравнение 5; сплошная линия: вклад растяжения углеводородных радикалов, уравнение 4; отталкивания голов, уравнение 3. Параметры: пунктирная линия: , . вклад стерического Как упоминалось выше, оптимальный радиус агрегата определялся минимизацией свободной энергии агрегации, уравнение 7. Варьирование радиуса ядра агрегата вызывает изменение эффективной площади на молекулу, уравнение 18. Изменение площади может быть силами связано с эффективными (поверхностным давлением), , обусловленными различными вкладами в свободную энергию, уравнения 7 и 9 ( ) [100]. Для равновесного агрегата эти силы сбалансированы: Не зависящие от природы соли вклады в свободную энергию , , , . , и и соответствующие силы изображены на рисунке 4. Стерическое отталкивание, , раздвигает головы и увеличивает площадь , причем тем сильнее, чем больше размер головы . В рамках аналитического приближения Семенова [66], уравнение 4, упругость цепи дает и действует так, чтобы уменьшить растяжение углеводородных радикалов, уменьшая радиус мицеллы и, следовательно, увеличивая площадь на молекулу ПАВ. Отметим, что в рамках более строгой версии теории самосогласованного поля учитывается влияние геометрии 41 Рисунок 5. Зависимости электростатического потенциала и локальной концентрации (моль/л) точечных подвижных ионов от расстояния от поверхности углеводородного ядра сферического агрегата. Радиус агрегата и поверхностная плотность заряда составляют 1.5нм и 0.113Кл/м2 , соответственно. Синяя линия (1): дисперсионное отталкивание противоионов ( ); черная линия (2): без дисперсионных взаимодействий между противоионами и мицеллой ( ); красная линия (3): дисперсионное притяжение противоионов ( – ). Расстояния максимального приближения ионов к поверхности мицеллы и концентрация электролита в объеме раствора обозначены на графиках; . Символы (г): расчеты Бострѐма с сотр. [13] , , 42 ядра на конформационную статистику цепей, и теория может предсказывать отталкивание или притяжение в зависимости от радиуса и формы ядра [106, 108]. Силы стерического отталкивания и деформации углеводородных радикалов способствуют диспергированию агрегатов и особенно быстро возрастают, когда площадь становится малой. Уравнение 5 дает : поверхностное натяжение сближает головы и уменьшает площадь на молекулу ПАВ, т.е. способствует агрегации. дает весьма существенный вклад в общую силу, который сдвигает минимум свободной энергии в область сильно растянутых углеводородных радикалов. Свободная электростатическая энергия и двумерное ионное давление содержат вклады от энтропии и энергий электростатических и дисперсионных взаимодействий, которые зависят от пространственного распределения ионов, уравнения 9-12 [100]. Дисперсионные взаимодействия оказывают существенное влияние на профили электрического потенциала и на распределение подвижных ионов вблизи мицеллы, рисунок 5. Дисперсионное отталкивание противоионов ( ) приводит к уменьшению концентрации вблизи мицеллы. Дисперсионное притяжение противоионов ( их ) может привести к перезарядке [14] поверхности мицеллы при высокой концентрации соли, рисунок 5д (кривая 3). Сокращение расстояния максимального приближения иона усиливает эффективное притяжение подвижных зарядов к мицелле, приводя к более сильному экранированию электрического потенциала, рисунок 5a. Эффект изменения расстояния максимального приближения сказывается сильнее для притягивающих, чем для отталкивающих дисперсионных потенциалов противоионов. На рисунке 6 изображены зависимости ионного вклада в свободную энергию сферической мицеллы и соответствующей силы от площади на молекулу ПАВ при низкой (а,б) и высокой (в,г) концентрациях соли. Интервал площадей, показанный на оси абсцисс, отвечает мицеллярным ядрам, содержащим радикалы , растянутые на 50-97% максимально возможной длины. Расчеты выполнены в предположении дисперсионного притяжения ( ) и отталкивания ( ) между мицеллой и противоионом, а также при отсутствии дисперсионного взаимодействия ( ). Константы дисперсионного взаимодействия взяты из работы Тавареса и сотр. [12] Электрический слой вокруг мицеллы можно рассматривать как сферический (или цилиндрический) конденсатор, одной обкладкой которого служит поверхность фиксированного заряда мицеллы, а другой – слой подвижных ионов, неоднородно распределенных в пространстве. В отсутствии дисперсионных взаимодействий свободная электростатическая энергия такого конденсатора определяется двумя составляющими – электростатической 43 Рисунок 6. Зависимость ионного вклада в свободную энергию агрегации (а,в) и силу (б,г) от площади на голову для сферического агрегата при низкой (0.01M) и высокой (4M) концентрациях соли. Сплошные противоионами и мицеллой ( – ); линии: без дисперсионных взаимодействий ); пунктирные линии: дисперсионное притяжение ( точечные линии: дисперсионное отталкивание ). Все кривые рассчитаны для точечных ионов: , , , энергией и энтропией подвижных ионов, уравнение 9. Первый вклад, быть разбит на две составляющие: энергию . , также может диффузного слоя и энергию слоя Штерна. При низкой концентрации соли существенный вклад в , рисунок 7. ( . Прочие параметры модели: высокой – между плотного вносит , а при не зависит от присутствия соли, определяется падением потенциала в плотном слое Штерна и с увеличением кривизны агрегата убывает. Энергия диффузного слоя определяется градиентом потенциала (см. первое слагаемое в правой части уравнения 20) вокруг мицеллы, а также эффективной толщиной электрического слоя подвижных ионов. С увеличением кривизны поверхностная плотность заряда , уравнение 17, падает, поэтому уменьшается – электрическое поле вблизи мицеллы ослабевает. Ослабление электрического поля мицеллы приводит к тому, что электрический слой подвижных ионов становится все более размытым, и его эффективная толщина увеличивается. При низкой концентрации соли возрастает с ростом , рисунок 7, что объясняется увеличением эффективной толщины электрического слоя. При высокой концентрации соли электрическое поле мицеллы существенно экранировано, поэтому эффективная толщина 44 Рисунок 7. Зависимость энергии электростатических взаимодействий и и ее составляющих от площади на голову ПАВ для сферического агрегата при низкой (0.01M, черные линии) и высокой (4M, красные линии) концентрациях соли. Толстые сплошные линии: тонкая сплошная линия: ; пунктирные линии: . Все энергии рассчитаны в единицах в расчете на молекулу ПАВ для точечных ионов: , , , электрического слоя невелика, и ; . Прочие параметры модели: . с увеличением кривизны убывает, рисунок 7. Энтропийный вклад представляет собой потерю энтропии свободных ионов в связи с их концентрированием у заряженной поверхности агрегата за счет электростатического притяжения. Добавление соли приводит к экранированию поверхностного заряда агрегата, и, следовательно, ослаблению его электрического поля и электростатического притяжения ионов. Ослабление электрического поля вызывает уменьшение электростатической энергии , рисунок 7, а ослабление притяжения – уменьшение потери энтропии подвижных ионов. Следовательно, падает, ср. рисунки 6а и в. Увеличение площади также вызывает ослабление поля мицеллы и притяжения ионов к агрегату в связи с уменьшением приводит к убыванию , что как при низкой, так и при высокой концентрациях соли, рисунки 6а,в. Однако скорость убывания с увеличением , а значит, сила , ощущаемая как стремление голов ПАВ к расталкиванию, при низкой концентрации соли выше, чем при высокой, ср. рисунки 6б и г. При низкой концентрации соли скорость убывания 45 определяется скоростью убывания потери энтропии и электростатической энергии с ростом . При высокой концентрации соли ионы слабо притягиваются к агрегату, величина потери энтропии мала, поэтому скорость убывания убывания с ростом определяется в основном скоростью . Дисперсионные взаимодействия вносят дополнительный вклад энергию , уравнение 9, и изменяют профили распределения подвижных ионов. Притяжение ) понижает, а отталкивание ( ( в свободную ) повышает как при низкой, так и при высокой концентрациях соли, рисунок 6а,в. Однако влияние дисперсионных сил на при низкой концентрации соли противоположно таковому при высокой концентрации соли. Так, при низкой концентрации соли дисперсионное притяжение уменьшает, рисунок 6б, а при высокой концентрации соли – увеличивает, рисунок 6г, расталкивание голов ПАВ. Дисперсионное отталкивание дает противоположный эффект. Притяжение сильнее сказывается на и , чем отталкивание, рисунок 6. Дисперсионное притяжение противоионов способствует экранированию заряда мицеллы, а значит, уменьшает электростатическую энергию, но увеличивает потерю энтропии подвижных ионов, привлекая их к мицелле; дисперсионное отталкивание увеличивает электростатическую энергию и уменьшает потерю энтропии. Противоположные эффекты , оказываемые дисперсионными взаимодействиями на две составляющие свободной энергии , в значительной степени компенсируют друг друга, поэтому влияние дисперсионных взаимодействий на величину в основном определяется энергией этих взаимодействий, , как объясняется ниже. С учетом дисперсионных взаимодействий выражение для плотности ионов 1:1 электролита на поверхности агрегата малой кривизны имеет вид (30) где – контактная плотность иона , – локальная плотность иона. Третье слагаемое в правой части уравнения 30 зависит от потенциала дисперсионного взаимодействия. При отсутствии дисперсионных взаимодействий это слагаемое исчезает, а уравнение 30 принимает вид, известный в литературе как уравнение теоремы о контактной величине плотности ионов [47]. С помощью теоремы о среднем третье слагаемое уравнения 30 можно представить в виде (31) где – эффективная толщина адсорбционного слоя вблизи мицеллы. Расчеты показывают, что для мицеллярных систем составляет ~0.01нм, что существенно меньше, чем ван дер вальсов 46 радиус иона, поэтому значение интеграла в уравнении 30 определяется контактными величинами плотностей ионов. Уравнение 30 принимает вид (32) В отсутствии дисперсионных взаимодействий второе слагаемое в круглых скобках в левой части уравнения 32 равно нулю. Этим слагаемым определяется изменение контактной плотности при включении дисперсионных взаимодействий. Оно практически не зависит от концентрации соли (при изменении концентрации соли на 2 порядка изменяется на доли процента). С помощью теоремы о среднем выражение для энергии дисперсионных взаимодействий , уравнение 11, можно представить в виде (33) где - эффективная толщина адсорбционного слоя, по смыслу сходная с . Как и , величина не превосходит ван дер ваальсова радиуса иона и пренебрежимо мало зависит от концентрации соли, поэтому энергия дисперсионных взаимодействий практически определяется контактной плотностью ионов. При низкой и умеренной концентрациях соли контактная плотность ионов определяется поверхностной плотностью заряда (первое слагаемое в правой части уравнения 32) и слабо зависит от концентрации соли в объеме раствора. Искривление поверхности агрегата уменьшает поверхностную плотность заряда убывает , уравнение 32, и дисперсионного притяжения ( расталкивания и, следовательно, контактная плотность ионов , уравнение 33. Таким образом, энергия ) растет с увеличением , что приводит к ослаблению голов ПАВ, рисунок 6б. Дисперсионное отталкивание ( ) вызывает усиление расталкивания голов ПАВ. При высокой концентрации соли контактная плотность ионов все слабее зависит от кривизны агрегата, поскольку все более существенный вклад вносит постоянное второе слагаемое правой части уравнения 32. Так, для агрегата, параметры которого даны в подписи к рисунку 6 (а радиус отвечает полностью растянутой цепи ), первое слагаемое в правой части уравнения 32 составляет 13.6M, а второе – 0.02 и 8M при концентрациях соли 0.01 и 4M, соответственно. Предполагая контактную плотность не зависящей от , из уравнения 33 имеем: . Таким образом, зависимость энергии дисперсионного взаимодействия от кривизны в пределе высокой концентрации соли становится качественно иной, чем при низкой концентрации соли. Дисперсионное притяжение усиливает, а дисперсионное отталкивание ослабляет расталкивание голов ПАВ, что и видно на рисунке 6г. 47 Наблюдаемый при высокой концентрации соли эффект усиления электростатического отталкивания полярных голов может привести к за счет дисперсионного притяжения ослабляет рост мицелл и диспергированию длинных Диспергирование цилиндрических мицелл цилиндрических при повышении агрегатов концентрации [100-101]. соли было обнаружено в эксперименте для ряда систем [109]. Влияние дисперсионных взаимодействий на ионную составляющую свободной эн ергии мицеллообразования сильнее при высокой концентрации соли, ср. рисунки 6a и в, а на силу поверхностного ионного давления – при низкой концентрации соли, ср. рисунки 6б и г. Благодаря этому радиус агрегата более чувствителен к дисперсионным взаимодействиям при низких концентрациях соли, а ККМ и числа агрегации – при высоких концентрациях соли. Эффект конечных размеров ионов для и иллюстрируется на рисунке 8. При включении конечных размеров ионов доступного пространства у поверхности мицеллы становится меньше, в результате чего понижается их концентрация, и экранирование ослабляется. Понижение концентрации подвижных ионов приводит к уменьшению потери энтропии и абсолютной величины , а ослабление экранирования – к увеличению низкой концентрации соли величины всех составляющих . При изменяются более существенно для агрегатов малой кривизны, поскольку объем пространства, доступного для размещения Рисунок 8. Зависимость ионного вклада в свободную энергию (а) и силу (б) от площади на голову для сферического агрегата при низкой (0.01M) и высокой (4M) концентрациях соли. Сплошные линии рассчитаны с учетом исключенного объема ионов , уравнение БМКСЛ; пунктирные линии рассчитаны для точечных ионов: параметры модели: , , . , , . Прочие , – 48 ионов вблизи таких поверхностей меньше, чем вблизи сильно искривленных. Эффекты конечных размеров ионов для следовательно, , и потери энтропии нивелируются, поэтому и, остаются практически неизменными при низкой концентрации соли, рисунок 8. При высокой концентрации соли конечных размеров ионов для увеличивается за счет , поскольку эффекты и потери энтропии хотя достаточно сильны, но в значительной степени компенсируют друг друга. Сильные эффекты, оказываемые конечными размерами ионов для и потери энтропии, связаны с более значительным изменением концентраций ионов вблизи агрегата при высокой концентрации соли, чем при низкой. По мере уменьшения кривизны увеличивается быстрее для слабо искривленных поверхностей, чем для сильно искривленных. Это приводит к усилению (в сравнении со случаем точечных ионов) расталкивания голов ПАВ при высокой концентрации соли. Таким образом, введение конечных размеров ионов вызывает небольшое возрастание ККМ, сдвиг равновесной площади в область бóльших величин, при этом числа агрегации уменьшаются, а переход сфера-цилиндр сдвигается в область более высоких концентраций соли. Рисунок 9 иллюстрирует эффекты дисперсионного притяжения ( размеров ионов ( ) для ионного вклада ) и собственных и энергии дисперсионных взаимодействий для сферического агрегата в зависимости от концентрации добавленной соли. Вклад дисперсионных взаимодействий в свободную энергию агрегации существен, что согласуется с результатами расчетов в работах [3, 36, 99]. Энергия дисперсионных взаимодействий почти не зависит от концентрации соли вплоть до ~0.5M растворов, давая почти постоянный вклад в lnККМ [100], уравнение 23. Слабая зависимость от концентрации соли объясняется с помощью контактной теоремы [47], уравнение 32. При низких и умеренных концентрациях соли контактная плотность определяется первым слагаемым в правой части уравнения 32, которое постоянно при добавлении соли. Таким образом, контактная плотность ионов вблизи мицеллы, а значит, и энергия дисперсионных взаимодействий , уравнение 33, не зависят от концентрации соли вплоть до 0.5-1M растворов. Для разных противоионов дисперсионный вклад дает параллельный сдвиг кривых ККМ – ионная сила в логарифмических координатах в согласии с экспериментом. Включение притягивающих дисперсионных взаимодействий приводит к понижению ККМ, а отталкивающих – к повышению ККМ. Дисперсионные взаимодействия существенно влияют на зависимости чисел агрегации от концентрации соли и переходы сфера-цилиндр. Дисперсионное притяжение противоионов к агрегату способствует образованию цилиндров, солевой порог перехода сфера-цилиндр понижается, а числа агрегации возрастают. Потеря ионной гидратной оболочки, проникновение 49 Рисунок 9. Зависимости ионного вклада в свободную энергию (пунктирная и сплошные линии) и энергии дисперсионных взаимодействий (штрих-пунктирная линия) от логарифма концентрации соли (моль/л) для сферических агрегатов. Радиус ядра агрегата соответствует длине полностью растянутого углеводородного радикала ПАВ с Дисперсионные силы притягивают противоионы: линия: точечные подвижные ионы, ионов, . . Серая сплошная ; черная сплошная линия: конечные размеры ; серая пунктирная линия: ионный вклад для точечных ионов , и без дисперсионных взаимодействий между подвижными ионами и мицеллой; . Остальные модельные параметры даны в подписи к рисунку 6. ионов в мицеллярную корону и подход ионов к мицеллярному ядру в рамках модели передается малыми . Уменьшение понижению ККМ. При усиливает дисперсионное притяжение ионов и приводит к высокой концентрации уменьшение может привести к немонотонному изменению чисел агрегации от концентрации соли, т.е. диспергированию длинных цилиндрических мицелл, что объяснялось выше. Сильная гидратация (большие ) подавляет образование цилиндров и сдвигает порог перехода сфера-цилиндр в область более высоких концентраций соли. Рисунок 9 показывает, что эффект исключенного объема ионов (который противодействует агрегации) невелик при низких и умеренных концентрациях соли, но может стать существенным при высоких концентрациях. Эффекты высаливания имеют значение только при высоких концентрациях соли, приводя к изгибу вниз кривой ККМ-ионная сила в логарифмических координатах, рисунок 10. Высаливание не оказывает влияния на рассчитанные переходы сфера-цилиндр, поскольку в рамках предлагаемой модели гидрофобный вклад предполагается не зависящим от формы агрегата, уравнение 8. 50 Рисунок 10. Зависимость ККМ от концентрации соли (моль/л) в логарифмических координатах. Черная сплошная линия: без учета высаливания ( с учетом высаливания ( , 4.2 , ); серая пунктирная линия: =-0.05л/моль на CH2 группу). Прочие параметры модели: , , , , Т=298К. Влияние противоионов на образование и рост мицелл На рисунках 11-16 и в таблице 4 показаны экспериментальные и рассчитанные ККМ, переходы сфера-цилиндр и числа агрегации алкилпиридиниев-X (Cn PX, n=10, 12, 14, 16; X=Cl-, Br-, I-), алкилтриметиламмониев-X (Cn TAX, n=12, 14, 16; X= OH-, Cl-, Br-, NO 3-), додецилдиметиламмониев-X (С12 DAX, X= Cl-, Br-), X-додецилсульфатов (XDS, X=Li+, Na+, K+, Cs+) и X-нонилкарбоксилатов (С 9 COOX, X=Na+, K+) в широком интервале концентраций солей [100]. ККМ в бессолевых растворах существенно зависит от противоиона [36]: понижение ККМ описывается рядом [28, 36, 110-111] OH-<Cl-<Br-<NO 3-<I- для анионов, рисунки 11-13, и Li+<Na+<K+<Cs+ для катионов, рисунки 14а и 16а, что верно передается моделью. Например, ККМ C12 PI [44, 112-114] значительно ниже, чем ККМ C12 PCl [36, 115] и C12 PBr [36, 115], рисунок 12а. Модель верно воспроизводит обращение ряда Na+>K+ для понижения ККМ алкилкарбоксилатов по сравнению с рядом Li+<Na+<K +<Cs+ для алкилсульфатов, рисунок 16. При низкой и средней концентрациях соли как рассчитанные, так и экспериментальные кривые ККМ (в логарифмических координатах) почти параллельны для ПАВ с разными противоионами, рисунки 11а, 12а, 13а, 14а. При более высоких концентрациях (0.5-1M) 51 появляются отклонения от линейности, которые зависят от природы соли. В настоящей модели более сильные отклонения описываются более отрицательными константами Сеченова, таблица 2. Такие отклонения хорошо видны для децилпиридиний бромида (C10 PBr) [41] и додецилдиметиламмоний хлорида (C12 DACl) [39], рисунки 12a и 13a. На рисунке 13a видно, что ККМ для C12 DACl и C12 DABr становятся примерно одинаковыми при ~2M соли. При концентрациях выше 2M расчетные кривые пересекаются, что означает обращение ряда Гофмейстера: ККМ C12 DACl становится ниже, чем ККМ C12 DABr. Сферические мицеллы додецилпиридиний иодида (C12 PI) больше, чем мицеллы C12 PBr, а мицеллы цетилпиридиний бромида (С 16 PBr) больше, чем мицеллы С 16PCl, рисунок 12б. В ряде систем, в частности, в растворах C 14 TAX, C16 TAX и NaDS, при низких концентрациях соли могут образовываться глобулярные или достаточно крупные сферические мицеллы [116]. В данной работе не предусматривается возможность упаковки углеводородных радикалов ПАВ со сдвигом полярных голов друг относительно друга в радиальном направлении в ядре [116], за счет которых эффективный размер агрегата мог бы увеличиться; поэтому наибольшее возможное число агрегации сфер – это то, которое может получиться в предположении жидкоподобной упаковки полностью растянутых радикалов в ядре с четкой границей раздела, уравнение 21. Настоящая модель не учитывает образование глобулярных агрегатов, поэтому рассчитанные числа агрегации получаются заниженными по сравнению с экспериментальными, рисунки 11б, 12б и 14б. Однако последовательность изменения значений чисел агрегации Рисунок 11. Зависимости ККМ (a) и чисел агрегации (б) от ионной силы (моль/л) для водных растворов алкилтриметиламмониевого ПАВ Cn TAX + NaX (X=Br¯, Cl¯, NO3 ¯, OH¯). Линии: расчет настоящей работы; незакрашенные символы: модель МФ [24]; закрашенные символы: эксперимент (см. Приложение 1). Числа агрегации были рассчитаны при концентрации ПАВ, равной 0.01M, T=298.15K. 52 Рисунок 12. Зависимости ККМ (a) и чисел агрегации (б) от ионной силы (моль/л) для водных растворов алкилпиридиниевого ПАВ Cn PX + NaX (X= Br¯, Cl¯, I¯). Линии: расчет; символы: эксперимент (см. Приложение 1). Числа агрегации были рассчитаны при концентрации ПАВ, равной ККМ, или выше (указано), T=298.15K. Рисунок 13. Зависимости ККМ, переходов сфера-цилиндр (a) и чисел агрегации (б) от ионной силы (моль/л) в водном растворе додецилдиметиламмониевого ПАВ С12 DAX + NaX (X=Br¯, Cl¯). Линии: расчет; символы: эксперимент. (a) Рассчитанные переходы сфера-цилиндр (пунктирные линии) и ККМ (сплошные линии). (б) Средневесовые (сплошная линия) и zсредние (штрих-пунктирная линия) числа агрегации в 0.01M растворе ПАВ. Ссылки на эксперимент даны в Приложении 1. 53 Рисунок 14. ККМ (а) и рост (б) агрегатов в водно-солевых растворах X-додецилсульфата (XDS + XCl, X=Li+, Na+, K+, Cs+). Символы: эксперимент; сплошные линии: расчет настоящей работы; пунктирные линии: расчет по модели МФ [24]; I – ионная сила (моль/л); показаны числа агрегации в 69mM растворе ПАВ. Ссылки на эксперимент даны в Приложении 1. Рисунок 15. Зависимости ККМ (кружки и сплошная линия) и переходов сфера-цилиндр (треугольники и пунктирная линия) от ионной силы (моль/л) в растворе CsDS + CsCl. Линии: расчет; символы: эксперимент (см. Приложение 1). 54 Рисунок 16. ККМ в бессолевых растворах (а) X-додецилсульфатов (XDS, X=Li+, Na+, K+, Cs+) и (б) X-нонилкарбоксилатов (C9 COOX, X=Na+, K+). Крестики: эксперимент; незакрашенные символы: модель МФ [24]; закрашенные символы: настоящая работа. Ссылки на эксперимент даны в Приложении 1. сферических мицелл, рассчитанных в присутствии разных ионов, согласуется с наблюдаемой в эксперименте последовательностью Гофмейстера. Бромиды алкилтриметиламмониев, алкилпиридиниев и додецилдиметиламмониев обнаруживают более сильный одномерный рост, чем хлориды, рисунки 11б, 12б, 13б, а иодиды додецилпиридиния растут сильнее, чем бромиды, рисунок 12б, причем для разных галогенидов этих ПАВ пороговые концентрации солевых переходов сфера-цилиндр могут отличаться более чем на порядок величины. Модель удовлетворительно описывает эти переходы и верно предсказывает системы, в которых указанные переходы отсутствуют, например, для DPBr [41], рисунки 11б, 12б, 13б, таблица 4. Так, на рисунках 13а и 15 помимо кривых ККМ показаны кривые пороговых переходов сферацилиндр для додецилдиметиламмоний хлорида и бромида и додецилсульфата цезия в зависимости от ионной силы раствора в логарифмических координатах. Как видно, рассчитанные кривые хорошо согласуются с экспериментальными данными. Рассчитанные числа агрегации также удовлетворительно согласуются с экспериментом при умеренном и высоком содержании соли, рисунки 11б, 12б, 13б, 14б. При одинаковых концентрациях добавленного электролита цилиндры, образующиеся в растворах бромидов, длиннее, чем цилиндры, образующиеся в растворах хлоридов. Цилиндрические мицеллы иодида начинают образовываться при особенно низкой ионной силе раствора (~0.007M [44]) и растут чрезвычайно интенсивно. В эксперименте такое поведение DPI объясняется сильным специфическим взаимодействием противоиона с полярной головой и образованием комплекса с переносом заряда с иодид-иона на пиридиниевую группу [44, 112-113, 117]. В рамках предлагаемой модели бромид-противоионы подходят ближе к гидрофобной 55 Таблица 4. Концентрации соли (моль/л) в точках перехода сфера-цилиндр. ПАВ CnTAX ПАВ Cn PX DDAX Соль NaX, X=Br¯ NaX, X=Cl¯ nC=12 nC=14 nC=16 XDS nC=10,12 nC=14 nC=16 Эксп. 1.8 0.12 0.06 0.07 н.п.* 0.19 0.03, 0.05 Расч. 1.8 0.25 0.035 0.1 н.п. 0.15 0.05 Эксп. н.п. 2.7 1.18 0.8 * >0.7 Расч. н.п. 2.3 1.2 0.8 н.п. 4.0 0.9 Соль XCl, X=Na+ 0.45 0.40 XCl, X=Li+ 1.16 0.75 *н.п. – нет перехода; прочерк означает отсутствие данных поверхности, чем хлориды, таблица 3, и имеют более отрицательные дисперсионные константы, таблица 2, что свидетельствует о более сильном притяжении. Более сильное притяжение противоионов вызывает понижение ККМ, способствует образованию сферических мицелл большего размера, сдвигает солевой порог перехода сфера-цилиндр в область низких концентраций соли и стимулирует образование длинных цилиндрических мицелл. Так, найденные комбинации параметров и обеспечивают сдвиг солевых порогов переходов сфера-цилиндр в присутствии бромид- и хлорид-противоионов друг относительно друга не менее, чем на порядок, что согласуется с экспериментом, таблица 4. Меньшие отражают образование более компактных пар бромид-противоиона с полярной головой ПАВ. Это согласуется с недавним экспериментальным подтверждением частичной дегидратации бромида в мицеллярных растворах алкилтриметиламмония [118]. Сильное специфическое связывание иодид-ионов с пиридиниевыми головами в алкилпиридиниевых мицеллах [44] отражается еще меньшим, чем для бромида, расстоянием дисперсионной константы и более высокой абсолютной величиной , таблицы 2-3. Такое связывание уменьшает электростатическое отталкивание между полярными головами, сильно понижает ККМ и приводит к раннему и интенсивному росту стержнеобразных мицелл в соответствии с экспериментом, рисунок 12б. Большие расстояния для гидроксид-иона, таблица 3, могут быть связаны с низкой поляризуемостью и сильной гидратацией этого иона [92-93]. Полученные значения расстояний максимального приближения ионов согласуются с концепцией Коллинса [2]: хаотропная голова (например, пиридиниевая или сульфатная) не образует компактных пар с сильно гидратированными космотропными ионами, но образует такие пары с хаотропами (например, Br¯, I¯, NO3 ¯ или K+ и Cs+, соответственно). Космотропная 56 карбоксильная голова сильнее связывает более космотропный Na+, чем менее космотропный K+, таблица 3, что отражается неравенством . Неравенство обращается для хаотропной сульфатной головы алкилсульфатов. В соответствии с этим, модель предсказывает более сильный мицеллярный рост при добавлении соли для растворов KDS+KCl и CsDS+CsCl, чем для раствора NaDS+NaCl. Это согласуется с экспериментом, как видно на рисунке 14б. Напомним, что анионы оказывают более сильное влияние на ККМ, числа агрегации и переходы сфера-цилиндр, чем катионы, рисунки 11-14. В настоящей модели этот факт описывается более отрицательными значениями дисперсионных констант, таблица 2. В целом предлагаемая модель удовлетворительно предсказывает ККМ, рисунки 11а-14а, 15-16, мицеллярный рост, рисунки 11б-14б, и пороговые концентрации солевых переходов сфера-цилиндр, таблица 4, рисунки 13а, 15 [100]. 4.3 Влияние коионов на образование и рост мицелл Замена коиона лишь незначительно сказывается на ККМ и переходах сфера-цилиндр [46- 47]. Однако при высокой концентрации соли обнаружено удивительно сильное влияние коиона на мицеллярный рост и вязкоупругие характеристики растворов. В частности, добавление KCl [48] вместо NaCl [49] к водному раствору катионного ПАВ эруцилбис(гидроксиэтил)метиламмоний хлорида (EHAC) смещает положение наблюдаемого пика вязкости по оси концентраций соли почти в 5 раз. До настоящего времени отсутствовало объяснение таким существенным эффектам коиона [46, 48-49]. Теоретически эффект коиона был описан на примере равновесия перехода одной формы протеина в другую, адсорбированных на поверхности субстрата [35]. Объяснение роли коионов в указанном равновесии было дано благодаря учету потенциалов дисперсионного взаимодействия ионповерхность в уравнении мПБ для плоскости. При высоких концентрациях соли теория [35] предсказывает сильную специфическую адсорбцию коионов (анионов) на отрицательно заряженной поверхности за счет притягивающих дисперсионных взаимодействий ион-субстрат. Привлечение протонов к отрицательно заряженной поверхности и, следовательно, понижение поверхностного значения pH и приводит к смещению равновесия. Для мицеллярных растворов ПАВ эффект коионов ранее не моделировался. Предлагаемая в настоящей работе модель была применена в растворах эруцилбис(гидроксиэтил)метиламмоний хлорида (EHAC) и цетилтриметиламмоний бромида (C16 TABr) с коионами X (X=Na+, K +) и додецилсульфата натрия (NaDS) с коионами X (X=F¯, Cl¯, Br¯, I¯, SCN¯). 57 Рисунок 17. Зависимость ККМ при T=298.15K (a) и чисел агрегации (б) от ионной силы (моль/л) для водного раствора додецилсульфата натрия NaDS + NaX (X=F¯ , Cl¯, Br¯, I¯, SCN¯). Линии: расчет; символы: эксперимент [46-47]. (б): Штрих-пунктирная линия, незакрашенные символы и все данные на графике с левой стороны от разрыва на оси абсцисс при T=298.15K; другие линии и точки при T=308.15K. Вертикальные штрихи: положение экспериментального перехода сфера-цилиндр. При низких и умеренных концентрациях соли модель предсказывает несущественное влияние коиона на ККМ, рисунок 17a, переходы сфера-цилиндр и числа агрегации, рисунки 17б и 18, в согласии с экспериментом [46-47]. Это не удивительно, т.к. коионы находятся вдали от мицеллы и практически не влияют на мицеллярную структуру. Зависимость ККМ от природы коиона начинает проявляться, хотя и слабо, при высоких концентрациях соли. Эта зависимость может быть объяснена различным высаливающим действием солей, описываемым гидрофобным вкладом в свободную энергию агрегации и различием в собственных размерах ионов. Из рисунков 17б и 18 видно, что в области высоких концентраций соли числа агрегации становятся чувствительны к природе коиона. При этом заряд поверхности экранирован, и электростатическое отталкивание недостаточно сильно, чтобы удерживать коионы далеко от мицеллы. Дисперсионное притяжение способствует проникновению коионов в корону, что ведет к увеличению предпочтительной кривизны агрегата. Глубокое проникновение коионов (малые ) может ослабить мицеллярный рост, а затем привести к диспергированию агрегатов. Максимумы чисел агрегации и диспергирование длинных червеобразных мицелл при добавлении соли наблюдались экспериментально для ряда систем [33]. Рассчитанные зависимости электрического потенциала и концентраций коионов Na + и K + 58 Рисунок 18. Зависимость чисел агрегации от концентрации соли (моль/л) в водных растворах катионных ПАВ в присутствии солей с различными коионами. (а) Цетилтриметиламмоний бромид C16 TABr + XBr (X=Na+, K +); (б) эруцилбис (гидроксиэтил)метиламмоний хлорид (EHAC) + XCl (X=Na+, K +). Линии: расчет; символы: эксперимент [48-49, 119-120]. Пунктирными линиями с символами (б) показаны экспериментальные зависимости вязкости при нулевой скорости сдвига от концентрации соли (правая шкала). Вертикальные штрихи: положение экспериментальных переходов сфера-цилиндр; вертикальные сплошные линии: положение пиков вязкости [48-49, 121]. и противоионов Br- вблизи сферической мицеллы в 1M растворе соли показаны на рисунке 19. Как видно, в связи с возможностью K+ проникать в мицеллярную корону, его концентрация за плоскостью фиксированного заряда полярных голов при приближении к поверхности ядра увеличивается, повышая электрический потенциал, в сравнении с Na+. На кривой распределения ионов K+ в районе плоскости максимального приближения бромид-ионов наблюдается разрыв. Этот разрыв, однако, исчезает при уменьшении собственных размеров ионов до точечных, рисунок 20. При учете собственных размеров ионов с помощью уравнения БМКСЛ (см. Приложение 3) коэффициенты активности для разных ионов отличны от единицы (в отличие от случая, когда ионы точечные) и связаны между собой: когда изменяется значение коэффициента активности одного иона, значение другого также меняется. Так, при переходе через плоскость максимального приближения бромид-ионов в направлении из объема раствора к ядру мицеллы концентрация бромид-ионов падает до нулевого значения, а коэффициент активности возрастает от некоторого конечного значения до бесконечности, что вызывает скачок коэффициента активности коионов калия и, следовательно, их концентрации. Следует отметить, что активность коионов калия справа и слева от скачка остается постоянной, 59 Рисунок 19. (a) Зависимости локальной концентрации ионов локального электрического потенциала , моль/л, и (б) приведенного от расстояния от поверхности гидрофобного ядра сферической мицеллы в присутствии 1M электролита XBr с разными коионами X (X=Na+, K +) при T=298.15K. Радиус ядра и поверхностная плотность заряда мицеллы составляют 2нм и 0.233Кл/м2 , собственные размеры ионов: =0.266нм, =0.240нм, =0.390нм. Другие параметры =0.28нм, =0.190нм, =0.600нм, =0.325нм. Вертикальные сплошные линии указывают положение плоскостей максимального приближения ионов к ядру мицеллы. Вертикальные штриховые линии обозначают положение плоскости фиксированного заряда полярной головы. поскольку значение электрохимического потенциала ионов калия постоянно всюду, где они могут проникать. Рисунок 18a показывает, что добавление слабо притягивающегося и непроникающего в корону Na+ способствует мицеллярному росту, тогда как сильно притягивающийся и проникающий K+ ослабляет рост, а затем вызывает укорочение мицелл при высокой концентрации соли. Рисунок 18б показывает числа агрегации, рассчитанные для системы EHAC+хлориды щелочных металлов. Для данной системы в области высоких концентраций соли интенсивный рост червеобразных мицелл сопровождается ростом вязкости и переходом в вязкоупругое состояние, при котором вязкость, достигнув максимума, начинает уменьшаться. До настоящего времени появление пика на кривых реологического эксперимента и 60 Рисунок 20. Зависимость локальной концентрации ионов, моль/л, от расстояния от гидрофобного ядра сферической мицеллы в присутствии 1M электролита XBr с разными коионами X (X=Na+, K +) при T=298.15K. Сплошные линии: расчет с учетом собственных размеров ионов: =0.190нм, =0.266нм, =0.390нм; пунктирные линии: расчет с точечными ионами. Обозначения и остальные параметры такие же, как на рисунке 19. дальнейшее уменьшение вязкости объясняли образованием ветвлений червеобразных мицелл, которые увеличивают текучесть пространственной мицеллярной сетки [37, 48-49], что эквивалентно уменьшению эффективной длины червеобразных агрегатов в растворе [33]. Моделирование мицеллярных ветвлений [104] выходит за рамки настоящего исследования, тем не менее полученные в настоящей работе результаты, рисунок 18б, позволяют объяснить влияние коионов на положение пика вязкости на качественном уровне. Малая предпочтительная кривизна агрегатов приводит к интенсивному росту и способствует образованию ветвлений. Следовательно, для непроникающего Na+, в присутствии которого рост мицелл сильнее, ветвления должны образовываться при более низкой концентрации соли, чем для глубоко проникающего K +, рисунок 18б. При высоких концентрациях K+ увеличение предпочтительной кривизны агрегатов приводит к диспергированию длинных цилиндрических мицелл и, следовательно, уменьшению их средней длины. 4.4 Сопоставление результатов настоящей работы с результатами предыдущих работ Как отмечалось в Главе 1, специфические эффекты ионов могут быть учтены в рамках модели МФ [24], пригодной для описания ККМ и размеров небольших агрегатов в растворах 61 Таблица 5. Рассчитанные и экспериментальные ионных ПАВ, и модели ШБ [21-22], в ККМ Сn TACl при T=298.15K. рамках которой в согласии с экспериментом были описаны ККМ, числа агрегации и ККМ, мM ПАВ концентрационные пороги переходов сфера- Данная работа Модель ШБ[22] Эксп.[22] C12 TACl 22.40 13.14 20.3 C14 TACl 4.70 2.73 4.47 C16 TACl 0.94 0.57 1.30 цилиндр для анионных ПАВ. Ниже дано сопоставление результатов, полученных с помощью настоящей модели, с результатами расчетов по указанным моделям. Таблица 5 содержит рассчитанные (с помощью настоящей модели и модели ШБ) и экспериментальные ККМ алкилтриметиламмоний хлоридов с разной длиной цепи в бессолевой системе. Видно, что модель, предлагаемая в настоящей работе, описывает ККМ существенно точнее, чем модель ШБ [21-22]. Для раствора NaDS+NaCl настоящая модель и модель ШБ дают почти идентичные кривые ККМ-концентрация соли, рисунок 21а. Для сферических мицелл в бессолевой системе NaDS настоящая модель и модель ШБ дают близкие значения электрических потенциалов на расстоянии 0.358нм от положения эффективного заряда полярной головы (-94мВ в настоящей модели и -92мВ в модели ШБ), рисунок 21б, которые соответствуют имеющимся Рисунок 21. Зависимости (a) ККМ, (б) приведенного поверхностного электрического потенциала и (в) чисел агрегации от ионной силы (I) и концентрации соли (С NaCl) в водном растворе NaDS+NaCl (298.15K). Символы: эксперимент (см. Приложение 1); расчеты с помощью модели настоящей работы (сплошные жирные линии), модели ШБ (тонкие сплошные линии) [21] и модели МФ (пунктирные линии) [24]. 62 экспериментальным оценкам -потенциалов: -80мВ и -101.2мВ [22]. На рисунке 22 даны экспериментальные и рассчитанные значения параметров роста мицелл XDS+XCl (X= Li+, Na+, K+, Cs+). Как видно, модель ШБ точнее для Cs+ и K+, а настоящая модель – для Na+ и Li+. Параметры роста, показанные на рисунке 22, получены при концентрации соли, близкой к порогу перехода сфера-цилиндр, где цилиндрические мицеллы еще не испытывают интенсивного роста. При более высоких концентрациях солей щелочных металлов в растворах додецилсульфатов, где цилиндрические мицеллы длинные, модель ШБ не опробована. В отличие от модели ШБ, которая протестирована только для нескольких фиксированных концентраций противоионов хлорида и салицилата, настоящая модель успешно применена в широком интервале концентраций целого ряда анионов, в т.ч. выступающих в роли коионов. Модель МФ и предлагаемая модель одинаково хорошо описывают ККМ в бессолевых растворах, как видно из рисунков 11а и 16. Обе модели воспроизводят обращение ряда катионов щелочных металлов для алкилкарбоксилатов в сравнении с алкилсульфатами. Сопоставление рассчитанных результатов с экспериментом показывает, что при низких и умеренных концентрациях соли модель МФ и предлагаемая модель удовлетворительно описывают кривые ККМ-концентрация соли для катионного ПАВ С n TAX+NaX (X=Cl-, Br-), рисунок 11а, однако для анионного ПАВ XDS+XCl (Li+, Na+, K+, Cs+) настоящая модель дает лучшее описание кривых ККМ при умеренных и высоких концентрациях электролита, рисунок 14a. В системах C16 TABr, NaDS, для которых при низких и умеренных концентрациях соли характерно образование глобулярных мицелл [122-123], более точна модель МФ, рисунок 21в. Однако для предсказания роста мицелл модель МФ не может быть использована в отличие от Рисунок 22. Параметр роста + + + + , уравнение 25, мицелл XDS в 0.45M растворах XCl (X=Li , Na , K , Cs ), T=313.15K. Ссылки на эксперимент даны в Приложении 1. 63 настоящей модели, которая дает удовлетворительные результаты в растворах с длинными цилиндрическими мицеллами. В целом, предложенная в настоящей работе модель существенно расширяет возможности описания специфических эффектов ионов в мицеллярных системах: 1) Использование набора величин параметров, оцененных в рамках предложенной модели, позволяет удовлетворительно описывать как образование сферических, так и рост цилиндрических мицелл для любой системы ПАВ+соль, рассматриваемой в работе. 2) Модель применима в широких интервалах концентраций (от бессолевых систем до концентраций насыщенных растворов) электролитов. 3) Модель позволяет описывать не только влияние противоионов, но и коионов на образование, переходы сфера-цилиндр и рост мицелл. 4) Предложенный на основе модели механизм влияния природы ионов на агрегацию ПАВ позволил на качественном уровне интерпретировать данные реологического эксперимента о влиянии природы коиона на положение пика на кривой вязкости. 5) Предложенный механизм влияния природы ионов на агрегацию ПАВ позволил при высоких концентрациях электролита объяснить эффект диспергирования длинных цилиндрических мицелл, наблюдаемый в эксперименте для ряда систем. 6) Найденная совокупность комбинаций параметров модели может быть использована для предсказания поведения не рассмотренных в данной работе систем: - в системах ионное ПАВ+соль, где в роли компонентов системы могут выступать либо другое ПАВ, либо другая соль, не рассмотренные в настоящей работе; - в водно-солевом растворе смеси двух ионных ПАВ; - в водных растворах ионного ПАВ в смеси солей. Для этого часть найденных в настоящей работе параметров может быть использована без изменения, а часть должна быть оценена дополнительно. 4.5 Значения параметров модели и структура агрегата Для описания поведения каждой из рассматриваемых в настоящей работе систем ПАВ+соль найдены соответствующие комбинации параметров модели, величины которых представлены в таблицах 1-3 [100-101]. В таблице 6 для сравнения помещены значения некоторых параметров настоящей модели и значения аналогичных характеристик, измеренных экспериментально или используемых в более ранних молекулярно-термодинамических моделях агрегации. В целом, из таблицы 6 видно, что для аммониевых, пиридиниевых, сульфатных и карбоксилатных ПАВ значения 64 Таблица 6. Сопоставление экспериментальных и используемых в моделях величин и . , нм2 Эффективная площадь поперечного сечения полярной головы ПАВ Настоящая работа МФ[24] ШБ [21-22] БВК[23] Эксп. Эксп. метод Сn TAX 0.58 0.09’0.21а) 0.32 - 0.39’0.43б) SANS, SAXS, [47] Сn PX 0.52 0.14’0.28а) - 0.375 0.22 SANS, SAXS[47] XDS 0.45 0.23’0.25а) 0.25 - Cn COOX 0.50 0.18’0.20а) - - 0.19’0.30в) SANS, SAXS[123-124] - - Расстояние от гидрофобного ядра до плоскости заряда полярных голов , нм ПАВ Настоящая работа МФ [24] ШБ [21-22] Шилоач и Бланкштейн [20] Эксп. Эксп. метод Сn TAX 0.28 0.10 0.26 0.25 0.18 SAXS[107] Сn PX 0.3 0.10 - 0.26 0.22’0.43г) SAXS[107] XDS Cn COOX 0.65 0.65 0.40 0.20 0.37 - 0.38 - SAXS[107], 0.36, 0.565, 0.4’1.25д) SANS[123], cryo-TEM 0.21 SAXS[107] и SAXS[116] Наличие интервалов в таблице означает, что: а) в модели МФ – это средняя эффективная площадь поперечного сечения полярной головы ПАВ, которая зависит не только от размера полярной головы, но и от размера гидратированного противоиона, присутствующего в растворе; в присутствии галогенидов или катионов щелочных металлов значения б) изменяются в указанных интервалах; нижняя граница площади поперечного сечения триметиламмониевой головы рассчитана из ее ван дер ваальсова радиуса, а верхняя граница – из радиуса головы в гидратированном состоянии. в) размер сульфатной группы был рассчитан [124] из данных о дифракции рентгеновских лучей и нейтронов на кристаллах XDS∙nH2 O, где n=0’1; г) на пиридиниевой группе имеется несколько активных для взаимодействия с противоионом центров, отстоящих от α-углеродного атома на расстояниях, попадающих в указанный интервал; д) нижняя граница соответствует толщине гидрофильного слоя (мицеллярной короны) для мицелл NaDS, а верхняя граница – для мицелл CsDS, для которых образуются глобулярные агрегаты или укрупненные сферические мицеллы. 65 эффективной площади настоящей работы в 1.5-2 раза превосходят соответствующие величины, рассчитанные из экспериментальных данных о молекулярном строении полярных групп [47, 107, 123-124] и используемые в предложенных ранее молекулярно- термодинамических моделях мицеллообразования [17, 20-24]. Для аммониевых, пиридиниевых и сульфатных голов значения настоящей работы изменяются симбатно экспериментальными оценками эффективных размеров полярных групп, с полученными различными экспериментальными методами, в т.ч. методами малоуглового рассеяния рентгеновских лучей (SAXS) и нейтронов (SANS) на кристаллах и в растворах, содержащих ПАВ: триметиламмониевая группа немного объемнее пиридиниевой [47, 124], а гидратированная карбоксильная группа объемнее сульфатной при близких ван дер ваальсовых размерах [47, 123]. В соответствии с дифракционными данными работы [124], полученными для кристаллов NaDS∙nH2 O, где n=0’1, =0.19’0.30нм2 . Хотя сульфатная группа считается слабо гидратированной, тем не менее найдено [123], что, имея площадь =0.22нм2 (рассчитанную из объема полярной группы, найденного на основе данных SAXS и SANS для растворов CsDS), она окружена ~10 молекулами воды в мицеллярной короне, что должно существенно отразиться либо на значении параметра (в случае, если молекулы воды сильно связаны с полярной группой), либо на эффективной площади , приходящейся на молекулу ПАВ в мицелле. Для большинства полярных голов используемые в настоящей работе значения оказываются ближе к экспериментальным размерам голов в гидратированном состоянии, чем к площадям Значения , рассчитанным на основе ван дер ваальсовых радиусов атомов. (расстояния от гидрофобного ядра мицеллы до эффективного заряда полярных голов) настоящей работы близки к значениям соответствующего параметра, используемого в работе Шилоача и Бланкштейна [20], в случае аммониевого и пиридиниевого ПАВ, таблица 6. Значения настоящей работы для аммониевой, пиридиниевой и сульфатной голов в 1.5 раза превосходят экспериментальные [107], рассчитанные из данных о рассеянии рентгеновских лучей, но, в общем, изменяются симбатно с ними. Для сульфатов и карбоксилатов разница между значениями настоящей работы и экспериментальными величинами больше, чем для аммониевых и пиридиниевых голов. Это может быть связано с тем, что при низких и умеренных концентрациях электролитов алкилсульфаты [122], рисунок 21в, и алкилкарбоксилаты [125] уже с довольно короткими углеводородными радикалами с склонны образовывать глобулярные [24, 50, 122-123] или укрупненные сферические мицеллы [116]. В поверхностном слое глобулярных и укрупненных сферических мицелл полярные головы могут быть сдвинуты друг относительно друга в радиальном направлении [116], что делает границу раздела углеводородное ядро-раствор более размытой. Сдвиг голов 66 друг относительно друга увеличивает эффективную толщину мицеллярной короны, которая, как было найдено методами SANS и SAXS в растворах CsDS, составляет 0.565нм в работе [123] или 1.10’1.25нм в работе [116]. В настоящей работе для додецилсульфатов было получено значение =0.65нм, попадающее в интервал изменения экспериментальных величин [123], таблица 6. Оцененные в настоящей работе дисперсионные константы изменяются в соответствии с последовательностями Гофмейстера для ККМ, чисел агрегации и солевых порогов перехода сфера-цилиндр. Более сильный специфический эффект для анионов по сравнению с катионами в предлагаемой модели отражается немного бóльшими абсолютными значениями дисперсионных констант , таблица 2. Исходя из литературных данных, можно сделать вывод, что знаки и величины рассчитываемых в рамках теории Лифшица дисперсионных констант существенно зависят от точности оценки поляризуемостей и диэлектрических проницаемостей (показателей преломления в видимой области частот) как функций частот поглощения и определяются экспериментально, рассчитываются квантовомеханически или с применением одно- или мультимодальных приближений. В связи с этим, значения дисперсионных констант , рассчитанные для одной и той же системы, могут колебаться в широких пределах. Оцененные в настоящей работе силовые константы дисперсионного взаимодействия катионов и анионов на порядок выше, чем рассчитанные Таваресом и сотр. [12] с помощью одномодального приближения для взаимодействий Рисунок 23. Силовые константы дисперсионного взаимодействия анионов с макроионом (молекулой протеина или мицеллой); Незакрашенные кружки [12]: =1.4; закрашенные кружки [12]: преломления; звездочки – настоящая работа. (а) катионов и (б) , (нм3 ) – поляризуемость. =1.6, где – показатель 67 гидратированных ионов с протеином, имеющим показатель преломления близкий к показателю преломления углеводорода. В этой же работе [12] с помощью одномодального приближения были рассчитаны зависимости дисперсионных констант от поляризуемостей ионов при разных показателях преломления протеина, рисунок 23. Из рисунка 23 видно, что изменение показателя преломления протеина от до увеличивает силовые константы в несколько раз [12]. В настоящей работе дисперсионные константы в 3 раза для анионов и в 5 раз для катионов выше констант, полученных из квантово-механических расчетов, и в 6 раз выше констант, рассчитанных с помощью одномодального приближения для границы раздела воздух-вода в работе [3]. При квантово-механическом описании появляются дополнительные по сравнению с одно- и мультимодальным приближениями высокочастотные вклады в поляризуемость для всех ионов. Появление этих вкладов приводит к возрастанию абсолютной величины силовой константы дисперсионного взаимодействия на 10-120% [3]. Эффективные расстояния эффекты и гидратации-дегидратации в соответствии с концепцией Коллинса [2] учитывают в мицеллярной короне. Они, как отмечалось, индивидуальны для каждой пары подвижный ион-голова ПАВ и различаются для противоиона и коиона. Два параметра и взаимодействий иона с ядром позволяют описать сложную структуру электрического слоя вокруг мицеллы и учесть локальное накопление ионов вблизи поверхности мицеллы при общей отрицательной адсорбции соли на границе раздела. Такая сложная структура обнаружена при моделировании методом молекулярной динамики границы раздела углеводород-водный раствор соли [91], рисунок 24б. На распределениях подвижных ионов вблизи незаряженной углеводородной границы четко видны локальные пики ионов брома и натрия. При моделировании было обнаружено, что более поляризуемые ионы подходят к поверхности углеводорода ближе, создавая более высокие приповерхностные концентрации (бромид на рисунке 24б), при этом адсорбция более поляризуемых ионов по всему электрическому слою может оставаться отрицательной. Более высокая адсорбция сильно поляризуемых ионов также подтверждается данными о рассеянии рентгеновских лучей на границах раздела [91]. Рисунок 24а показывает, что распределения брома и натрия, рассчитанные с помощью настоящей модели, обнаруживают сходные пики на границе раздела раствор соли – капля углеводорода с низкой отрицательной зарядовой плотностью. Адсорбции ионов на рисунке 24а определяются разницей закрашенных/заштрихованных областей, расположенных выше и ниже нулевой линии. В соответствии с уравнением изотермы адсорбции Гиббса имеем (34) 68 где - поверхностное натяжение на границе раздела; – объемная концентрация соли; – адсорбция иона , где , – площадь границы раздела, и – локальная концентрация и концентрация ионов в объеме, соответственно; - эффективная толщина слоя растворителя, обедненного ионами. адсорбция соли В случае если отрицательна, знак положителен, что означает обеднение ионами приповерхностных слоев растворителя. В настоящей работе для слабо заряженной углеводородной сферы в растворах NaBr и NaCl были получены величины , равные 0.37 и 0.57нм, рисунок 24а, соответственно, что означает отрицательную адсорбцию обеих солей. Рисунок 24. (а) Зависимости концентрации распределения расстояния от углеводородной данной работе: ионов слабо сферы, локальной заряженной рассчитанные =0.6нм, =0.19, =0.39нм. в =0.009Кл/м2 , 2нм, =0.28нм, от На рисунке зависимости расстояний приближения изображены 25 максимального галогенид-ионов , используемых в настоящей работе в ряде систем ПАВ+соль, от эффективных толщин =0.325нм, слоев растворов вблизи границы раздела Профили воздух-раствор, обедненных ионами [91]. распределения ионов Na и Br и молекул Видно, что в случае галогенид-ионов на воды вблизи границы раздела воздух-раствор незаряженной границе раздела углеводород- соли, рассчитанные методом молекулярной раствор динамики [91]. указанные параметры изменяются симбатно. (б) + и Изменение в мицеллярных от иона к иону зависит от природы полярной головы ПАВ. Так, в присутствии пиридиниевых голов сильнее, чем в случае триметиламмониевых, в случае диметиламмониевых всего. Более резкое изменение растворах изменяется изменяется слабее от иона к иону приводит к более существенной разнице в солевых порогах перехода сфера-цилиндр, ср. рисунки 11б, 12б и 13б. Расстояния близки к радиусам гидратированных или частично дегидратированных 69 Рисунок 25. Зависимость расстояний максимального приближения галогенид-ионов в мицеллярных растворах алкилпиридиния, алкилтриметиламмония и алкилдиметиламмония, используемых в настоящей работе, от толщин слоев растворителя вблизи незаряженной границы раздела воздух-вода, обедненной ионами, в водных растворах солей NaX (X= Cl¯, Br¯, I¯), оцененных из работы [91]. ионов [47], которые были получены из данных о вязкости, диффузии, сжимаемости, проводимости, растворимости и различных термодинамических и спектроскопических свойствах растворов электролитов. Например, радиус гидратированного бромид-иона ~0.32 нм [118], а расстояние максимального приближения бромида как противоиона колеблется в пределах 0.325’0.390нм. На рисунке 26 изображены контактные значения потенциалов дисперсионного взаимодействия, т.е. величины дисперсионных потенциалов в точках максимального приближения ионов к углеводородному ядру, рассчитанные в настоящей работе и значения аналогичных величин, используемых в модели Иванова и сотр. [36] Из рисунка 26 видно, что значения, используемые в настоящей работе, в целом согласуются со значениями из работы [36]. Поскольку для ряда солей отсутствуют экспериментально измеренные константы Сеченова, то в настоящей работе константы , таблица 2, получались подгонкой рассчитанных ККМ к экспериментальным при высоких концентрациях соли. Найденные величины констант согласуются с имеющимися экспериментальными оценками. Так, в соответствии с данными о растворимости н-гексана [126] л/моль на CH2 группу как для NaCl, так и для KCl, 70 Рисунок 26. Контактные значения потенциалов дисперсионного взаимодействия для (а) анионов и (б) катионов. Кружки – модель настоящей работы, крестики – модель Иванова и сотр. [36] и л/моль для NaBr, а в настоящей работе = -0.05, -0.04, -0.03л/моль для NaCl, KCl и NaBr, соответственно. Как отмечалось в Главе 1, при использовании уравнения 2 получаются завышенные л/моль для NaCl, KCl и NaBr, соответственно. В то же время не исключается, что значительно бóльшие величины для LiCl и NaOH, найденные в настоящей работе, являются артефактом. Собственные диаметры ионов оценены [47] по экспериментальным данным о рассеянии рентгеновских лучей и нейтронных потоков на кристаллах и о растворимости газов, вязкости и диффузии в жидкостях. ЗАКЛЮЧЕНИЕ 1. Для описания эффектов специфики ионов в мицеллярных растворах ионных ПАВ разработан новый вариант молекулярно-термодинамической модели агрегации. 2. Найденные в настоящей работе наборы величин параметров модели позволяют описывать образование и (алкилтриметиламмониевых, рост мицелл в растворах катионных и алкилдиметиламмониевых, анионных ПАВ алкилпиридиниевых, эруцилбис(гидроксиэтил) метиламмония, алкилсульфатов и алкилкарбоксилатов) в присутствии целого ряда 1:1 электролитов: галогенидов, нитратов, тиоцианатов и гидроксидов щелочных металлов в широком диапазоне концентраций. 3. Предложенная модель удовлетворительно описывает влияние природы противоиона на ККМ, солевой порог переходов сфера-цилиндр и рост мицеллярных агрегатов при дальнейшем добавлении соли. 71 4. В рамках предложенной модели при высоких концентрациях одних солей предсказывается интенсивный рост цилиндрических агрегатов, а других – диспергирование. 5. С помощью разработанной модели впервые описано существенное влияние коионов на рост мицелл при высокой концентрации соли. Предложенный механизм влияния природы ионов на агрегацию ионных ПАВ позволил дать качественное объяснение наблюдаемым в реологическом эксперименте эффектам специфики коиона на вязкоупругость растворов ионных ПАВ. 6. Гидратация ионов в мицеллярной короне передается эффективными параметрами ( ), в величинах которых отражается степень компактности ионных пар голова ПАВ – подвижный ион. От компактности указанных пар существенно зависит предпочтительная кривизна агрегатов. Предложенная модель представляет количественную формулировку общей концепции Коллинса в применении к растворам ионных ПАВ. Дальнейшее развитие предлагаемой модели может состоять в переходе к описанию агрегатов более сложных морфологий : мицеллярных ветвлений, пространственных сеток червеобразных мицелл, бислоев и везикул. Благодаря сходству в агрегативном поведении классических ионных ПАВ и других самоорганизующихся флюидных систем, содержащих амфифильные молекулы (молекулы блоксополимеров, фосфолипидные мембраны и др.), ожидается, что сформулированный в настоящей работе подход окажется полезным при моделировании эффектов специфики ионов в этих сложных объектах. Другим интересным направлением приложения развитого подхода является его применение к растворам ПАВ в присутствии смеси 1:1 электролитов. Заметим, что развитие подхода в указанном направлении не требует оценки дополнительных параметров – все расчеты могут быть проведены на основе набора значений параметров для ПАВ и 1:1 электролитов, оцененных в настоящей работе. Важной перспективой развития модели является ее распространение для водно-солевых растворов смесей ПАВ и для растворов ПАВ, содержащих добавки низкомолекулярных органических ионов, что представляется важным в практическом отношении (например, при подборе составов смесей в системах с управляемой вязкостью для различных технологических приложений). 72 СПИСОК ЛИТЕРАТУРЫ 1. Kunz, W. Specific Ion Effects //Singapore, World Scientific Publishing Co. Pte. Ltd., 2010.- 346p. 2. Collins, K.D. Charge Density-Dependent Strength of Hydration and Biological Structure //Biophys. J.-1997.-V.72,P.65-76. 3. Parsons, D.F., Deniz, V., Ninham, B.W. Nonelectrostatic Interactions between Ions with Anisotropic ab initio Dynamic Polarisabilities //Colloids Surf., A-2009.-V.343,P.57-63. 4. Boström, M., Kunz, W., Ninham, B.W. Hofmeister Effects in Surface Tension of Aqueous Electrolyte Solution //Langmuir-2005.-V.21,P.2619-2623. 5. Boström, M., Williams, D.R.M., Ninham, B.W. Surface Tension of Electrolytes : Specific Ion Effects Explained by Dispersion Forces //Langmuir-2001.-V.17,P.4475-4478. 6. Boström, M., Ninham, B.W. Dispersion Self-Free Energies and Interaction Free Energies of Finite-Sized Ions in Salt Solutions //Langmuir-2004.-V.20,P.7569-7574. 7. Karraker, K.A., Radke, C.J. Disjoining Pressures, Zeta-Potentials and Surface Tensions of Aqueous Non-Ionic Surfactant/Electrolyte Solutions: Theory and Comparison to Experiment //Adv. Colloid Interface Sci-2002.-V.96,P.231. 8. Santos, A.P.d., Levin, Y. Ions at the Water−Oil Interface: Interfacial Tension of Electrolyte Solutions //Langmuir-2012.-V.28,P.1304-1308. 9. Horinek, D., Netz, R.R. Specific Ion Adsorption at Hydrophobic Solid Surfaces //Phys. Rev. Lett.-2007.-V.99,P.226104. 10. Parsons, D.F., Ninham, B.W. Importance of Accurate Dynamic Polarizabilities for the Ionic Dispersion Interactions of Alkali Halides //Langmuir-2010.-V.26,N.3,P.1816-1823. 11. Tavares, F.W., Bratko, D., Prausnitz, J.M. The Role of Salt–Macroion van der Waals Interactions in the Colloid–Colloid Potential of Mean Force //Curr. Opin. Colloid Interface Sci.-2004.V.9,P.81-86. 12. Tavares, F.W., Bratko, D., Blanch, H.W., Prausnitz, J.M. Ion-Specific Effects in the Colloid- Colloid or Protein-Protein Potential of Mean Force: Role of Salt-Macroion van der Waals Interactions //J. Phys. Chem. B-2004.-V.108,P.9228-9235. 13. Boström, M., Tavares, F.W., Bratko, D., Ninham, B.W. Specific Ion Effects in Solutions of Globular Proteins: Comparison between Analytical Models and Simulation //J. Phys. Chem. B-2005.V.109,P.24489. 14. Boström, M., Williams, D.R.M., Ninham, B.W. Specific Ion Effects: Why the Properties of Lysozyme in Salt Solutions Follow a Hofmeister Series //Biophys. J.-2003.-V.85,P.686–694. 73 15. Curtis, R.A., Ulrich, J., Montaser, A., Prausnitz, J.M., Blanch, H.W. Protein-Protein Interactions in Concentrated Electrolyte Solutions Hofmeister-Series Effects //Biotechnol. Bioeng.2002.-V.79,N.4,P.367-380. 16. Boström, M., Ninham, B.W. Energy of an Ion Crossing a Low Dielectric Membrane: the Role of Dispersion Self-Free Energy //Biophys. J.-2005.-V.114,P.95–101. 17. Nagarajan, R., Ruckenstein, E. Theory of Surfactant Self- Assembly: A Predictive Molecular Thermodynamic Approach //Langmuir-1991.-V.7,P.2934-2969. 18. Goldsipe, A., Blankschtein, D. Molecular-Thermodynamic Theory of Micellization of pH- Sensitive Surfactants //Langmuir-2006.-V.22,P.3547-3559. 19. Puvvada, S., Blankschtein, D. Theoretical and Experimental Investigations of Micellar Properties of Aqueous Solutions Containing Binary Mixtures of Nonionic Surfactants //J. Phys. Chem.-1992.-V.96,N.13,P.5579-5592. 20. Shiloach, A., Blankschtein, D. Prediction of Critical Micelle Concentrations and Synergism of Binary Surfactant Mixtures Containing Zwitterionic Surfactants //Langmuir-1997.-V.13,P.3968-3981. 21. Srinivasan, V., Blankschtein, D. Effect of Counterion Binding on Micellar Solution Behavior: 1. Molecular-Thermodynamic Theory of Micellization of Ionic Surfactants //Langmuir-2003.V.19,P.9932-9945. 22. Srinivasan, V., Blankschtein, D. Effect of Counterion Binding on Micellar Solution Behavior: 2. Prediction of Micellar Solution Properties of Ionic Surfactant-Electrolyte Systems //Langmuir2003.-V.19,P.9946-9961. 23. Bauer, A., Woelki, S., Kohler, H.-H. Rod Formation of Ionic Surfactants: Electrostatic and Conformational Energies //J. Phys. Chem. B-2004.-V.108,P.2028-2037. 24. Moreira, L., Firoozabadi, A. Molecular Thermodynamic Modeling of Specific Ion Effects on Micellization of Ionic Surfactants //Langmuir-2010.-V.26,N.19,P.15177-15191. 25. Самойлов, О.Я. Структура водных растворов электролитов и гидратация ионов //Москва, АН СССР, 1957.-184c. 26. Vlachy, N., Jagoda-Cwiklik, B., Vácha, R., Touraud, D., Jungwirth, P., Kunz, W. Hofmeister Series and Specific Interactions of Charged Headgroups with Aqueous Ions //Adv. Colloid Interface Sci.-2009.-V.146,P.42-47. 27. Kunz, W., Lo Nostro, P., Ninham, B.W. The Present State of Affairs with Hofmeister Effects //Curr. Opin. Colloid Interface Sci.-2004.-V.9,P.1-18. 28. Missel, P.J., Mazer, N.A., Carey, M.C., Benedek, G.B. Influence of Alkali- Metal Counterion Identity on the Sphere-to-Rod Transition in Alkyl Sulfate Micelles //J. Phys. Chem.-1989.V.93,N.26,P.8354-8366. 74 29. Cuccovia, I.M., Agostinho-Neto, A., Wendel, C.M.A., Chaimovich, H., Romsted, L.S. Determination of Interfacial Co-ion Concentration in Ionic Micelles by Chemical Trapping: Halide Concentration at the Interface of Sodium Dodecyl Sulfate Micelles //Langmuir -1997.-V.13,P.50325035. 30. Romsted, L.S. Do Amphiphile Aggregate Morphologies and Interfacial Compositions Depend Primarily on Interfacial Hydration and Ion-Specific Interactions? The Evidence from Chemical Trapping //Langmuir-2007.-V.23,P.414-424. 31. Geng, Y., Romsted, L.S., Froehner, S., Zanette, D.J., Magid, L.J., Cuccovia, I.M., Chaimovich, H. Origin of the Sphere-to-Rod Transition in Cationic Micelles with Aromatic Counterions: Specific Ion Hydration in the Interfacial Region Matters //Langmuir-2005.-V.21,P.562-568. 32. Abezgauz, L., Kuperkar, K., Hassan, P.A., Ramon, O., Bahadur, P., Danino, D. Effect of Hofmeister Anions on Micellization and Micellar Growth of the Surfactant Cetylpyridinium Chloride //J. Colloid Interface Sci.-2010.-V.342,P.83-92. 33. Magid, L.J., Han, Z., Li, Z. Tuning Microstructure of Cationic Micelles on Multiple Length Scales: The Role of Electrostatics and Specific Ion Binding //Langmuir-2000.-V.16,P.149-156. 34. Brady, J.E., Evans, D.F., Warr, G.G., Grieser, F., Ninham, B.W. Counterion Specificity as the Determinant of Surfactant Aggregation //J. Phys. Chem.-1986.-V.90,N.9,P.1853-1859. 35. Bostrom, M., Williams, D.R.M., Ninham, B.W. Specific Ion Effects: The Role of Co-Ions in Biology //Europhys. Lett.-2003.-V.63,N.4,P.610-615. 36. Ivanov, I.B., Slavchov, R.I., Basheva, E.S., Sidzhakova, D., Karakashev, S.I. Hofmeister Effect on Micellization, Thin Films and Emulsion Stability //Adv. Colloid Interface Sci. -2011.-V.168 (Suppl.),P.93-104. 37. Oelschlaeger, C., Suwita, P., Willenbacher, N. Effect of Counterion Binding Efficiency on Structure and Dynamics of Wormlike Micelles //Langmuir-2010.-V.26,N.10,P.7045-7053. 38. Kuperkar, K., Abezgauz, L., Prasad, K., Bahadur, P. Formation and Growth of Micelles in Dilute Aqueous CTAB Solutions in the Presence of NaNO 3 and NaClO 3 //Journal of Surfactants and Detergents-2010.-V.13,P.293-303. 39. Okuda, H., Ozeki, S., Ikeda, S. Adsorption of Ions on Aqueous Surfaces of NaBr Solutions of Dodecyldimethylammonium Chloride //J. Colloid Interface Sci.-1987.-V.115,N.1,P.155-166. 40. Ozeki, S., Ikeda, S. The Difference In Solubilization Power between Spherical and Rodllke Micelles of Dodecyldimethylammonlum Chloride in Aqueous Solutions //J. Phys. Chem.-1985.V.89,P.5088-5093. 41. Fujio, K., Mitsui, T., Kurumizawa, H., Tanaka, Y., Uzu, Y. Solubilization of a Water-Insoluble Dye in Aqueous NaBr Solutions of Alkylpyridinium Bromides and Its Relation to Micellar Size and Shape //Colloid Polym. Sci.-2004.-V.282,P.223-229. 75 42. Ozeki, S., Ikeda, S. The Viscosity Behavior of Aqueous NaCl Solutions of Dodecyldimethylammonium Chloride and the Flexibility of Its Rod- Like Micelle //J. Colloid Interface Sci.-1980.-V.77,N.1,P.219-231. 43. Ozeki, S., Ikeda, S. The Sphere-Rod Transition of Micelles of Dodecyldimethylammonium Bromide in Aqueous NaBr Solutions, and the Effects of Counterion Binding on the Micelle Size, Shape and Structure //Colloid Polym. Sci.-1984.-V.262,P.409-417. 44. Ikeda, S., Fujio, K. The Salt-Induced Sphere-Rod Transition of Micelles of Dodecylpyridinium Iodide in Aqueous NaI Solutions //Colloid Polym. Sci.-1992.-V.270,P.1009-1017. 45. Ikeda, S. Sphere-Rod Transition of Surfactant Micelles and Size Distribution of Rodlike Micelles //J. Phys. Chem.-1984.-V.88,P.2144-2149. 46. Ikeda, S., Hayashi, S., Imae, T. Rodlike Micelles of Sodium Dodecyl Sulfate in Concentrated Sodium Halide Solutions //J. Phys. Chem.-1981.-V.85,P.106-112. 47. Israelachvili, J.N. Intermolecular and Surface Forces //3rd ed.-Elsevier Inc., 2011.-674p. 48. Crose, V., Cosgrove, T., Maitland, G., Hughes, T., Karlsson, G. Rheology, Cryogenic Transmission Electron Spectroscopy, and Small-Angle Neutron Scattering of Highly Viscoelastic Wormlike Micellar Solutions //Langmuir-2003.-V.19,P.8536-8541. 49. Raghavan, S.R., Edlund, H., Kaler, E.W. Cloud-Point Phenomena in Wormlike Micellar Systems Containing Cationic Surfactant and Salt //Langmuir-2002.-V.18,P.1056-1064. 50. Israelachvili, J.N., Mitchell, D.J., Ninham, B.W. Theory of Self- Assembly of Hydrocarbon Amphiphiles into Micelles and Bilayers //Faraday Transactions-1976.-. 51. Verbruggen, E.M.J., Hermens, J.L.M., Tolls, J. Physicochemical Properties of Higher Nonaromatic Hydrocarbons: A Literature Study //J. Phys. Chem. Ref. Data-2000.-V.9,N.6,P.14351446. 52. Tolls, J., van Dijk, J., Verbruggen, E.J.M., Hermens, J.L.M., Loeprecht, B., Schuurmann, G . Aqueous Solubility-Molecular Size Relationships: A Mechanistic Case Study Using C10- to C19Alkanes //J. Phys. Chem. A-2002.-V.106,P.2760–2765. 53. Plyasunov, A.V., Shock, E.L. Thermodynamic Functions of Hydration of Hydrocarbons at 298.15 K and 0.1 MPa //Geochimica Cosmochimica Acta-2000.-V.64,P.439–468. 54. Ferguson, A.L., Debenedetti, P.G., Panagiotopoulos, A.Z. Solubility and Molecular Conformations of n-Alkane Chains in Water //J. Phys. Chem.-2009.-V.113,P.6405-6414. 55. McAuliffe, C. Solubility in Water of C1 -C9 Hydrocarbons //Nature-1963.-V.200,P.1092-1093. 56. Franks, F. Solute-Water Interactions and the Solubility Behavior of Long-Chain Paraffin Hydrocarbons //Nature-1966.-V.210,P.87-88. 57. Tanford, C. The Hydrophobic Effect //New York, Wiley, 1973.-233p. 76 58. Xie, W.H., Shiu, W.Y., Mackay, D. A Review of the Effect of Salts on the Solubility of Organic Compounds in Seawater //Marine Environmental Research-1997.-V.44,N.4,P.429-444. 59. Ni, N., Yalkowsky, S.H. Prediction of Setschenow constants //Int. J. Pharm.-2003.- V.254,P.167–172. 60. Baldwin, R.L. How Hofmeister Ion Interactions Affect Protein Stability //Biophys. J.-1996.- V.71,P.2056. 61. Morrison, T.J. The Salting-out of Non-electrolytes. Part I. The Effect of Ionic Size, Ionic Charge, and Temperature //J. Chem. Soc.-1952.-P.3814-3818. 62. Morrison, T.J., Billet, F. The Salting-out of Non-electrolytes. Part II. The Effect of Variation in Non-Electrolyte //J. Chem. Soc.-1952.-P.3819-3822. 63. Long, F.A., McDevit, W.F. Activity Coefficients of Nonelectrolyte Solutes in Aqueous Salt Solutions //Chem. Rev.-1951.-P.119-169. 64. Mukerjee, P., Chan, C.C. Effects of High Salt Concentrations on the Micellization of Octyl Glucoside: Salting-Out of Monomers and Electrolyte Effects on the Micelle-Water Interfacial Tension //Langmuir-2002.-V.18,P.5375-5381. 65. Franchini, M.K., Carstensen, J.T. Confirmation of the Mukerjee Term in an Extended Corrin- Harkins Relation Using an Anionic Amphiphilic Drug //J. Pharm. Sci.-1996.-V.85,N.2,P.220-227. 66. Semenov, A.N. Contribution to the Theory of Microphase Layering in Block-Copolymer Melts //J. Exp. Theor. Phys.-1985.-V.61,N.4,P.733-742. 67. Onsager, L., Samaras, N.N.T. The Surface Tension of Debye-Huckel Electrolytes //J. Chem. Phys.-1934.-V.2,P.528-536. 68. Manciu, M., Ruckenstein, E. Specific Ion Effects via Ion Hydration: I.Surface Tension //Adv. Colloid Interface Sci.-2003.-V.105,P.63-101. 69. Ninham, B.W., Yaminsky, V. Ion Binding and Ion Specificity: The Hofmeister Effect and Onsager and Lifshitz Theories //Langmuir-1997.-V.13,N.7,P.2097-2108. 70. Davies, B., Ninham, B.W. Van der Waals Forces in Electrolytes //J. Chem. Phys.-1972.- V.56,N.12,P.5797-5801. 71. Netz, R.R. Static van der Waals Interactions in Electrolytes //Eur. Phys. J. E-2001.-V.5,P.189- 205. 72. Lifshitz, E.M. The Theory of Molecular Attractive Forces between Solids //Zh. Eksp. Teor. Fiz. (JETP)-1955.-V.29,P.94. 73. Dzyaloshinskii, I.E., Lifshitz, E.M., Pitaevskii, L.P. The General Theory of van der Waals Forces //Adv. Phys.-1961.-V.10:38,P.165-209. 74. Derjaguin, B.V., Dzyaloshinsky, N.E., Koptelova, M.M., Pitayevsky, L.P. Molecular-Surface Forces in Binary Solutions //Discuss. Faraday Soc.-1965.-V.40,P.246-252. 77 75. Куни, Ф.М., Русанов, А.И. Микроскопическая теория дисперсионных взаимодействий в капиллярных системах. In В книге: Русанов, А. И., Гудрич, Ф. Ч. Современная теория капиллярности. К 100-летию теории капиллярности Гиббса //Ленинград: Химия, 1980, c.162213. 76. McLachlan, A.D. Retarded Dispersion Forces in Dielectrics at Finite Temperatures //Proc. Roy. Soc. Lond. A-1963.-V.274,P.80-90. 77. Davies, B. Lifshitz Theory — Exact or Approximate Statistical Mechanics? //Phys. Lett.-1974.- V.48,N.4,P.298-300. 78. Renne, M.J., Nijboer, B.R.A. Van der Waals Interaction between Two Spherical Dielectric Particles //Chem. Phys. Lett.-1970.-V.6,N.6,P.601-604. 79. Langbein, D. Retarded Dispersion Energy between Macroscopic Bodies //Phys. Rev. B-1970.- V.2,N.8,P.3371-3383. 80. Marvin, A.M., Toigo, F. Van der Waals Interaction between a Point Particle and a Metallic Surface. I. Theory. //Phys. Rev. A-1982.-V.25,N.2,P.782-802. 81. Nabutovskii, V.M., Belosludov, V.R., Korotkikh, A.M. Interaction Potential between Small Neutral Particles and Spherical or Cylindrical Surfaces //J. Exp. Theor. Phys.-1979.-V.50,N.2,P.352355. 82. Parsegian, V.A. Long-Range Physical Forces in the Biological Milieu //Ann. Rev. Biophys. Bioeng.-1973.-V.2,P.221-255. 83. Schmeits, M., Lucas, A.A. Physical Adsorption on Small Spherical Particles and Spherical Pores //J. Chem. Phys.-1976.-V.65,P.2901-2906. 84. McLachlan, A.D. Effect of the Medium on Dispersion Forces in Liquids //Discuss. Faraday Soc.-1965.-V.40,P.239-245. 85. Renne, M.J. Microscopic Theory of Retarded Van der Waals Forces between Macroscopic Dielectric Bodies //Physica-1971.-V.56,P.125-137. 86. McLachlan, A.D. Three-Body Dispersion Forces //Mol. Phys.-1963.-V.6,N.4,P.423-427. 87. Nijboer, B.R.A. On the Many-Body Van der Waals Binding Energy of a Dense Fluid //Physica- 1975.-V.79A,P.420-432. 88. Renne, M.J., Nijboer, B.R.A. Comment on ‗Effective van der Waals Interaction between Rare Gas Atoms in a Dense Classical Fluid‘ //J. Phys. C-1973.-V.6,P.L10-L12. 89. Jungwirth, P., Winter, B. Ions at Aqueous Interfaces: From Water Surface to Hydrated Proteins //Annu. Rev. Phys. Chem.-2008.-V.59,P.343–366. 90. Petersen, P.B., Saykally, R.J. Evidence for an Enhanced Hydronium Concentration at the Liquid Water Surface //J. Phys. Chem. B-2005.-V.109,P.7976–7980. 78 91. Jungwirth, P., Tobias, D.J. Molecular Structure of Salt Solutions: A New View of the Interface with Implications for Heterogeneous Atmospheric Chemistry //J. Phys. Chem. B-2001.- V.105,P.10468-10472. 92. Manciu, M., Ruckenstein, E. On the Interactions of Ions with the Air/Water Interface //Langmuir-2005.-V.21,N.24,P.11312–11319. 93. Jungwirth, P., Tobias, D.J. Specific Ion Effects at the Air/Water Interface //Chem. Rev.-2006.- V.106,P.1259-1281. 94. Lima, E.R.A., Horinek, D., Netz, R.R., Biscaia, E.C., Tavares, F.W., Kunz, W., Boström, M. Specific Ion Adsorption and Surface Forces in Colloid Science //J. Phys. Chem. B-2008.V.112,P.1580-1585. 95. dos Santos, A.P., Diehl, A., Levin, Y. Surface Tensions, Surface Potentials, and the Hofmeister Series of Electrolyte Solutions //Langmuir-2010.-V.26,N.13,P.10778–10783. 96. Parsons, D.F., Boström, M., Maceina, T.J., Salis, A., Ninham, B.W. Why Direct or Reversed Hofmeister Series? Interplay of Hydration, Non-Electrostatic Potentials, and Ion Size //Langmuir2010.-V.26,N.5,P.3323–3328. 97. Boström, M., Williams, D.R.M., Ninham, B.W. Specific Ion Effects: Why DLVO Theory Fails for Biology and Colloid Systems //Phys. Rev. Lett. -2001.-V.87,P.168103-168111. 98. Boström, M., Williams, D.R.M., Ninham, B.W. Ion Specificity of Micelles Explained by Ionic Dispersion Forces //Langmuir-2002.-V.18,P.6010-6014. 99. Lukanov, B., Firoozabadi, A. Specific Ion Effects on the Self- Assembly of Ionic Surfactants: A Molecular Thermodynamic Theory of Micellization with Dispersion Forces //Langmuir-2014.V.30,P.6373−6383. 100. Koroleva, S.V., Victorov, A.I. Modeling of the Effects of Ion Specificity on the Onset and Growth of Ionic Micelles in a Solution of Simple Salts //Langmuir-2014.-V.30,P.3387-3396. 101. Koroleva, S.V., Victorov, A.I. The Strong Specific Effect of Coions on Micellar Growth from Molecular-Thermodynamic Theory //Phys. Chem. Chem. Phys.-2014.-V.16,P.17422-17425. 102. Skjold-Jørgensen, S. Gas Solubility Calculations. II. Application of a New Group-Contribution Equation of State //Fluid Phase Equilib.-1984.-V.16,P.317-351. 103. Lue, L., Zoeller, N., Blankschtein, D. Incorporation of Nonelectrostatic Interactions in the Poisson-Boltzmann Equation //Langmuir-1999.-V.15,P.3726-3730. 104. Andreev, V.A., Victorov, A.I. Molecular Thermodynamics for Micellar Branching in Solutions of Ionic Surfactants //Langmuir-2006.-V.22,P.8298-8310. 105. Русанов, А.И. Мицеллообразование в растворах поверхностно-активных веществ //Санкт-Петербург, Химия, 1992.-280с. 79 106. May, S., Ben-Shaul, A. Ch. 2 Molecular Packing in Cylindrical Micelles. In Zana, R., Kaler, E. W. Giant Micelles. Properties and Applications. //CRC Press: Taylor & Francis Group, 2007. 107. Stigter, D. Micelle Formation by Ionic Surfactants. II. Specificity of Head Groups, Micelle Structure //J. Phys. Chem.-1974.-V.78,N.24,P.2480-2485. 108. Srinivasan, V., Blankschtein, D. Prediction of Conformational Characteristics and Micellar Solution Properties of Fluorocarbon Surfactants //Langmuir-2005.-V.21,N.4,P.1647-1660. 109. Magid, L.J. The Surfactant-Polyelectrolyte Analogy //J. Phys. Chem. B-1998.-V.102,P.4064- 4074. 110. Anacker, E.W., Ghose, H.M. Counterions and Micelle Size. II. Light Scattering by Solutions of Cetylpyridinium Salts //J. Am. Chem. Soc.-1968.-V.90,N.12,P.3161-3166. 111. Goddard, E.D., Harva, O., Jones, T.G. The Effect of Univalent Cations on the Critical Micelle Concentration of Sodium Dodecyl Sulphate //Trans. Faraday Soc.-1953.-V.49,P.980-984. 112. Mukerjee, P., Ray, A. The Specificity of Counterion Adsorption to Micelles of Dodecylpyridinium Iodide and Their Critical Concentrations //J. Phys. Chem.-1966.-V.70,N.7,P.21502157. 113. Jones, M.N., Piercy, J. Light Scattering Studies on n-Dodecyltrimethylammonium Bromide and n-Dodecylpyridinium Iodide //J. Chem. Soc., Faraday Trans. 1-1972.-V.68,P.1839-1848. 114. Fujio, K., Maruyama, Y., Uzu, Y. Degree of Counterion Binding of Alkylpyridinium Halide Micelles: Its Relation to Micelle Shape and Size //J. Oleo Sci.-2005.-V.54,N.7,P.375-382. 115. Gonzalez-Perez, A., Varela, L.M., Garcia, M., Rodriguez, J.R. Sphere-to-Rod Transitions in Homologous Alkylpyridinium Salts: A Stauff-Klevens-Type Equation for the Second Critical Micelle Concentration //J. Colloid Interface Sci.-2006.-V.293,P.213–221. 116. Lee, H.S., Arunagirinathan, M.A., Vagias, A., Lee, S., Bellare, J.R., Davis, H.T., Kaler, E.W., McCormick, A.V., Bates, F.S. Almost Fooled Again – New Insights into CsDS Micelle Structures //Langmuir-2014.-V.30,N.43,P.12743–12747. 117. Mukerjee, P., Ray, A. Charge- Transfer Interactions and the Polarity at the Surface of Micelles of Long-chain Pyridinium Iodides //J. Phys. Chem.-1966.-V.70,N.7,P.2144-2149. 118. Harada, M., Satou, H., Okada, T. Hydration Structures of Bromides on Cationic Micelles //J. Phys. Chem. B-2007.-V.111,N.42,P.12136-12140. 119. Weican, Z., Ganzuo, L., Jianhai, M., Qiang, S., Liqiang, Z., Haojun, L., Chi, W. Effect of KBr on the micellar properties of CTAB //Chin. Sci. Bull.-2000.-V.45,N.20,P.1854-1857. 120. Imae, T., Kamiya, R., Ikeda, S. Formation of Spherical and Rod- like Micelles of Cetyltrimethylammonium Bromide in Aqueous NaBr Solutions //J. Colloid Interface Sci.-1985.V.108,N.1,P.215-225. 80 121. Khatory, A., Lequeux, F., Kern, F., Candau, S.J. Linear and Nonlinear Viscoelasticity of Semidilute Solutions of Wormlike Micelles at High Salt Content //Langmuir-1993.-V.9,P.1456-1464. 122. Missel, P.J., Mazer, N.A., Benedek, G.B., Carey, M.C. Influence of Chain Length on the Sphere-to-Rod Transition in Alkyl Sulfate Micelles //J. Phys. Chem.-1983.-V.87,P.1264-1277. 123. Liu, Y.C., Ku, C.Y., LoNostro, P., Chen, S.H. Ion Correlations in a Micellar Solution Studied by Small-Angle Neutron and X-Ray Scattering //Physical Review E-1995.-V.51,N.5,P.4598-4607. 124. Smith, L.A., Hammond, R.B., Roberts, K.J., Machin, D., McLeod, G. Determination of the Crystal Structure of Anhydrous Sodium Dodecyl Sulphate Using a Combination of Synchrotron Radiation Powder Diffraction and Molecular Modelling Techniques //J. Mol. Struct.-2000.V.554,P.173-182. 125. Rodrı´guez-Pulido, A., Casado, A., Munoz-Ubeda, M., Junquera, E., Aicart, E. Experimental and Theoretical Approach to the Sodium Decanoate-Dodecanoate Mixed Surfactant System in Aqueous Solution //Langmuir-2010.-V.26,N.12,P.9378–9385. 126. Aquan-Yuen, M., Mackay, D., Shiu, W.Y. Solubility of Hexane, Phenanthrene, Chlorobenzene, and p-Dichlorobenzene in Aqueous Electrolyte Solutions //J. Chem. Eng. Data-1979.-V.24,N.1,P.3034. 127. Emerson, M.F., Holtzer, A. On the Ionic Strength Dependence of Micelle Number. II //J. Phys. Chem.-1967.-V.71,N.6,P.1898-1907. 128. Nomura, H., Koda, S., Matsuoka, T., Hiyama, T., Shibata, R., Kato, S. Study of Salt Effects on the Micelle–Monomer Exchange Process of Octyl-, Decyl-, and Dodecyltrimethylammonium Bromide in Aqueous Solutions by Means of Ultrasonic Relaxation Spectroscopy //J. Colloid Interface Sci. 2000.-V.230,P.22-28. 129. Imae, T., Ikeda, S. Sphere-Rod Transition of Micelles of Tetradecyltrimethylammonium Halides in Aqueous Sodium Halide Solutions and Flexibility a nd Entanglement of Long Rodlike Micelles //J. Phys. Chem.-1986.-V.90,P.5216-5223. 130. Holmberg, K., Jönsson, B., Kronberg, B., Lindman, B. Surfactants and Polymers in Aqueous Solutions //Chichester, UK, John Wiley and Sons, Ltd., 1999.-545p. 131. Imae, T., Ikeda, S. Characteristics of Rodlike Micelles of Cetyltrimethylammonium Chloride in Aqueous NaCI Solutions: Their Flexibility and the Scaling Laws in Dilute and Semidilute Regimes //Colloid Polym. Sci.-1987.-V.265,P.1090-1098. 132. Kuperkar, K., Abezgauz, L., Danino, D., Verma, G., Hassan, P.A., Aswal, V.K., Varade, D., Bahadur, P. Viscoelastic micellar water/CTAB/NaNO 3 solutions: Rheology, SANS and cryo-TEM analysis //J. Colloid Interface Sci.-2008.-V.323,P.403-409. 81 133. Maeda, H., Ozeki, S., Ikeda, S., Okabayashi, H., Matsushita, K. Carbon-13 Nuclear Magnetic Resonance Study of Dodecyldimethylammonium Chloride Solutions //J. Colloid Interface Sci.-1980.V.76,N.2,P.532-540. 134. Chung, M.I., Tak, I.J., Lee, K.M. Light Scattering Studies on the Second CMC of the Aqueous Solution of Dodecylpyridinium Chloride and Tetradecylpyridinium Chloride //J. Korean Chem. Soc. 1975.-V.19,N.6,P.398-402. 135. Causi, S., Lisi, R.D., Milioto, S. Thermodynamic Properties of N-octyl-, N-decyl- and N- dodecylpyridinium Chlorides in Water //J. Solution Chem.-1991.-V.20,N.11,P.1031-1058. 136. Fujio, K., Ikeda, S. Size of Spherical Micelles of Dodecylpyridinium Bromide in Aqueous NaBr Solutions //Langmuir-1991.-V.7,N.12,P.2899-2903. 137. Škerjanc, J., Kogej, K., Cerar, J. Equilibrium and Transport Properties of Alkylpyridinium Bromides //Langmuir-1999.-V.15,N.15,P.5023-5028. 138. Youn, Y.W., Lee, K.M. Viscosity and Density Studies on the Second CMC of the Aqueous Solution of Dodecyl Pyridinium Chloride //J. Korean Chem. Soc.-1975.-V.19,N.5,P.289-293. 139. Porte, G., Appell, J., Poggi, Y. Experimental Investigations on the Flexibility of Elongated Cetylpyridinium Bromide Micelles //J. Phys. Chem.-1980.-V.84,P.3105-3110. 140. Varade, D., Joshi, T., Aswal, V.K., Goyal, P.S., Hassan, P.A., Bahadur, P. Effect of Salt on the Micelles of Cetylpyridinium Chloride //Colloids Surf., A-2005.-V.259,P.95-101. 141. Vlasov, A.Y., Savchuk, K.R., Starikova, A.A., Smirnova, N.A. Effects of Counterion Specificity in Salt-Aqueous Solutions of Dodecylsulfates of Alkali Metals //Zhidk. Krist. Ikh Prakt. Ispol'z.-2011.-V.4,N.38,P.90–102. 142. Mukerjee, P., Mysels, K., Kapauan, P. Counterion Specificity in the Formation of Ionic Micelles - Size, Hydration, and Hydrophobic Bonding Effects //J. Phys. Chem.-1967.V.71,N.13,P.4166-4175. 143. Bendedouch, D., Chen, S.H., Koehler, W.C. Determination of Interparticle Structure Factors in Ionic Micellar Solutions by Small Angle Neutron Scattering //J. Phys. Chem. -1983.-V.87,P.26212628. 144. Hayashi, S., Ikeda, S. Micelle Size and Shape of Sodium Dodecyl Sulfate in Concentrated NaCl Solutions //J. Phys. Chem.-1980.-V.84,N.7,P.744-751. 145. Nguyen, D., Bertrand, G.L. Calorimetric Observations of the Sphere-Rod Transition of Sodium Dodecyl Sulfate: Effects of Electrolytes and Nonelectrolytes at 25 O C //J. Phys. Chem.-1992.V.96,P.1994-1998. 146. Corrin, M.L., Harkins, W.D. The Effect of Salts on the Critical Concentration for the Formation of Micelles in Colloidal Electrolytes //J. Am. Chem. Soc.-1947.-V.69,N.3,P.683-688. 82 147. Herzfeld, S.H. Critical Concentrations of Potassium n-Alkanecarboxylates as Determined by the Change in Color and Spectrum of Pinacyanole //J. Phys. Chem.-1952.-V.56,P.953–959. 148. Herzfeld, S.H. Effect of Salt on the Critical Concentrations of Potassium n-Alkanecarboxylates as Determined by the Change in Color of Pinacyanole //J. Phys. Chem. -1952.-V.56,P.959-963. 149. Huang, J.-B., Mao, M., Zhu, B.-Y. The Surface Physico-Chemical Properties of Surfactants in Ethanol–Water Mixtures //Colloids Surf., A-1999.-V.155,P.339-348. 150. Maczynski, A., Shaw, D.G., Goral, M., Wisniewska-Goclowska, B., Skrzecz, A., Owczarek, I., Blazej, K., Haulait-Pirson, M.-C., Hefter, G.T., Kapuku, F., Maczynska, Z., Young, C.L. IUPAC-NIST Solubility Data Series. 81. Hydrocarbons with Water and Seawater—Revised and Updated. Part 4. C6H14 Hydrocarbons with Water //J. Phys. Chem. Ref. Data -2005.-V.34,P.709 (745 pages). 151. Mukerjee, P. Salt Effects on Nonionic Association Colloids //J. Phys. Chem.-1965.- V.69,P.4038-4040. 83 Приложение 1. Ссылки на экспериментальные данные о критической концентрации мицеллообразования, солевых порогах перехода сфера-цилиндр и числах агрегации ККМ Система Числа агрегации Переходы сферацилиндр Ссылки Катионные ПАВ n-алкилтриметиламмоний-X [36, 38, 113, Cn TAX+NaX 120, 127-131] (X= Cl¯, Br¯, OH¯, NO3 ¯, n=12,14,16) n-алкилдиметиламмоний-X Сn DAX+NaX [39, 133] (X= Cl¯, Br¯, n=12) n-алкилпиридиний-X [36, 41, 44, Cn PX+NaX 110, 112, 114(X= Cl¯, Br¯, I¯, n=10,12,14,16) 115, 134-138] эруцилбис(гидроксиэтил)метиламмоний хлорид EHAC+XCl + + (X= Na , K , n=22) Анионные ПАВ [21, 24, 28, 47, X-додецилсульфат 116, 122, 127, XDS+XCl (X= Li+, Na+, K+, Cs+, n=12) 141-144] X-алкилкарбоксилат [24, 107, 130, Cn COOX+XCl 146-149] (X= Na+, K+, n=9,11) [37-38, 113, 119-120, 127129, 131-132] [37-38, 43, 119-120, 129, 131-132] [42-43] [43] [41, 44, 110, 114, 136-137, 139-140] [41, 136, 138140] [48-49] [48-49] [24, 28, 46, 122, 127, 141, 143144] [116, 145] - - Приложение 2. Дисперсионное взаимодействие иона со сферическим и цилиндрическим агрегатами Энергия дисперсионного взаимодействия атома с макроскопической частицей (сферой, цилиндром, плоскостью) в вакууме может быть записана как разложение по кривизне частицы [81] (2.1) Здесь – дисперсионный потенциал атома вблизи плоской поверхности, поправка на кривизну, кривизны , для плоскости, цилиндра и сферы, соответственно; – – радиус – расстояние между атомом и поверхностью частицы. Мы используем уравнение 2.1 для описания дисперсионного взаимодействия в воде. Эффект среды может быть учтен введением частотно-зависимой диэлектрической функции [47], для и . Для малых расстояний без учета запаздывания имеем , в выражениях 84 (2.2) (2.3) где – избыточная поляризуемость иона в среде, а – диэлектрические функции углеводородного ядра и воды, соответственно, при характеристической мнимой частоте остальных , – постоянная Планка; для и при всех . Комбинируя уравнения 2.1-2.3, получаем (2.4) Второе слагаемое в скобках – поправка на кривизну, а множитель перед скобками совпадает с общим членом суммы в выражении силовой константы для плоского случая [3, 12] (2.5) В приближении одночастотного гармонического осциллятора для диэлектрических функций и имеем [47] (2.6) где – главная частота поглощения; (углеводород, вода), , – показатель преломления диэлектрика для углеводородов и , для воды [47]. Поправка на кривизну в уравнении 2.4 будет (2.7) Подставляя уравнение 2.7 в уравнение 2.4, получаем уравнение, которое содержит первые два вклада разложения по кривизне такого выражения (2.8) Таким образом, для сферического и цилиндрического агрегатов дисперсионный потенциал дается выражением (2.9) где дисперсионная константа рассчитывается для плоскости, уравнение 2.5. 85 Приложение 3. Свободная энергия, энтропия и химические потенциалы для смеси твердых сфер Плотность свободной энергии смеси твердых сфер дается выражением [102] (3.1) где (3.2) есть плотность свободной энергии идеального газа, а – плотность остаточной энтропии, связанной с исключенным объемом во флюиде твердых сфер. Последняя плотность рассчитывается в рамках теории БМКСЛ [102] (3.3) , (3.4) (3.5) Здесь – длина тепловой волны де Бройля и – твердосферный диаметр иона . Для химического потенциала сфер имеем (3.6) где первое слагаемое – вклад идеального газа, а остаточная часть, , дается выражением (3.7) Здесь , плотности ; штрихи означают частные производные относительно частичной . Приложение 4. Экспериментальное определение растворимости н-гексана и констант Сеченова в растворах солей NaCl, NaBr, NaNO3 , KCl Настоящее приложение содержит результаты экспериментального определения растворимости н-гексана и констант Сеченова в растворах неорганических 1:1 электролитов. Метод и материалы. Чистота н-гексана и солей NaCl, NaBr, NaNO 3 , KCl составляла >99%; чистота н-октана составляла >99.99%. Все реактивы использовались без дальнейшей очистки и подготовки. 86 Растворы солей приготовлялись в дважды дистиллированной воде. Аликвоты углеводородов и растворов солей помещались в стеклянные емкости (~20мл), которые герметично закрывались алюминиевыми крышками с отверстиями, с вложенными в них политетрафторэтиленовыми прокладками (для предотвращения потери летучих компонентов раствора). Для отбора проб использовались стеклянные шприцы различного объема с металлическими иглами. Измерения оборудованного выполнялись неполярной с помощью капиллярной газового колонкой хроматографа Agilent (30м×530μм×1μм, 6890A, HP-INNOWax полиэтиленгликоль, Agilent 19095N-123) и пламенно-ионизационным детектором. Средняя скорость потока газов (смеси N 2 , H2 и воздуха) через колонку составляла 34.5мл/мин; температуры колонки, детектора и инжектора составляли 100, 250 и 200o C, соответственно. Газовый хроматограф был откалиброван с помощью стандартных растворов н-октана, содержащих аналит (н-гексан) в области концентраций, ожидаемых в исследуемых образцах. Все анализы выполнялись в области линейного отклика детектора. Линейность отклика детектора проверялась периодически. Аликвота ~18мл одного из приготовленных растворов солей и аликвота н-гексана ~1.52мл помещались в стеклянную емкость с крышками и закрывалась герметично. Для ускорения достижения равновесия в системе в течение по меньшей мере недели растворы взбалтывались вручную несколько раз в день в течение 1 минуты и помещались обратно в термостат при 25o C, а после последнего взбалтывания оставлялись в термостате самое малое на 24ч до окончательного расслоения фаз раствора. В термостате емкости находились вниз крышками, чтобы позволить провести дальнейший отбор пробы без контакта с н-гексаном. После насыщения водно-солевых растворов гексаном из склянок в нижнем положении путем прокалывания тефлоновой прокладки отбирались аликвоты (~16мл) водной фазы. Масса аликвоты фиксировалась на весах после ее переноса в новую стеклянную емкость с октаном известной массы (объем ~2мл), необходимым для экстракции растворенного гексана. Следует отметить, что в процессе экстракции растворенного гексана из водной фазы объемом ~16мл в октановую объемом ~2мл происходит его концентрирование ~8раз, что желательно в связи с необходимостью измерения низких значений растворимости углеводородов. Для минимизации потерь гексана ввиду его летучести, перенос содержимого шприца производился под слоем октана; после переноса раствора склянка быстро и герметично закрывалась алюминиевыми крышками с тефлоновыми прокладками. Для каждой пробы использовался новый шприц. Стеклянные емкости с октаном также периодически подвергались ручному взбалтыванию и оставлялись в термостате при 25oC на 4-5 дней до полного расслоения фаз. 87 Рисунок 4.1. Зависимость логарифма растворимости н-гексана [моль/л] от концентрации солей: черные кружки – NaCl; красные крестики – KCl; зеленые квадраты – NaBr; синие звездочки – NaNO 3; линии – линейная аппроксимация экспериментальных данных методом наименьших квадратов; экспериментальные данные (а) настоящей работы; (б) работы [126]. Октановые пробы с экстрагированным и сконцентрированным гексаном помещались в небольшие стеклянные емкости (~2мл) с пластиковыми крышками с тефлоновыми прокладками, через которые с помощью микролитровых шприцев отбирались пробы (~2μл) для хроматографического анализа. Результаты измерений Значение растворимости гексана в воде, измеренное в настоящей работе logS=-3.92 ( , попадает в интервал экспериментальных значений logS=-(3.96’3.81), полученных ранее другими методами [51, 150]. Результаты измерения растворимости н-гексана в растворах различных солей (NaCl, KCl, NaBr, NaNO3 ) при разных концентрациях показаны на рисунке 4.1а. Как видно, полученные в настоящей работе зависимости логарифма растворимости от концентрации соли могут быть удовлетворительно аппроксимированы линейными представляет собой константу высаливания функциями, угол наклона которых . Таким образом, логарифм растворимости гексана линейно убывает вплоть до высоких концентраций соли (~3М), что наблюдалось и в более ранних экспериментах [126], поскольку для слабо растворимых веществ (какими являются углеводороды) чаще всего линейность логарифма растворимости соединения соблюдается вплоть до высоких концентраций соли [63]. Для сравнения на рисунке 4.1б изображены экспериментальные данные работы [126] в присутствии солей NaCl, KCl и NaBr до концентраций2.5М. Видно, что в общем растворимость гексана, измеренная в настоящей работе, ниже, чем измеренная в работе [126]. Заниженные значения растворимости гексана 88 Таблица 4.1. Величины наклонов кривых могут logS – Cs. концентрированием гексана в октане. Для более значительного *, л/моль (эксп.) Соль Настоящая работа быть связаны с недостаточным концентрирования необходимо увеличить объемы аликвот солевых растворов с Работа[126] растворенным гексаном. В таблице 4.1 NaCl 0.30 0.27 сравниваются значения тангенсов углов наклона KCl 0.29 0.27 (константы Сеченова), измеренные в настоящей NaBr 0.25 0.16 NaNO3 0.21 - *) работе и в работе [126]. По результатам работы [62] Мукерджи Для всех измерений погрешность определения [151] было найдено выражение для константы составляет порядка 0.01 л/моль (метод наименьших квадратов). Сеченова в растворе NaCl, применимое для C1-C4 углеводородов: (4.1) При =6 уравнение 4.1 дает =0.29л/моль, что ближе к результату настоящей работы для NaCl, чем к результату работы [126]. Для было также получено выражение, применимое для C3-C6 углеводородов [64]: (4.2) где и – число групп и в углеводороде. В соответствии с уравнением 4.2 =0.30л/моль для гексана в водном растворе NaCl, что согласуется с результатом настоящей работы. В соответствии с данными настояшей работы для NaCl и KCl почти неразличимы, как и в работе [126], таблица 4.1. В настоящей работе NaCl высаливает гексан немного сильнее, чем KCl (т.е. катионы могут быть упорядочены в ряд Гофмейстера по уменьшению эффекта высаливания Na+>K+), что качественно согласуется с данными работы [62] для бутана. В соответствии с измеренными данными настоящей работы анионы можно расположить в следующей последовательности Гофмейстера в порядке уменьшения эффекта высаливания Cl-> Br->NO 3 -, что совпадает с последовательностью Гофмейстера, наблюдаемой при высаливании бензола [63]. В пересчете на один углеродный атом измеренные в настоящей работе константы высаливания составляют -0.050, -0.048, -0.042 и -0.035л/моль для NaCl, KCl, NaBr и NaNO3 (ср. с величинами параметра , используемого при расчетах с помощью предлагаемой модели: -0.05, -0.04, -0.03 и -0.03л/моль для соответствующих солей).