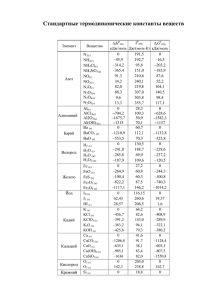
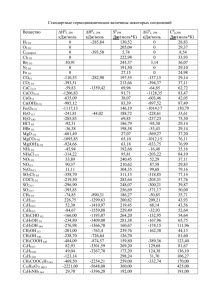
МОЛЕКУЛЯРНОЕ МОДЕЛИРОВАНИЕ И КВАНТОВО- ХИМИЧЕСКИЕ РАСЧЁТЫ В ИЗУЧЕНИИ ПРОЦЕССОВ НЕФТЕПЕРЕРАБОТКИ И НЕФТЕХИМИИ
advertisement

На правах рукописи ЛЮБИМЕНКО ВАЛЕНТИНА АЛЕКСАНДРОВНА МОЛЕКУЛЯРНОЕ МОДЕЛИРОВАНИЕ И КВАНТОВОХИМИЧЕСКИЕ РАСЧЁТЫ В ИЗУЧЕНИИ ПРОЦЕССОВ НЕФТЕПЕРЕРАБОТКИ И НЕФТЕХИМИИ 05.17.07 – Химическая технология топлива и высокоэнергетических веществ АВТОРЕФЕРАТ диссертации на соискание ученой степени доктора химических наук Москва – 2015 Работа выполнена в ФГБОУ ВПО «Российский государственный университет нефти и газа имени И. М. Губкина» Научный консультант: доктор химических наук, профессор Винокуров Владимир Арнольдович Официальные оппоненты: Немухин Александр Владимирович доктор химических наук, профессор ФГБОУ ВО «МГУ имени М.В. Ломоносова», химический факультет, кафедра физической химии, заведующий лабораторией химической кибернетики Орешенков Александр Владимирович доктор технических наук ФАУ «25 ГосНИИ химмотологии Минобороны России», ведущий научный сотрудник лаборатории физических методов исследования Хавкин Всеволод Артурович доктор технических наук, профессор ОАО «Всероссийский научноисследовательский институт по переработке нефти», заместитель генерального директора Ведущая организация: ФГБУН Институт нефтехимического синтеза им. А. В. Топчиева Российской академии наук (ИНХС РАН) Защита состоится «28» января 2016 г. в 14.00 в аудитории № 541 на заседании диссертационного совета Д 212.200.04 при ФГБОУ ВПО «Российский государственный университет нефти и газа им. И. М. Губкина» по адресу:119991, Москва, ГСП-1, Ленинский проспект, 65 С диссертацией можно ознакомиться в научно-технической библиотеке ФГБОУ ВПО «Российский государственный университет нефти и газа им. И. М. Губкина» и на сайте http://gubkin.ru Автореферат разослан «8 » октября 2015 г. Ученый секретарь диссертационного совета, кандидат технических наук, доцент Л. Ф. Давлетшина 2 ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ Актуальность темы. Современная наука располагает мощными инструментами проведения исследований физико-химических процессов – методами компьютерной химии, которые позволяют оценивать направление протекания химических реакций, рассчитывать распределение зарядов в молекулах, определять равновесную геометрию сложных молекул и молекулярных комплексов, вычислять энергию межмолекулярных взаимодействий, оказывающих решающее влияние на результаты процессов нефтепереработки. Однако методы компьютерной химии до сих пор не находили широкого применения для изучения физико-химических процессов, протекающих при добыче и переработке углеводородного сырья, а также в нефтехимии. Добываемое углеводородное сырье – это сложная смесь химических соединений различной структуры и состава. Выделение из нефти, газоконденсата, битума индивидуальных компонентов и экспериментальное изучение их превращений, взаимодействий с другими компонентами в ходе процессов нефтегазодобычи и переработки представляет значительные трудности. Поэтому актуальным является применение вычислительных методов компьютерной химии, не требующих затрат на разделение смесей, на экспериментальное оборудование и реактивы для проведения исследований. Так, неэмпирические и полуэмпирические квантово-химические методы позволяют проводить расчеты параметров, которые сложно, а в некоторых случаях невозможно, определить в ходе экспериментов. К таким параметрам относятся, например, энтальпия образования, энтропия, теплоёмкость многих сложных гетероатомных химических соединений, присутствующих в нефти и битумах, дипольные моменты (не для всех известных соединений их можно найти в справочной литературе), энергия взаимодействия молекул в однокомпонентных и многокомпонентных системах. Актуальность темы определяется также тем, что применение методов компьютерной химии для изучения механизма физико-химических процессов, протекающих при добыче и переработке нефти, значительно ускоряет получение результатов, по сравнению с экспериментальными исследованиями, часто не уступая им в точности. Это даёт возможность сократить время, необходимое для оптимального планирования экспериментов, проверки и подтверждения результатов расчётов, а затем и для принятия решений о направлении совершенствования технологий в нефтегазодобывающей и перерабатывающей промышленности. Актуальным также является компьютерное изучение механизма действия присадок различного назначения к топливам, поскольку процессы, протекающие в их присутствии, до сих пор остаются неизученными, а подбор и создание присадок к дизельным топливам и бензинам осуществляется на эмпирическом уровне. Понимание механизма действия присадок различной функциональности в топливах позволит прогнозировать их влияние на эксплуатационные характеристики топлив и целенаправленно разрабатывать присадки для придания топливам эксплуатационных свойств, отвечающих современным требованиям. Целью работы является создание нового направления в исследовании физико-химических явлений, сопровождающих процессы нефтепереработки и 3 нефтехимии, основанного на применении методов молекулярного моделирования и квантово-химических расчётов. Для реализации поставленной цели решены следующие задачи: Рассчитаны термодинамические характеристики химических реакций, которые могут протекать в условиях паротепловой обработки пластов при добыче тяжёлых нефтей и битумов в интервале температур 298-800К, по стандартным значениям энтальпий образования и энтропий веществ, участвующих в реакциях, вычисленным полуэмпирическим квантово-химическим методом PM6. Методом Меллера-Плессе MP2/6-311++G(d, p) рассчитана энергия межмолекулярных взаимодействий бензола с другими ароматическими углеводородами, присутствующими в узких бензиновых фракциях, а также энергия межмолекулярных взаимодействий индивидуальных ароматических углеводородов. Рассчитана энергия межмолекулярных взаимодействий лёгких меркаптанов с ароматическими углеводородами узких бензиновых фракций методом MP2/6311++G(d, p). Полуэмпирическим методом PM6 рассчитана энтальпия образования межмолекулярных комплексов депрессорно-диспергирующих присадок к дизельным топливам с входящими в состав этих топлив парафиновыми углеводородами. Рассчитаны (методом PM6) значения энергий возбуждения в низшее триплетное состояние компонентов композиционной присадки к дизельным топливам. Неэмпирическими квантово-химическими методами рассчитана энергия взаимодействия диспергирующих присадок на основе сукцинимида и малеинимида с ароматическими углеводородами, присутствующими в дизельных топливах (на примере бензола). Осуществлено компьютерное моделирование и квантово-химические расчёты механизма антидетонационного действия присадок к бензинам на основе ароматических аминов. Получены результаты молекулярного моделирования и квантовохимических расчётов взаимодействия инициатора горения дизельных топлив с УВ, входящими в состав этих топлив. Выведено уравнение для расчёта цетанового числа ДТ в зависимости от концентрации инициатора горения и цетанового числа исходного топлива. Создано уравнение зависимости периода индукции при окислении бензиновой фракции от концентрации антиокислительной присадки 2,4-ди-трет-бутил6-метилфенола. Рассчитано распределение спиновой плотности в 2,6-ди-третбутилфеноксидном радикале полуэмпирическим методом PM6. Осуществлено компьютерное моделирование взаимодействия катализаторов – алкилхлорсиланов – с пропиленом. Получены результаты экспериментов по каталитическим превращениям метилбензолов, риформингу бензиновой фракции и крекингу н-гексадекана. Научная новизна работы. Научная новизна работы заключается в создании нового направления – изучение механизма физико-химических явлений в процессах нефтепереработки и нефтехимии методами компьютерной химии. Этому на4 правлению можно дать следующее название: «Молекулярное моделирование и квантово-химические расчёты в изучении процессов нефтепереработки и нефтехимии». Главные научные достижения, полученные в диссертации, включают следующие конкретные результаты проведённых исследований. Впервые осуществлены расчёты термодинамики реакций акватермолиза компонентов тяжёлых нефтей и выявлены условия их протекания. Впервые установлен механизм наложения узких бензиновых фракций друг на друга на основе расчётов энергии взаимодействия бензола с ароматическими углеводородами. Впервые создано параметрическое уравнение, связывающее температуры кипения узких бензиновых фракций с их плотностью. Впервые рассчитаны энергии взаимодействия лёгких меркаптанов с ароматическими углеводородами (УВ) прямогонных бензиновых фракций, установлены закономерности распределения низкокипящих меркаптанов между узкими бензиновыми фракциями. Полуэмпирическими квантово-химическими методами впервые выявлен механизм действия депрессорно-диспергирующих присадок к дизельным топливам (ДТ) на их агрегативную и седиментационную устойчивость. Впервые определены структуры межмолекулярных комплексов парафиновых УВ с присадками и рассчитаны тепловые эффекты взаимодействия компонентов депрессорнодиспергирующих присадок с углеводородными соединениями ДТ. Впервые создано уравнение для описания зависимости цетанового числа ДТ от концентрации в нём инициатора горения и проведён квантово-химический расчёт механизма влияния аналогичных присадок на цетановое число ДТ. Впервые разработан метод определения стабильности присадок к ДТ по значениям энергий их возбуждения в низшее триплетное состояние. Впервые создано параметрическое уравнение для расчёта периода индукции окисления бензинов в присутствии антиокислительной присадки в зависимости от ее концентрации и дано квантово-химическое обоснование механизма действия присадки АГИДОЛ-1 на качество бензинов. Впервые молекулярным моделированием и квантово-химическими расчётами выявлен механизм антидетонационного действия присадок к бензинам на основе ароматических аминов. Впервые осуществлены компьютерное моделирование и квантовохимические расчёты структуры и состояния алкилхлорсиланов, определён механизм изменения их каталитической активности в реакции алкилирования бензола пропиленом. Впервые квантово-химическими расчётами выявлены закономерности каталитического превращения метилбензолов в присутствии цеолиталюмосиликатных катализаторов, Практическая ценность работы. Разработано новое направление исследования физико-химических явлений, возникающих при переработке углеводородного сырья, на основе применения молекулярного моделирования и квантовохимических расчётов. В рамках разработанного направления можно моделиро5 вать структуру индивидуальных соединений и межмолекулярных комплексов, образующихся из компонентов, входящих в состав топлив и других нефтепродуктов, рассчитывать их термодинамические характеристики и энергию межмолекулярных взаимодействий. Результаты расчётов позволяют прогнозировать поведение присадок различного функционального назначения как при их индивидуальном присутствии в топливах, так и в смеси с другими присадками. Методы молекулярного моделирования и квантово-химические расчёты, используемые в рамках разработанного нового направления исследований, дают возможность изучать механизмы явлений в процессах нефтепереработки, механизмы превращений веществ в нефтехимии на молекулярном уровне, что обычно недоступно из-за сложности состава нефти и нефтепродуктов, а также трудностей выделения из них индивидуальных соединений. На основе выявленного механизма наложения составов узких бензиновых фракций друг на друга предложен метод снижения содержания бензола в риформате, который применен на ОАО «Московский НПЗ» и ОАО «Новокуйбышевский НПЗ». Уравнение, связывающее температуру кипения узких бензиновых фракций с их плотностью, может быть использовано для контроля за процессом. По полученным данным о механизме распределения меркаптанов между узкими бензиновыми фракциями разработан метод снижения содержания меркаптановой серы в риформате. Этот метод применяется на ОАО «Новокуйбышевский НПЗ». На основе установленного квантово-химическими методами механизма действия депрессорно-диспергирующих присадок к дизельным топливам предложен метод подбора эффективных комплексных присадок. Уравнение зависимости цетанового числа дизельного топлива от концентрации инициатора горения с экспериментальной точностью позволяет определять необходимую концентрацию этой присадки без проведения испытаний, что может быть использовано в НИИ и на НПЗ России. Метод определения стабильности присадок к ДТ может быть использован для выбора присадок, обладающих оптимальной стабильностью. Параметрическое уравнение, связывающее период индукции окисления бензинов в присутствии антиокислительной присадки в зависимости от её концентрации, позволяет быстро оценивать концентрацию антиокислителя, обеспечивающую необходимую стабильность топлива. Это уравнение было использовано в Сирийской Арабской Республике для расчёта периода индукции бензинов, хранящихся в подземных хранилищах. Компьютерное моделирование и квантово-химические расчёты структуры и состояния алкилхлорсиланов в реакции алкилирования бензола пропиленом позволили выявить механизм реакции и выбрать наиболее эффективный катализатор. Неэмпирическим квантово-химическим методом Хартри–Фока установлен механизм каталитического превращения метилбензолов в присутствии цеолиталюмосиликатных катализаторов. 6 Достоверность полученных результатов расчётов определяется их соответствием экспериментальным и литературным данным. Личное участие автора. Автором выполнены постановка задач, создание моделей изучаемых молекулярных систем, квантово-химические расчёты и анализ полученных результатов. Апробация работы. Основные положения и результаты диссертационной работы доложены и обсуждены на XVIII Международном Конгрессе по химической технологии (18th International Congress of Chemical and Process Engineering, 24-28 August 2008. Praha, Czech Republic), на XV Симпозиуме по межмолекулярному взаимодействию и конформациям молекул (14-18 июня 2010 г. в г. Петрозаводске), на VI международной научно-технической конференции «Глубокая переработка нефтяных дисперсных систем» (16 декабря 2011 г., Москва), на VIII Всероссийской научно-технической конференции «Актуальные проблемы развития нефтегазового комплекса России» (1-3 февраля 2010 г., Москва), на международной научно-практической конференции «Высоковязкие нефти и природные битумы: проблемы и повышение эффективности разведки и разработки месторождений» 5-7 сентября 2012 года, на XXI Международном Конгрессе по химической технологии (21th International Congress of Chemical and Process Engineering, 23-27 August 2014. Praha, Czech Republic), на III Международной научно-практической конференции «Научные перспективы XXI века. Достижения и перспективы нового столетия», Россия, г. Новосибирск, 15-16.08.2014 г. Публикации. По результатам диссертации опубликовано 39 научных работ, в том числе 16 статей в журналах, рекомендованных ВАК, 1 патент и 1 монография и 2 учебных пособия. Объем и структура работы. Диссертация изложена на 324 страницах, включает 68 таблиц, 91 рисунок и состоит из введения, 5 глав, выводов, списка литературы из 307 наименований и приложения. ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ Во введении отмечено, что квантово-химические расчёты, основанные на уравнениях квантовой механики и расчётах с помощью ЭВМ, позволяют дать точное математическое описание любых экспериментально наблюдаемых внутри- и межмолекулярных взаимодействий с учётом структуры и состояния различных соединений, определяет возможность исчерпывающего объяснения любых экспериментальных данных о реакционной способности органических и неорганических соединений и предсказания возможных направлений протекания их химических превращений. Однако до сих пор методы молекулярного моделирования и квантовой химии не находили широкого применения для детального изучения взаимодействий между компонентами нефтей и нефтепродуктов, выявления направления и механизма их превращения в нефтехимических процессах и в процессах переработки углеводородного сырья. В первой главе представлен литературный обзор, в котором рассмотрены методы расчёта структуры и свойств химических соединений: методы молекулярной механики, полуэмпирические и неэмпирические квантово-химические 7 методы. Обсуждаются области применения этих методов, виды расчётов, которые можно проводить с помощью их различных вариантов на ЭВМ, точность выполняемых расчётов. Особое внимание уделено методам, которые позволяют рассчитывать термодинамические свойства индивидуальных химических соединений (энтальпию образования и энтропию) и энергию межмолекулярных взаимодействий. Отмечено, что методы молекулярной механики используются для поиска равновесной геометрии и устойчивых конформаций органических молекул, содержащих тысячи атомов. Расчеты с применением полуэмпирических методов доступны для молекул и молекулярных комплексов, состоящих из сотен атомов, а неэмпирические квантово-химические методы расчета структуры и свойств химических соединений применимы для соединений из десятков атомов. В зависимости от размеров молекул изучаемого индивидуального вещества или комплекса и требуемой точности расчётов значений необходимых параметров (стандартная энтальпия образования, энтропия, теплоемкость, распределение зарядов, энергия взаимодействия и др.) можно подобрать наиболее подходящий вычислительный метод. Во второй главе подробно рассмотрено выполнение расчётов различными методами компьютерной химии с помощью пакетов программ ChemBioOffice 2008 (для Windows), разработанного компанией СambridgeSoft Corporation; ChemBio3D 11.0 Ultra, включающего набор инструментов для создания трехмерных моделей молекул химических соединений, а также различные методы расчета их структуры и свойств; Firefly А. Грановского, GAMESS (US). Третья глава посвящена применению методов компьютерной химии для термодинамического расчёта направления превращений некоторых компонентов тяжёлых нефтей при нефтедобыче методом паротеплового воздействия (ПТВ) на пласт. При ПТВ могут протекать реакции термического крекинга углеводородов и гетероатомных компонентов, высокотемпературного гидролиза (акватермолиза) гетероатомных соединений, уплотнения, переноса водорода дегидрирования одних соединений при одновременном насыщении и гидрогенолизе других. В литературе приводятся предполагаемые реакции, протекающие в пласте, но экспериментально возможность их протекания не изучена. Поэтому задачей данной главы было определение термодинамической возможности реакций превращения некоторых компонентов тяжелых нефтей при температурах, характерных для ПТВ. С этой целью полуэмпирическим методом PM6, с использованием программного пакета MOPAC2007, были рассчитаны термодинамические функции − f HT и ST химических веществ, рассматриваемых в работе в качестве модельных соединений тяжелых нефтей,и их реакций, и вычислено изменение энергии Гиббса реакций по главному уравнению термодинамики r GTo r H To T r STo . При ПТВ на пласт возможны следующие реакции термического крекинга углеводородов и гетероатомных соединений: 8 (1) (4) (2) (5) (3) (6) Изменение энергии Гиббса r GTo для реакций (1-3) и (4-6) приведено на рис. 1а и 1б, соответственно. В условиях ПТВ на пласт температура может достигать значений близких 350С (723К). 150 2 -50 -150 3 -250 -350 1 300 400 500 600 700 800 T, K GT , кДж GT , кДж 50 80 60 40 20 0 -20 -40 -60 300 400 500 600 T, K 700 5 5(лит.) 4(лит.) 4 6 800 а б Рисунок 1 – Изменение энергии Гиббса с ростом температуры: а – для реакций (1) – (3); б – для реакций (4) – (6) Как видно из рис. 1а и 1б, с повышением температуры r GTo реакций 1-5 уменьшается и переходит в область отрицательных значений, что говорит о возможности протекания реакций крекинга при температурах выше 400-450К. На рисунке 1б приведены результаты расчета r GTo для реакций (4) и (5) методом PM6 в сравнении с литературными данными, откуда видно, что результаты расчета близки литературным значениям. Следовательно, квантовохимические методы расчета термодинамических параметров индивидуальных соединений и химических реакций между ними применимы для определения термодинамической возможности химических превращений при ПТВ на пласт. Высокотемпературный гидролиз компонентов тяжелых нефтей – циклогексилфенилсульфида, 2-феноксинафталина, тиофана и N-циклогексиланилина может протекать по следующим реакциям: 9 (7) (8) (9) (10) (11) rGT, кДж Рассчитанное методом PM6 изменение энергии Гиббса ( r GTo ) реакций (7)(11) с ростом температуры (рис. 2) отражает невозможность протекания реакции (9) в условиях ПТВ, остальные реакции – (7), (8), (10) и (11), возможны во всем исследованном температурном интервале. Акватермолиз молекул смолистых веществ по связям С–S и C-O можно представить следующими реакциями, для которых зависимость рассчитанных значений r GTo от температуры при120 ведены на рис. 3. 100 По данным расчёта методом 9 80 PM6 изменения энергии Гиббса при стандартных условиях реакции аква60 термолиза (12)-(14) с разрывом свя40 зей C-S и C-O осуществимы во всем 20 8 интервале температур ПТВ (рис. 3). 7 0 Следовательно, возможно преобра11 -20 зование смол и асфальтенов с отры10 -40 вом боковых фрагментов и образо300 400 500 600 700 800 ванием углеводородов и гетероатомT, K ных компонентов меньшей молекуРисунок 2 - Зависимость изменения лярной массы. энергии Гиббса реакций (7)-(11) от температуры 10 (12) (13) (14) 50 12 GT, кДж 0 13 -50 14 -100 -150 -200 300 400 500 600 T, K 700 800 Рисунок 3 – Зависимость r GTo от температуры для реакций (12)-(14) 11 Реакции гидрогенолиза в условиях ПТВ могут протекать за счёт таких источников H2 как тетралин или муравьиная кислота, большие объёмы которых получаются при переработке растительного сырья. Образование водорода в присутствии тетралина или муравьиной кислоты может происходить по следующим реакциям: 80 60 40 20 0 -20 -40 -60 -80 (15) 15 GT , кДж . (16) По данным расчёта методом PM6, 16 установлено, что протекание реакции (15) возможно при температурах выше 500К, 16а реакция (16) может проходить во всем температурном диапазоне ПТВ (рис. 4). Из рис. 4 следует, что значения r GTo , 300 400 500 600 700 800 T, K рассчитанные методом PM6 для реакции (16), близки литературным данным. Для Рисунок 4 – Зависимость r GTo реакции (15) литературные данные для реакций (15) и (16) от температуo G расчёта отсутствуют. r T ры: (16а – литературные данные Получающийся в реакциях (15) и (16) для реакции (16)) водород далее может участвовать в реакциях гидрогенолиза гетероатомных и полициклических соединений. Гидрогенолиз в присутствии муравьиной кислоты можно представить следующими реакциями: (17) (18) (19) (20) (21) Рассчитанные значения r GTo для интервала температур 300-800К позволили определить, что реакции гидрогенолиза (17)-(21) могут протекать во всем диапазоне пластовых температур при ПТВ (рис. 5). 12 GT, кДж 0 -50 20 -100 1 18 19 21 -150 -200 300 400 500 600 T, K 700 800 Рисунок 5 – Зависимость r GTo от температуры для реакций гидрогенолиза (17)-(21) с участием муравьиной кислоты как источника водорода Если источником водорода будет служить тетралин, то реакции гидрогенолиза могут быть представлены следующими уравнениями: (22) (23) (24) Кроме реакций гидрогенолиза при ПТВ возможны реакции с уплотнением структуры, например: . (25) Расчёты для реакций гидрогенолиза компонентов тяжёлых нефтей в присутствии тетралина показали, что реакции (22)-(24) могут протекать при температурах выше 500К, реакция (24) во всей области температур близка к состоянию равновесия, а реакция уплотнения (25) может идти до конца во всём исследованном интервале температур (рис. 6). r GTo 13 По результатам проведенных расчётов можно заключить, что при извлечении 50 тяжёлых нефтей и природных 0 битумов паротепловым мето24 дом наиболее вероятны сле-50 дующие химические реакции: 22 гидролиз эфирных, 23 -100 сульфидных и аминовых -150 «мостиков» с отрывом более 24 низкомолекулярных фраг-200 ментов: парафиновых, арома300 400 500 600 700 800 тических, гетероатомных; T, K термический крекинг гетеРисунок 6 – Зависимость r GTo от температу- роатомных компонентов и ры для реакций гидрогенолиза (22)-(24) в при- полициклической ароматики сутствии тетралина как источника H2 и для ре- (ПЦА), в том числе с образоакции уплотнения (25) ванием газов, низкомолекулярных соединений на основе углеводородов и продуктов уплотнения; перераспределение водорода от полициклических нафтеноароматических углеводородов к сераорганическим соединениям и ПЦА с образованием соответственно сероводорода и низкомолекулярных углеводородов и других соединений; гидрогенолиз гетероатомных соединений и гидрокрекинг ПЦА с участием водорода, образующегося в реакции конверсии оксида углерода водяным паром. При использовании муравьиной кислоты в качестве донора водорода возможно протекание реакций гидролиза азот- и серосодержащих соединений, гидрокрекинга и гидрирования ПЦА. Результаты термодинамических расчётов, с целью определения условий протекания реакций с разрывом гетероатомных связей, гидрокрекинга ПЦА, образования низкомолекулярных и газообразных продуктов экспериментально подтверждены в работе Петрухиной Н.Н. с соавторами. Четвертая глава посвящена применению методов квантовой химии в исследовании различных соединений и физико-химических процессов, применяемых широко в нефтепереработке, для решения некоторых её практических задач на теоретической научной основе. Одной из таких задач является снижение наложения бензиновых фракций друг на друга - низкокипящих на высококипящие и наоборот. Решение этой задачи является одним из важнейших условий повышения эффективности осуществления процессов перегонки нефти, газовых конденсатов и их смесей на установках АВТ, так как наложение углеводородного (УВ) состава одной фракции на УВ состав другой фракции ведёт к неоднозначности свойств производимых на НПЗ товарных продуктов, получаемых из одноrGT , кДж 100 14 го и того же сырья и при одних и тех же технологических режимах работы установок перегонки. Так, товарные бензины АИ-92 и АИ-95, в соответствии с требованиями стандартов ЕВРО-4 и ЕВРО-5, должны содержать не более 1,0 % об. бензола и до 35 % об. и ниже ( 25% об.) АрУВ. Ограничение содержания бензола обеспечивает экологическую безопасность применения автомобильного топлива за счет снижения содержания бенз-α-пирена в выхлопных дымовых газах при сгорании бензина в ДВС. Требование ограничения содержания АрУВ связано с тем, что они, с одной стороны, повышают детонационную стойкость бензинов, а с другой – при больших концентрациях в товарном бензине увеличивают нагарообразование в камерах сгорания и на клапанах двигателей, что ухудшает эксплуатационные показатели ДВС (кпд и мощность), а также их экономические и экологические параметры. Бензиновые фракции, подаваемые в реакторы установки риформинга, содержат определенное количество сернистых соединений, которые являются ядами для катализатора типа Pt,Re/γ−Al2O3. Поэтому качество исходной бензиновой фракции, подаваемой на гидроочистку в реактор, с загруженным Al-Co-MoO катализатором, оказывает влияние на качество гидрогенизата, которое характеризуется его фракционным составом, соотношением суммы углеводородов с числом атомов C в молекулах УВ от 4 до 9. Качество гидрогенизата затем отражается в качестве риформата, и, что особенно важно, в содержании бензола в риформате. Плотность исходной бензиновой фракции и гидрогенизата является одним из параметров, отражающих их качество. Для выявления связи между плотностью сырья и качеством гидрогенизата, поступающего на установки риформинга ОАО «МНПЗ», в лабораторных условиях были проведены экспериментальные исследования качества узких бензиновых фракций, выделенных из нефти, содержащей до 10 % масс. газового конденсата. Для этих фракций были определены следующие физико-химические свойства: плотность, пределы температур выкипания и сумма углеводородов состава С4 С9 . По результатам хроматографического анализа было установлено наложение бензола на состав каждой из выделенных узких бензиновых фракций. Зависимость температур кипения узких бензиновых фракций, выкипающих в интервале от н.к. до 180C, от их плотности приведена на рис. 7, а содержание в этих фракциях бензола – на рис. 8. Температура кипения узких бензиновых фракций растёт с ростом их плотности по полиномиальным кривым второго порядка (рис. 7), причем наиболее резкое возрастание tкип фракций происходит, начиная с плотности 750 кг/м2, что связано с ростом содержания в них ароматических УВ. 15 6 5 160 4 3 140 2 120 1 100 Выход бензола, об. % Температура кипения, о С 180 0,05 0,045 0,04 0,035 0,03 0,025 0,02 0,015 0,01 730 80 730 735 740 745 750 755 740 Плотность, кг/м3 Рисунок 7 – Зависимость температур кипения фракций бензина, испаряющегося в блоке гидрогенизации, от их плотности: 1 – н.к., 2 – 30%, 3 – 50%, 4 – 70%, 5 – 90%, 6 – к.к. 750 Плотность, 760 760 кг/м3 Рисунок 8 – Зависимость содержания бензола в гидрогенизате от его плотности Для описания зависимости tкип узких бензиновых фракций от их плотности () выведено уравнение, адекватно отражающее экспериментальные данные: − , (26) где – температура кипения фракции, имеющей плотность ; - температура кипения фракции с плотностью 0; k – константа (kнк=0,0222; k30= 0,0356; k50=0,0367; k70=0,0456; k90=0,0533; kкк=0,0500). Адекватность уравнения (26) опытной зависимости температур кипения от плотности на основе сопоставления результатов расчёта температур кипения бензиновых фракций по этому уравнению с экспериментальными данными следует из табл. 1. Таблица 1 – Связь температуры кипения узких бензиновых фракций с плотностью по данным экспериментов и расчетов по уравнению (26) tк, °С , фракции, выкипающей в об.% ρ, 3 нк 30 50 70 90 кк кг/м 730 735 740 750 760 Эксп. Расч. Эксп. Расч. Эксп. Расч. Эксп. Расч. Эксп. Расч. Эксп. Расч. 98 99 100 107 118 98,0 98,6 100,2 106,9 118,0 102 103 106 116 134 102,0 102,9 105,6 116,2 134,0 108 109 112 123 141 108,0 108,9 111,7 122,7 141,0 110 111 115 128 151 110,0 111,1 114,6 128,2 151,0 120 121 125 141 168 120,0 121,3 125,3 141,3 168,0 134 135 139 154 179 134,0 135,3 139,3 155,3 182,0 Бензол, имеющий tкип.= 80,1С, должен был бы выкипеть при этой температуре полностью в процессе разгонки БФ на узкие фракции. Однако бензол присутствует во всех реальных узких бензиновых фракциях, в том числе и во фракциях, выкипающих при t > 100 С (рис. 8). Из рис. 8 следует, что с возрастанием плотности БФ от 730 до 745 кг/м3 включительно содержание бензола в высококипящих фракциях сначала снижается от 0,05 до 0,018 % об., а затем повышается до уровня 0,02 % об. при увеличении плотности до 760 кг/м3. Факт присутствия бензола во всех узких фракциях 16 является ярким примером наложения углеводородного состава фракций при переходе от низкокипящих к высококипящим БФ. Удерживание молекул бензола в высококипящих бензиновых фракциях можно объяснить образованием межмолекулярных комплексов за счёт нековалентных (ван-дер-ваальсовых) взаимодействий бензола в основном и в возбужденном состоянии с ароматическими соединениями и другими УВ. Для того, чтобы доказать возможность образования межмолекулярных комплексов бензола с УВ, входящими в состав бензиновых фракций, необходимо было рассчитать энергию межмолекулярного взаимодействия веществ, входящих в состав таких комплексов и сравнить её с энергией теплового движения молекул. Если энергия межмолекулярного взаимодействия компонентов комплекса превысит энергию теплового движения, то можно будет сделать вывод, что за наложение бензиновых фракций ответственны ван-дер-ваальсовы межмолекулярные взаимодействия. В бензине, по данным хроматографического анализа содержатся ароматические углеводороды C7, C8, C9, C10, а также парафиновые и нафтеновые УВ. В качестве представителей ароматических углеводородов, которые могут образовать межмолекулярные комплексы с бензолом за счет нековалентных взаимодействий, нами были выбраны толуол (С7); о-, м- и п-ксилол и 1,2,4-триметилбензол (С9), в качестве представителей других классов углеводородов служили н-гексан, циклогексан и метилциклогексан. Для оценки энергии взаимодействия молекул бензола с молекулами нгексана (tкип=68,74 С), циклогексана (tкип= 80,73 С), метилциклогексана (tкип=100,93 С), толуола (tкип=110,62 С), этилбензола (tкип=136,19С), п-ксилола (tкип=138,348С), м-ксилола (tкип=139,103С), о-ксилола (tкип=144,414С), и 1,2,4триметилбензола (tкип=163,9 С), содержащихся в соответствующих бензиновых фракциях, были проведены расчеты энергий молекул перечисленных ароматических УВ и их комплексов с бензолом неэмпирическим квантово-химическим методом MP2/++6-311G(d,p), с использованием пакетов программ GAMESS (ChemOffice Ultra 2008, CambridgeSoft Corp.) и Firefly А.А. Грановского. Использованный квантово-химический метод расчёта позволяет учесть дисперсионные взаимодействия между молекулами углеводородов, которые и являются ответственными за силы притяжения, возникающие между молекулами углеводородов. Базисный набор атомных орбиталей, применённый в расчётах, включает диффузионные орбитали на всех атомах, что позволяет более полно учитывать дисперсионные взаимодействия молекул на больших расстояниях. Модели комплексов бензола с ароматическими углеводородами узких бензиновых фракций, созданные с помощью программы моделирования ChemBio3D 11.0 Ultra и оптимизированные методом MP2/++6-311G(d,p) показаны на рис. 912. Молекулы бензола и ароматических углеводородов в комплексах ориентированы относительно друг друга различным образом. Рассчитанные энергии взаимодействия молекул бензола с ароматическими УВ в комплексах, показанных на рис. 9-12, даны в табл. 2. 17 1 Рисунок 9 – Комплексы бензола и этилбензола расстояние между молекулами: 1 –2,8 Ǻ, (ароматические кольца расположены под углом 90); 2 – 2,9 Ǻ (угол между ароматическими кольцами 30 2 Рисунок 10 – Трехмерные модели межмолекулярных комплексов бензола и толуола: 1 - расстояние между плоскостями ароматических колец 3,4 Ǻ. Центр молекулы бензола смещен по горизонтали на 1,2 Ǻ от центра кольца толуола (находится над связью С(5)-С(6)); 2 - расстояние H(24) до центра молекулы толуола 3,9 Ǻ. Молекулы расположены под углом 10 18 1 2 3 4 Рисунок 11 – Модели комплексов бензола с п- и о-ксилолом: 1 – расстояние от атомов H(22) и H(23) до плоскости п-ксилола 3,3Ǻ; угол между ароматическими кольцами 50; 2 – расстояние от атомов С(10) и С(11) до плоскости пксилола 3,4Ǻ; молекула бензола перпендикулярна молекуле п-ксилола; 3 - расстояние от атома H до плоскости бензола 2,7 Ǻ, угол между плоскостями циклов 20; 4 – расстояния от атомов С(7) и С(8) до плоскости о-ксилола 2,8 Ǻ до атомов С(17) и С(14) 3Ǻ (показаны пунктиром), угол между плоскостями циклов 36 1 2 3 Рисунок 12 – Трехмерные модели комплекса бензола с м-ксилолом и 1,2,4триметилбензолом: 1 – атомы H(26) и H(27) расположены над плоскостью мксилола на расстоянии 3,3 Å; угол между плоскостями колец 50; 2 – атом H (10) находится над плоскостью кольца м-ксилола на расстоянии 2,7 Å, угол между плоскостями ароматических колец 30; 3 - расстояния H(33) и Н(28) до плоскости кольца 1,2,4-триметилбензола равны 2,7 Å (показаны пунктиром); угол между плоскостями молекул 56 Для сравнения энергий взаимодействия бензола с ароматическими УВ и энергий его взаимодействия с представителями других классов УВ, присутствующими в узких БФ, были созданы модели комплексов бензола с гексаном, циклогексаном и метилциклогексаном (рис. 13), оптимизирована их геометрия и рассчитаны энергии взаимодействия бензола с этими УВ (табл. 2). 19 Таблица 2 – Энергия взаимодействия молекул бензола с некоторыми УВ узких бензиновых фракций (дипольный момент бензола – 0,00 Д, углеводорода - УВ) Комплекс Бензол/толуол 1 2 Бензол/п-ксилол 1 2 Бензол/этилбензол 1 2 Бензол/о-ксилол 1 2 Бензол/м-ксилол 1 2 Бензол/1,2,4-триметилбензол Бензол/н-гексан Бензол/циклогексан 1 2 Бензол/метилциклогексан 1 2 УВ, Д лит./расч. 0,37-0,40/0,37 0,06/0,04 0,35-0,40/0,35 0,62/0,64 0,37/0,32 отсут. /0,36 0/0,00 0/0,00 0/0,00 0/0,00 0/0,00 Eвзаимод. ккал/моль кДж/моль -2,87 -12,02 -2,81 -11,74 -2,78 -11,64 -2,23 -9,34 -2,97 -12,43 -2,53 -10,57 -5,43 -22,71 -4,85 -20,28 -2,78 -11,64 -3,12 -13,07 -3,08 -12,88 -1,99 -8,32 -1,35 -5,65 -1,46 -6,13 -1,11 -4,65 -1,54 -6,43 1 2 3 4 Рисунок 13 – Трехмерная модель комплекса бензола с н-гексаном, циклогексаном (ЦГ) и метилциклогексаном (МЦГ): 1 - расстояние между плоскостью бензольного кольца и молекулой н-гексана 3,2 Ǻ. Расстояние между атомом H и бензольным кольцом: 2 – 3,5 Ǻ; 3 – 3,5 Ǻ; 4 – 3,2 Ǻ Результаты расчетов (табл. 2) показали, что энергии взаимодействия бензола и ароматических УВ значительно выше энергии его взаимодействия с УВ других классов - н-гексаном, циклогексаном и метилциклогексаном. Отсюда можно сделать вывод о возможности удержания бензола в высококипящих узких бензиновых фракциях за счет взаимодействия молекул бензола с ароматическими углеводородами. Энергия теплового движения равна: Е=RT или 20 E=8,314293=2436 Дж/моль. Эта величина в несколько раз меньше энергии ММВ бензола с ароматическими УВ. Небольшое количество молекул ароматических углеводородов, например, бензола и толуола, в бензиновых фракциях могут находиться в возбужденном состоянии уже при комнатной температуре и при взаимодействии образовывать комплексы в низшем триплетном состоянии. Структура комплекса бензола и толуола и его энергия в низшем триплетном состоянии рассчитана ограниченным методом Хартри-Фока (ROHF/6-311++G(d,p)), этим же методом рассчитаны энергии низших триплетных состояний бензола и толуола, а также их структура. Для определения энергии возбуждения бензола и толуола рассчитаны энергии основных состояний этих соединений методом Хартри-Фока (RHF/6311++G(d,p)). Комплекс бензола и толуола в низшем триплетном состоянии имеет структуру, приведенную на рис. 14. Расстояние между ароматическими кольцами молекул в низшем триплетном комплексе бензола и толуола 5,2 Ǻ, что значительно больше, чем в случае основного состояния комплекса. В комплексе бензол находится в низшем триплетном, толуол – в основном состоянии. Как следует из результатов расчетов, триплетный комплекс бензола и толуола может образоваться только из молекул бензола и толуола, находящихся в низшем триРисунок 14 – Комплекс бензола и то- плетном состоянии. Энергия взаимолуола в низшем триплетном состоя- действия молекул в комплексе нии: расстояние между ароматически- 308,33 кДж/моль (-73,69 ккал/моль). В комплексе на рис. 14 молекула ми кольцами 5,2 Å(Молекулы расплобензола характеризуется двумя уколожены параллельно роченными C-C-связями (1,325 Ǻ) и четырьмя удлиненными (1,478 Ǻ), в основном состоянии длина C-C-связи в бензоле составляет 1,394 Ǻ, неспаренные электроны находятся у противоположных атомов углерода С(2) и С(5). При выделении узких бензиновых фракций из прямогонного бензина возможно образование бимолекулярных комплексов ароматических, нафтеновых и парафиновых углеводородов. Для сравнения энергии взаимодействия бензола с УВ узких бензиновых фракций и энергии ММВ индивидуальных УВ были проведены расчёты энергии взаимодействия молекул в бимолекулярных комплексах углеводородов различных классов (табл. 3). Структура бимолекулярных комплексов углеводородов различных классов, полученных оптимизацией их геометрии методом MP2/6-311++G(d,p), представлена на рис. 15. 21 2а 1 3а 2б 3б 4а 4б 5 6 7а 7б Рисунок 15 - Бимолекулярные комплексы: 1 – толуола; 2а, б – о-ксилола; 3а, б – м-ксилола; 4а, б – метилциклогексана; 5 – бензола; 6 - п-ксилола; 7а, б – этилбензола Таблица 3 – Энергия взаимодействия углеводородов в бимолекулярных комплексах Eвзаимод Молекулы УВ R, Å ккал/моль кДж/моль в комплексе 1 Толуол -2,85 -11,92 3,4 2а о-Ксилол -7,25 -30,33 3,7 2б о-Ксилол -7,09 -29,67 3,4 3а м-Ксилол -1,23 -5,15 4,5 3б м-Ксилол -4,61 -19,28 3,4 4а Метилциклогексан -1,76 -7,36 2,5 4б Метилциклогексан -1,33 -5,56 3,0 5 Бензол -2,09 -8,76 3,4 6 п-Ксилол -3,14 -13,14 3,5 7а Этилбензол -2,72 -11,37 2,9 7б Этилбензол -2,94 -12,29 2,8 Энергии взаимодействия молекул УВ зависят от их ориентации в бимолекулярных комплексах (рис. 15, табл. 3) и возрастают при переходе от метилциклогексана к этилбензолу и толуолу, п-, м- и о-ксилолу. По литературным данным 22 экспериментально измеренная энергия взаимодействия молекул бензола составляет -2,4±0,4 ккал/моль. Полученное методом MP2/6-311++G(d,p) значение -2,09 ккал/моль близко экспериментальному значению. В литературе приводятся данные расчётов энергий взаимодействия бензола с ароматическими УВ и молекул ароматических УВ в бимолекулярных комплексах (димерах). Однако расчёты были выполнены разными методами с разными базисными наборами АО, при этом некоторые авторы учитывают суперпозиционную ошибку базисного набора (СОБН), другие пренебрегают этой ошибкой. Поэтому невозможно провести сопоставление расчётов, выполненных разными авторами для одних и тех же бимолекулярных комплексов. В данной работе все расчёты энергий межмолекулярных взаимодействий проведены одним и тем же методом MP2/6-311++G(d,p) с учетом СОБН по формулам: − − , (27) − , (28) − − , (29) где - энергия бимолекулярного комплекса; , – энергии индивидуальных молекул А и B; - энергия взаимодействия молекул в бимолекулярном комплексе без учета СОБН; - энергия взаимодействия молекул A и B в бимолекулярном комплексе; – суперпозиционная ошибка базисного набора; , - энергия молекулы A и молекулы В в базисном наборе бимолекулярного комплекса, соответственно; , энергия молекулы А и молекулы B в геометрии, которую они имеют в бимолекулярном комплексе. Применение квантово-химических методов для расчёта межмолекулярных взаимодействий низших меркаптанов с ароматическими УВ узких бензиновых фракций, выделенным из нефтей разного состава и их смесей с газовыми конденсатами, позволило выявить механизм, лежащий в основе распределения меркаптанов C1-C5 по узким бензиновым фракциям прямогонного бензина. В прямогонных бензиновых фракциях, направляемых на установки риформинга содержание общей серы строго регламентировано и должно лежать в пределах 0,07-0,08 % масс., что позволяет получать в блоках гидроочистки установок риформинга гидрогенизат с содержанием общей серы не более 0,5 ppm. Такой гидрогенизат, направляемый в блок реакторов установки риформинга, вызывает минимальную деактивацию катализатора. В современных условиях в переработку вовлекаются как сырьевые нефтесмеси, так и газовые конденсаты с высоким содержанием меркаптанов. Бензиновые фракции со значительным содержанием меркаптанов могут стать причиной нарушений работы технологического процесса в блоке гидроочистки установок риформинга БФ. Поэтому анализ содержания в них меркаптановой серы, а также распределения меркаптанов по узким бензиновым фракциям представлял научный и практический интерес. В ходе мониторинга подготовки сырья процесса риформинга на ОАО «Новокуйбышевский НПЗ» было определено содержание меркаптанов в бензиновых фракциях, выделенных из нефти и ее смесей с газоконденсатом. В качестве об23 разцов нефтей служило сырье установки АВТ-11 и нефти, отобранные с коммерческих узлов учёта ОАО «Новокуйбышевский НПЗ». Анализ распределения низкокипящих меркаптанов с длиной УВ цепи С1– С5 по узким бензиновым фракциям, выделенным из образцов смеси Самарской нефти с Оренбургским газоконденсатом, показал присутствие этих меркаптанов во всех узких бензиновых фракциях (рис. 16). Рисунок 16 - Распределение меркаптанов УВ с длиной УВ цепи С1–С5 по узким бензиновым фракциям, выделенным из смеси Самарской нефти и Оренбургского газоконденсата в ОАО «Новокуйбышевский НПЗ» Исключением являются меркаптаны с длиной УВ цепи C5, которые отсутствуют во фракции н.к.-62С. Температуры кипения лёгких меркаптанов приведены в табл. 4. Таблица 4 – Температуры кипения лёгких меркаптанов Меркаптан Метилмеркаптан Этилмеркаптан Изопропилмеркаптан Пропилмеркаптан трет-Бутилмеркаптан Т. кип., С 6,00 35,04 52,56 67,82 64,22 Меркаптан втор-Бутилмеркаптан Изобутилмеркаптан н-Бутилмеркаптан 2,2-Диметилпропилмеркаптан-1 трет-Амилмеркаптан Т. кип., С 84,98 88,72 98,46 64,22 99,00 Данные табл. 4 и рис. 16 отражают тот факт, что меркаптаны состава С1-С5 распределяются по всем узким бензиновым фракциям, независимо от их температур кипения. Так, метил- и этилмеркаптан, имеющие, соответственно, температуры кипения 6,00 и 35,04С, обнаружены не только во фракции н.к.-62С, но и во всех остальных фракциях. Пропилмеркаптан, входящий в сумму меркаптанов C3, присутствует во фракции н.к.-62С, что ниже его температуры кипения, а также и во всех фракциях с более высокими температурами выкипания. Присутствие пропилмеркаптана во фракции с пределами выкипания ниже его температуры кипения можно объяснить образованием азеотропных смесей с н-, изоалканами и нафтенами C6. По литературным данным все лёгкие меркаптаны образуют азеотропные смеси с минимумом температуры кипения с н-, изоалканами и нафтенами. 24 Присутствие лёгких меркаптанов во фракциях с пределами выкипания выше температуры кипения этих меркаптанов можно объяснить только межмолекулярными взаимодействиями этих соединений с ароматическими углеводородами соответствующих узких бензиновых фракций. Для проверки данного заключения из анализа распределения меркаптанов между узкими бензиновыми фракциями были проведены расчеты энергии межмолекулярного взаимодействия меркаптанов с ароматическими углеводородами. Расчеты выполнены неэмпирическим квантово-химическим методом МеллераПлессе MP2/6-311++G(d,p), учитывающим энергию корреляции электронов. В литературе приводятся данные по энергии взаимодействия H2S и метилмеркаптана с бензолом. Энергия взаимодействия H2S с бензолом, рассчитанная методом CCSD[T]/aug-cc-pVTZ, по литературным данным составила -2,64 ккал/моль, методом CCSD[T]/aug-cc-pVQZ было получено значение -2,74 ккал/моль. По литературным данным энергия взаимодействия метилмеркаптана с бензолом, рассчитанная методом MP2/aug-cc-pVDZ, составляет -3 ккал/моль. Поскольку авторы используют различные расчетные методы энергии взаимодействия H2S и метилмеркаптана с бензолом, а для других меркаптанов и ароматических углеводородов расчеты энергии взаимодействия отсутствуют в литературе, оценить зависимость энергии взаимодействия от длины углеводородной цепи меркаптана и строения ароматического углеводорода невозможно. Для выявления закономерностей, определяющих взаимодействие легких меркаптанов с ароматическими углеводородами узких бензиновых фракций, нами выполнены расчёты энергии взаимодействия этих меркаптанов с бензолом и другими ароматическими углеводородами методом MP2/6-311++G(d,p). Модели комплексов H2S с бензолом даны на рис. 17, модели легких меркаптанов с некоторыми ароматическими углеводородами приведены на рисунке 19. I II Рисунок 17 - Модели комплексов H2S с бензолом: R1, R2 – расстояние между атомом S и центром бензольного кольца: R1=3,7 Å; R2=3,8 Å. Комплекс I: длина связи S-H 1,331 Å, 1=92,4; длина связи C-H в бензоле 1,395 Å. Комплекс II: длина связи (S-H)1 1,332 Å, (S-H)2 1,330 Å, 2=93,2; длина связи C-H в бензоле a=1,396 Å, b=1,395 Å Полученные нами значения энергии взаимодействия H2S и бензола близки к литературным данным и составляет для модели I -9,22 кДж/моль (-2,20 ккал/моль), для модели II эта величина равна -8,41 кДж/моль (2,01 ккал/моль). Расстояния между молекулами H2S и бензола в комплексах совпадают с литературными данными (рис. 17). Рассчитанные энергии взаимодействия легких меркаптанов с ароматическими углеводородами для комплексов на рис. 18 сведены в табл. 5. 25 Данные результатов расчётов энергии взаимодействия лёгких меркаптанов с ароматическими углеводородами узких бензиновых фракций, выделяемых из нефти, показали, что между меркаптанами и ароматическими углеводородами существуют достаточно сильные межмолекулярные взаимодействия, энергия которых лежит в пределах от -9,20 кДж/моль для взаимодействия метилмеркаптана с бензолом до -29,59 кДж/моль для взаимодействия пропилмеркаптана с мксилолом. Таблица 5 - Энергия взаимодействия легких меркаптанов с ароматическими углеводородами (рис. 18) № п/п Молекулярный комплекс № п/п Eвзаимод ккал/ моль кДж/ моль 1a Бензол+CH3SH -2,20 -9,20 1б 2a 2б 3a 3б 4a 4б 4в 5a Бензол+CH3SH -2,47 -10,33 Толуол+CH3SH -2,72 -11,40 Толуол+CH3SH -2,67 -11,17 М-ксилол+CH3SH -3,04 -12,70 М-ксилол+CH3SH -3,38 -14,12 О-ксилол+CH3SH -2,41 -10,06 О-ксилол+CH3SH -4,29 -17,95 О-ксилол+CH3SH -4,02 -16,83 М-ксилол+C2H5SH -3,51 -14,68 5б 6a 6б 7a 7б 8а 8б 9а 9б 26 Молекулярный Комплекс Eвзаимод ккал/ моль кДж/ моль М-ксилол+C2H5SH -3,58 -14,97 М-ксилол+C3H7SH М-ксилол+C3H7SH -3,66 -15,32 -4,92 -20,59 М-ксилол+i-C3H7SH -3,86 -16,16 М-ксилол+i-C3H7SH -4,27 -17,85 М-ксилол+C4H9 SH -3,67 -15,33 М-ксилол+C4H9SH -4,50 -18,85 М-ксилол+C4H9-2-SH -4,26 -17,81 М-ксилол+C4H9-2-SH -4,60 -19,23 Рисунок 18 - Модели комплексов легких меркаптанов с ароматическими углеводородами узких бензиновых фракций: расстояние атома S от плоскости бензольного кольца для всех комплексов не превышает 4 Å, атом H расположен над бензольным кольцом на расстоянии 2,7-2,8 Å Из проведённых расчётов можно сделать следующие выводы. Несмотря на то, что расчёты энергий взаимодействия лёгких меркаптанов с ароматическими углеводородами бензиновых фракций, ввиду их сложности и больших затрат времени, выполнены не для всех ароматических соединений, присутствующих в бензинах, полученные результаты подтверждают, что энергии взаимодействия с 27 Прирост цетанового числа (ЦЧ) ароматикой достаточно для удержания лёгких меркаптанов в высококипящих бензиновых фракциях, так как она превышает энергию теплового движения молекул при температурах выкипания высококипящей фракции 140-180С, составляющую около 2,4 кДж . В четвертой главе представлено также применение квантово-химических методов для расчёта взаимодействия инициатора горения с компонентами дизельных топлив (ДТ). Для повышения цетанового числа ДТ применяют присадки – инициаторы воспламенения, которыми могут служить гидропероксиды алкилов и алкилнитраты. За рубежом в качестве инициатора воспламенения топливовоздушных смесей (ДТ+воздух) широко применяется этилгексилнитрат (ЭГН) CH3-CH(C2H5)-CH2-CH2-CH2-CH2-NO2, производство которого осваивается и в России. Зависимость цетанового числа (ЦЧ) ДТ от концентрации 10 Ц0=55 ЭГН при различных значениях 9 ЦЧ исходного топлива приведена 8 Ц0=45 7 на рис. 19. Ц0=35 6 5 Рисунок 19 - Прирост цетанового числа ДТ с ростом концентрации ЭГН при различных значениях начального цетанового числа (ЦЧ0) исходных топлив Ц0=25 4 3 2 1 0 0 0,05 0,1 0,15 Концентрация ЭГН, % об. 0,2 Для описания зависимости цетанового числа ДТ от концентрации ЭГН (СЭГН) выведено уравнение ЦЧ=ЦЧ0+ЦЧ, где 2 ΔЦЧ 0,1307 ЦЧ02 6,9362 ЦЧ0 156,42 СЭГН 0,0183 ЦЧ02 0,0597 ЦЧ0 21,7710 СЭГН. (30) Для объяснения механизма действия ЭГН в ДТ проведены расчеты полуэмпирическим квантово-химическим методом PM6, входящим в пакет программ MOPAC2007, свойств ЭГН и его взаимодействия с молекулами н-ПрУВ и АрУВ. Механизм действия ЭГН на УВ ДТ может быть представлен следующими основными стадиями. 1. Возбуждение молекул ЭГН. При возбуждении молекулы ЭГН в триплетное состояние легко происходит разрыв связи O-NO2 CH3CH(C2H5)-(CH2)4ONO2 CH3CH(C2H5)-(CH2)4O + NO2 Энтальпия образования ЭГН в основном состоянии f H 298 = -307,583 кДж/моль (73,514 ккал/моль), в возбужденном f H 298 =-172,393 кДж/моль (41,203 ккал/моль). Изменение энтальпии при возбуждении молекул ЭГН, рассчитанное по закону Гесса, составляет 135,19 кДж/моль (32,311 ккал/моль), что близко литературному значению энергии разрыва связи O-N в алкилнитратах – 150 кДж/моль (35,89 ккал/моль). 28 Образовавшиеся в результате возбуждения молекулы ЭГН радикалы могут вступать в реакции с УВ ДТ. 2. На следующей стадии после распада молекулы ЭГН возможно взаимодействие радикала CH3CH(C2H5)-(CH2)4O с АрУВ. Реакция с толуолом, как показало компьютерное моделирование с использованием полуэмпирического метода, PM6, приводит к образованию 2-этилгексилового спирта, за счёт отрыва атома водорода от метильной группы толуола: CH3CH(C2H5)-(CH2)4O + C6H5CH3 CH3CH(C2H5)-(CH2)4OH + C6H5CH2 . Молекула толуола превращается в относительно стабильный радикал C6H5CH2, так как неспаренный электрон делокализован в бензольном кольце между атомами углерода в пара- и орто-положении по отношению к метиленовой группе (рис. 20, табл. 6). Рассчитанное методом PM6 значение теплового эффекта реакции CH3CH(C2H5)-(CH2)4O с толуолом составляет –90,81 кДж/моль (21,704 ккал/моль). Распределение спиновой плотности в бензильном радикале представлено в табл. 6. Таблица 6 – Распределение спиновой плотности в бензильном радикале, расчет методом PM6 (MOPAC2007) Атом Рисунок 20 – Бензильный радикал C(1) C(2) C(3) C(4) C(5) C(6) C(7) Спиновая плотность -0,4268 0,4924 -0,3975 0,4806 -0,3975 0,4926 0,8343 Атом H(8) H(9) H(10) H(11) H(12) H(13) H(14) Спиновая плотность -0,0170 0,0138 -0,0165 0,0138 -0,0170 -0,0276 -0,0276 Свободный электрон в бензильном радикале (табл. 6) в основном локализован на атоме C(7) метиленовой группы (спиновая плотность 0,8343), хотя и наблюдается частичная делокализация электронов на атомах углерода С(2), С(4) и С(6) бензольного кольца (спиновая плотность на атомах С(2) С(4) и С(6) равна, соответственно: 0,4924; 0,4926 и 0,4806). По литературным данным спинавая плотность на атоме С(7), по разным источникам составляет 0,6-0,7. В составе ДТ содержатся полиароматические УВ. Для компьютерного моделирования протекания реакции 2-этилгексильного радикала с полиароматическими УВ в качестве последнего был выбран -метилнафталин. Компьютерное моделирование показало, что взаимодействия радикала CH3CH(C2 H5)-(CH2)4O с -метилнафталином приводит к образованию 2этилгексилового спирта, за счет отрыва атома водорода от метильной группы метилнафталина. При этом молекула -метилнафталина превращается в стабильный радикал C10H7CH2. Тепловой эффект реакции составляет -105,25 кДж/моль (25,155 ккал/моль). 29 Расчет распределения плотности спина в -метиленнафталиновом радикале показал, что свободный электрон в нем делокализован в большей степени, чем в бензильном радикале, т.е. -метиленнафталиновый радикал более стабилен, чем бензильный. Структура -метиленнафталинового радикала приведена на рисунке 21, распределение спиновой плотности – в табл. 7. Таблица 7 – Распределение спиновой плотности в -метиленнафталиновом радикале (расчет методом PM6, пакет MOPAC2007) Рисунок 21 – Структура метиленнафталинового радикала Атом Спиновая плотность Атом Спиновая плотность C(1) C(2) C(3) C(4) C(5) C(6) C(7) C(8) C(9) C(10) 0,3914 -0,3708 0,4061 -0,3927 0,4247 -0,3933 0,5729 -0,4509 0,5901 -0,4578 C(11) H(12) H(13) H(14) H(15) H(16) H(17) H(18) H(19) H(20) 0,7560 -0,0135 0,0128 -0,0141 0,0138 -0,0196 0,0157 -0,0202 -0,0252 -0,0253 Наиболее высокая спиновая плотность, равная 0,7560, отмечена у атома C(11) метиленовой группы. Неспаренный электрон делокализован в сопряженной π-системе нафталинового ядра, спиновая плотность на атомах углерода С(1), С(3), С(5), С(7) и С(9) составляет, соответственно: 0,3914; 0,4061; 0,4247; 0,5729 и 0,5901. Спиновая плотность на атоме углерода метиленовой группы в метиленнафталиновом радикале ниже, чем в бензильном, что отражает его меньшую реакционноспособность по сравнению с бензильным радикалом. Радикал CH3CH(C2 H5)-(CH2)4O проявдяет высокую реакционную способность, спиновая плотность на атоме O в этом радикале, рассчитанная методом PM6 (MOPAC2007), равна 0,923. Этот радикал может вступать в реакции с алканами, например, с додеканом: CH3CH(C2H5)-(CH2)4O +C12H26 CH3CH(C2H5)-(CH2)4OH + C11H23CH2, изменение энтальпии этой реакции по данным расчета составило -59,401 кДж/моль (-14,197 ккал/моль). Радикал NO2, реагируя с н-алканами, превращается с тепловым эффектом 70,57 кДж/моль (-16,87 ккал/моль) в молекулу HONO, которая распадается на радикалы HO и NO, а н-алканы превращаются при этом в н-алкильный радикал (PM6): C12H26 + NO2 C11H23CH2 + HONO, HONO HO + NO. По результатам расчётов можно сделать выводы, что при возбуждении молекул 2-ЭГН происходит их распад с образованием радикалов NO 2 и RO (R – алкил), которые инициируют воспламенение топливо-воздушной смеси (ТВС) за 30 счёт образования реакционноспособных радикалов при взаимодействии с молекулами УВ. Степень делокализации неспаренного электрона в арильных радикалах в 1,31,4 раза превышает степень делокализации неспаренного электрона в н-алкильных радикалах, что отражает большую стабильность арильных радикалов. Следовательно, присутствие АрУВ менее благоприятно для инициирования и спокойного горения ТВС. Установленные закономерности объясняют известную из практики более высокую эффективность присадок типа алкилнитратов в топливах с более высоким цетановым числом (то есть, содержащих меньше ароматических углеводородов). В четвертой главе с помощью квантово-химических методов расчета свойств антиокислительной присадки 2,6-ди-трет-бутил-4-метилфенола выявлен механизм ее действия и выведено уравнение зависимости периода индукции окисления бензина при его хранении от концентрации присадки. Товарные бензины с ОЧ (ИМ) 80, 92, 95 и 98 состоят из бензиновых фракций первичной перегонки нефти и фракций вторичных процессов – риформинга низкооктановой бензиновой фракции (БФ) 85÷180 ºС и каталитического крекинга вакуумного газойля, мазута или их смесей, и содержат н- и изо-парафиновые, нафтеновые, алкилароматические и непредельные углеводороды. При хранении товарных бензинов они подвергаются окислению кислородом воздуха с образованием кислородсодержащих соединений и смол. Для предотвращения окислительных процессов в воздушной «подушке» над бензином, хранящемся в резервуаре, используют антиокислительные присадки типа Агидол-1 или Агидол-12. Агидол-1 (2,6-ди-трет-бутил-4-метилфенол) относится к экранированным фенолам и эффективен до начала окисления и на начальных его стадиях, когда количество гидропероксидов в топливе невелико. При взаимодействии с пероксидными радикалами 2,6-ди-трет-бутил-4-метилфенол образует стабильный радикал по реакции ROO· + CH3C6H5(C4H9)2OH ROOH + CH3C6H5(C4H9)2O·. При рекомбинации радикала CH3C6H5(C4H9)2O· с другими радикалами образуются неактивные продукты. О стабильности радикала (рис. 22), получающегося при отрыве атома H от OH-группы 2,6-ди-трет-бутил-4метилфенола можно судить по распределению спиновой плотности, рассчитанному методом PM6/UHF (MOPAC2007) для открытых электронРисунок 22 – Радикал, образующий- ных оболочек: на атоме O спиновая ся при взаимодействии 2,6-ди-трет- плотность составляет 0,2351; на атомах углерода в орто-положении по отношебутил-4-метилфенола с пероксидами нию к кислороду – 0,5300, а на атоме углерода в пара-положении – 0,6147. Такое распределение спиновой плотности свидетельствует о высокой стабильности радикала CH3C6H5(C4H9)2O· за счет делокализции неспаренного электрона в -системе его ароматического кольца. 31 Величина индукционного периода окисления зависит от природы БФ и присадок. На рис. 23 приведены данные по ингибированию процесса окисления бензина крекинга при изменении концентрации присадки 2,4-ди-трет-бутил-6метилфенола от 0 до 0,67 масс. %. Без присадки индукционный период (ИП) окисления бензина составляет 200 мин. В присутствии антиокислительной присадки ИП резко возрастает уже при Cпр. = 0,05 масс. % и растет с повышением концентрации присадки в БФ. Зависимость ИП окисления бензиновой фракции от концентрации антиокислительной присадки 2,6-ди-трет-бутил-4-метилфенола адекватно описывается уравнением 1 1 τ τ0 k , C0 C Период индукции, мин которое выведено на основе предположения, что обрыв 1800 цепей при окислении УВ бен1600 зиновой фракции проходит по 1400 бимолекулярному механизму (0 и C0 – начальные значения 1200 ИП и концентрации присадки, 1000 k – константа). 800 В четвертой главе так600 же представлены результаты квантово-химических расчё400 тов энергии взаимодействия 200 между компонентами дизель0 ных топлив, которые позволи0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 ли сформулировать механизм Концентрация присадки, масс. % действия депрессорноРисунок 23 – Зависимость периода индук- диспергирующих присадок. ции окисления бензиновой фракции от кон- Дизельные топлива с присадцентрации антиокислительной присадки 2,6- ками представляют собой ди-трет-бутил-4-метилфенола многокомпонентные растворы сложного химического состава, в которых возможно образование молекулярных ассоциатов и сложных молекулярных комплексов между соединениями различной природы, структуры и молекулярной массы. При температурах выше 30 С ДТ – это истинный раствор, а с понижением температуры в них начинается кристаллизация парафинов и истинный раствор переходит в дисперсную систему. Все процессы, протекающие в ДТ: ассоциация молекул, образование комплексов, кристаллизация парафинов, снижение температуры застывания при введении депрессорных присадок, обусловлено межмолекулярными взаимодействиями компонентов ДТ. Образование межмолекулярных комплексов присадок с компонентами ДТ может проис32 ходить за счет дисперсионных сил, водородных связей и диполь-дипольного взаимодействия. Непосредственное изучение межмолекулярных взаимодействий в ДТ экспериментальными методами представляет собой неразрешимую задачу вследствие их сложного химического состава, поэтому для изучения взаимодействий компонентов в ДТ были привлечены методы компьютерной химии. С помощью пакета ChemBio3D Ultra 11.0 были созданы модели межмолекулярных комплексов парафинов с депрессорной присадкой на основе сополимера этилена с винилацетатом (ЭВА) и диспергирующими присадками на основе алкилсукцинимида и алкиламина итаконовой кислоты (в качестве моделей молекул диспергирующих присадок были выбраны N-додецилсукцинимид и ундециламин итаконовой кислоты). Моделью депрессорной присадки служил небольшой фрагмент сополимера этилена с винилацетатом, а моделью парафинового углеводорода – додекан (C12H26). На рис. 24 показана трехмерная модель комплекса молекулы додекана с участком молекулы депрессора ЭВА и с N-додецилсукцинимидом. Как видно из рисунка, молекула депрессора образует комплекс с молекулой парафинового углеводорода, который может служить центром кристаллизации при понижении температуры ДТ, а молекулы диспергатора взаимодействуют с полярными группами молекул депрессора, создавая препятствия для увеличения размеров кристалликов парафина и их срастания. Расчеты показали, что энергия 2 взаимодействия в тройном комплексе алкилсукцинимида с С12H26 и ЭВА 1 превышает энергию суммарную энер3 гию взаимодействия в двойных комРисунок 24 – Комплекс додекана (1) с плексах алкилсукцинимида с С12H26, депрессором ЭВА (2) и диспергатором ЭВА с С12H26 и алкилсукцинимида с ЭВА. Рассчитанные значения энтальN-додецилсукцинимидом (3) пий образования индивидуальных присадок и их комплексов представлены в табл. 8. Таблица 8 – Энтальпии образования индивидуальных соединений и комплексов парафиновых углеводородов с компонентами депрессорно-диспергирующих присадок (PM6, MOPAC2007) , Eвзаимод. Индивидуальное соединение или молекуляр f H 298 , ный комплекс кДж/моль кДж/моль ЭВА -1414,78 C12H26 -276,35 33 N-додецилсукцинимид -623,75 Ундециламин итаконовой к-ты -871,28 ЭВА+ C12H26 -1727,87 -36,74 ЭВА+N-додецилсукцинимид -2093,51 -54,98 ЭВА+ ундециламин итаконовой к-ты -2465,67 -179,61 Ундециламин итаконовой к-ты+ C12H26 * -1177,65 -30,02 N-додецилсукцинимид + C12H26* -928,97 -28,87 N-додецилсукцинимид + C12H26+ЭВА -2393,58 -78,70 Ундециламин итаконовой к-ты+ C12H26+ЭВА -2647,64 -85,23 Димер N-додецилсукцинимида -1271,98 -24,48 Димер ундециламина итаконовой к-ты -1782,64 -40,08 *При параллельном расположении УВ цепи молекулы C12H26 и алкила депрессорной присадки По данным расчетов энергия взаимодействия ЭВА с C12H26 выше, чем с депрессорными присадками N-додецилсукцинимидом и ундециламином итаконовой кислоты (табл. 8), поэтому можно сделать вывод о преимущественном образовании комплексов углеводородов с метиленовой цепью депрессора ЭВА. Энергия взаимодействия ЭВА и ундециламина итаконовой кислоты значительно выше энергии взаимодействия ЭВА и N-додецилсукцинимида, однако энергия взаимодействия компонентов в комплексах ЭВА с C12H26 и диспергаторами отличается всего на 6,53 кДж/моль, что отражает стабильность обоих комплексов, но комплекс ЭВА с C12H26 и ундециламином итаконовой кислоты обладает большей стабильностью, что и объясняет высокую эффективность композиционной депрессорно-диспергирующей присадки, отмеченную в литературе. Показана применимость квантово-химических методов расчета для оценки стабильности присадок к ДТ (табл. 9). В современных условиях применяют многофункциональные композиционные присадки, компонентами которых являются органические соединения с функциональными группами разной полярности. Пример композиционной присадки (литературные данные): этилгексилнитрат (ЭГН) – цетаноповышающая присадка; АМА-АН – депрессорная присадка, сополимер алкилакрилата СН2=СНCOОR(где R – алкильный радикал, содержащий не менее 16 атомов углерода) и акрилонитрила Таблица 9 – Свойства компонентов (АН); композиционной присадки (E*- энергия алкиламин (R = C10–C19) возбуждения в триплетное состояние). Расчет итаконовой кислоты (дисперметодом PM6 гатор А); Дипольный * E , кДж/моль Компоненты СМ-1 – противоизносная момент, Д присадка, 50%-ный конценЭГН 3,72 135,1 трат высших алкиламидов (R = А 5,86 150,2 C10–C20) ненасыщенных жирАМА-АН 2,19 213,8 ных кислот в углеводородном АС 5,65 297,1 растворителе; СМ-1 4,56 260,2 34 алкилсульфонат кальция (АС, R =C5–C12) – антидымная присадка; Расчёт стабильности компонентов присадки проводилась в предположении, что при воспламенении ДТ могут протекать процессы возбуждения молекул присадок в триплетное состояние и их последующего распада на радикалы. Как следует из таблицы, энергия возбуждения компонентов композиционной присадки в низшее триплетное состояние возрастает в ряду E*ЭГН<E*A<E*АМА-АН <E*СМ-1 <E*AC, что свидетельствует об увеличении стабильности присадок в ряду ЭГН<А< АМА-АН< СМ-1< AC. Квантово-химические методы использованы для расчёта энергии взаимодействия молекул парафиновых углеводородов C16-C21 с депрессорной присадкой на основе сополимеров алкилакрилатов и -олефинов. Парафиновые углеводороды (н-алканы) в ДТ при понижении температуры кристаллизуются, образуя пространственную структуру, что приводит к потере текучести топлива. Температура кристаллизации парафиновых углеводородов повышается по мере увеличения их молекулярной массы и температуры кипения. Наиболее высокую температуру кристаллизации имеют углеводороды с симметричным линейным строением молекул (табл. 10). Таблица 10 - Температуры кипения и кристаллизации нормальных парафиновых углеводородов, входящих в состав ДТ Парафиновый углеводород Декан (C10H22) Ткип, С 174,1 Ткрист, С −30,0 Парафиновый углеводород Гексадекан (C16H34) Ткип, С 287,1 Ткрист, С 18,1 Ундекан (C11H24) 195,9 −25,6 Гептадекан (C17H36) 302,6 22,0 Додекан (C12H26) 216,3 −9,7 Октадекан (C18H38) 317,4 28,0 Тридекан (C13H28) 235,5 −6,0 Нонадекан (C19H40) 331,6 32,0 Тетрадекан (C14H30) 253,6 5,5 Эйкозан (C20H42) 345,1 36,4 Пентадекан (C15H32) 270,7 10,0 Генэйкозан (C21H44) 356,0 40,4 Для снижения температуры застывания ДТ за рубежом применяют депрессорные присадки на основе полимеров - высших метакрилатов. Эффективность товарных полиметакрилатов в качестве депрессорных присадок обусловлена строением молекул этих присадок: длиной и степенью разветвленности углеводородной цепи, природой и расположением алкильных заместителей. Очевидно, что решающую роль в кристаллизации н-алканов в присутствии депрессоров при понижении температуры ДТ играют межмолекулярные взаимодействия как между молекулами н-алканов, так и н-алканов с молекулами депрессора. С целью выявления механизма действия присадок в данной работе были рассчитаны энергии межмолекулярных взаимодействий молекул депрессора с высшими н-алканами ДТ и энергии взаимодействия между молекулами налканов. Расчеты выполнены полуэмпирическим квантово-химическим методом PM6, входящим в пакет MOPAC2009 (табл. 11). 35 Таблица 11 – Энтальпии образования н-алканов С16-С21, рассчитанные методом PM6, в сравнении с литературными данными (единицы измерения ккал/моль использованы для удобства сравнения с литературными значениями) н-Алкан C16H34 C17H36 C18H38 o , f H 298 o , f H 298 o , f H 298 ккал/моль (PM6) -86,07 -91,07 -96,07 ккал/моль Лит. -89,23 -94,15 -99,08 кДж/моль (PM6) -360,12 -381,04 -401,96 н-Алкан C19H40 C20H42 C21H44 o , f H 298 ккал/моль (PM6) -101,08 -106,08 -111,08 o , f H 298 o , f H 298 ккал/моль кДж/моль Лит. (PM6) -104,00 -422,92 -108,93 -443,84 -464,76 В расчётах энергии взаимодействия высших н-алканов с молекулами депрессора был использован фрагмент молекулы присадки на основе сополимера метилакрилата с олефинами с молярной массой 454,74 (рис. 25). Энтальпия образования этого фрагмента по данным расчета методом PM6 равна 1169,06 кДж/моль. Рисунок 25 – Структура фрагмента депрессорЭнтальпии образования ной присадки, использованного для расчета бимолекулярных комплексов энергии взаимодействия с высшими н-алканами фрагмента депрессора и молекулами н-алканов и энергии межмолекулярного взаимодействия компонентов этих комплексов приведены в табл. 12. Таблица 12 – Значения энтальпий образования бимолекулярных комплексов депрессора с н-алканами C16-C21 и энергии взаимодействия молекул в комплексах (PM6) o o f H 298 f H 298 ,компл , ,компл , Eвзаимод, Eвзаимод, н-Алкан н-Алкан кДж/моль кДж/моль кДж/моль кДж/моль (PM6) (PM6) C16H34 -1568,69 -39,53 C19H40 -1639,94 -47,97 C17H36 -1593,62 -43,52 C20H42 -1661,24 -48,34 C18H38 -1615,18 -44,15 C22H44 -1682,53 -48,71 Энергия взаимодействия н-алканов с депрессором растет с удлинением цепи углеводорода (табл. 12), причем наибольшее возрастание энергии взаимодействия происходит при переходе от чётного н-алкана к нечётному, что связано, по-видимому, с изменением симметрии молекул. По данным табл. 12 энергия взаимодействия н-алканов с депрессором значительно выше энергии теплового движения молекул при 40С, равной 2,43 кДж/моль, следовательно комплексы молекул н-алканов и депрессора образуются при температуре введения присадки в дизельное топливо. Об этом свидетельствует и тот факт, что обычно производители рекомендуют вводить депрессоры 36 при температуре выше температуры помутнения топлива, но при этом отмечают, что наивысшая эффективность действия присадки достигается при её введении в ДТ, в котором парафины находятся в полностью растворенном состоянии, т.е. при температурах выше 30C. На рис. 26 приведена структура бимолекулярного комплекса нC21H44, полученная оптимизацией геометрии методом PM6. В этом комплексе молекулы н-алкана Рисунок 26 – Бимолекулярный комплекс расположены параллельно друг из молекул н-С21H44: расстояние между другу. Аналогичное строение имеют бимолекулярные комплекмолекулами 0,15 нм сы остальных н-алканов. Таблица 13 – Энергия межмолекулярного Расчёты позволили уставзаимодействия н-алканов С16–С21 в бимоле- новить, что энергия взаимокулярных комплексах (PM6) действия н-алканов с молекуo Состав бимолеEвзаимод, лами депрессора выше, чем н f H 298(компл) , кулярного комкДж/моль алканов C16-C21 между собой кДж/моль плекса (табл. 12 и 13). Это отражает, C16H34 -756,62 -36,41 что при растворении депресC17H36 -799,24 -37,16 сорной присадки в ДТ происC18H38 -845,03 -41,08 ходит взаимодействие молекул C19H40 -887,65 -41,84 высших н-алканов с молекулаC20H42 -933,44 -45,76 ми депрессора с образованием C21H44 -976,06 -46,52 комплексов, устойчивых при положительных температурах топлива. С понижением температуры топлива такие комплексы могут являться, как отмечалось ранее, центрами кристаллизации высших парафинов. По результатам проведенных расчётов энергии взаимодействия н-алканов с молекулами депрессоров выявлен механизм действия депрессорнодиспергирующих присадок в ДТ. Показано, что центрами кристаллизации налканов в ДТ в присутствии депрессорных присадок являются комплексы молекул высших н-алканов с фрагментами полимолекулярной цепи депрессора. Наличие большого числа таких центров ведет к уменьшению размеров и изменению формы кристаллов н-алканов, выделяющихся при понижении температуры топлива. Разветвления и полярные группы в молекулах депрессоров препятствуют агрегированию кристаллов н-парафинов, снижая температуру застывания топлива. Из выявленного механизма действия депрессорно-диспергирующих присадок следует, что при низком содержании н-алканов в ДТ и достаточном количестве центров для образования комплексов с н-алканами в молекулах депрессора возможно снижение не только температуры застывания топлива, но и температуры его помутнения. Снижение температуры помутнения ДТ в присутствии депрессорно-диспергирующих присадок отмечалось в работах И.Н. Гришиной, Р.А. Тертеряна. 37 В четвертой главе по результатам квантово-химических расчетов методом PM6 свойств ароматических аминов выявлен механизм антидетонационного действия этих соединений при добавлении к бензинам. В России выпускаются следующие антидетонационные присадки к бензинам на основе ароматических аминов: N-метиланилин (монометиланилин), экстралин, АДА, ДАКС. Так, экстралин представляет собой технический Nметиланилин, содержащий 90% основного вещества и 10% смеси анилина и диметиланилина, присадка АДА – это монометиланилин, стабилизированный антиокислителем. Ароматические амины проявляют различную антидетонационную эффективность, что зависит от их строения и состава. Антидетонационные свойства ароматических аминов, определенные для автомобильного бензина с ОЧИ=86, приведены в табл. 14. Таблица 14 - Антидетонационные свойства ароматических аминов, использованных в качестве присадки к бензину с ОЧИ=86 Концентрация, Прирост ОЧ Наименование аминов % масс. ОЧМ ОЧИ Анилин 2,0 3,7 4,6 N-метиланилин 2,0 4,6 6,2 Смесь толуидинов 2,0 4,1 5,1 Орто-ксилидин 1,0 1,2 1,8 Пара-ксилидин 1,0 1,1 1,7 По мнению некоторых исследователей, антидетонационные свойства аминов определяются наличием атомов водорода, присоединенных к атому азота в молекуле. Эти атомы водорода взаимодействуют с радикалами, образующимися в бензино-воздушной смеси при горении и обрывают цепные реакции, приводящие к детонации смеси. Молекулы аминов, не содержащие атомов водорода у атома азота, проявляют слабые антидетонационные свойства. Другие авторы предполагают, что амины являются «ловушками» для радикалов, и процесс улавливания этих частиц аминами обусловлен ион-дипольным взаимодействием и возникновением водородных связей между протонами у атома азота амина и атомами кислорода гидроперекисей или протонами гидроперекисей и атомами азота в аминах. Были попытки установить корреляцию между энергией разрыва связи N-H в молекуле амина и его антидетонационными свойствами. Несмотря на большое число работ, посвященных исследованиям антидетонационных свойств ароматических аминов, механизм реакций, протекающих в присутствии этих антидетонаторов, полностью не выяснен, поэтому представляло интерес изучение реакций, которые могут протекать в процессе окисления бензино-воздушной смеси в присутствии ароматических аминов в качестве антидетонаторов. Для определения реакционной способности ароматических аминов проведен расчет энергии гомолитического разрыва связи N-H в ароматических аминах по реакции RNH2 RNH2 H2 методом PM6. Наибольшая энергия разрыва связи N38 H получена для анилина (356,1 кДж/моль) и м-толуидина (346,8 кДж/моль). В Nметиланилине эта величина составляет 327,3 кДж/моль, для 3,4-ксилидина и птолуидина расчетное значение энергии разрыва связи N-H равно 319,3 и 319,1 кДж/моль, соответственно, для 2,5-ксилидина – 318,7 и о-толуидина 315,3 кДж/моль. Энергии разрыва связей N-H в молекулах всех ароматических аминов имеют значения выше 300 кДж/моль. Столь большие значения отражают малую вероятность разрыва N-H-связей в молекулах ароматических аминов в процессах предпламенного окисления топливо-воздушной смеси. Детонационный режим горения топлива в двигателях внутреннего сгорания обычно связывают с разложением алкилпероксидов, которые образуются в больших концентрациях при окислении углеводородов, и, будучи неустойчивыми, разлагаются, приводя к взрыву топливо-воздушной смеси. Для определения наиболее вероятных направлений распада перекисных соединений были рассчитаны изменения энтальпии следующих реакций на примере распада пероксида C6H13OOH: r H 298 312,082 кДж C6H13OOH C6H13OO + H , (31) r H 298 149,216 кДж C6H13OOH C6H13O + HO , (32) r H 298 217,382 кДж C6H13OOH C6H13 + HOO . (33) Наименьшие затраты энергии, как следует из результатов расчета, требуются для распада перекиси C6H13OOH на гидроксильный радикал HO и радикал C6H13O (реакция 32). По-видимому, это направление разложения пероксидов в предпламенных процессах окисления углеводородов топлив и будет наиболее вероятным. Неспаренный электрон в радикале C6H13O локализован на атоме O, а в радикале C6H13 - на атоме углерода концевой CH2-группы, что отражается в высокой реакционной способности этих радикалов, реакционная же способность атома водорода хорошо известна. В пероксидных радикалах C6H13OO и HOO спиновая плотность неспаренного электрона распределена между атомами кислорода таким образом, что на концевом атоме O в радикале HOO она составляет 0,67, а в радикале C6H13OO эта величина равна 0,64. Таким образом, результатами расчётов установлено, что пероксидные радикалы являются более устойчивыми или, другими словами, менее реакционноспособными по сравнению с другими радикалами, которые могут образоваться при распаде пероксидов. Поскольку в публикациях высказано мнение о том, что антиоксиданты на основе ароматических аминов являются «ловушками» для радикалов, были проведены расчёты изменения энтальпии реакций между ароматическими аминами и наиболее реакционноспособными радикалами, лавинообразный рост концентрации которых в ходе распада пероксидов может вызывать детонацию. Изменение энтальпии реакций ароматических аминов с наиболее активными радикалами представлены в табл. 15. 39 Таблица 15 - Изменение энтальпий реакций ароматических аминов с радикалами № Реакция Δ r H 298 , кДж 1 C H NHCH + HO C H N CH + H O –173,648 6 2 3 4 5 5 3 6 5 3 2 C6H5NH2 + HO C6H5NH + H2O C6H5NHCH3 + C6H13O C6H5 N CH3 + C6H13OH C6H5NH2 + C6H13O C6H5NH + C6H13OH –144,871 – 88,935 – 60,158 -181,811 6 -97,098 7 –182,258 8 –97,545 Из данных табл. 15 следует, что энтальпия реакций ароматических аминов с радикалами HO и C6H13O лежит в пределах от –60 кДж до –182 кДж, причём при взаимодействии радикалов HO с ароматическими аминами изменение энтальпии реакции почти в 2 раза превышает изменение энтальпии реакций аминов с радикалами C6H13O. Все реакции, представленные в табл. 15, приводят к образованию радикалов ароматических аминов вида или , где R – углеводородная часть радикала. Кроме того, продуктами реакций 1-8 (табл. 15) являются вода или спирт C6H13OН. Определение стабильности радикалов и проведено по данным расчётов распределения спиновой плотности неспаренного электрона на атоме азота (табл. 16). Таблица 16 - Спиновая плотность неспаренного электрона на атоме N в ароматических аминах (расчет методом PM6) Радикал арома- Спиновая плотРадикал ароматичеСпиновая плоттического ами- ность неспаренского амина ность неспаренного на ного электрона электрона на атоме на атоме N N 0,727 0,685 м− C6H5NH 0,691 0,669 40 − 0,675 п− 0,677 0,673 Спиновая плотность неспаренного электрона на атоме азота радикалов ароматических аминов значительно ниже плотности неспаренного электрона в радикалах HO и C6H13O, что отражает низкую реакционную способностиь этих радикалов. Пониженная спиновая плотность на атоме азота в радикалах ароматических аминов обусловлена делокализацией неспаренного электрона в ароматическом кольце. На основании проведенных расчётов можно сделать вывод о том, что механизм антидетонационного действия ароматических аминов заключается в связывании ими наиболее реакционноспособных радикалов HO и C6H13O с образованием нейтральных молекул воды и спирта. Радикалы аминов, образующиеся в ходе реакции, обладают пониженной реакционной способностью и не участвуют в реакциях продолжения цепи при окислении бензина, что ведет к повышению его детонационной стойкости. Радикал , у которого спиновая плотность на атоме азота наиболее высокая (табл. 16) может участвовать в реакциях рекомбинации с активными радикалами при столкновении с третьей частицей. В главе 5 приведены результаты квантово-химических расчётов для исследования процессов нефтехимии: гомогенной реакции алкилирования бензолом пропиленом в присутствии алкилхлорсиланов, реакции дисмутации толуола и трансалкилирования толуола м-ксилолом. Экспериментальными исследованиями каталитической активности ряда органохлорсиланов: CH3SiCl3, (CH3)2SiCl2, (CH3)3SiCl, C2H5SiCl3, (C2H5)2SiCl2, (C2H5)3SiCl и C6H5SiCl3 при их концентрации 0,8 мол. % и мольном отношении C6H6:C3H6=3:1 показано, что наибольшей каталитической активностью обладает этилтрихлорсилан C2H5SiCl3. Каталитическая активность этилхлорсиланов резко уменьшается с уменьшением числа атомов хлора при их замене на этильный радикал у атома кремния. Выход изопропилбензола (ИПБ) при температуре 150 С и времени контакта 30 мин в присутствии этилтрихлорсилана составляет 25,2 % масс., а в присутствии триэтилхлорсилана – 7,5 % масс. При замене этильного радикала у атома кремния на метильный каталитическая активность органохлорсиланов в реакции алкилирования снижается незначительно. Выход ИПБ в присутствии метилтрихлорсилана при температуре 150С составляет 20,1 % масс. В ряду метилхлорсиланов, также как и в ряду этилхлорсиланов каталитическая активность резко снижается при замещении атомов хлора на метильную группу. Каталитическая активность фенилхлорсилана ниже активности этилхлорсилана и активности метилхлорсилана. Моделирование осуществлялось полуэмпирическим методом PM6, реализованным в программном пакете MOPAC2007. Создавалась модель системы из молекул алкилхлорсилана и пропилена, далее проводилась минимизация её энергии, а затем осуществлялось запрограммированное уменьшение расстояния между первым атомом углерода молекулы пропилена и атомом кремния молеку41 лы катализатора до величины, близкой к средней длине химической связи Si—C (0,19 нм): Данные по заселённостям внешних энергетических уровней атомов C(1) и C(2) молекулы пропилена приведены в табл. 17. Таблица 17 - Изменение заселённостей внешних орбиталей атомов С(1) и С(2) в молекуле пропилена при сближении с молекулами катализаторов Расстояние Si—C(1), нм 0,45 0,40 0,35 0,30 0,25 0.22 0,20 0,19 0,55 0,50 0,45 0,35 0,25 0,22 0,19 Заселенность орбиталей Расстояние по Малликену Si—C(1), нм C(1) C(2) Трихлорэтилсилан 3,27 3,27 3,29 3,31 3,37 3,43 3,48 3,49 Хлортриэтилсилан 3,28 3,28 3,28 3,26 3,33 3,38 3,46 Заселенность орбиталей по Малликену C(1) C(2) 2,93 2,93 2,92 2,89 2,78 2,64 2,53 2,48 0,40 0,35 0,30 0,25 0,22 0,20 0,19 Дихлордиэтилсилан 3,26 3,28 3,28 3,34 3,40 3,47 3,48 2,93 2,93 2,92 2,84 2,83 2,81 2,72 0,45 0,40 0,30 0,25 0,22 0,20 0,19 Трихлорметилсилан 3,28 3,28 3,29 3,31 3,36 3,39 3,41 2,93 2,93 2,92 2,81 2,77 2,71 2,72 2,93 2,93 2,91 2,83 2,71 2,61 2,56 Из табл. 17 следует, что в случае сближения с трихлоралкилсиланами уменьшение заселённости внешних орбиталей второго атома углерода молекулы пропилена проходит в большей степени, чем в случае ди- и монохлорпроизводных соединений. Таким образом, вероятно, трихлоралкилсиланы, в силу своего электронного строения и геометрических характеристик, активнее способствуют образованию изопропильного катион-радикала, вступающего в дальнейшие превращения. Это, в свою очередь, позволяет предположить, что органохлорсиланы могут проявлять каталитические свойства и в других подобных реакциях с участием непредельных соединений. В пятой главе представлен также механизм реакции дисмутации толуола в смесь ксилолов, предложенный на основе квантово-химических расчётов и результатов экспериментальных исследований кинетики реакции в присутствии катализатора EMCAT-100 при температурах 475, 500 и 525С. Экспериментально было установлено, что в ксилольной фракции содержание индивидуальных ксилолов располагается в ряду: м-> о- п-ксилол. С повы42 шением температуры это соотношение не меняется, но содержание смеси ксилолов в катализате увеличивается. Результаты расчётов состава равновесной смеси для реакции дисмутации толуола при температурах 748, 773 и 798 K приведены в табл. 18 Таблица 18 – Состав равновесной смеси реакции дисмутации толуола в диапазоне температур 743-798 K Толуол, мол. доли 0,4703 0,4708 0,4712 T, K 743 773 798 орто0,0663 0,0671 0,0678 Ксилолы, мол. доли метапара0,1370 0,0616 0,1363 0,0613 0,1356 0,0610 Бензол, мол. доли 0,2649 0,2646 0,2644 Расчёты состава равновесной смеси для реакции дисмутации толуола показали, что в диапазоне температур 743-798 K состав смеси практически не зависит от температуры, в ней содержание м-ксилола немного выше, и в 2 раза превышает содержание о- и п-ксилола (табл. 18). При этом следует отметить, что проведение реакции дисмутации толуола в потоке, вдали от равновесия, приводит к более низкому выходу м-ксилола по сравнению с равновесными условиями: выход м-ксилола снижается почти в 2 раза (табл. 19). Таблица 19 - Выход ксилолов от мольной скорости подачи толуола при разных температурах Мольная скорость подачи толуола, 103 моль/см3ч 11,29 14,10 18,00 11,40 14,10 18,00 11,40 14,10 18,00 Выход ксилолов, мольные доли o– м– п– t=475С 0,024 0,053 0,026 0,021 0,050 0,025 0,015 0,047 0,020 Среднее значение t=500С 0,039 0,090 0,036 0,032 0,076 0,031 0,028 0,066 0,026 Среднее значение t=525С 0,040 0,096 0,038 0,034 0,079 0,034 0,029 0,071 0,028 Среднее значение Константы скорости, 10-3 моль/см3ч ko– kм– kп– 0,285 0,297 0,292 0,291 0,663 0,789 0,767 0,739 0,323 0,362 0,319 0,334 0,478 0,489 0,548 0,522 1,223 1,252 1,280 1,253 0,437 0,461 0,458 0,456 0,680 0,700 0,720 0,690 1,500 1,740 1,650 1,630 0,740 0,760 0,750 0,750 Тем не менее, предсказываемое термодинамикой преобладание м-ксилола в равновесной смеси, выполняется и при проведении реакции в неравновесных поточных условиях. Значения энергии активации превращения толуола в ксилолы, E, и предэкспоненциального множителя k0 уравнения Аррениуса, полученные по данным табл. 19, указывают на бὁльшую скорость превращения толуола в м-ксилол (табл. 20). 43 Таблица 20 - Кинетические константы реакции дисмутации толуола Константа o-ксилол 83871 77,9 E, Дж/моль k 0 , моль/см3ч м-ксилол 71010 227,2 п-ксилол 74391 54,0 Предложен механизм дисмутации толуола в присутствии алюмосиликатного катализатора с участием иона H+: CatH + C6H5CH3 Cat¬(H+C6H5CH3) Cat¬(H+C6H5CH3) + C6H5CH3 Cat¬( C6H5CH3) + C6H6 +CH3+ Cat¬( C6H5CH3) + CH3+ CatH + C6H4(CH3)2. В пятой главе приведены также результаты атмосферного риформинга бензиновой фракции на цеолиталюмосиликатных катализаторах, модифицированных Al- и Zn-фенилсилоксанами. Установлена более высокая активность модифицированных катализаторов по сравнению с немодифицированными, что отражается в увеличении ОЧ риформата. Исследованиями крекинга н-гексадекана в присутствии алюмосиликатных катализаторов, содержащих молибден и никель, установлена изомеризующая и крекирующая способность этих катализаторов. На таких катализаторах проходят только реакции крекинга, изомеризации и диспропорционирования углеводородов и отсутствуют реакции дегидроциклизации с образованием нафтеновых и ароматических углеводородов. Отсутствие ароматики определяет ценность эксплуатационных свойств такого катализата в качестве высокооктанового компонента товарного бензина. Алюмосиликатные катализаторы, содержащие Mo и Ni могут быть использованы для переработки н-парафиновых концентратов в изопарафины с МОЧ=82-88 или ИОЧ=92-88 пунктов. В кристаллической решётке образцов катализаторов, прокалённых при 500С, присутствуют полосы поглощения 1600-1640 см-1, характерные для ОН-групп, связанных с кремнием или алюминием. Эти OH-группы играют важную роль в процессах каталитического превращения н-гексадекана за счёт подвижности атома H и участия катиона H+ в образовании промежуточных соединений. ВЫВОДЫ 1. Создано новое направление изучения процессов нефтепереработки и нефтехимии, основанное на компьютерном моделировании структуры молекул, межмолекулярных комплексов, квантово-химических расчетах их термодинамических параметров и энергии межмолекулярных взаимодействий. 2. Впервые установлен механизм наложения узких бензиновых фракций, заключающийся во взаимодействии бензола с другими ароматическими углеводородами и получено параметрическое уравнение, связывающее температуру начала кипения фракций с их плотностью. Разработан метод снижения содержания бензола в риформате до стандартов, соответствующих ЕВРО-4 и ЕВРО-5. 44 3. Впервые рассчитаны энергии взаимодействия легких меркаптанов с ароматическими углеводородами (УВ) бензиновых фракций, установлены закономерности распределения низкокипящих меркаптанов между узкими бензиновыми фракциями, разработан метод снижения содержания меркаптановой серы в риформате. 4. Полуэмпирическими квантово-химическими методами впервые изучен механизм действия депрессорно-диспергирующих присадок к дизельным топливам (ДТ) на их агрегативную и седиментационную устойчивость. Механизм включает сольватацию депрессорных присадок молекулами парафиновых УВ с ориентацией полярных групп в объем ДТ и защитой полярных групп в сольвате молекулами диспергирующих присадок. 5. Впервые определены структуры комплексов парафиновых УВ с присадками и рассчитаны тепловые эффекты при взаимодействии компонентов депрессорно-диспергирующих присадок с углеводородными соединениями ДТ. 6. Впервые создано уравнение для описания зависимости цетанового числа от концентрации инициатора горения дизельного топлива и проведен квантовохимический расчет механизма действия аналогичных присадок на цетановое число ДТ. 7. Впервые на основе квантово-химических расчетов выявлен механизм антидетонационного действия присадок на основе ароматических аминов. 8. Впервые создано параметрическое уравнение для расчета периода индукции окислении бензинов в присутствии анитиокислительной присадки в зависимости от ее концентрации и дано квантово-химическое обоснование действия присадки АГИДОЛ-1 на качество бензинов. 9. Исследована структура и состояние алкилхлорсиланов и определён механизм изменения их каталитической активности в реакции алкилирования бензола пропиленом и выбран оптимальный катализатор. 10. Впервые квантово-химическими методами исследовано каталитическое превращение метилбензолов в присутствии цеолиталюмосиликатных катализаторов и предложен механизм превращения. 45 ПУБЛИКАЦИИ 1. Любименко В.А., Петрухина Н.Н., Туманян Б.П., Колесников И.М. / Термодинамические параметры реакций превращения некоторых компонентов тяжелых нефтей при паротепловом воздействии// ХТТМ, 2012. - №4. – С. 27-33. 2. Любименко В.А., Петрухина Н.Н., Туманян Б.П., Колесников И.М. Термодинамические аспекты превращений гетероатомных соединений в условиях паротеплового воздействия. В сб. «Высоковязкие нефти и природные битумы: проблемы и повышение эффективности разведки и разработки месторождений: материалы Международной научно-практической конференции». – Казань: Изд-во «Фэн», 2012. – 380 с. (С. 85-89). 3. Любименко В.А., Виноградов В.М., Колесников И.М., Винокуров В.А. Методы молекулярной механики и квантовой химии в химических расчетах: Учебное пособие по курсу «Молекулярное моделирование и анализ». – М.: РГУ нефти и газа им. И.М. Губкина, 2007. –173 с. 4. Любименко В.А., Колесников И.М., Колесников С.И., Кильянов М.Ю. /Атмосферный риформинг бензиновой фракции на цеолиталюмосиликате //ХТТМ, 2008. – №6. – С. 27-29. 5. Любименко В.А., Гришина И.Н., Гизатуллин Р.А., Колесников И.М. / Связь индукционного периода окисления бензиновой фракции с концентрацией антиокислителя // ХТТМ, 2008. – №1. – С 34-35. 6. Любименко В.А., Дела Торре А.Р., Колесников И.М., Левинбук М.И., Алхимов С.Н. / Трансалкилирование толуола м-ксилолом //Технологии нефти и газа, 2009. – №2. – С. 26-27. 7. Любименко В.А., Василенко П.А., Петров С.И., Жалнина Т.И., Якубсон К.И. Патент RU 2164024, 2001. Способ определения содержания нефтепродуктов в воде. 8. Любименко В.А., Широков Д.В., Колесников И.М., Лапидус А.Л./ Квантовохимический анализ активности катализаторов процессов переработки компонентов попутных нефтяных и нефтезаводских газов // Нефтепромысловое дело, 2010. – №5. – С. 62-64. 9. Любименко В.А., Широков Д.В., Колесников И.М., Лапидус А.Л. / Квантовохимический подход к анализу активности катализаторов ароматизации пропана и алкилирования бензола пропиленом // Газохимия, 2010. – № 4-5. – С. 62-65. 10. Любименко В.А., Данилов А.М., Колесников С.И., Колесников И.М. / Математическая модель для расчета прироста цетанового числа дизельных топлив в присутствии инициатора воспламенения// ХТТМ, 2010. – №5. – С. 11-17. 11. Борщ В.Н., Любименко В.А., Кильянов М.Ю., Колесников И.М., Винокуров В.А. / Квантово-химическое исследование комплексообразования малеинимида с молекулами бензола и воды // Химическая физика, 2011. – Т. 30, №8. – С.11-21. 12. Колесников С.И., Любименко В.А., Кильянов М.Ю., Колесников И.М. Винокуров В.А., Иванов Е.В. / Взаимосвязь четкости ректификации и распределения бензола в бензиновых фракциях // ХТТМ, 2011. – Т.47, №3. – С. 27-31. 46 13. Любименко В.А., Колесников И.М., Зубер В.И., Дибров С.Н., Колесников С.И., Винокуров В.А. / Связь температур выкипания фракций углеводородов с плотностью исходного бензина // Труды РГУ нефти и газа им. И.М. Губкина, 2009. – .№ 2. – С. 103-112. 14. Борщ В.Н., Любименко В.А., Гришина И.Н., Колесников И.М. / Квантовохимическое исследование комплексообразования сукцинимида с углеводородами // Труды РГУ нефти и газа им. И.М. Губкина, 2009. - № 2. – С. 112-119. 15. Lyubimenko V.A., Kolesnikov I.M., Kolesnikov S.I., Kil’yanov M. Yu. Atmospheric reforming of benzene fractions on the zeolitealuminosilicate catalyst. 18th International Congress of Chemical and Process Engineering. 24-28 August 2008. Praha, Czech Republic. Summaries 4. PRES 2008 and System Engineering. Praha, Czech Republic, 2008. - P.1512-1513. 16. Lyubimenko V.A., Dela Torre A.R., Levinbuk M.I., Kolesnikov I.M. Toluene dismutation kinetics. 18th International Congress of Chemical and Process Engineering. 24-28 August 2008. Praha, Czech Republic. Summaries 4. PRES 2008 and System Engineering. Praha, Czech Republic, 2008. - P. 1514-1515. 17. Lyubimenko V.A., Dela Torre A.R., Levinbuk M.I., Kolesnikov I.M. Transalkylation of toluene with meta-xylene. 18th International Congress of Chemical and Process Engineering. 24-28 August 2008. Praha, Czech Republic. Summaries 4. PRES 2008 and System Engineering. Praha, Czech Republic, 2008. - P. 1516-1517. 18. Любименко В.А., Колесников И.М. Роль межмолекулярных взаимодействий в распределении общей и меркаптановой серы между фракциями углеводородов, выделенными из нефти. Тезисы XV Симпозиума по межмолекулярному взаимодействию и конформациям молекул. 14-18 июня 2010 г. Петрозаводск. – С. 30. 19. Любименко В.А., Колесников И.М., Зубер В.И., Дибров С.Н., Колесников С.И., Винокуров В.А. Межмолекулярные взаимодействия бензола с ароматическими углеводородами узких бензиновых фракций. Тезисы XV Симпозиума по межмолекулярному взаимодействию и конформациям молекул. 14-18 июня 2010 г. Петрозаводск. – С.122. 20. Широков Д. В., Любименко В.А., Колесников И. М. Квантово-химический анализ активности катализаторов процессов ароматизации пропана и алкилирования бензола пропиленом. Тезисы докладов VIII Всероссийской научнотехнической конференции «Актуальные проблемы развития нефтегазового комплекса России». 1-3 февраля 2010 г. – Ч. 1. – М.: РГУ нефти и газа им. И. М. Губкина, 2010. – С. 292. 21. Любименко В.А., Колесников И.М., Данилов А.М. Математическая модель для прироста октанового числа в зависимости от начального его значения и концентрации присадки // Тезисы докладов VIII Всероссийской научнотехнической конференции «Актуальные проблемы развития нефтегазового комплекса России». 1-3 февраля 2010 г. – Ч. 1 – М.: РГУ нефти и газа им. И. М. Губкина, 2010. – С. 278. 47 22. Любименко В.А., Колесников И.М., Олтырев А.Г. Связь между общим содержанием серы в сырье и содержанием меркаптановой серы во фракциях, выделенных из нефти. Тезисы докладов VIII Всероссийской научнотехнической конференции «Актуальные проблемы развития нефтегазового комплекса России». 1-3 февраля 2010 г. – Ч. 1 – М.: РГУ нефти и газа им. И. М. Губкина, 2010. – С. 291. 23. Любименко В.А., Колесников И.М. Оптимизация физико-химических свойств бензиновых фракций для производства риформата с минимальным содержанием бензола. Глубокая переработка нефтяных дисперсных систем. Материалы VI международной научно-технической конференции . – М.: Издательство «Техника», ТУМА ГРУПП, 2011. – С. 57-59. 24. Любименко В.А., Гришина И.Н., Колесников И.М., Колесников С.И. Связь индукционного периода окисления бензиновой фракции с концентрацией антиокислительной присадки. Глубокая переработка нефтяных дисперсных систем. Материалы VI международной научно-технической конференции. – М.: Издательство «Техника», ТУМА ГРУПП, 2011. – С. 54-57. 25. Любименко В.А., Колесников И.М., Колесников С.И. Распределение серы по узким бензиновым фракциям. Глубокая переработка нефтяных дисперсных систем. Материалы VI международной научно-технической конференции . – М.: Издательство «Техника», ТУМА ГРУПП, 2011. – С. 59-61. 26. Гришина И.Н., Любименко В.А., Колесников И.М., Винокуров В.А. Механизм действия депрессорно-диспергирующих присадок к дизельным топливам. Глубокая переработка нефтяных дисперсных систем. Материалы VI международной научно-технической конференции . – М.: Издательство «Техника», ТУМА ГРУПП, 2011. – С. 118-120. 27. Гришина И.Н., Любименко В.А., Колесников И.М., Винокуров В.А. Выявление механизма действия депрессорно-диспергирующих присадок к дизельным топливам. Актуальные проблемы развития нефтегазового комплекса России. IX Всероссийская научно-техническая конференция. 30 января – 1 февраля 2012 г. Тезисы докл. Ч. 1. – М.: Издательский центр РГУ нефти и газа им. И.М. Губкина. – С. 241. 28. Любименко В.А., Колесников И.М., Колесников С.И. Содержание серы в узких бензиновых фракциях. Актуальные проблемы развития нефтегазового комплекса России. IX Всероссийская научно-техническая конференция. 30 января – 1 февраля 2012 г. Тезисы докл. Ч. 1. – М.: Издательский центр РГУ нефти и газа им. И.М. Губкина. – С. 249. 29. Любименко В.А., Гришина И.Н., Колесников И.М., Колесников С.И. Зависимость индукционного периода окисления бензина от концентрации антиокислительной присадки. Актуальные проблемы развития нефтегазового комплекса России. IX Всероссийская научно-техническая конференция. 30 января – 1 февраля 2012 г. Тезисы докл. Ч. 1. – М.: Издательский центр РГУ нефти и газа им. И.М. Губкина. – С. 250. 30. Boucenna A., Dekkar S., Gherbia A., Колесников И.М., Колесников С.И., Любименко В.А./ Каталитический крекинг н-гексадекана в присутствии металлосиликатов Al, Ni и Mo // Нефтепереработка и нефтехимия. Научно48 технические достижения и передовой опыт. ЦНИИТЭНефтехим, 2012. №12. – С. 23-25. 31. Любименко В.А., Гришина И.Н., Колесников И.М., Колесников С.И. Оптимизация условий производства композиционной присадки //ХТТМ, 2013. №5. – С.15-18. 32. Lyubimenko V.A., Kolesnikov I.M. Intermolecular interactions of mercaptans with aromatic hydrocarbons of close-cut gasoline fractions. 21th International Congress of Chemical and Process Engineering. 23-27 August 2014. Praha, Czech Republic. Summaries 4. PRES 2014 and System Engineering. Praha, Czech Republic, 2014. - P. 832. 33. Lyubimenko V.A., Kolesnikov I.M., Kolesnikov S.I., Esipova E.V., Vinokurov V.A. Thermodynamic calculation of optimum structure of metal oxide catalysts. 21th International Congress of Chemical and Process Engineering. 23-27 August 2014. Praha, Czech Republic. Summaries 4. PRES 2014 and System Engineering. Praha, Czech Republic, 2014. - P. 814. 34. Любименко В.А., Колесников И.М. /Распределение общей и меркаптановой серы по узким бензиновым фракциям нефтяного сырья различного состава //ХТТМ, 2013. - №2. – С. 29-32. 35. Любименко В.А., Колесников И.М. /Межмолекулярные взаимодействия легких меркаптанов с ароматическими углеводородами узких бензиновых фракций прямогонного бензина // Ежемесячный научный журнал Международного Научного Института «Educatio», 2014. - №3. - С. 149-153. (ISSN 345671769) 36. Любименко В.А. / О механизме антидетонационного действия присадок к бензинам на основе ароматических аминов // Ежемесячный научный журнал Международного Научного Института «Educatio», 2014. - №3. - С. 154-157. (ISSN 34567-1769). 37. Любименко В.А. / Компьютерное моделирование структуры и свойств межмолекулярных комплексов в дизельных топливах в присутствии депрессорно-диспергирующих присадок //Труды РГУ нефти и газа им. И.М. Губкина», 2014. - №2. – С. 43-51. 38. Любименко В.А. / Взаимодействие депрессорных присадок с парафиновыми углеводородами в дизельных топливах //Труды РГУ нефти и газа им. И.М. Губкина», 2014. - №3. – С. 88-95. 39. Любименко В.А. Механизм действия присадок к дизельным топливам. Компьютерное моделирование и квантово-химические расчеты. – Изд-во Lambert Academic Publishing, 2015. – 56 c. Автор выражает искреннюю и глубокую благодарность научному консультанту д.х.н., профессору Винокурову Владимиру Арнольдовичу и д.х.н., профессору Колесникову Ивану Михайловичу за ценные рекомендации и советы при выполнении диссертационной работы. 49