Диссертация Нгуен Ван Бо размещено 24.10.2014 г., 6.39 МБ
advertisement

2
РЕФЕРАТ
165 с., 29 рис., 42 табл., 232 использованных источника
МЕХАНИЗМ
ГАЗОФАЗНОГО
НИТРОСОЕДИНЕНИЯ,
МОНОМОЛЕКУЛЯРНОГО
N-НИТРОСОЕДИНЕНИЯ,
РАСПАДА,
C-
O-НИТРОСОЕДИНЕНИЯ,
АНИОН-РАДИКАЛЫ, КАТИОН-РАДИКАЛЫ, МАСС-СПЕКТРЫ, НИТРОМЕТАН,
1-НИТРОПРОПАН, НИТРОБЕНЗОЛ, О-НИТРОТОЛУОЛ, ДИНИТРОТОЛУОЛ,
2,4,6-ТРИНИТРОТОЛУОЛ, ДИМЕТИЛНИТРАМИН, ОКТОГЕН, ЭТИЛНИТРАТ,
ТЭН,
СТРОЕНИЕ
МОЛЕКУЛ,
ПЕРЕХОДНОЕ
АКТИВАЦИИ, КВАНТОВО-ХИМИЧЕСКИЕ МЕТОДЫ
СОСТОЯНИЕ,
ЭНЕРГИЯ
3
СПИСОК УСЛОВНЫХ СОКРАЩЕНИЙ
В тексте диссертационной работы использованы следующие условные сокращения:
АР
Анион-радикал
ВВ
Взрывчатые вещества
ВЭС
Высокоэнергетические соединения
Гескаген 1,3,5-тринитро-1,3,5-триазациклогексан
ДНТ
Динитротолуол
КР
Катион-радикал
НБ
Нитробензол
НМ
Нитрометан
ННП
Нитро-нитритная перегруппировка
НС
Нитросоединения
НТ
Нитротолуол
Октоген
1,3,5,7-тетранитро-1,3,5,7-тетраазациклооктан
ППЭ
Поверхность потенциальной энергии
ПС
Переходное состояние
ТНТ
2,4,6-тринитротолуол
ТЭН
Пентаэритриттетранитрат
D(C–N)
Энтальпия диссоциации связи C-NO2
D(N–N)
Энтальпия диссоциации связи N-NO2
D(O–N)
Энтальпия диссоциации связи O-NO2
E
Энергия активации химической реакции
e
Заряд электрона
EA
Сродство к электрону
DFT
Density Functional Method
HF
Hartree-Fock Methods
IP
Потенциал ионизации
4
MP
Moller-Plesset
T
Абсолютная температура, К
R
Универсальная газовая постоянная
∆fH0
Энтальпия образования из элементов при стандартных
условиях (P = 1 атм, T = 298.15 K)
∆H≠
Энтальпия активации химической реакции
5
Оглавление
Оглавление ............................................................................................................................5
Введение................................................................................................................................7
Глава 1. Использование квантово-химических методов для изучения механизма
распада ион-радикалов нитросоединений (литературный обзор)................................ 12
1.1 Распад нитроалканов................................................................................................... 12
1.2 Механизмы газофазного мономолекулярного распада нитробензола и его
производных ...................................................................................................................... 17
1.3 Механизм распада ион-радикалов С-нитросоединений.......................................... 21
1.4 Кинетика и механизм распада алифатических нитроэфиров ................................. 30
1.5 Кинетика и механизм термического распада тетранитрат пентаэритрита ........... 36
1.6 Механизм распада ион-радикалов нитроэфиров ..................................................... 38
1.7 Механизм распада нитраминов.................................................................................. 39
1.8 Механизм распада ион-радикалов нитраминов ....................................................... 44
1.9 Некоторые выводы по обзору литературы и постановка задач исследования ..... 45
Глава 2. Исследование геометрической и электронной структуры молекул и ионрадикалов нитрометана, диметилнитрамина, этилнитрата и нитробензола ............... 48
2.1 Нитрометан .................................................................................................................. 49
2.2 Нитробензол................................................................................................................. 57
2.3 Диметилнитрамин ....................................................................................................... 68
2.4 Этилнитрат................................................................................................................... 75
2.5 Основные выводы по Главе 2 .................................................................................... 83
Глава 3. Особенности механизмов распада ион-радикалов тринитротолуола ........... 85
3.1 Основные каналы термодеструкции молекул нитротолуолов ............................... 85
3.2 Основные каналы фрагментации анион-радикала тринитротолуола .................... 94
3.3 Основные каналы фрагментации катион-радикала тринитротолуола ................ 101
6
3.4 Основные выводы по Главе 3 .................................................................................. 104
Глава 4. Особенности механизмов распада ион-радикалов этилнитрата и
тетранитропентаэритрита ............................................................................................... 107
4.1 Основные каналы фрагментации катион-радикалов этилнитрата и
тетранитропентаэритрита, .............................................................................................. 107
4.2 Основные каналы фрагментации анион-радикалов этилнитрата и
тетранитропентаэритрита ............................................................................................... 113
4.3 Основные выводы по Главе 4 .................................................................................. 117
Глава 5. Особенности механизмов распада ион-радикалов диметилнитрамина и
1,3,5,7-тетранитро-1,3,5,7-тетраазациклооктана (октогена) ....................................... 119
5.1 Основные каналы фрагментации катион- и анион-радикалов
диметилнитрамина .......................................................................................................... 119
5.2 Основные каналы фрагментации катион- и анион-радикалов октогена ............. 125
5.3 Основные выводы по данной Главе 5 ..................................................................... 132
Заключение и основные выводы.................................................................................... 134
Список используемой литературы ................................................................................ 137
7
Введение
Актуальность работы. Нитросоединения (НС) – большой класс органических
соединений, широко используются в качестве взрывчатых веществ (ВВ) и высоко
энергетических материалов (ВЭМ). В связи с этим реакционная способность и ее
зависимость от молекулярной структуры отдельных представителей данного класса
соединений является объектом большого числа экспериментальных и теоретических
работ и, тем не менее, продолжает привлекать внимание исследователей и в наши
дни. Одним из наиболее эффективных методов изучения механизмов распада НС
является
использование
масс-спектрометрии.
Однако
очень
часто
при
интерпретации данных масс-спектрометрии исследователю приходится полагаться
на свою интуицию и опыт для воссоздания полной цепочки многостадийной реакции
распада заряженной молекулы. В связи с этим использование методов квантовой
химии становится в последнее время важной частью масс-спектрометрических
исследований. Применение квантово-химических расчетов в области химии ионов
позволяет с высокой точностью определять механизмы реакции распада ионрадикалов, рассчитывать кинетические параметры, а также предсказывать наличие
интермедиатов, образование которых невозможно в реакциях молекул [1].
В последнее время значительно возрос интерес к изучению механизмов
реакций распада оптически-возбужденных [2-5] и ионизированных молекул
нитросоединений [6-17]. Изучение возбужденных состояний в основном связано с
возможностью прямого инициирования ВВ оптическим излучением, что позволит
отказаться от высокочувствительных к удару и нагреву инициирующих соединений
[18]. Изучение механизмов и кинетики реакций распада ионизированных молекул
представляет принципиальный интерес для разработки и совершенствования
устройств-анализаторов,
позволяющих
обнаруживать
мельчайшие
частицы
взрывчатых веществ, а также выделять их из состава других нитросоединений, в
основе которых лежат принципы масс-спектрометрии [12-17,19,20]. Надежная
8
идентификация взрывчатых веществ приобретает особую актуальность в связи с
возросшей угрозой совершения террористических актов.
Нетривиальность
механизмов
и
этой
задачи
кинетических
объясняется,
параметров
прежде
реакций
всего,
распада
различием
нейтральных
и
ионизированных молекул [21-27]
Целью настоящей работы выявление закономерностей распада ионрадикалов
нитросоединений,
относящихся
к
классу
высокоэнергетических
материалов, а также их модельных соединений на основе использования результатов
современных квантово-химических методов.
Конкретные задачи включают в себя:
1.
Определение возможностей квантово-химических методов передавать
геометрические параметры молекул нитрометана, этилнитрата, диметилнитрамина,
их ион-радикалов, а также энергии ионизации и сродства к электрону.
2.
радикалов
Изучение механизмов реакций газофазного распада молекул анионэтилнитрата
и
диметилнитрамина,
модельных
соединений
высокоэнергетических материалов.
3.
Исследование основных каналов фрагментации катион- и анион-
радикалов пентаэритриттетранитрата (ТЭНа), октогена и тринитротолуола.
В качестве объектов исследования были выбраны ион-радикалы этилнитрата,
диметилнитрамина,
(ТЭНа),
1-нитропропана,
нитробензола,
1,3,5,7-тетранитро-1,3,5,7-тетраазациклооктана
пентаэритриттетранитрата
(октогена)
и
2,4,6-
тринитрометилбензола (ТНТ).
Для достижения поставленной цели необходимо было:
На основе анализа возможностей квантово-химических методов предсказывать
геометрические
параметры
ион-радикалов
нитрометана,
этилнитрата,
диметилнитрамина, а также энергии ионизации и сродства к электрону, выбрать ряд
9
методов
для
дальнейшего
изучения
каналов
фрагментации
ион-радикалов
нитросоединений.
Исследовать
механизмы
газофазного
распада
модельных
соединений
высокоэнергетических материалов.
Изучить
основные
и
альтернативные
каналы
распада
молекул
–
представителей высокоэнергетических материалов.
С использованием имеющихся экспериментальных данных по масс-спектрам
изучаемых
соединений
рассмотреть
особенности
конкуренции
различных
механизмов газофазного мономолекулярного распада.
Научная новизна работы определяется тем, что в ней впервые выполнено
систематическое изучение основных механизмов газофазного распада ион-радикалов
высокоэнергетических материалов и их модельных соединений с использованием
различных квантово-химических методов. При этом:
1.
получены данные по основным стадиям элиминирования молекулы воды
от катион-радикала 1-нитропана;
2.
для катион- и анион-радикала ТНТ изучены реакции отрыва гидроксила
и нитрогруппы;
3.
обнаружено, что в случае ион-радикалов ТНТ основным первичным
актом распада является образование аци-формы соответствующих соединений;
4.
проведено
подробное
теоретическое
исследование
основных
альтернативных механизмов распада ион-радикалов диметилнитрамина и октогена;
5.
установлена важная роль процесса образования аци-формы при распаде
катион-радикалов С- и N-нитросоединений;
6.
изучена корреляция между потенциалом ионизации пара-замещенных
нитробензолов и значениями энергий диссоциаций связи C-N (D(C-N)).
Практическая значимость работы связана с тем, что полученные в ней
результаты
могут
быть
использованы
для
интерпретации
данных
масс-
спектрометрического анализа распада отрицательно и положительно заряженных
10
ионов нитросоединений. Сведения по механизмам распада анион-радикалов
представителей важнейших классов высокоэнергетических материалов могут быть
использованы при идентификации взрывчатых веществ с использованием методов
масс-спектроскопии отрицательно заряженных ионов.
Положения, выносимые на защиту:
1. Результаты изучения геометрии и электронной структуры катион-радикалов и
анион-радикалов нитросоединений.
2. Результаты изучения механизма разложения катион-радикалов и анионрадикалов в модельных соединениях.
3. Результаты изучения механизма разложения катион-радикалов и анионрадикалов 2,4,6-тринитротолуола, ТЭНа, октогена.
Достоверность
представленных
результатов
подтверждается
сопоставлением результатов расчёта с данными масс-спектрометрии, кинетики
термического разложения, по геометрии и термохимическим характеристикам
молекул.
Апробации
докладывались
работы.
на
Основные
Международной
результаты
научной
диссертационной
конференции
работы
«Химическая
термодинамика и кинетика» (Донецк, Украина, 2012 г); «XXX Всероссийском
симпозиуме молодых ученых по химической кинетике» (Московская область, 2012
г);
Всероссийской
молодежной
конференции
«Химия
под
знаком
Сигма:
исследования, инновации, технологии» (Казань, 2012 г); научной конференции
«Исследование прочности материалов и технического обеспечения» (Ханой,
Вьетнам, 2012 г.); III Международной научной конференции «Химическая
термодинамика и кинетика» (Великий Новгород, 2013 г.); Международной научнопрактической конференции «Технические науки: Теоретические и прикладные
аспекты» (Уфа, 2014 г.); XIV Российской конференции (с международным участием)
по теплофизическим свойствам веществ (Казань, 2014 г). Кроме того, результаты
11
работы представлялись на итоговых научно-технических конференциях КНИТУ в
2013 и 2014 гг.
Работа
выполнена
на
кафедре
катализа
Казанского
национального
исследовательского технологического университета (КНИТУ).
Публикации:
По
материалам
диссертационной
работы
имеются
18
публикаций, в том числе 9 статей в журналах, входящих в перечень ВАК; 1 статья,
опубликованная в международном журнале, 8 тезисов докладов на Международных
и Всероссийских конференциях.
Структура диссертационной работы: Диссертация изложена на 165
страницах, содержит 42 таблицы, 29 рисунков, список литературы включает 232
ссылки. Работа содержит введение, 5 глав, раздел «Заключение и выводы», список
литературы.
В
первой
главе
проводится
обзор
работ,
посвящённых
экспериментальному и квантово-химическому изучению кинетики и механизмов
распада молекул различных классов нитросоединений и их ион-радикалов. Во
второй главе рассмотрены некоторые методические особенности проводимых
квантово-химических исследований. В третьей, четвертой и пятой главах приводятся
основные данные по механизму и кинетике распада молекул, анион- и катионрадикалов этилнитрата, диметилнитрамина, пентаэритриттетранитрата, 1,3,5,7тетранитро-1,3,5,7-тетраазациклооктана и 2,4,6-тринитрометилбензола.
Автор
выражает
глубокую
благодарность
своим
учителям:
научному
руководителю, профессору Храпковскому Г.М., начальнику УИО КНИТУ Шамову
А.Г., доценту Цышевскому Р.В. за помощь и поддержку на всех этапах выполнения
работы. В обсуждении результатов расчётов в ряде случаев принимали участие
доценты Гарифзянова Г.Г, Егоров Д.Л. и Николаева Е.В., за что я им искренне
признателен.
12
Глава 1. Использование квантово-химических методов для изучения
механизма распада ион-радикалов нитросоединений (литературный
обзор)
В
литературном
обзоре
обсуждаются
основные
результаты
изучения
механизма распада анион-радикалов и катион-радикалов С-, N-, O-нитросоединений.
Учитывая тематику нашей работы, основное внимание уделяется результатам
квантово-химических исследований. В то же время следует иметь в виду, что при
изучении механизма распада ион-радикалов нитросоединений всегда учитываются
результаты, полученные для соответствующих молекул. Кроме того, необходимо
иметь ввиду, что одно из основных направлений использования данных по
молекулярной
структуре
и
механизму
распада
ион-радикалов
связано
с
использованием методов масс-спектрометрии для изучения механизма термического
разложения взрывчатых веществ (ВВ). Поэтому в литературном обзоре мы кратко
обсуждаем и результаты теоретического изучения механизма термического
разложения некоторых основных классов ВВ (нитроалканов, нитроаренов, нитратов
алифатических спиртов, алифатических нитраминов), ион-радикалы которых
исследуются нами. В ряде случаев мы приводим минимально необходимые сведения
и о важнейших экспериментальных данных по кинетике термического разложения
нитросоединений. Подробный анализ этих результатов приводится в монографиях и
статьях, ссылки на которые имеются в соответствующих разделах литературного
обзора.
1.1 Распад нитроалканов
В настоящее время накоплен богатый материал по кинетике термического
распада различных классов нитросоединений, в первую очередь, алифатических и
ароматических
С-нитросоединений
[28-33].
нитроалканов является наиболее изученной [30-33].
При
этом
термодеструкция
13
На основе результатов экспериментального изучения кинетики газофазного
разложения нитроалканов были установлены два механизма первичного акта
термического их распада [28-32]:
1. Радикальный распад, первой лимитирующей стадией которого является
гомолитический разрыв связи C-NО2:
R-NO2 → R• + NO2•
(1.1.1)
2. Элиминирование HNO2. Этот механизм впервые был предложен в 1951 г.
[34] для реакции распада нитроэтана:
HC
CNO2
H
C
O
N
O
C
C
C
+ HNO2
(1.1.2)
Гомолитический разрыв связи C-N (1.1.1) обычно рассматривается как один из
основных механизмов распада С-нитросоединений. По радикальному механизму
распадается мононитросоединения (не имеющие атомов водорода в β-положении),
все гем-динитросоединения и 1,1,1-тринитросоединения и их галогенпроизводные, а
также кое-какие α-галогенмононитросоединения [35]. Результаты работ [35,36]
позволяют предположить, что изменения энергии активации реакции (1.1.1) в ряду
нитроалканов связаны в основном с стерическими напряжениями, вызванными
объемными заместителями (NO2, Cl, Br, J).
Результаты квантово-химического расчета [35-39] энергий диссоциации
большого числа мононитроалканов являются существенным дополнением к
имеющимся экспериментальным данным. Их использование позволяет гораздо
детальнее обсудить основные закономерности изменения энергии активации
радикального распада мононитрозамещенных алканов при введении в молекулу
нитрометана (НМ) различных заместителей. При этом нужно учитывать, что, за
исключением НМ и его производных, большинство исследованных нитроалканов
при
умеренных
температурах
(300–350°С)
распадаются
в
основном
по
14
молекулярному механизму (1.1.2) [35]. Поэтому значения D(C-N), рассчитанные с
использованием квантово-химических методов, приобретают особую значимость, в
том числе и при обсуждении конкуренции многообразных механизмов первичного
акта мономолекулярного распада. В свою очередь, при определении значений D(CN) термохимическим методом на основе энтальпий образования продуктов реакции
и исходных реагентов встает вопрос о надежности имеющихся данных по
энтальпиям образования данных соединений, а также о зависимости этого
термодинамического параметра от молекулярной и электронной структур [40-47].
Впервые
исследование
механизмов
молекулярного
распада
С-
нитросоединений с использованием полуэмпирических методов были проведены в
80-ые прошлого века Фаустовым и др. [48,49]. Они рассчитали структуру
переходного
состояния
реакции
элиминирования
азотистой
кислоты
из
нитроалканов и определили его неполярный характер. Начиная с 90-х годов
количество
теоретических
работ,
посвященных
исследованию
реакций
мономолекулярного распада С-нитросоединений, резко возросло. Примечательно,
что
в
подавляющем
большинстве
случаев
вычисления
выполнялись
с
использованием методов функционала плотности (DFT-методов) и неэмпирических
методов [50-54]. Применение этих методов позволяет увеличить надежность
получаемых оценок энергий активации реакций, что имеет принципиальное
значение при обсуждении механизмов газофазного мономолекулярного распада.
Расчетное
значение
энтальпии
активации
(175-190
кДж/моль)
реакции
β-
элиминирования азотистой кислоты из нитроэтана, полученное в работах [51-57],
хорошо согласуются с экспериментальной оценкой. Это доказывает, что нитроэтан
разлагается в основном по этому механизму. Об этом свидетельствует и тот факт,
что энтальпия активации процесса (1.1.2) является минимальной из всех возможных
альтернативных вариантов первичного акта мономолекулярного распада этого
соединения.
15
Как альтернатива радикальному распаду в литературе также активно
обсуждается еще один механизм термодеструкции нитроалканов, связанный с их
изомеризацией в нитриты, так называемая нитро-нитритная перегруппировка (ННП).
Например,
экспериментальное
исследование
термического
разложение
нитрометана (НМ) методом полевой масс-спектроскопии [35] показало наличие NO•
в продуктах его высоковакуумного пиролиза. Этот факт объяснили следующей
цепочкой реакций:
CH3NO2→ CH3ONO
(1.1.3)
CH3ONO → CH3O• + NO•
(1.1.4)
В ходе проведенного в 1986 году изучения ННП НМ методом инфракрасной
мультифотонной диссоциации (ИКМФД) [58] были обнаружены радикалы NO• и
CH3O•, которые в условиях эксперимента могли образовываться только по реакции
(1.1.4). Значения энергии активации для реакции (1.1.3) авторы работы [58] показали
в интервале 215.5-238.5 кДж/моль.
Результаты первой теоретической работы [59], выполненной с использованием
полуэмпирического метода MINDO/3, свидетельствуют о том, что ННП НМ имеет
существенно более низкий барьер, чем D(C-N). Данные более поздних работ,
полученных в рамках неэмпирических квантово-химических методов, а также
методов теории функционала плотности [60-69] указывают на преобладание
радикального распада в молекулах нитрометана и других нитроалканов над нитронитритной перегруппировкой.
Подводя итоги анализа результатов теоретического изучения ННП, следует
отметить, что имеющиеся в литературе результаты позволяют однозначно
утверждать о том, что нитро-нитритная перегруппировка, протекающая с
небольшими изменениями геометрических параметров, не может конкурировать с
альтернативными механизмами первичного акта термического разложения НМ и
других нитроалканов.
16
В определенных условиях важную роль при термическом разложении
нитросоединений может играть механизм образования их аци-форм на первичной
стадии реакции распада. Так, например, изучение каталитического разложения
нитрометана показало образование его аци-формы по реакции [30,31]:
O
CH3 NO2
H 2C
N
+
H O-
H 2C N
+
OOH
(1.1.5)
Надежно установлено, что через образование аци-форм на первой стадии
протекают жидкофазные распады нитрометана и нитроэтана. Этот механизм
реализуется и при газофазном распаде НМ при больших давлениях и температурах
600-8000С [70-73]. В работах [35,74,75] был предложен механизм элиминирования
воды от аци-формы нитрометана и динитрометана на основе расчета методом
MINDO/3. По этим данным образование аци-формы и дальнейший ее распад
является одним из основных каналов распада динитрометана. При этом надо
учитывать, что метод MINDO/3 дает колоссальные ошибки в значениях энтальпий
образования нитрометана и динитрометана. В дальнейшем изучение механизма
(1.1.5) проводилось методами HF, MP2, MP3, MP4(DQ), CCSD и B3LYP/6-31G(d)
[70,74].
Механизм образования аци-форм представляет, несомненно, интерес для
выявления закономерностей термораспада нитросоединений. Однако абсолютные
значения энтальпии активации для реакции (1.1.5) относительно большие. По этой
причине ее протекание в процессах мономолекулярного газофазного распада
нитросоединений является маловероятным [30,31]. Например, энергетический
барьер реакции образования аци-формы НМ несколько меньше, энтальпия
активации реакции (1.1.3). При этом он все же достаточно велик по сравнению с
энергией диссоциации связи C-N в НМ. В то же время известным фатом является то,
что при термораспаде нитрометана в жидкой фазе наблюдается значительное
снижение энергии активации первичного акта по сравнению с газофазным
17
процессом (на 80-100 кДж/моль-1) [30]. Поэтому в этих условиях реализация
механизмов разложения, связанных с образованием аци-форм нитроалканов является
вполне возможной.
1.2 Механизмы газофазного мономолекулярного распада
нитробензола и его производных
Систематические экспериментальные исследования, начатые еще в 60-ые
годы прошлого века, позволили
установить, что термическое
разложение
нитроаренов в газообразном состоянии протекает достаточно сложно. Это вызывает
затруднения
получение
в
вызывающих
доверие
кинетических
данных,
соответствующих первичному акту реакции [34,35]. Термохимических оценок
энтальпий образования соединений и энергий диссоциации разных связей в ряду
нитробензолов мало по сравнению с нитроалканами [36], что также затрудняет
интерпретацию результатов кинетического эксперимента. Поэтому применение
квантово-химических
методов
для
исследования
строения
и
реакционной
способности нитроаренов в процессах газофазного мономолекулярного разложения
представляет несомненный интерес [36].
На основе результатов экспериментального и теоретического исследования
для
нитроаренов
можно
предположить
существовании
трех
основных
альтернативных механизмов газофазного распада:
1. гомолитический разрыв связи C-NO2:
O
R
R
C
C
N
C
C
R
O
C
C
C
R
R
R
C
R
C
C
R
C
C
R
+ NO2
;
(1.2.1)
R
2. нитро-нитритная перегруппировка:
O
R
R
C
C
N
C
C
R
O
N
O
C
C
R
R
R
R
C
C
O
C
C
R
C
C
R
R
;
(1.2.2)
18
3. образование бициклического интермедиата:
O
R
R
C
C
N
C
C
R
O
O
C
C
R
R
N O R
C C
C R
R C
.
(1.2.3)
C C
R R
Установлено [76], что при сравнительно высоких температурах (~ 700-800K)
нитробензол (НБ), а также большинство его монофункциональных производных:
галоген-нитробензолы,
м-
и
п-нитроанилины,
нитротолуолы,
нитрофенолы
разлагаются по радикальному механизму (1.2.1). Вероятно, преимущественно по
механизму (1.2.1) протекает газофазный распад м- и п-динитробензолов, 1,3,5тринитробензола и ряда других нитроаренов с акцепторными заместителями.
Согласно условиям эксперимента [76] на это указывают кинетические особенности
протекания реакции по радикальному механизму, а также близость полученных
значений энергии диссоциации связи D(С-N). Подобные сведения об изменение
прочности и длины связи r(C-N) в ряду нитроаренов были получены в работе [77].
Прочность связи C-N в молекулах моно- и полинитроаренов, а также ее
зависимость от наличия различных заместителей, стали предметом изучения многих
теоретических работ [36,37,78-81], выполненных с использованием различных
методов теории функционала плотности и многоуровневых композитных методов.
Так, например, в работе [81] были рассчитаны энтальпии разрыва связи C-N в
мононитробензоле и 27 монозамещенных нитробензолов, содержащих различные
донорные и акцепторные заместители. Было установлено [81], что наличие
донорных заместителей, таких как CH3, NH2 и OH приводит к увеличению значения
D(C-N), тогда как введение в молекулу электрон акцепторных заместителей (NO2,
COOH, COH) снижает значение D(C-N).
Теоретическое изучение газофазного распада и влияния молекулярной
структуры на конкуренцию разных механизмов нитробензола проводилось
различными методами [37,82,83]. В работе [83] с использованием метода B3LYP/631G(d) были исследованы все основные механизмы мономолекулярного распада
19
нитробензола. Полученная с использованием этого метода оценка величины D(C-N)
для НБ составляет 292.6 кДж/моль, что хорошо согласуется с наиболее надежным
значением энергии активации газофазного распада НБ 291.6 кДж/моль [84]. На
основе этих результатов было отмечено, что участвовать в конкуренции с реакцией
радикального отрыва NO2 могут два процесса: ННП (1.2.2) (барьер активации 263.6
кДж/моль-1) и образования бициклического интермедиата (1.2.3) – 6(S)-7-окса-8азабицикло[4.2.0]-1,2,4-триен-8-оксида (227.4 кДж/моль-1).
Изучение механизма процесса (1.2.2) в ряду нитроаренов проводились с
использованием различных неэмпирических и DFT-методов [37,83,85], результаты
которых достаточно хорошо согласуются между собой. В работе [57] приведены
сведения о геометрических параметрах переходного состояния (ПС) и энтальпиях
активации для нитробензола и его монофункциональных производных, а также 1,2,5тринитробензола. Результаты многочисленных исследований указывают на то, что
энтальпия активации ННП меньше энергии диссоциации связи C-N, что делает
возможной конкуренцию процесса (1.2.2) с гомолитическим разрывом связи C-NO2
(1.2.1) [36]. Следует также отметить, что увеличение числа групп NO2 приводит к
незначительному снижению барьера реакции ННП (на 6.7 кДж/моль-1 по данным
метода B3LYP/6-31G(d)) [36]. Поэтому можно предполагать, что вклад ННП в
суммарную константу скорости газофазного распада для мононитробензолов должен
быть больше, чем для их полинитропроизводных.
В работах [35,86,87] было показано, что о-динитробензол распадается по
нерадикальному механизму – на это указывают как экспериментальное значение
энергии активации (Еа = 205 кДж/моль-1), так и результаты масс-спектрометрических
исследований.
В
работе
[83]
результаты
теоретического
исследования
c
использованием методов PBE/L1 и B3LYP/6-31G(d,p) показали, что вклад в процесс
термического распада о-динитробензола могут вносить ННП и образование
бициклического интермедиата. Вклад реакции изомеризации о-динитробензола в
(6S)-7-окса-8-азабицикло[4.2.0]-1,2,4-триен-8-оксида превалирует и вторая стадия
20
этого процесса – синхронное раскрытие четырехчленного цикла в бицикле и
элиминирование группы NO2 протекает с меньшей энтальпией активации, чем
бициклизация. По данным метода PBE/L1:
O
O
O
N
N
C
C
C
O
C
C
H
C
C
ON
ONO
H
O C
H
C
O N C
C
C
H C H
H
194.4
H
174.1
O
O
O N
C C
C H
HC
C C
H
H
В газообразном состоянии по аналогичному механизму распадаются другие
полинитробензолы,
например,
1,2,4-
и
1,2,3-тринитробензолы,
имеющие
нитрогруппы у соседних атомов углерода.
Кроме
рассмотренных
выше,
альтернативные
механизмы
нитросоединений.
Например,
в
литературе
термического
нередко
обсуждаются
разложения
описывается
и
другие
ароматических
механизм,
связанный
с
образованием нитрозоароматических соединений и атома кислорода на первой
стадии [88].
В работах [76,84,89] при изучении процесса термического распада онитротолуола, о-нитроанилина, о-нитрофенола было установлено, что энергии
активации первичного акта для них значительно ниже, чем у других соединений с
одинаковым числом нитрогрупп. Использование масс-спектрометрии показало
присутствие радикала ОН•, а также Н2О в продуктах разложения этих соединений
[35,86,87,90].
Было
высказано
предположение,
что
термолиз
указанных
ароматических нитросоединений начинается с изомеризации, сопровождающейся
внутримолекулярным переносом водорода к кислороду нитрогруппы по схеме:
R1H O
2
C
N
R
C
O
C
H
C
C
R3
C
Первые
H
R1 H O
R2
H
C
C
C
C
R3
результаты
C
C
N
H
O
.
квантово-химического
(1.2.4)
изучения
процесса
(1.2.4),
выполненные с использованием полуэмпирических методов MINDO/3 и MINDO,
21
были опубликованы в работе [91]. Было установлено, что барьер реакции
внутримолекулярного переноса атома водорода ниже, тем барьер ННП. В
дальнейшем в работе [64] этот процесс был рассмотрен для о-нитротолуола, онитроанилина и о-нитрофенола, а также для 2,4- и 2,6-динитро- и 2,4,6тринитропроизводных
толуола,
анилина
и
фенола
с
использованием
неэмпирического метода HF/6-31G(d). В работах [37,92,93] данная реакция
изучалась с использованием различных полуэмпирических, неэмпирических и DFTметодов.
1.3 Механизм распада ион-радикалов С-нитросоединений
При исследовании термодеструкции нитросоединений анализ продуктов
реакции проводится преимущественно с использованием хроматографических и
масс-спектрометрических методов [28,30-33]. Методы масс-спектрометрии дают
информацию о различных процессах фрагментации и перегруппировок [94].
Существенным является то, что эти методы могут применяться при высоких
температурах.
Поэтому
масс-спектрометрические
исследования
широко
используются для обсуждения механизма газофазного распада С-нитросоединений
[95,96,97]. При этом иногда результаты этого изучения расходятся с результатами
кинетического и квантово-химического исследований.
Так,
например,
исследование
продуктов
высоковакуумного
пиролиза
нитрометана методами масс-спектрометрии показывает на начальных стадиях
разложения присутствие интенсивной линии
m/z 30 (NO+), что исследователи
связывают с протеканием процесса его изомеризации в метилнитрит. Однако ни
результаты
как
квантово-химического,
так
и
кинетического
изучения
не
подтверждают этого вывода [30,52,59-61].
Одним из возможных объяснений этих противоречий может являться тот факт,
что частицы, первоначально появляющиеся в масс-спектрометре, являются ионами,
а, следовательно, могут распадаться по иным механизмам. В то же время, в
22
литературе преимущественно используется предположение о схожем химическом
поведении молекулы и ее ион-радикала [98].
Экспериментальные исследования термического распада катион-радикалов
нитрометана и его изомеров в основном активно проводились в 80-90-х годах
прошлого века. При этом систематических квантово-химических расчетов в
доступной нам литературе не обнаружено. Одной из таких работ является работа М.
Polášek и его соавторов [99] где исследовались изомерные продукты [C,H3,N,O]+•
распада
катион-радикалов
различными
нитрометана,
неэмпирическими
и
DFT
аци-нитрометана
методами,
а
и
метилнитрита
также
методами
нейтрализационно-реионизационной масс-спектрометрии.
В работе [100] авторы, исследуя реакции мономолекулярного распада
нитрометана и метилнитрита методом фотоионного-фотоэлектронного согласования
показали, что преимущественно эти процессы приводят к образованию катионов
CH3+,
NO2+,
NO+,
объясняя
это
протеканием
процесса
нитро-нитритной
перегруппировки. В работе [100] приводятся оценки энергии активации и энергии
реакции указанной реакции, равные 67.5 и -57.9 кДж/моль соответственно.
Соавторы работ [101,102], проводя исследование распада катион-радикалов
нитрометана и метилнитрита методом фотоионной-фотоэлектронной согласованной
масс-спектрометрии, показали, что нитро-нитритная перегруппировка катионрадикала нитрометана является самым энергетически выгодным процессом из всех
исследованных ими. По их данным для этой реакции энергия активации составляет
61.8 кДж/моль, а энтальпия реакции равна -60.8 кДж/моль.
В более поздней работе [103] проводили исследования изомеризации катионрадикала нитрометана в аци-нитрометан и метилнитрит методом изучения
метастабильных ионов и электронно-ударной масс-спектрометрии. О протекании
реакции образования аци-формы катион-радикала нитрометана говорило наличие в
масс-спектрах нитрометана и его аци-формы масс m/z 44 (M-HO•) и m/z 43 (M-H2O).
Появление массы, отвечающей воде, авторы работы [103] объяснили механизмом,
23
аналогичным механизму элиминирования H2O, предложенным в работах [35,70,75]
для объяснения механизма термического разложения динитрометана. Оценки
энергии активации и реакции процесса изомеризации катион-радикала нитрометана
в аци-форму, приведенные в работе [103] составляют 48.2 кДж/моль и -87.1
кДж/моль соответственно.
Как и другие исследователи, присутствие в масс-спектрах массы m/z 30
соавторы работы [103] связали с протеканием процесса (1.1.3). При этом было
показано, что CH3O+ образуется лишь в незначительных количествах, а полученный
метоксикатион мгновенно перегруппировывается в более устойчивую форму
CH2OH+. Из этого был сделан вывод, что катион CH3O+ может получаться при
распаде катион-радикала метилнитрита только из возбужденных электронных
состояний.
В работе [104] было изучена мономолекулярная фрагментация катионрадикала нитрометана с использованием метода фотоионизационной массспектрометрии, а также различными методами MS/MS, например, методом CIDI –
Collision induced dissociative ionization of neutral fragments. Результаты, полученные в
данной работе, хорошо согласуются с результатами работ [100] и [103]. Однако
авторы работы [104]
высказали предположение, что появление в масс-спектре
линии m/z 30 может быть связано не только с образованием NO+, но и с получением
CH2O+•.
Одна из первых попыток систематического изучения газофазного распада
катион-радикалов простейших нитроалканов с использованием современных
квантово-химических методов была предпринята в работе [24]. Расчет показал, что
процесс разрыва связи C-N в катион-радикале нитрометана является сложным
многостадийным процессом с барьером активации лимитирующей стадии 74.5
кДж/моль. Нитро-нитритная перегруппировка в катион-радикале нитрометана не
является первичным актом реакции, а протекает через образование промежуточного
интермедиата с практически разорванной связью C-N (422.3 пм). Энтальпия
24
активации
лимитирующей
стадии
этого
процесса
близка
к
барьеру
гетеролитического разрыва связи C-N (74.8 кДж/моль). При распаде катион-радикала
нитрометана образование аци-формы проходит в одну стадию. По сравнению с
реакциями молекулы нитрометана, для его катион-радикала образование аци-формы
является самым энергетически невыгодным процессом из всех рассмотренных. Его
энтальпия активации примерно в два раза превышает барьер ННП в катион-радикале
нитрометана
и
равна
116.5
кДж/моль,
что
хорошо
согласуется
с
экспериментальными оценками (96 кДж/моль [100]). Разброс экспериментальных
значений энтальпии реакции образования аци-формы катион-радикала нитрометана
лежит в пределах от -29.0 кДж/моль [100] до -87.1 кДж/моль [103]. Поэтому
полученное в работе [24] расчетное значение этой величины -51.4 кДж/моль можно
считать вполне удовлетворительным. Таким образом, по данным квантовохимического
расчета,
механизм
образования
аци-форм
в
катион-радикале
нитрометана, так же как и в нитрометане, при обычных условиях реализоваться не
может. Этот вывод подтверждается и экспериментальными исследованиями [103].
Изучение распада нитрометана методом диссоциативного присоединения
электрона [105] указывает на наличие следующих основных продуктов распада:
NO2−, O−, OH−, CN− и CNO−. По данным неэмпирического исследования
многоконфигурационным методом возмущений второго порядка MS-CASPT2 барьер
активации отрыва NO2− составляет 75.3 кДж/моль [11], что существенно меньше,
чем в молекуле (~60 ккал/моль). Следует отметить, что реакция отрыва NO2− от
анион-радикала нитрометана имеет четко локализованное переходное состояние,
тогда как обратная реакция отрывы NO2• от молекулы нитрометана является
безбарьерной реакцией.
Как и для катион-радикала нитрометана, разрыв связи С-N является
доминирующим каналом распада катион-радикала 1-нитропрпана [106]. Большой
интерес также представляет механизм распада, связанный с образованием аци-
25
формы катион-радикала 1-нитропропана в результате переноса γ-водорода (Рисунок
1.3.1).
Рисунок 1.3.1. Каналы распада катион-радикала 1-нитропропана
Дальнейший распад аци-формы катион-радикала 1-нитропропана может
происходить по трем основным каналам.
Первый канал соответствует отщеплению молекулы этилена, содержащей α- и
β-метиленовые группы, и образованием аци-формы катион-радикала нитрометана
[107-109] (канал А, Рисунок 1.3.1). Этот механизм широко известен в области массспектрометрии, как перегруппировка F.W. McLafferty. Впервые данная реакция была
предложена как один из возможных каналов распада ионизированных молекул
алифатических альдегидов [110]. Впоследствии механизм перегруппировки F.W.
McLaffert стал предметом изучения многих экспериментальных и теоретических
работ. Несмотря на то, что результаты некоторых экспериментальных работ
[111,112] указывали на протекание данной реакции в одну стадию, многие
исследователи больше склонялись к многостадийному каналу распада [113-115].
Наличие нескольких стадий также подтверждалось результатами теоретических
работ [116-123] по изучению распада различных классов органических соединений.
26
Второй возможный канал распада аци-формы катион-радикала 1-нитропропана
– отщепление гидроксила, содержащего γ-водород (канал В, Рисунок 1.3.1). [107109], и образование иона с m/z 72. Последующий распад иона m/z 72 может
протекать по нескольким возможным многостадийным механизмам, включая
элиминирование молекулы αH2O, содержащей два α-атома водорода и молекулы
γ
CH2O, содержащей два атома γ-водорода [106]. Результаты неэмпирического
квантово-химического исследования [123] указывают на существование двух
возможных циклических изомеров, соответствующих иону m/z 72. При этом процесс
отщепления гидроксила является сложным многостадийным процессом.
Третий возможный механизм распада аци-формы катион-радикала 1нитропропана – отрыв монооксида азота [106] (канал С, Рисунок 1.3.1) и
образование иона m/z 59. По данным работы [106], дальнейший распад иона m/z 59
происходит через отрыв молекулы водорода.
Механизм
распада
молекулярных
ионов
нитробензола
(НБ)
и
его
производных изучен гораздо хуже, чем реакции разложения нитроалканов. Впервые
подобные исследования были представлены в работах [124-127].
Для катион-радикала нитробензола в литературе приводят следующие
основные механизмы разложения [128,129]:
C6H6NO2+• → C6H5O+ + NO•
(1.3.1)
C6H6NO2+• → C6H5+ + NO2•
(1.3.2)
C6H6NO2+• → C5H5+ + CO + NO•
(1.3.3)
C6H6NO2+• → C4H3O+ + (NO•+ C2H2O)/(NO2• + C2H2)
(1.3.4)
C6H6NO2+• → C3H3+ + NO•+ CO + C2H2
(1.3.5)
C6H6NO2+• → C6H5O• + NO+
(1.3.6)
В масс-спектрах отсутствуют линии массы m/z 46, отвечающие катиону NO2+.
Это подтверждается теоретическими исследованиями [24,130], которые указывают
на высокое значение энтальпии реакции отрыва катиона NO2+ от катион-радикала
27
нитробензола (320.8 кДж/моль). Для сравнения, отрыв от молекулы нитробензола
радикала NO2 требует энергетических затрат почти в два раза меньших (148.5
кДж/моль).
По мнению большинства исследователей, реакции (1.3.1) и (1.3.6) должны
протекать
через
промежуточную
стадию,
связанную
с
нитро-нитритной
перегруппировкой, а появлению продуктов элементарной стадии (1.3.3) вероятнее
всего предшествуют, не только ННП, но и реакция (1.3.6). Не смотря на то, что, как
показывают многие исследователи [128], константа скорости разрыва связи С-N
намного превышает константу скорости перегруппировок, процессы образования
C6H5O+, C6H5+ и NO+ конкурируют друг с другом. Объяснение этого факта, по
мнению авторов работы [129] заключается в том, что реакции фрагментации и ННП
протекают через один и тот же промежуточный ион-дипольный комплекс, после
образования которого указанные процессы идут со сравнимыми скоростями:
O
O
N+
N+
–
O
O
N
O–
O
+ NO2
O
+ NO
O
+ NO
C5H5 + CO
Надо при этом отметить, что это предположение было сделано на
качественном уровне.
Образование катиона PhO+ происходит, вероятно, по двум каналам, о чем
говорит
бимодальность
линии
в
масс-спектрах
разложения
нитробензола,
28
отвечающему этому иону. Основной из них, вероятно, связан с нитро-нитритной
перегруппировкой.
По данным теоретического исследования [24] энтальпия активации ННП в
катион-радикале
нитробензола
составляет
114.2
кДж/моль,
что
ниже
термодинамической оценки гетеролитического разрыва связи C-N в катион-радикале
нитробензола с отрывом радикала NO2 на 34.3 кДж/моль.
Энергетические барьеры вторичных процессов разложения катион-радикала
НБ, связанных с образованием NO и NO+, рассчитанные в предположении
отсутствия
барьеров
обратных
реакций
равны
108.7
и
242.9
кДж/моль
соответственно [24]. Из этих значений видно, что наиболее энергетически выгоден
распад катион-радикала фенилнитрита на радикал NO и фенилоксикатион [24].
Энтальпия активации этой реакции меньше барьера ННП катион-радикала НБ, и,
следовательно, она не лимитирует весь процесс в целом. Однако этот результат не
объясняет появление в масс-спектрах катиона NO+.
Множество работ по исследованию распада замещенных нитробензолов [35]
появилось в середины 70-х годов и позднее. Все они дают в основном
согласующиеся результаты.
При исследовании разложения на начальных стадиях о- и п-изомеров
динитробензола и иоднитробензола методами дефокусировки поля [131] и
высоковакуумного пиролиза с использованием масс-спектрометров [132] было
установлено, что первичным актом распада катион-радикалов этих п-изомеров
является процесс ННП. Для о-динитробензола – вероятным первичным актом, по
мнению
авторов
работ
[131,132],
является
образование
циклического
активированного комплекса с последующим отрывом трех атомов кислорода, а для
о-иоднитробензола – разрыв связи С-N. Однако этот вывод можно сделать, опираясь
только на исследования методом высоковакуумного пиролиза [132].
Немного иные результаты показывает электронно-ударная фрагментация
[131]. Например, было обнаружено, что нитро-нитритная перегруппировка может
29
реализовываться только для нитробензола, а для о- и п-динитробензолов на первой
стадии происходит отщепление либо O• либо NO2•:
O
N
+O
–
O
N+
O–
-O
[M-O]
-NO2
–
O
N+
O
–
O
N+
O
-NO2
–
O
N+
O
-CO [C H ]
5 4
-NO
O
N
O
O
Таким образом, при исследовании механизмов термического разложения
различными методами (как в приведенных выше примерах, под действием
электронного удара без нагревания образца до высокой температуры и в случае
такого
нагревания)
могут
возникать
различные
их
интерпретации.
При
интерпретации результатов эксперимента, описанного в работе [131], возникает
вопрос о механизме термодеструкции при совместном воздействии электронной
ионизации и высокой температуры. Во многих случаях провести строгое разделение
факторов очень сложно.
Масс-спектр тринитротолуола (ТНТ) [133], полученный в рамках метода
электронной ионизации, имеет ярко выраженную линию m/z 210, соответствующую
отрыву гидроксильного радикала. Две линии с массовыми числами m/z 46 и m/z 181,
соответствующие отрыву групп NO2+ и NO2 имеют очень малые интенсивности.
Результаты изучения распада анион-радикалов трех изомеров динитробензола
[15] методом диссоциативного присоединения электрона указывают на одинаковый
механизм распада данных соединений. Масс-спектры анион-радикалов о-, м-, и пдинитробезолов имеют три линии наивысшей интенсивности с массовыми числами
m/z 168 (родительский ион), m/z 46 (NO2-) и m/z 138 (NO-). Анализ масс-спектров о-,
м-, и п-нитротолуолов также указывает на то, что отрыв NO2- будет доминирующей
реакцией при распаде данных производных нитробензола при диссоциативном
30
присоединении электрона [14]. Однако масс-спектры трех изомеров нитротолуола в
отличие от динитробензола имеют линии относительно высокой интенсивности с
массовыми числами m/z 119 и m/z 120, свидетельствующие о наличие гидроксила и
воды в продуктах распада. Результаты работы по диссоциативному присоединению
электрона к тринитротолуолу (ТНТ) указывают на то, что отрыв иона ОН будет
доминирующей первичной стадией распада анион-радикала ТНТ, тогда как отрыв
NO2-, судя по низкой интенсивности соответствующего пика, имеет очень низкий
выход.
1.4 Кинетика и механизм распада алифатических нитроэфиров
Анализ исследований по кинетике термического разложения нитратов
алифатических спиртов, выполненных в 50-60 годах прошлого века, детально
выполнен в работе [134]. И хотя позднее были получены новые результаты по
кинетике и механизму термического разложения этих соединений, следует иметь в
виду, что кинетика термодеструкции О-нитросоединений изучена не столь
подробно, как, например, кинетика термического распада С-нитросоединений.
Наиболее надёжные данные по кинетике и механизму термического распада
нитратов алифатических спиртов приводятся в работах [28, 30,33,135].
Проанализировав экспериментальные данные, авторы работы [135] сделали
вывод, что первичный акт термического разложения нитратов алифатических
спиртов связан с гомолитическим разрывом связи O-NO2.
Влияние
строения
молекул
на
величину
энергии
активации
и
предэкспоненциального множителя (А-фактора) реакции радикального распада
позволяют оценить результаты, полученные для реакций, протекающих в
газообразном состоянии. Так как в газовой фазе участвуют свободные молекулы, то
различные особенности влияния строения молекул на изменение в ряду
кинетических параметров должны проявляться в наиболее простой и наглядной
форме. Многие закономерности влияния молекулярной структуры на барьеры
31
реакций и предэкспоненциальные множители процессов радикального газофазного
распада нитроалканов и нитроаренов были установлены в ходе экспериментальных и
теоретических (квантово-химических) исследований [24,31,32,57,136-139].
В
отличие
от
нитроалканов
и
нитроаренов,
количество
нитратов
алифатических спиртов, распад которых изучался в газообразном состоянии,
небольшое (Таблица 1.4.1). Причем в работах разных авторов наблюдается большие
различия в значениях энергии активации и предэкспоненциального множителя
реакции, полученных для одного соединения, что препятствует сделать какие-либо
однозначные выводы. Так, например, изменение А-фактора в ряду представленных в
таблице 1.4.1 соединений превышает 2.8 логарифмические единицы. Тем не менее,
очень сложно связать это изменение со строением молекул, т.к. в ряде случаев оно
сопоставимо
с
разбросом
значений
предэкспоненциальных
множителей,
приводимых разными авторами для одного соединения. Например, для реакции
газофазного распада этилнитрата А-фактор в разных работах различается на 2.3
логарифмические единицы.
Таким
образом,
вопреки
тому,
что
опубликовано
множество
экспериментальных данных по кинетике термического разложения алифатических
нитратов, множество вопросов остаются спорными, а надежность некоторых
результатов эксперимента вызывает сомнения.
Мы
уже
термодеструкции
говорили,
что
экспериментальные
нитросоединений
объясняют
данных
радикальным
по
кинетике
механизмом
первичного акта реакции. Об этом говорят данные по составу продуктов на
начальных стадиях реакции, близость кинетических параметров в ряду изученных в
газообразном состоянии нитроалканов. Кроме того значения предэкспоненциальных
множителей для большинства реакций колеблются от 1014 до 1016 с-1, что характерно
для реакций гомолитического разрыва связи. Эти значения А-фактора наблюдаются
для нитрометана, нитроэтана, нитропропанов и ряда других мононитроалканов, для
которых радикальный механизм доказан при проведении процессов при достаточно
32
высоких температурах. Однако для реакций газофазного распада алифатических
нитратов эти результаты не являются бесспорным утверждением реализации
радикального механизма реакции.
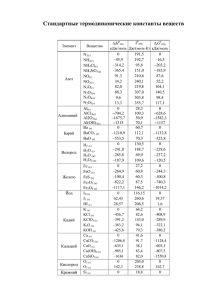
Таблица 1.4.1. Основные кинетические параметры начальной стадии термораспада
нитроэфиров в газовой фазе [135]
Соединение
Метилнитрат
Этилнитрат
Пропилнитрат
Т, °C
Е, кДж/моль
lgA c-1
211-240
162
14.33
166
15.5
169
15.7
240-290
151
13.7
180-214
170
16.0
175-209
159
14.74
182-219
158
14.37
151
13.86
165
15.5
161-181
164
15.1
100-170
168
15.78
171
16.3
181
168
100-150
176
15.27
168
16.4
300-530
167
16.5
110-150
171
16.4
260-360
169
16.5
150-200
176
15.93
Циклогексилнитрат
130-150
145
13.7
Изооктилнитрат
198-244
157
15.4
Нитрогликоль
140-170
147
13.67
Тринитроглицерин
140-165
163
16.4
150-160
150
15.0
171
160
2-Пропилнитрат
Пентаэритриттетранитрат
33
Связано это в первую очередь вероятно с тем, что, отсутствуют независимые
экспериментальные оценки барьеров различных альтернативных механизмов
мономолекулярного распада нитроалканов и алифатических нитратов.
Термохимические
оценки
D(C-N),
как
мы
уже
отмечали,
для
мононитроалканов немногочисленны и не отличаются высокой точностью. К тому
же при сравнительно невысоких температурах (~ 300°C) термодеструкция
нитроэтана и других мононитроалканов в газовой фазе, протекает в основном по
механизму элиминирования азотистой кислоты [28]:
C2H5NO2 → C2H4 + HNO2
(1.4.1)
С увеличением температуры преимущественно реализуется радикальный
механизм за счет больших значений предэкспоненциальных множителей реакции
(1016,0 с-1 для радикального распада, 1012,1 с-1 для реакции элиминирования HNO2).
Подобная схема термораспада возможна и для алифатических нитратов,
например, для метилнитрата:
CH3ONO2 → CH2O + HNO2
(1.4.2)
При наличии нескольких альтернативных механизмов первичного акта
реакции эффективная константа скорости, измеряемая экспериментально, равна
сумме констант элементарных процессов:
kэф. = Ak1 + Bk2 + Ck3…
(1.4.3)
где A, B, C – относительная доля соответствующих реакций.
Из этого следует, что в экспериментальную константу скорости могут давать
вклад не один, а несколько альтернативных процессов. Поэтому для правильной
интерпретации результатов эксперимента необходимо иметь оценки констант
скоростей основных альтернативных реакций, полученные независимым путем.
Применение
методов
квантовой
химии
для
анализа
бесспорности
экспериментальных данных, а также для исследования влияния строения молекул на
34
изменение в ряду кинетических параметров первичного акта термического
разложения нитроэфиров открывают новые возможности.
Сначала теоретическое изучение механизма термодеструкции нитроэфиров
проводилось с использованием полуэмпирических квантово-химических методов
MINDO/3 и CNDO/2 [139] (по утверждению авторов работы [139] метод MINDO/3
позволяет получить расчетные оценки энергии диссоциации связи O-NO2,
достаточно близкие к экспериментальным значениям).
Как
уже
говорилось,
для
нитроэфиров
могут
реализовываться
как
радикальный, так и различные нерадикальные механизмы первичного акта
термического разложения. Поэтому работа [59], посвященная теоретическому
изучению механизма термодеструкции метилнитрата, представляет значительный
интерес.
В
этой
работе
были
исследованы
две
альтернативных
схемы
нерадикального распада указанного соединения:
1.реакция элиминирования HNO2:
CH3ONO2 → CH2O + HNO2
(1.4.4)
2.бимолекулярная реакция:
CH3ONO2 + CH3ONO2→ CH3ONO + CH2OHONO2
(1.4.5)
По данным квантово-химического расчета реакция (1.4.4) имеет существенно
более низкое значение энтальпии активации (135 кДж/моль), чем процесс
радикального распада.
Структура переходного состояния и механизм реакции (1.4.5) напоминают
процесс бимолекулярного распада нитрометана [35], который протекает через
трехчленное переходное состояние и, вероятно, начинается с межмолекулярного
переноса водорода. При этом наблюдаются значительные различие в энтальпии
активации (∼30 кДж/моль) двух близких по механизму процессов. Одно из
возможных объяснений этого факта состоит в том, что прочность С-N и N=O связей
35
в нитрометане выше, чем O-NO2 и N=О связей в метилнитрате. Поэтому разрыв N=O
связи в нитрометане требует больших затрат энергии.
Излишне говорить, что реакция (1.4.5) является крайне маловероятной при
разложении нитроэфиров в парах. Однако она вполне может реализовываться при
жидкофазном
распаде
обсуждаемых
соединений.
Скорость
распада
по
бимолекулярному механизму может быть выше скорости радикального распада
только при сравнительно низких температурах. Это связано с более низкими в
сравнении с радикальным распадом нитроэфиров значениями энтальпий активации и
характерными для подобных процессов отрицательными значениями энтропии
активации. В то же время возможность протекания реакции 1.4.5 при термораспаде
алифатических нитратов вызывает серьезные сомнения [135].
Существенно дополняют информацию о возможных альтернативных вариантах
термического распада нитроэфиров данные, полученные в работе [140] с
использованием
методов
функционала
плотности.
Так,
например,
расчеты,
выполненные методом B3LYP/6-31G(d) указывают на то, что энтальпия активации
реакции элиминирования азотистой кислоты (164.3 кДж/моль) от метилнитрата
почти на 10 кДж/моль выше, чем оценка D(O-N) (156.3, кДж/моль), полученная в
рамках данного метода. В целом результаты, работы [140] предсказывают, что
энтальпия активации реакции газофазного элиминирования HNO2 от алифатических
нитроэфиров превышает D(O-N).
Для
газофазного
распада
нитроэфиров
также
представляет
интерес
нерадикальный механизм, связанный с β-элиминированием НNO3, изученный в
работе [139] с использованием полуэмпирического метода MINDO/3. Процесс
элиминирования НNO3 из нитроэфиров [141] начинается с внутримолекулярного
переноса водорода. Расчеты, проведенные с использованием MINDO/3, показывают,
что для этилнитрата энтальпия активации этого процесса на 22 кДж/моль ниже, чем
энтальпия реакции β-элиминирования НNO3, хотя и в этом случае энтальпия
активации (213 кДж/моль) выше D(O-N). При этом также следует отметить, что
36
расчетное
значение
энтальпии
активации,
учитывая
тенденцию
MINDO/3
переоценивать энтальпию образования переходного состояния, может быть
существенно завышенным. Поэтому нельзя полностью исключить этот процесс в
качестве альтернативы радикальному распаду. Более поздняя оценка энтальпии
активации реакции элиминирования HNO3 от этилнитрата, полученная в рамках
метода B3LYP/6-31G(d), на ~30 кДж/моль выше D(O-N), и на ~60 кДж/моль ниже,
чем энтальпия активации реакции β-элиминирования НNO3.
Данные работы [140] в целом указывают на то, что в условиях газофазного
распада при невысоких давлениях скорость радикального распада должна быть
значительно выше за счет большей величины предэкспоненциальных множителей.
Однако
ситуация
может
коренным
образом
измениться
при
проведении
термодеструкции при высоких и сверхвысоких давлениях, например, при детонации.
В этих условиях распад по радикальному механизму становится невыгодным,
поскольку ему отвечает значительно больший объем активации и реакции.
Протекание процесса по альтернативному, молекулярному механизму, в котором
реализуются относительно более компактные переходные состояния, может стать
основным каналом термического распада.
1.5 Кинетика и механизм термического распада тетранитрат
пентаэритрита
Среди нитратов алифатических спиртов в настоящее время наибольшее
внимание исследователей привлекает тетранитрат пентаэритрита (ТЭН). Он широко
применяется
при
промышленности.
проведении
Также
данное
взрывных
вещество
работ
в
используется
горнодобывающей
в
медицине
при
профилактике стенокардии и ишемической болезни мышц, как и нитроглицерин
относится к категории веществ, называемых вазодилататорами. Из-за относительно
низкой
термической
стабильности
и
чувствительности
к
механическим
воздействиям, ТЭН обычно рассматривают как самое чувствительное вторичное
37
взрывчатое вещество; очень часто вещества с более низкой чувствительностью к
тепловому и механическому воздействию по сравнению с ТЭН относят к первичным
взрывчатым веществам [142-145]. Широкое применение ТЭН в качестве вторичного
взрывчатого
вещества
послужило
причиной
для
большого
количества
экспериментальных и теоретических исследований его термической стабильности.
Разброс экспериментальных значений энергий активаций достаточно велик и
составляет ~170 кДж/моль (125-293 кДж/моль) [146-155]. Наиболее надежным
считается интервал 136.4-197.9 кДж/моль [151-155]. Самые последние оценки
энергии
активации
и
предэкспоненциального
множителя,
полученные
с
использованием метода дифференциальной сканирующей калориметрии в 2012 году,
составляют соответственно 136.4 кДж/моль и 7.47×1014 с-1. Как и в случае
алифатических спиртов, гомолитический разрыв связи O-NO2 обычно считается
основным первичным каналом распада ТЭН [146,148].
В работах [156,157] были изучены несколько каналов распада ТЭНа (1.5.11.5.7), включая реакции разрыва связей О-N (1.5.1), С-О (1.5.2), С-С (1.5.3),
элиминирования HNO2 (1.5.4), нитро-нитритной перегруппировки (1.5.5), а также
реакция (1.5.6), механизм которой впервые был рассмотрен на примере кремнийорганических соединений [158]. Так же была рассмотрена реакция одновременного
отщепления HONO и CO (1.5.7), сопровождающаяся разрывом одной из связей С-С
и миграцией водорода к центральному атому углерода.
C(CH2ONO2)4 → (CH2ONO2)3C-CH2-O• + •NO2
(1.5.1)
C(CH2ONO2)4 → (CH2ONO2)3C- H2C• + •ONO2
(1.5.2)
C(CH2ONO2)4 → (CH2ONO2)3C• + CH2O + •NO2
(1.5.3)
C(CH2ONO2)4 → (CH2ONO2)3C-CHO + HONO
(1.5.4)
C(CH2ONO2)4 → (CH2ONO2)3C-CH2-O-ONO
(1.5.5)
C(CH2ONO2)4 → (CH2ONO2)3C-O-CH2-ONO
(1.5.6)
C(CH2ONO2)4 → (CH2ONO2)3CH + HONO + CO
(1.5.7)
38
Приведенные
в
работе
[156]
данные
по
барьерам
активации
и
предэкспоненциальным множителям указывают на то, что радикальный отрыв NO2
следует считать доминирующим каналом распада ТЭН как в газовой фазе, так и на
поверхности кристалла. Расчетные значения энергии разрыва связи O-N (~146.4
кДж/моль) в газовой фазе, полученные в рамках методов PBE0 и wB97XD с базисом
6-31+G(2df,p), хорошо согласуются с другими теоретическими оценками (149.8
[157], 157.7 [159], 163.2 [160], 164.0 [161], 166.9 [160] кДж/моль). Эти оценки
приблизительно на 20-30 кДж/моль меньше, чем барьер активации реакции
элиминирования азотистой кислоты. Если учесть, что предэкспоненциальный
множитель
реакции
радикального
распада
значительно
превышает
соответствующую величину для реакции элиминирования азотистой кислоты,
можно утверждать, что возможность экспериментального наблюдения этого
механизма исключается.
1.6 Механизм распада ион-радикалов нитроэфиров
Анализ
литературы указывает на отсутствие результатов систематического
изучения каналов распада ион-радикалов нитроэфиров. Гораздо большее внимание
уделялось изучению фотохимических свойств молекул алифатических нитратов
[162,163]. Прежде всего, это связано с тем, что метилнитрат и этилнитрат являются
вторичными
загрязнителями
атмосферы,
образующиеся
в
результате
фотохимических реакций с участием углеводородов и оксидов азота. Основными
продуктами фотолиза этилнитрата являются NO2 и HONO [164], тогда как отрыв NO2
считается основным каналом фотохимического распада метилнитрата и этилнитрата
[165-167].
Анализ масс-спектров метилнитрата и этилнитрата [134] свидетельствуют о
наличии интенсивной линии m/z 46, которая соответствует образованию катиона
NO2+. Второй по интенсивности линией в масс-спектре этилнитрата с массовым
числом
m/z
76
соответствует
реакции
отщепления
метильного
радикала.
39
Примечательным является отсутствие данной линии в масс-спектре метилнитрата.
Также интересным является наличие линии с массовым числом m/z 29, которая
наблюдается в масс-спектрах этих двух соединений. Анализируя масс-спектр
этилнитрата, можно предположить, что данная линия соответствует реакции разрыва
связи O-NO2, сопровождающейся отрывом радикального фрагмента ONO2. Однако
подобный механизм не объясняет наличие данной линии в масс-спектре
метилнитрата. Мы можем только предположить, что линия с массовым числом m/z
29 может соответствовать иону с молекулярной структурой COH.
Для
подтверждения данной гипотезы необходимо проведение квантово-химических
расчетов.
Масс-спектр ТЭН, как и масс-спектр этилнитрата, имеет две ярко выраженные
линии с массовыми числами m/z 46 и m/z 76, что соответствует отщеплению
положительно заряженных частиц NO2+ и CH2ONO2+. Интересно отметить, что в
масс-спектре ТЭН отсутствует линия с m/z 29, наблюдаемая в масс-спектрах
метилнитрата и этилнитрата. Также следует отметить, что в масс-спектрах ТЭН и
метилнитрата отсутствует линия с массовым числом m/z 62, указывающая на
наличие ONO2+ в продуктах распада катион радикалов данных веществ, тогда как в
масс-спектре этилнитрата данная линия имеет практически нулевую интенсивность.
Данный факт является очень интересным, так как образование ONO2- наряду с NO2считаются основными каналами распада анион-радикала ТЭН [13,165].
1.7 Механизм распада нитраминов
Кинетика термического разложения первичных алифатических N-нитраминов
экспериментально изучена крайне слабо [30]. При этом выводы о механизме
процессов,
сделанные
различными
авторами,
в
значительной
мере
носят
гипотетический характер. В литературе отсутствуют подробные сведения о составе
продуктов на начальных стадиях разложения. Гораздо более детально исследованы
процессы термодеструкции вторичных N-нитраминов. В ряде обзоров [166-170]
40
проанализированы наиболее важные результаты экспериментальных исследований
термодеструкции этих соединений, выполненных в ведущих научных центрах
России
и
зарубежных
стран.
Ряд
интересных
обобщений
кинетических
закономерностей термодеструкции содержится в монографии [30]. Для нескольких
простейших вторичных N-нитраминов, прежде всего для диметилнитрамина
(ДМНА), имеются детальные данные по составу продуктов на начальных стадиях
разложения.
Кинетика газофазного термораспада ДМНА изучалась различными методами,
в том числе и современными физическими. В ряде работ приводятся подробные
схемы
механизма
разложения
этого
соединения.
Полученные
результаты
представляют значительный интерес для понимания основных закономерностей
термодеструкции первичных нитраминов. Особенно учитывая, что в большинстве
работ в качестве первичного акта газофазного распада всех алифатических
нитраминов предполагается гомолитический разрыв связи N-NO2.
Как уже говорилось, в большинстве случаев изучение кинетики термического
распада проводилось для вторичных нитраминов. Поэтому пристального внимания
заслуживает работа [171], в которой впервые были получены сведения по
газофазному
распаду
нескольких
простейших
первичных
нитраминов.
Как
указывают авторы, термораспад первичных алкилнитраминов в интервале 220-280°С
протекает до глубины превращения 80–90% по реакции первого порядка.
Параметры
первичных
уравнения
алкилнитраминов
Аррениуса
реакции
CH3NHNO2,
термического
C2H5NHNO2,
разложения
C3H7NHNO2,
изо-
C3H7NHNO2, C4H9NHNO2, изо-C4H9NHNO2, представлены в таблице 1.7.1.
Авторы работы [171] полагают, что положительные значения энтропии
активации и величины активационных параметров свидетельствуют о том, что
разложение первичных алкилнитраминов в газовой фазе протекает по радикальному
механизму с разрывом связи N–N аналогично вторичным нитраминам. В то же время
при анализе экспериментальных данных возникает ряд вопросов. Например, почему
41
предэкспоненциальный множитель реакции газофазного распада н-бутилнитрамина
более чем на 1.5 логарифметических единиц превышает соответствующую величину
в реакции газофазного распада этилнитрамина? Объяснить такие резкие колебания в
ряду аррениусовских параметров с позиций единого механизма реакции сложно.
Кроме того, предположить возможность изменения механизма реакции для
соединений близких по химическому строению трудно.
Таблица 1.7.1. Константы скорости (k×104, с-1) при различных температурах и
параметры уравнения Аррениуса термического разложения алкилнитраминов.
240°
250°
270°
280°
Ea, кДж/моль
Lg A,c-1
CH3NHNO2
1.41
3.35
14.5
–
171.6
13.65
C2H5NHNO2
0.75
1.77
7.78
14.4
174.9
13.69
C3H7NHNO2
0.48
1.03
5.47
11.5
189.0
14.89
изо-C3H7NHNO2
1.02
2.14
9.03
18.6
172.6
13.55
C4H9NHNO2
0.71
1.64
8.72
17.6
191.6
15.34.
изо-C4H9NHNO2
1.08
2.33
-11.1
23.7
182.7
14.61
Соединение
В ряду первичных N-нитраминов, скорее всего, имеет место конкуренция двух
параллельно реализуемых механизмов первичного акта реакции: радикального и
нерадикального
с
предэкспоненциального
более
низким
множителя.
значением
В
работах
энергии
[172-175],
активации
и
выполненных
с
использованием различных квантово-химических методов было показано, что
оценки D(N-N) в ряду первичных нитраминов значительно (на 30-40 кДж/моль)
превышают энергии активации газофазного распада. Это ставит под сомнение
принимаемый большинством исследователей радикальный механизм первичного
акта реакций этих соединений в газообразном состоянии.
В работах [168-171] был подробно изучен другой, более выгодный, механизм
распада первичных нитраминов, связанный с внутримолекулярным переносом
водорода от азота аминогруппы к кислороду нитрогруппы. По данным расчета с
использованием неэмпирических и DFT-методов энтальпия активации этой реакции
42
на 25–30 кДж/моль ниже D(N-N). Так, например, по данным метода B3LYP/6-31G(d)
величина D(N-N) в молекуле метилнитрамина составляет 195.2 кДж/моль, тогда как
энтальпия активации реакции образования аци-формы, полученная в рамках этого
метода, равна 149.4 кДж/моль. Исследование дальнейшего развития данного
механизма
до
элементарных
продуктов
показало,
что
один
из
наиболее
энергетически выгодных вторичных процессов распада аци-форм первичных Nнитраминов является реакция элиминирования воды. Этот процесс является
лимитирующей стадией данной многостадийной реакции. Энтальпия активации
указанного процесса составляет 166.8 кДж/моль по данным метода B3LYP/6-31G(d).
Существенно,
что
расчетные
оценки
барьера
реакции
согласуются
с
экспериментальными значениями энергии активации газофазного распада (Таблица
1.7.1), что указывает на возможный вклад обсуждаемого механизма в константу
скорости первичного акта газофазного распада.
N,N-диалкилнитрамины
самоускорением.
В
этом
в
газовой
отношении
фазе
они
распадаются
отличаются
от
с
небольшим
большинства
нитросоединений других классов: нитроэфиров, алифатических и ароматических
нитросоединений, которые в газовой фазе распадаются мономолекулярно, т.е.
процесс лимитируется лишь одной стадией. Первичным актом распада нитраминов
следует
считать
гомолитический
разрыв
связи
N-NO2,
поскольку
энергия
диссоциации этой связи в пределах ошибки опыта совпадает с энергией активации
реакции [176,177].
В таблице 1.7.2 приведены активационные параметры первой стадии (разрыв
связи N–NO2) процесса термического разложения N,N-диалкилнитраминов.
Впервые
теоретическое
изучение
механизмов
термодеструкции
N,N-
диметилнитрамина была предпринято в работе [178], в которой было установлено,
что DFT-методы позволяют получить расчетные оценки энергии диссоциации связи
N-NO2, достаточно близкие к экспериментальным значениям. Однако, как замечают
авторы барьер реакции отщепления азотистой кислоты по данным всех методов
43
выше
энергии
диссоциации,
что
исключает
возможность
протекания
термодеструкции N,N-диметилнитрамина по этому каналу.
Таблица
1.7.2.
Активационные
параметры
термического
разложения
N,N-
диалкилнитраминов в газовой фазе
Соединение
∆T, OC
Ea, Дж/моль
А, с-1
Лит-ра
N,N-диметилнитрамин
180-240
(162.8)
1013.7
[176]
N,N-диэтилнитрамин
180-240
(192.9)
1015.1
[177]
N-нитропиперидин
200-240
(176.1)
1014.8
[177]
(162.8)
13.6
[177]
N-нитроформолин
200-245
10
В работах [179-182] подробно рассматриваются альтернативные реакции
распада N,N-диметилнитрамина. По результатам расчетов, выполненных методом
B3LYP/6-31G(d), активационный барьер реакции элиминирование HNO2 составляет
190 кДж·моль-1, что на 13 кДж·моль-1 больше по сравнению с энергозатратами для
распада ДМНА по радикальному механизму (177 кДж·моль-1). Однако, как считают
авторы трудов [169], канал элиминирования HNO2 вносит незначительный вклад в
общий
механизм
невыгодной
деструкции
реакцией
является
N,N-диметилнитрамина.
процесс
Самой
нитро-нитритной
энергетически
перегруппировки.
Энтальпия активации ННП составляет 282 кДж·моль-1, что свидетельствует о
невозможности реализации термодеструкции по данному каналу [179].
Зачастую простейшие молекулы нитросоединений рассматриваются как
модельные соединения для изучения реакций
применяемых на практике
энергонасыщенных материалов или взрывчатых веществ. Так, например, молекулу
ДМНА
довольно
часто
рассматривают
как
прототип
таких
широко
распространенных материалов как гексоген и октоген. Как показывает анализ
литературы, данные по аррениусовским параметрам реакций термического
разложения гексогена и октогена существенно различаются в разных работах. Так,
например, значения энергии активации для термического распада октогена в
конденсированной фазе лежат в интервале 54.4 – 280.3 кДж/моль [183]. Для распада
44
в жидкой и газовой фазе интервалов изменения энергии активации несколько уже и
составляет 197.1-270.7 кДж/моль и 134.3-221.3 кДж/моль [183] соответственно.
Результаты теоретических работ по распаду октогена в газовой [184,185] и твердой
фазах [186-188] свидетельствуют о том, что разрыв связи N-N будет доминирующей
первичной реакцией с энергией активацией ~167.4-188.3 кДж/моль. Процессы
элиминирования HONO и ННП, скорее всего, не смогут конкурировать с
радикальным распадом из-за более высоких значений барьеров активации и более
низких значений предэкспоненциального множителя. Более того, следует отметить,
что наличие различных дефектов, например, вакансий на поверхности октогена
может дополнительно на 20.9 кДж/моль снижать энергию разрыва связи N-N в
молекуле октогена.
Результаты теоретического изучения распада гексогена в газовой фазе [189]
также указывают на то, что радикальный распад будет доминирующим механизмом
при распаде данного соединения.
1.8 Механизм распада ион-радикалов нитраминов
По распаду ион-радикалов нитраминов имеются только очень скудные данные.
Практический интерес в основном представляет распад анион-радикалов гексогена и
октогена.
Прежде
экспериментальных
всего,
такие
результатов,
данные
необходимы
полученных
в
для
результате
интерпретации
диссоциативного
присоединения электрона. Так, по данным работ [16,190] одним из основных
каналов распада анион-радикалов октогена и гексогена является отрыв аниона NO2-.
Помимо отрыва NO2-, анион-радикалы октогена и гексогена могут распадаться по
механизмам, основными стадиями которых является образование ионов C3H6N5O4− и
C2H4N3O2− соответственно. Об образовании данных ионов в продуктах распада
октогена и гексогена при диссоциативном присоединении электрона свидетельствует
наличие в их масс-спектрах линий высокой интенсивности с массовыми числами m/z
176 и m/z 102.
45
В работе [191] была осуществлена попытка изучить механизм распада анионрадикала гексогена с использованием квантово-химических методов. В этой работе
приводятся исключительно относительные энергии продуктов и возможных
интермедиатов, полученных в рамках метода MP2/AUG-ccpVDZ//B3LYP/6-31G(d).
Отсутствие
результатов
квантово-химического
изучения
существенно
затрудняет интерпретацию результатов масс-спектрометрического исследования
гексогена и октогена и других вторичных нитраминов. Линии наибольшей
интенсивности, наблюдаемые в масс-спектре ДМНА [133], полученные методом
электронной ионизации, соответствуют C2H5N2O+ (m/z 73), C2H4N+ (m/z 42) и C2H5N+
(m/z 43), образующихся в результате отрыва нейтральных фрагментов OH, HONOH
и HONO. Следует отметить, что в масс-спектре ДМНА отсутствует линия m/z 46,
соответствующая отрыву NO2+, тогда как линия, соответствующая отрыву молекулы
NO2, имеет относительно низкую интенсивность.
В отличие от ДМНА, масс-спектры электронной ионизации гексогена и
октогена [133] имеют две ярко выраженные линии с массовыми числами m/z 30 и
m/z 46. Они, скорее всего, соответствуют NO+ и NO2+. Далее в нашей работе мы
подробно рассмотрим механизмы распада ион-радикалов нитраминов и предложим
свой вариант интерпретации имеющихся экспериментальных данных.
1.9 Некоторые выводы по обзору литературы и постановка задач
исследования
Анализ изложенного выше материала позволяет сделать следующие выводы.
В
настоящее
время
в
литературе
имеются
достаточно
подробные
экспериментальные и расчетные данные по механизму термического распада
молекул различных классов нитросоединений, тогда как механизмы распада ионрадикалов изучены гораздо в меньшей степени. Здесь также стоит отметить, что
интерпретация масс-спектров и каналов распада ион-радикалов нитросоединений
зачастую проводятся интуитивно. В связи с этим, применение квантово-химических
46
методов
имеет
большое
значение
для
интерпретации
результатов
масс-
спектроскопического эксперимента. Тем более, как показывают уже имеющиеся в
литературе
данные
квантово-химических
исследований
каналов
распадов
заряженных молекул различных классов органических соединений, фрагментация
ион-радикалов очень часто протекает по отличным от молекул и зачастую более
сложным схемам.
Из приведенного выше материала можно также сделать вывод о том, что
исследование распада ион-радикалов требует индивидуального подхода. Так,
например, если в случае молекул этилнитрата основным каналом газофазного
распада является гомолитический разрыв связи O-NO2, то с большой степенью
вероятности можно предположить, что данный механизм будет иметь место и для
другого представителя нитроэфиров – молекулы ТЭН. Правильность данного
предположения подтверждается результатами многочисленных экспериментальных
и теоретических исследований. Та же тенденция наблюдается в ряду ДМНА,
гексоген, октоген. Однако масс-спектр этилнитрата содержит линии, которых нет в
масс-спектре ТЭН, а распад катион-радикала ДМНА будет проходить по совсем
иному сценарию, чем распад ионизированных молекул октогена и гексогена,
несмотря на наличие одинаковых функциональных групп в составе данных
соединений.
Более того, даже при сравнении масс-спектров нитрометана и 1-нитропропана
можно сделать вывод о разных каналах распада этих двух простейших
представителей класса нитроалканов. Так, например, если основным каналом
распада нитрометана является отрыв катион NO2+, то в случае 1-нитропропана
доминирующим механизмом будет отрыв молекулы NO2.
Также стоит отметить отсутствие теоретических исследований распада
отрицательно заряженных ионов нитросоединений за исключением работ по распаду
анион-радикала нитрометана. Тем временем, результаты подобных исследований
должны представлять большой практический интерес, учитывая применение
47
методов масс-спектроскопии отрицательных ионов при создании устройств
обнаружения взрывчатых веществ.
В нашей работе делаются попытки восполнить указанные выше пробелы.
48
Глава 2. Исследование геометрической и электронной структуры
молекул и ион-радикалов нитрометана, диметилнитрамина,
этилнитрата и нитробензола
Современные квантово-химические методы, эффективно используются для
изучения геометрической и электронной структуры молекул ВВ, а также влияния
этих характеристик
на
реакционную
способность. При
этом наибольшую
популярность приобрели гибридные методы теории функционала плотности,
позволяющие изучать механизмы реакций молекул с достаточно высокой
надежностью и со сравнительно небольшими затратами вычислительных ресурсов и
машинного
времени,
что
является
несомненным
преимуществом
перед
неэмпирическими методами [36,192]. В то же время, следует иметь в виду, что
выводы о механизмах могут существенно отличаться при использовании различных
квантово-химических методов. Например, данные работ [193,194] указывают на
существенное различие сведений по механизму реакции газофазного распада
катион-радикала бутана, полученных в рамках методов B3LYP и МР2.
Таким образом, выбор надежного квантово-химического метода для изучения
основных элементарных стадий распада ион-радикалов молекул нитросоединений
приобретает первостепенную важность. Существенное значение при выборе метода
теоретического
изучения
механизмов
реакций
является
правильная
оценка
параметров геометрической и электронной структуры молекул и ион-радикалов
нитросоединений. В литературном обзоре (глава 1) мы отмечали, что эта проблема
является очень важной и требует самостоятельного исследования.
В связи с вышесказанным в настоящей главе мы приводим сведения о
возможностях различных квантово-химических методов для расчета геометрических
параметров, зарядов на атомах, дипольных моментов, а также потенциалов
ионизации и энергий сродства к электрону молекул и ион-радикалов простейших
представителей различных классов ВВ [195]. В качестве объектов исследования
49
были
выбраны
нитрометан
диметилнитрамин
(ДМНА),
(НМ),
нитробензол
относящиеся
(НБ),
соответственно
этилнитрат
к
С-,
O-,
(ЭН)
и
и
N-
нитросоединениям.
В работе наибольшее внимание уделялось проведению
расчетов с
использованием методов теории функционала плотности (DFT). Прежде всего, это
связано с тем, что неэмпирические методы высокого уровня такие, как CCSD,
QCISD, CASSCF, являются ресурсоемкими, что делает их использование при
изучении свойств молекул реальных ВВ, состоящих обычно более чем из пятнадцати
атомов, и особенно механизмов реакций деструкции, в настоящее время
нецелесообразно. Поэтому для проведения расчетов использовались негибридные
(PBEPBE [196], PW91PW91 [197,198], OLYP [199,200]), гибридные (B3LYP
[200,202], BHandHLYP [202,201], M06 [203]) и дважды гибридные – B2PLYP [204] и
MPW2PLYP [205] методы теории функционала плотности [206,207].Также был
проведен расчет с использованием DFT метода wB97XD [208,209], учитывающего
дисперсионное взаимодействие. В дополнение к различным DFT методам были
также использованы неэмпирические методы HF и MP2 [210]. Все расчеты
проводились с использованием набора базисных функций 6-311+G(3df,p) и
программы Gaussian 09 [211].
2.1 Нитрометан
Геометрическая структура молекулы НМ представлена на рисунке 2.1.1(а).
Расчетные и экспериментальные данные длин связей и валентных углов приведены в
таблице 2.1.1.
Данные таблицы 2.1.1 показывают, что все методы с высокой надежностью
передают значения длин связей и валентных углов в молекуле НМ. Хорошее
согласие расчета с экспериментом наблюдается и для дипольного момента (Таблица
2.1.2). Наилучшие результаты достигаются при использовании негибридных методов
функционала плотности PW91PW91 и PBEPBE. В целом следует отметить, что
50
гибридные, дважды гибридные методы теории функционала плотности, а также
неэмпирические методы MP2 и HF незначительно завышают значения дипольного
момента в НМ. Хуже всего с экспериментом согласуются результаты метода MP2.
По данным работы [10] значения дипольного момента, рассчитанного методом
связанных кластеров с учетом тройных возбуждений CCSD(T) с базисом 6311++G(2d,2p), составляет 3.59 Д, что наиболее близко к оценкам метода B3LYP и
wB97XD.
Рисунок 2.1.1. Геометрическая структуры а) молекулы, б) анион- и в) катионрадикала нитрометана
Таблица
2.1.1.
Расчетные
и
экспериментальные
значения
геометрических
параметров структуры нитрометана (длины связей в Å, углы в градусах)
Методы
C1-H2 C1-N5 N5-O6 N5-O7
C1N5O6
C1N5O7
O6N5O7
H2C1N5
O6N5C1H2
HF
1.077
1.481
1.183
1.183
117.1
117.2
125.7
108.0
153.2
MP2
1.086
1.484
1.226
1.227
116.6
117.8
125.6
108.3
180.0
PBEPBE
1.093
1.504
1.229
1.228
116.7
117.3
126.0.
108.4
161.3
PW91PW91
1.091
1.503
1.228
1.228
116.5
117.6
125.9
108.5
170.1
OLYP
1.089
1.509
1.224
1.224
116.8
117.2
126.0
108.4
158.9
B3LYP
1.085
1.499
1.218
1.218
117.0
117.3
125.7
108.2
157.6
BHandHLYP
1.078
1.481
1.198
1.198
117.1
117.2
125.9
108.1
155.0
wB97XD
1.085
1.488
1.210
1.210
116.7
117.8
125.6
108.4
177.1
MPW2PLYP
1.082
1.490
1.219
1.219
116.6
117.7
125.7
108.3
180.0
B2PLYP
1.084
1.493
1.223
1.223
116.9
117.5
125.7
108.2
161.4
M06
1.086
1.492
1.207
1.208
116.3
117.7
126.0
108.7
177.8
Эксп. [212,213]
1.089
1.489
1.224
1.224
117.4
117.4
125.3
107.5
-
51
Таблица 2.1.2. Расчетные и экспериментальные значения дипольного момента (D, Д),
вертикальной энергии сродства к электрону (EAv, эВ), адиабатической энергии
сродства к электрону (EAad, эВ), вертикального потенциала ионизации (IPv, эВ),
адиабатического потенциала ионизации (IPad, эВ) и вертикального потенциала
ионизации, рассчитанного по теореме Купманса (IPK, эВ) в нитрометане
Методы
D
EAv
EAad
IPv
IPad
IPK
HF
3.95
1.05
1.46
11.32
9.34
12.63
MP2
4.12
0.80
0.21
11.74
11.97
12.50
PBEPBE
3.43
0.30
-0.23
11.05
10.75
6.97
PW91PW91
3.45
0.24
-0.30
11.13
10.83
7.05
OLYP
3.39
0.49
-0.03
10.93
10.62
6.86
B3LYP
3.61
0.24
-0.40
11.50
11.15
8.53
BHandHLYP
3.74
0.41
-0.32
11.77
10.94
10.60
wB97XD
3.63
0.43
-0.25
11.60
11.17
10.75
MPW2PLYP
3.84
0.29
-0.19
11.59
10.63
10.63
B2PLYP
3.84
0.29
-0.15
11.53
10.99
10.47
M06
3.54
0.38
-0.24
11.71
11.30
8.90
Эксперимент
3.46
-0.26±0.08
11.29; 11.80
11.08; 11.28
11.29; 11.80
[10]
[9]
[10]
[10]
[10]
Относительное изменение малликеновских зарядов на атомах [214] в
молекуле
НМ
передается
всеми
используемыми
методами
согласованно.
Исключение составляют заряды на атоме углерода. По данным большинства методов
на атоме углерода имеется небольшой отрицательный заряд, однако методы HF, MP2
и OLYP предсказывает наличие небольшого положительного заряда на углероде
(Таблица 2.1.3).
Анализ геометрических параметров анион-радикала нитрометана (АРНМ)
(Рисунок 2.1.1(б), Таблица 2.1.4) указывает на то, что присоединение электрона к
НМ приводит к незначительному изменению валентных углов C1N5O6, C1N5O7 и
O6N5O7 (Рисунок 2.1.1, Таблица 2.1.4). Среди
изменения длин связей, обращает
52
внимание сравнительно небольшое уменьшение связи C-N и увеличение связей N-O;
длины связей C-H остаются практически неизменными.
Таблица 2.1.3. Значения малликеновских зарядов на атомах нитрометана (е.э.)
C1
H2
H3
H4
N5
O6
O7
HF
0.025
0.217
0.217
0.215
0.810
-0.741
-0.743
MP2
0.029
0.219
0.224
0.223
0.665
-0.670
-0.690
PBEPBE
-0.023
0.182
0.189
0.192
0.439
-0.482
-0.497
PW91PW91
-0.019
0.178
0.191
0.192
0.393
-0.455
-0.481
OLYP
0.141
0.152
0.156
0.160
0.558
-0.578
-0.588
B3LYP
-0.040
0.190
0.193
0.196
0.455
-0.493
-0.501
BHandHLYP
-0.029
0.206
0.207
0.208
0.590
-0.589
-0.594
wB97XD
-0.091
0.204
0.214
0.214
0.461
-0.486
-0.517
MPW2PLYP
-0.013
0.203
0.211
0.211
0.538
-0.564
-0.586
B2PLYP
-0.016
0.204
0.209
0.209
0.536
-0.564
-0.577
M06
-0.150
0.201
0.210
0.210
0.393
-0.411
-0.453
Методы
Таблица 2.1.4. Расчетные значения геометрических параметров структуры анионрадикала нитрометана (длины связей в Å, углы в градусах)
C1-H2
C1-N5
N5-O6
N5-O7
C1N5O6
C1N5O7
O6N5O7
H2C1N5
HF
1.074
1.488
1.188
1.188
118.0
118.0
124.0
109.2
MP2
1.092
1.459
1.300
1.300
113.4
113.4
121.6
109.0
PBEPBE
1.099
1.458
1.303
1.303
114.0
114.0
122.3
109.3
PW91PW91
1.098
1.461
1.307
1.307
113.9
113.9
122.2
109.2
OLYP
1.095
1.463
1.304
1.304
114.3
114.3
122.3
109.3
B3LYP
1.092
1.446
1.286
1.286
114.7
114.7
122.2
109.2
BHandHLYP
1.084
1.472
1.307
1.308
114.2
114.2
122.4
109.3
wB97XD
1.092
1.471
1.308
1.308
114.1
114.1
122.1
109.2
MPW2PLYP
1.089
1.478
1.303
1.302
114.2
114.3
122.0
109.1
B2PLYP
1.091
1.461
1.307
1.307
113.9
113.9
120.0
109.1
M06
1.094
1.450
1.291
1.291
114.6
114.6
122.5
109.5
Методы
53
Также необходимо отметить, что фрагмент C1-N5-O6-O7 в случае АРНМ не
является плоским в отличие от молекулы НМ, что согласуется с результатами
теоретических
исследований,
выполненных
с
использованием
различных
неэмпирических методов высокого уровня [10,11].
Значения вертикального сродства к электрону (EAvert) (Таблица 2.1.2)
рассчитанные по формуле (2.1.1):
EAv = Etot(М•-) - Etot(Мeq)
(2.1.1)
где Etot(Мeq) – полная электронная энергия молекулы в равновесном состоянии,
Etot(М•-) – полная электронная энергия анион-радикала, геометрическая структура,
которого отвечает равновесной конфигурации молекулы Мeq, во всех случаях больше
нуля.
Оценки адиабатического сродства к электрону EAad, рассчитанные по
формуле (2.1.2):
EAad = Etot(Мeq •-) + ZPE(Мeq•-) - Etot(Мeq) – ZPE(Мeq)
(2.1.2)
где Etot(Мeq •-) и Etot(Мeq) – значения полной электронной энергии анион-радикала и
исходной молекулы в их равновесных конфигурациях, ZPE(Мeq•-) и ZPE(Мeq) –
энергии нулевых колебаний анион-радикала и молекулы, полученные в рамках
методов теории функционала плотности, в отличие от оценок методов HF и MP2
имеют
отрицательные
значения
и
находятся
в
хорошем
согласии
с
экспериментальным значением (Таблица 2.1.2). Наилучшее согласие достигается при
использовании методов wB97XD и M06. В целом следует отметить, что
негибридные и гибридные DFT методы систематически завышают значение EAad,
тогда как дважды гибридные методы занижают данное значение по сравнению с
экспериментом.
Результаты
неэмпирических
методов
не
согласуются
с
экспериментом и оценками DFT методов. Следует отметить, что оценки DFT
методов ближе к экспериментальному значению EAad по сравнению с результатами,
полученными с использованием неэмпирических методов высокого уровня [10].
54
По данным расчета в анион-радикале нитрометана (АРНМ) по сравнению с
молекулой НМ наблюдается увеличение абсолютного значения отрицательного
заряда на атомах кислорода и уменьшение величины положительного заряда на
атоме азота (Таблица 2.1.5). Также имеет место увеличение абсолютной величины
отрицательного заряда на атоме углерода и снижение положительного заряда на
атомах водорода. В то же время следует отметить, что метод MP2 предсказывает
увеличение положительного заряда на атоме углерода.
Таблица 2.1.5. Значения малликеновских зарядов на атомах анион-радикала
нитрометана (е. э.)
C1
H2
H3
H4
N5
O6
O7
HF
-1.867
0.434
0.434
0.416
0.973
-0.690
-0.700
MP2
0.064
0.143
0.143
0.142
0.258
-0.875
-0.875
PBEPBE
-0.252
0.141
0.141
0.142
0.067
-0.619
-0.619
PW91PW91
-0.256
0.142
0.142
0.140
0.002
-0.585
-0.585
OLYP
-0.106
0.116
0.116
0.114
0.065
-0.652
-0.652
B3LYP
-0.128
0.129
0.123
0.129
0.083
-0.669
-0.669
BHandHLYP
-0.041
0.138
0.138
0.130
0.165
-0.765
-0.765
wB97XD
-0.118
0.148
0.138
0.135
0.121
-0.699
-0.725
MPW2PLYP
-0.030
0.138
0.138
0.134
0.129
-0.754
-0.754
B2PLYP
-0.026
0.135
0.135
0.130
0.137
-0.756
-0.756
M06
-0.220
0.136
0.128
0.137
0.307
-0.744
-0.744
Методы
Как
и
в
случае
присоединения
электрона,
удаление
электрона
с
последующим образованием катион-радикала нитрометана (КРНМ) практически не
влияет
на
геометрическую
структуру
(Таблица
2.1.6,
Рисунок
2.1.1(в)).
Геометрические параметры катион-радикала НМ (КРНМ), как и в случае АРНМ,
незначительно отличаются от молекулы нитрометана.
Неэмпирический метод MP2, а также дважды гибридные методы теории
функционала плотности предсказывают незначительное увеличение длины связи N5O6 и уменьшение длины связи N5-O7, в то время как данные, полученные в рамках
55
негибридных и гибридных DFT методов, свидетельствуют об уменьшении длины
связи N5-O6 и увеличении длины связи N5-O7.
Таблица 2.1.6. Расчетные значения геометрических параметров структуры катионрадикала нитрометана (длины связей в Å, углы в градусах)
C1-H2
C1-N5
N5-O6
N5-O7
C1N5O6
C1N5O7
O6N5O7
H2C1N5
HF
1.081
1.498
1.233
1.240
122.1
120.6
117.2
106.1
MP2
1.089
1.488
1.345
1.105
110.6
128.1
121.3
105.5
PBEPBE
1.103
1.457
1.239
1.239
124.2
127.4
111.5
107.5
PW91PW91
1.100
1.456
1.238
1.238
124.4
126.4
111.5
107.4
OLYP
1.098
1.464
1.235
1.235
124.2
124.7
111.3
107.4
B3LYP
1.093
1.467
1.226
1.225
124.6
113.7
110.7
107.1
BHandHLYP
1.086
1.485
1.149
1.297
126.9
124.3
119.5
105.9
wB97XD
1.092
1.475
1.166
1.293
127.5
124.1
118.0
108.2
MPW2PLYP
1.091
1.488
1.307
1.167
113.0
124.4
119.6
105.4
B2PLYP
1.090
1.476
1.365
1.163
115.6
114.4
118.0
107.8
M06
1.095
1.476
1.292
1.164
113.5
127.8
118.7
106.0
Методы
Необходимо также отметить, что все используемые в данной работе методы
указывают на уменьшение значений валентных углов O6N5O7 и H2C1N5 в катионрадикале нитрометана по сравнению с исходной молекулой.
По
данным
большинства
используемых
нами
методов
(исключение
составляют результаты методов MP2 и HF) в КРНМ по сравнению с молекулой НМ
наблюдается увеличение положительных и уменьшение абсолютных величин
отрицательных зарядов на атомах (Таблица 2.1.7). При этом наибольший по
величине
положительный
заряд
сосредоточен
на
атоме
азота,
тогда
как
отрицательный заряд локализован на атомах кислорода. Метод MP2 предсказывает
уменьшение заряда на атоме углерода, тогда как по данным метода HF в КРНМ
наблюдается снижение заряда на атоме азота.
Значения вертикального (IPv) и адиабатического (IPad) потенциала ионизации
были рассчитаны на основе формул (2.1.3) и (2.1.4) соответственно:
56
IPv = Etot(М•+) - Etot(Мeq)
(2.1.3)
IPad = Eeq(М•+) - Etot(Мeq)
(2.1.4)
где Etot(Мeq)•- – полная электронная энергия молекулы в равновесном состоянии,
Etot(М•+) – полная электронная энергия катион-радикала, геометрическая структура,
которого отвечает равновесной конфигурации молекулы, Eeq(Мeq)•- – полная
электронная энергия катион-радикала в равновесном состоянии.
Таблица 2.1.7. Значения малликеновских зарядов на атомах катион-радикала
нитрометана (е. э.)
C1
H2
H3
H4
N5
O6
O7
HF
0.042
0.278
0.269
0.281
0.655
-0.281
-0.245
MP2
-0.002
0.280
0.284
0.284
0.864
-0.192
-0.518
PBEPBE
0.042
0.255
0.255
0.269
0.569
-0.196
-0.195
PW91PW91
0.052
0.248
0.257
0.268
0.558
-0.185
-0.199
OLYP
0.202
0.225
0.221
0.238
0.688
-0.289
-0.284
B3LYP
-0.003
0.261
0.258
0.273
0.575
-0.183
-0.179
BHandHLYP
-0.023
0.280
0.272
0.272
0.675
-0.377
-0.099
wB97XD
-0.053
0.259
0.284
0.284
0.613
-0.287
-0.100
MPW2PLYP
-0.006
0.282
0.256
0.282
0.657
-0.129
-0.342
B2PLYP
0.015
0.262
0.275
0.275
0.538
-0.118
-0.248
M06
-0.112
0.285
0.267
0.284
0.550
-0.053
-0.222
Методы
Как видно из таблицы 2.1.2 все методы с удовлетворительной точностью
передают значения IPv и IPad.
Оценки потенциала ионизации, полученные в рамках теоремы Купманса
(IPK) как абсолютное значение энергии высшей занятой молекулярной орбитали, с
использованием негибридных DFT методов достаточно плохо согласуются с
экспериментом (Таблица 2.1.2). Наиболее надежные расчетные оценки IPK были
получены при использовании методов BHandHLYP, wB97XD, MPW2PLYP и
B2PLYP.
57
2.2 Нитробензол
Нитробензол (НБ) является простейшим представителем класса ароматических
С-нитросоединений, к которому принадлежат такие известные взрывчатые вещества
как 2,4,6-тринитротолуол (ТНТ), 2,4,6-тринитрофенол (пикриновая кислота) и 1,3,5,триамино-2,4,6,-тринитробензол (ТАТБ).
Рисунок 2.2.1. Структуры: а) нитробензола; б) анион-радикала нитробензола и в)
катион-радикала нитробензола
Структура молекулы НБ представлена на рисунке 2.2.1(а). Расчетные и
экспериментальные геометрические параметры приведены в таблице 2.2.1.
Расчетные данные (Таблица 2.2.1) указывают на то, что геометрические
параметры молекулы НБ, предсказанные разными квантово-химическими методами,
находятся в хорошем согласии и между собой и с экспериментом. Следует отметить,
что эти результаты согласуются с расчетными данными, полученными в работе [24]
с использованием метода B3LYP и базиса 6-31G(d).
Хорошее согласие расчета с экспериментом наблюдается и для дипольного
момента (Таблица 2.2.2). Наилучшие результаты достигаются при использовании
негибридных методов функционала плотности PBEPBE, PW91PW91 и OLYP. В
целом следует отметить, что гибридные и дважды гибридные методы теории
функционала плотности незначительно завышают значения дипольного момента в
58
НБ. Хуже всего с экспериментом согласуются результаты методов HF, MP2,
mPW2PLYP и B2PLYP .
Таблица
2.2.1.
Расчетные
и
экспериментальные
значения
геометрических
параметров нитробензола (длины связей в Å, углы в градусах)
Метод
С6-C1 C6-N12 N12-O13 C1-H7 C1C6C5 C6C1C2 C1C2C3 C2C3C4 C6N12O13 O13N12O14
HF
1.378 1.464
1.185
1.071
122.4
118.5
120.1
120.4
117.6
124.8
MP2
1.389 1.471
1.227
1.084
122.8
118.1
120.6
119.9
117.5
124.9
PBEPBE
1.394 1.483
1.232
1.090
122.3
118.5
120.2
120.3
117.5
125.0
PW91PW91
1.393 1.481
1.232
1.088
122.3
118.5
120.2
120.3
117.5
124.9
OLYP
1.395 1.488
1.228
1.085
121.9
118.7
120.3
120.1
117.6
124.9
B3LYP
1.388 1.477
1.221
1.081
122.3
118.5
120.2
120.3
117.7
124.7
BHandHLYP
1.378 1.461
1.201
1.073
122.4
118.5
120.2
120.4
117.6
124.8
wB97XD
1.383 1.474
1.212
1.081
122.4
118.4
120.2
120.3
117.6
124.8
MPW2PLYP
1.386 1.471
1.221
1.079
122.5
118.4
120.3
120.2
117.6
124.8
B2PLYP
1.388 1.474
1.226
1.081
122.5
118.4
120.3
120.2
117.6
124.8
M06
1.381 1.475
1.211
1.083
122.4
118.5
120.1
120.4
117.5
125.0
Эксп. [212]
1.399 1.486
1.223
1.093
123.4
117.7
120.5
120.2
117.3
125.3
Данные по зарядам на атомах в молекуле НБ (Таблица 2.2.3), полученные в
рамках различных теоретических методов, также находятся в хорошем согласии
друг с другом. По результатам расчетов в молекуле НБ отрицательный заряд в
основном локализован на атомах углерода С1, С3, С5, О13 и О14. Также имеется
небольшой отрицательный заряд, сосредоточенный на атомах углерода С2 и С4,
величина
которого
составляет
всего
несколько
сотых
единиц
электрона.
Наибольший положительный заряд локализован на атомах водорода H7, H8, H9, H10 и
H11. На атоме азота N12 по данным методов HF, MP2, BHandHLYP и B2PLYP тоже
наблюдается незначительный положительный заряд, а согласно методам PBEPBE,
PW91PW91, OLYP, WB97XD и M06 – отрицательный заряд.
59
Таблица 2.2.2. Расчетные и экспериментальные значения дипольного момента (D, Д),
вертикальной энергии сродства к электрону (EAv, эВ), адиабатической энергии
сродства к электрону (EAad, эВ), вертикального потенциала ионизации (IPv, эВ),
адиабатического потенциала ионизации (IPad, эВ) и вертикального потенциала
ионизации, рассчитанного по теореме Купманса (IPK, эВ) нитробензола
Методы
D
EAv
EAad
IPv
IPad
IPK
HF
4.906
0.10
-0.73
10.72
8.54
10.08
MP2
5.160
0.65
0.36
11.17
10.55
10.07
PBEPBE
4.533
-0.84
-1.19
10.34
9.42
6.84
PW91PW91
4.569
-0.91
-1.26
10.40
9.48
6.91
OLYP
4.512
-0.61
-0.96
9.66
9.20
6.69
B3LYP
4.703
-0.83
-1.25
10.01
9.75
7.93
BHandHLYP
4.768
-0.58
-1.09
9.89
9.70
8.98
wB97XD
4.586
-0.64
-1.10
11.00
9.87
9.92
MPW2PLYP
4.903
-0.60
-0.98
9.63
9.45
8.97
B2PLYP
4.913
-0.59
-0.95
9.59
9.41
8.85
M06
4.601
-0.78
-1.19
10.00
9.83
8.19
Эксперимент [212]
4.220
-0.7
-
9.94
9.86
9.94
Таблица 2.2.3. Значения малликеновских зарядов на атомах нитробензола (е. э.)
Метод
C1
C2
C3
C4
C5
C6
H7
H8
H9
H10
H11
N12
O13
O14
HF
-0.451 -0.016 -0.425 -0.016 -0.451 1.654 0.188 0.134 0.135 0.134 0.188 0.282 -0.677 -0.677
MP2
-0.546 0.085 -0.508 0.085 -0.546 1.751 0.193 0.131 0.140 0.131 0.193 0.132 -0.621 -0.621
PBEPBE
-0.315 -0.037 -0.326 -0.037 -0.315 1.319 0.151 0.107 0.107 0.107 0.151 -0.104 -0.405 -0.405
PW91PW91
-0.356 -0.031 -0.313 -0.031 -0.356 1.441 0.143 0.096 0.096 0.096 0.143 -0.194 -0.366 -0.366
OLYP
-0.444 0.043 -0.312 0.043 -0.444 1.833 0.098 0.050 0.054 0.050 0.098 -0.053 -0.508 -0.508
B3LYP
-0.266 -0.072 -0.335 -0.072 -0.266 1.115 0.180 0.136 0.135 0.136 0.180 -0.050 -0.410 -0.410
BHandHLYP -0.340 -0.043 -0.381 -0.043 -0.340 1.287 0.189 0.141 0.141 0.141 0.189 0.077 -0.509 -0.509
wB97XD
-0.371 -0.056 -0.353 -0.056 -0.371 1.313 0.179 0.133 0.132 0.133 0.179 -0.052 -0.405 -0.405
MPW2PLYP -0.385 -0.018 -0.387 -0.018 -0.385 1.400 0.184 0.134 0.135 0.134 0.184 0.016 -0.496 -0.496
B2PLYP
-0.359 -0.027 -0.389 -0.027 -0.359 1.337 0.187 0.138 0.139 0.138 0.187 0.023 -0.494 -0.494
М06
-0.116 -0.163 -0.330 -0.163 -0.116 0.609 0.239 0.178 0.177 0.178 0.239 -0.004 -0.364 -0.364
60
Присоединение электрона к молекуле НБ приводит к образованию анионрадикала нитробензола (АРНБ). Структура, геометрические параметры и заряды на
атомах в АРНБ представлены на рисунке 2.2.1(б) и в таблицах 2.2.4, 2.2.5
соответственно.
Таблица 2.2.4. Расчетные значения геометрических параметров в анион-радикале
нитробензола (длины связей в Å, углы в градусах)
Метод
С6-C1 C6-N12 N12-O13 C1-H7 C1C6C5 C6C1C2 C1C2C3 C2C3C4 C6N12O13 O13N12O14
HF
1.412
1.367
1.266
1.071
118.9
120.1
121.0
119.0
119.0
122.0
MP2
1.393
1.379
1.268
1.083
118.9
119.8
122.0
117.7
118.8
122.3
PBEPBE
1.422
1.400
1.285
1.090
118.8
120.0
121.4
118.3
118.9
122.2
PW91PW91
1.420
1.398
1.285
1.089
118.9
120.0
121.4
118.4
118.9
122.2
OLYP
1.423
1.403
1.280
1.085
118.4
120.2
121.5
118.2
119.0
122.1
B3LYP
1.415
1.390
1.281
1.081
118.7
120.0
121.4
118.4
119.0
122.0
BHandHLYP
1.404
1.374
1.266
1.073
118.8
120.0
121.3
118.6
118.9
122.1
wB97XD
1.412
1.383
1.272
1.081
118.8
120.0
121.4
118.5
119.0
122.0
MPW2PLYP
1.412
1.384
1.278
1.079
119.0
119.9
121.5
118.4
118.9
122.2
B2PLYP
1.414
1.387
1.281
1.081
119.0
119.9
121.5
118.4
118.9
122.2
М06
1.410
1.386
1.266
1.084
118.8
120.0
121.4
118.4
119.0
122.1
Анализ расчетных значений геометрических параметров, приведенных в табл.
2.2.4, указывает на то, что присоединение электрона к молекуле НБ с последующим
образованием АРНБ приводит к незначительно увеличению длины связи С6-С1 на
0.034 A. При этом длина связи С6-N12 и валентные углы С1-С6-С5, С2-С3-С4 и О13-N12O14 уменьшаются соответственно на 0.097° и 3.9°, 2.2° и 2.9°. Остальные
геометрические параметры практически не изменяются. Из данных таблицы 2.2.4,
можно сделать о том, что расчетные значения геометрических параметров АРНБ,
полученные различными квантово-химическими методами, находятся в хорошем
согласии друг с другом.
По данным всех используемых методов в АРНБ по сравнению с молекулой
НБ наблюдается увеличение положительных и уменьшение отрицательных зарядов
61
на атомах (таблица 2.2.5). Положительный заряд локализован на атомах углерода С6
и водорода H7, H8, H9, H10, H11. По сравнению с молекулой НБ в АРНБ наблюдается
увеличение абсолютного значение отрицательного заряда на атомах С2, С4, О13 и О14.
Исключение составляют результаты, полученные методами MP2 и М06. Метод
MP2 предсказывает уменьшение абсолютного значения отрицательного заряда на
атомах углерода С2 и С4 и увеличение положительного заряда на атоме углерода С6.
Таблица 2.2.5. Значения малликеновских зарядов на атомах в анион-радикале
нитробензола (е. э.)
C1
Метод
C2
C3
C4
C5
C6
H7
H8
H9
H10 H11
N12
O13
O14
HF
-0.336 -0.188 -0.365 -0.188 -0.336 1.570 0.169 0.095 0.103 0.095 0.169 -0.057 -0.865 -0.865
MP2
-0.613 0.086 -0.609 0.086 -0.613 1.858 0.171 0.082 0.108 0.082 0.171 -0.087 -0.861 -0.861
PBEPBE
-0.262 -0.132 -0.417 -0.132 -0.262 1.120 0.128 0.063 0.075 0.063 0.128 -0.284 -0.544 -0.544
PW91PW91
-0.256 -0.144 -0.389 -0.144 -0.256 1.120 0.127 0.056 0.070 0.056 0.127 -0.378 -0.494 -0.494
OLYP
-0.346 -0.074 -0.402 -0.074 -0.346 1.600 0.073 0.003 0.024 0.003 0.073 -0.340 -0.597 -0.597
B3LYP
-0.218 -0.159 -0.407 -0.159 -0.218 0.943 0.157 0.090 0.099 0.090 0.157 -0.221 -0.577 -0.577
BHandHLYP
-0.308 -0.129 -0.434 -0.129 -0.308 1.188 0.168 0.097 0.106 0.097 0.168 -0.154 -0.682 -0.682
wB97XD
-0.329 -0.132 -0.422 -0.132 -0.329 1.084 0.166 0.100 0.107 0.100 0.166 -0.154 -0.613 -0.613
MPW2PLYP
-0.355 -0.093 -0.443 -0.093 -0.355 1.283 0.165 0.091 0.102 0.091 0.165 -0.188 -0.684 -0.684
B2PLYP
-0.340 -0.094 -0.453 -0.094 -0.340 1.243 0.166 0.093 0.105 0.093 0.166 -0.169 -0.688 -0.688
М06
0.169 -0.363 -0.256 -0.363 0.169 -0.177 0.240 0.163 0.157 0.163 0.240 0.189 -0.666 -0.666
Данные таблицы 2.2.2 показывают, что все используемые методы, за
исключением метода HF, достаточно близко передают значение адиабатического
сродства к электрону (ЕАad).
Данные таблицы 2.2.6 указывают на то, что геометрические параметры катионрадикала нитробензола (КРНБ) (Рисунок 2.2.1(в)) отличаются незначительно от
молекулы НБ, за исключением двугранного угла, отвечающего за поворот группы
NO2
относительно
плоскости
бензольного кольца.
При
этом
наблюдается
незначительное уменьшение длины связи N12-О13 в КРНБ по сравнению с НБ. Для
остальных параметров по данным методов HF, BHandHLYP, wB97XD, mPW2PLYP,
62
B2PLYP и M06 наблюдаются увеличение длин связи С1-С6, С6-N12 и значений
валентных углов С6-С1-С2, С1-С2-С3, О13-N12-O14 и уменьшение значений валентных
углов С1-С6-С5, С2-С3-С4, С6-N12-O13 в КРНБ по сравнению с НБ. По данным методов
MP2, PBEPBE, PW91PW91, OLYP и В3LYP, наоборот, наблюдаются уменьшение
длин связи С1-С6, С6-N12 и значений валентных углов С6-С1-С2, С1-С2-С3, О13-N12-O14
вместе с увеличением значений валентных углов С1-С6-С5, С2-С3-С4, С6-N12-O13 и
двугранного угла О14-N12-C5-C6.
Таблица 2.2.6. Расчетные значения геометрических параметров катион-радикала
нитробензола (длины связей в Å, углы в градусах)
Метод
С6-C1 C6-N12 N12-O13 C1-H7 C1C6C5 C6C1C2 C1C2C3 C2C3C4 C6N12O13 O13N12O14
HF
1.374 1.465
1.178 1.074
120.7
119.3
121.0
118.6
116.2
127.5
MP2
1.403 1.400
1.232 1.087
125.4
116.3
120.3
121.5
123.1
113.8
PBEPBE
1.417 1.451
1.227 1.092
124.2
117.3
119.8
121.6
117.6
124.9
PW91PW91
1.416 1.449
1.227 1.090
124.2
117.3
119.8
121.6
117.6
124.9
OLYP
1.418 1.459
1.221 1.087
123.9
117.5
119.9
121.4
117.2
125.6
B3LYP
1.411 1.452
1.218 1.083
124.0
117.3
119.8
121.7
114.5
127.1
BHandHLYP 1.370 1.470
1.193 1.077
120.9
119.3
120.9
118.7
116.2
127.7
wB97XD
1.359 1.484
1.205 1.084
121.1
118.4
121.5
118.9
116.0
127.8
MPW2PLYP 1.378 1.481
1.214 1.083
121.0
119.2
121.0
118.7
116.1
127.8
B2PLYP
1.380 1.484
1.218 1.084
121.0
119.2
121.0
118.7
116.1
127.8
M06
1.379 1.488
1.201 1.088
121.1
119.4
120.7
118.9
116.0
128.1
Анализ
зарядов на атомах КРНБ (Таблица 2.2.7) показывает, что
отрицательные заряды на атомах С1, С3, С5, N12 и О13 значительно уменьшаются по
сравнению с молекулой НБ. Положительные заряды в КРНБ локализованы на атомах
С2, С4, N12, С6, H7, H8, H9, H10 и H11.
Из
данных
таблицы
2.2.2
можно
сделать
вывод,
что
большинство
использованных нами методов с удовлетворительной точностью передает значения
IPv и IPad. Наилучшее согласие с экспериментом при оценке IPv и IPad достигается
при использовании методов M06 и wB97XD. Оценки потенциала ионизации,
63
полученные в рамках теоремы Купманса (IPK) как абсолютное значение энергии
высшей занятой молекулярной орбитали, с использованием негибридных DFT
методов достаточно плохо согласуются с экспериментом (Таблица 2.2.2). Как и в
случае с НМ, метод теории функционала плотности с учетом дисперсных
взаимодействий, wB97XD, наравне с неэмпирическими методами MP2, HF наиболее
близко к эксперименту передает значения потенциала ионизации IPK, определенное
по теореме Купманса, тогда как негибридные и гибридные DFT методы
систематически занижают значения IPK (Таблица 2.2.2).
Таблица 2.2.7. Значения малликеновских зарядов на атомах катион-радикала
нитробензола (е. э.)
C1
Метод
C2
C3
C4
C5
C6
H7
H8
H9
H10
H11
N12
O13
O14
HF
-0.458 0.106 -0.315 0.106 -0.460 1.940 0.237 0.192 0.188 0.192 0.237 0.310 -0.638 -0.638
MP2
-0.467 0.220 -0.367 1.173 -0.367 0.220 0.180 0.170 0.205 0.205 0.170 0.351 -0.347 -0.347
PBEPBE
-0.051 0.167 -0.321 0.167 -0.051 0.672 0.170 0.157 0.160 0.157 0.170 0.211 -0.304 -0.304
PW91PW91 -0.065 0.166 -0.312 0.167 -0.065 0.751 0.160 0.146 0.149 0.146 0.160 0.178 -0.291 -0.290
OLYP
0.043 0.292 -0.225 0.292 0.043 0.530 0.118 0.109 0.115 0.109 0.118 0.302 -0.423 -0.423
B3LYP
0.041 0.104 -0.313 0.109 0.011 0.515 0.201 0.189 0.192 0.188 0.202 0.232 -0.327 -0.343
BHandHLYP -0.340 0.058 -0.249 0.058 -0.339 1.545 0.242 0.203 0.195 0.203 0.242 0.113 -0.465 -0.465
wB97XD
-0.422 -0.041 -0.169 0.087 -0.388 1.710 0.218 0.182 0.174 0.178 0.222 -0.041 -0.357 -0.354
MPW2PLYP -0.372 0.072 -0.250 0.072 -0.372 1.649 0.236 0.196 0.189 0.196 0.236 0.051 -0.451 -0.451
B2PLYP
-0.342 0.057 -0.247 0.057 -0.342 1.586 0.240 0.200 0.193 0.200 0.240 0.050 -0.445 -0.445
M06
-0.373 -0.123 -0.009 -0.189 -0.348 1.510 0.279 0.213 0.202 0.214 0.278 -0.045 -0.304 -0.305
Интересная корреляция наблюдается между расчетными значениями IPK
(Таблица 2.2.8) и энтальпиями диссоциации связи С-NO2 (табл. 2.2.9) в молекулах
монозамещенных нитробензолов [81]. Как видно из рисунка 2.2.2 значения IPK и
D(С-N), рассчитанные методами B3LYP/6-31G(d'f,p') и wB97XD/6-31+G(2df,p) для
пара-замещенных нитробензолов достаточно хорошо коррелируют между собой.
Максимальное значение коэффициента корреляции (-0.965) наблюдается при
использовании результатов метода wB97XD/6-31+G(2df,p). Для орто- и мета-
64
замещенных соединений полученные значения среднеквадратичного отклонения R2
(Рисунок
2.2.2)
значительно
ниже
по
сравнению
с
пара-замещенными
нитробензолами. Отметим, что изменения в ряду потенциалов ионизации и
прочности связи C-NO2 связаны с изменениями сопряженной системы в ряду
изученных соединений. Замещение в молекуле нитробензола атома водорода на
донорные заместители в пара-положении к нитрогруппе приводит к уменьшению
потенциала ионизации и росту энергии диссоциации связи C-NO2. Основным
фактором, приводящим к отмеченным изменениям, является полярное сопряжение
донорных заместителей с сильным акцептором, нитрогруппой. Акцепторные
заместители в пара-положении к нитрогруппе увеличивают потенциал ионизации
молекулы и уменьшают величину D(C-N).
Таблица 2.2.8. Рассчитанные по теореме Купманса (IPK, эВ) и экспериментальные
значения потенциалов ионизации монозамещенных нитробензолов
Соединение
B3LYP/6-31G(d'f,p')
wB97XD/6-31+G(2df,p) Эксперимент
1
2
3
4
C6H5NO2
7.61
9.87
9.92a
o-F-C6H4-NO2
7.48
9.76
9.86a,c
m-F-C6H4-NO2
7.47
9.78
9.88a
p-F-C6H4-NO2
7.58
9.87
9.90a
o-Cl-C6H4-NO2
7.4
9.57
-
m-Cl-C6H4-NO2
7.46
9.63
9.90b
p-Cl-C6H4-NO2
7.56
9.71
10.0b
o-OH-C6H4-NO2
6.8
9.03
9.10a
m-OH-C6H4-NO2
6.78
9.06
9.0a
p-OH-C6H4-NO2
6.92
9.17
9.10a
o-NH2-C6H4-NO2
6.06
8.26
8.27a
m-NH2-C6H4-NO2
6.14
8.37
8.31a
p-NH2-C6H4-NO2
6.25
8.44
8.43a
o-CH3-C6H4-NO2
7.28
9.48
9.50a
65
Продолжение таблицы 2.2.8
1
2
3
4
m-CH3-C6H4-NO2
7.27
9.48
9.50a
p-CH3-C6H4-NO2
7.36
9.57
9.56a
o-COH-C6H4-NO2
7.32
9.87
-
m-COH-C6H4-NO2
7.49
10.02
-
p-COH-C6H4-NO2
7.55
10.07
10.27b
o-COOH-C6H4-NO2
7.57
9.94
-
m-COOH-C6H4-NO2
7.88
10.17
10.30a
p-COOH-C6H4-NO2
7.87
10.16
10.20a
o-NO2-C6H4-NO2
7.96
10.3
10.71a
m-NO2-C6H4-NO2
8.42
10.68
10.43a
p-NO2-C6H4-NO2
8.36
10.61
10.50a
o-CN-C6H4-NO2
7.93
10.17
10.52b
m-CN-C6H4-NO2
8.01
10.26
10.30b
p-CN-C6H4-NO2
8.09
10.34
10.20b
a
Экспериментальные значения из [215]
b
Экспериментальные значения из [216]
c
Экспериментальные значения соответствуют вертикальному потенциалу ионизации.
Таблица 2.2.9. Энтальпии реакции и экспериментальные значения энергии
диссоциации связи C-NO2 в монозамещенных нитробензолах, рассчитанные при
298К.
Соединение
1
B3LYP/6-31G(d'f,p') wB97XD/6-31+G(2df,p)
Эксперимент
2
3
4
C6H5NO2
290.4
305.9
291.2a; 295.8b; 285.3c
o-F-C6H4-NO2
273.2
285.3
-
m-F-C6H4-NO2
285.3
300.4
-
p-F-C6H4-NO2
293.3
308.4
-
o-Cl-C6H4-NO2
260.2
279.5
281.0a
m-Cl-C6H4-NO2
283.3
299.2
293.0a
p-Cl-C6H4-NO2
289.5
305.0
300.0a
66
Продолжение таблицы 2.2.9
1
2
3
4
o-OH-C6H4-NO2
323.0
331.4
m-OH-C6H4-NO2
288.3
303.3
297.9a
p-OH-C6H4-NO2
300.8
315.9
-
o-NH2-C6H4-NO2
309.2
320.9
-
m-NH2-C6H4-NO2
292.9
307.5
301.0a; 295.0b
p-NH2-C6H4-NO2
308.4
322.6
308.0a; 302.1b
o-CH3-C6H4-NO2
280.3
294.6
293.7b; 280.3c
m-CH3-C6H4-NO2
291.2
306.7
284.0a; 284.5c
p-CH3-C6H4-NO2
294.6
310.5
275.0a; 298.7b; 285.3c
o-COH-C6H4-NO2
263.6
280.3
-
m-COH-C6H4-NO2
284.9
301.2
-
p-COH-C6H4-NO2
282.0
297.5
-
o-COOH-C6H4-NO2
254.4
274.9
-
m-COOH-C6H4-NO2
285.8
301.2
-
p-COOH-C6H4-NO2
283.7
298.3
-
o-NO2-C6H4-NO2
243.9
264.8
-
m-NO2-C6H4-NO2
279.1
295.0
284.1a; 278.2b; 285.8c
p-NO2-C6H4-NO2
278.2
293.7
286.2a; 280.3b; 287.0c
o-CN-C6H4-NO2
264.8
280.7
-
m-CN-C6H4-NO2
280.3
296.2
-
p-CN-C6H4-NO2
280.7
295.4
-
a
Энергии активации в работе [217] приведены для температуры ~700K
b
Энергии активации в работах [78,218] приведены для температуры 298 K
c
В работе [88] энергии активации разрыва связи С-NO2 в НБ, м-ДНБ, п-ДНБ и м-НТ приведены для
T = 673-753 K; для o-НТ и п-НТ – 1100-1250 K
67
Рисунок 2.2.2. Корреляционные зависимости между энтальпией диссоциации связи
C-NO2 и IPK для а) орто-, б) мета- и в) пара-монозамещенных нитробензолов (I –
нитробензол, II – фторнитробензол, III – хлорнитробензол, IV – нитрофенол, V –
нитроанилин, VI – нитротолуол, VII – нитробензойный альдегид, VIII –
нитробензойная кислота, IX –динитробензол, X – нитробензонитрил)
68
2.3 Диметилнитрамин
Интерес исследователей к диметилнитрамину (ДМНА) [219-224] связан с тем,
что данное соединение является модельным для изучения реакций 1,3,5,7тетранитро-1,3,5,7-тетраазациклооктана
(октоген)
и
1,3,5-тринитро-1,3,5-
триазациклогексана (гексоген) – высокоэффективных ВВ.
Как и в случае с нитрометаном, расчетные методы надежно передают
значения геометрических параметров (Рисунок 2.3.1(а), Таблица 2.3.1) и дипольных
моментов (Таблица 2.3.2) молекулы ДМНА.
Рисунок 2.3.1. Геометрические структуры: а) молекулы, б) анион- и в) катионрадикала диметилнитрамина
Таблица
2.3.1.
Расчетные
и
экспериментальные
значения
геометрических
параметров структуры диметилнитрамина (длины связей в Å, углы в градусах)
Методы
1
N1-N10 N1-C2 N10-O11 C2-H3
H3C2N1
C6N1C2
C2N1N10
N1N10O11
O11N10O12
2
3
4
5
6
7
8
9
10
HF
1.345
1.453
1.189
1.080
107.0
119.5
115.7
117.4
125.1
MP2
1.379
1.453
1.227
1.090
107.0
118.1
114.0
117.0
126.0
PBEPBE
1.395
1.454
1.233
1.098
107.5
119.8
115.7
116.8
126.0
PW91PW91
1.393
1.454
1.233
1.096
107.5
120.0
115.8
116.8
126.2
OLYP
1.397
1.456
1.229
1.093
107.5
119.6
116.0
116.9
126.1
B3LYP
1.376
1.455
1.224
1.089
107.4
120.4
116.0
117.1
125.8
BHandHLYP
1.350
1.446
1.205
1.079
110.1
120.6
116.0
117.2
125.5
69
Продолжение таблицы 2.3.1
1
2
3
4
5
6
7
8
9
10
wB97XD
1.368
1.449
1.215
1.087
110.0
119.9
115.7
117.2
125.6
MPW2PLYP
1.375
1.453
1.223
1.084
110.0
119.4
115.1
117.1
125.8
B2PLYP
1.381
1.455
1.227
1.085
109.9
119.3
115.0
117.1
125.9
M06
1.368
1.445
1.213
1.090
107.5
121.9
116.3
116.8
126.2
Эксп. [213]
1.382
1.460
1.223
1.121
101.9
127.6
116.2
114.8
126.2
Таблица 2.3.2. Расчетные и экспериментальные значения дипольного момента (D, Д),
вертикальной энергии сродства к электрону (EAv, эВ), адиабатической энергии
сродства к электрону (EAad, эВ), вертикального потенциала ионизации (IPv, эВ),
адиабатического потенциала ионизации (IPad, эВ) и вертикального потенциала
ионизации, рассчитанного по теореме Купманса (IPK, эВ) в диметилнитрамине
Методы
D
EAv
EAad
IPv
IPad
IPK
HF
4.78
1.52
0.39
9.22
8.67
11.44
MP2
4.77
1.26
0.25
10.33
9.70
11.55
PBEPBE
4.54
0.47
-
10.18
9.08
6.52
PW91PW91
4.58
0.43
-
10.25
9.15
6.60
OLYP
4.50
0.66
-
9.55
8.91
6.38
B3LYP
4.74
0.55
-0.25
9.99
9.42
7.73
BHandHLYP
4.79
0.92
-0.10
10.04
9.54
9.28
wB97XD
4.67
0.93
-0.07
10.05
9.52
9.81
MPW2PLYP
4.75
0.75
-0.07
9.86
9.37
9.42
B2PLYP
4.75
0.75
-0.05
9.81
9.33
9.27
M06
4.65
0.83
-0.06
10.09
9.54
8.05
4.61 [226]
-
-
-
9.53[226]
-
Эксперимент
Следует отметить, что все используемые методы значительно занижают
значение валентного угла ∠C6N1C2 по сравнению с экспериментом.
70
Расчетные оценки дипольного момента ДМНА, полученные нами, находятся
в лучшем согласии с экспериментом, чем расчетные значения, приведенные в работе
[225].
Анализ
зарядов на атомах (Таблица 2.3.3) указывает на то, что
отрицательный заряд в основном локализован на атомах азота аминогруппы и
атомах кислорода нитрогруппы. Также следует отметить наличие небольшого
отрицательного заряда на атомах углерода. Положительный заряд сосредоточен на
атомах водорода и атоме азота нитрогруппы.
Таблица 2.3.3. Значения малликеновских зарядов на атомах ДМНА (е.э.)
Методы
N1
C2
H3
H4
H5
C6
H7
H8
H9
N10
O11
O12
HF
-0.428 -0.065 0.176 0.197 0.192 -0.065 0.197 0.176 0.192 0.899 -0.735 -0.735
MP2
-0.483 -0.067 0.180 0.200 0.196 -0.067 0.200 0.180 0.196 0.796 -0.665 -0.665
PBEPBE
-0.335 -0.117 0.156 0.180 0.165 -0.119 0.180 0.157 0.164 0.554 -0.492 -0.493
PW91PW91
-0.317 -0.123 0.155 0.179 0.164 -0.125 0.179 0.155 0.164 0.506 -0.468 -0.469
OLYP
-0.443 0.052 0.127 0.145 0.131 0.051 0.146 0.128 0.131 0.722 -0.595 -0.596
B3LYP
-0.307 -0.129 0.157 0.182 0.169 -0.129 0.182 0.157 0.169 0.538 -0.496 -0.496
BHandHLYP -0.341 -0.125 0.194 0.169 0.184 -0.125 0.169 0.194 0.183 0.675 -0.589 -0.589
wB97XD
-0.291 -0.191 0.200 0.174 0.190 -0.188 0.173 0.199 0.190 0.542 -0.498 -0.499
mPW2PLYP -0.381 -0.110 0.194 0.170 0.184 -0.110 0.170 0.194 0.184 0.644 -0.569 -0.569
B2PLYP
-0.380 -0.110 0.193 0.169 0.182 -0.110 0.169 0.193 0.182 0.636 -0.562 -0.562
M06
-0.203 -0.232 0.164 0.196 0.189 -0.236 0.196 0.165 0.189 0.462 -0.444 -0.445
Прежде чем приступить к обсуждению результатов, полученных для анионрадикала диметилнитрамина (АРДМНА), необходимо отметить, что геометрические
параметры, рассчитанные в рамках негибридных DFT методов PBEPBE, OLYP и
PW91PW91 не согласуются с результатами остальных методов и представляются
сомнительными.
Так по данным методов PBEPBE и PW91PW91 длина связи N-N в АРДМНА
составляют 4.774 и 4.680 Å соответственно. Более того, торсионный угол N10-N1-C2H4, изменение которого связано с вращением вокруг связи N1-С2, по данным методов
71
PBEPBE и PW91PW91 в анион радикале составляет -102.5°, тогда как в молекуле
ДМНА он равен -52.2°.
Попытки локализовать структуру АРДМНА, отвечающую минимуму на
поверхности потенциальной энергии методом OLYP закончились неудачно. В ходе
оптимизации геометрии молекулы длина связи N-N достигла значения 10.6 Å, после
чего процесс закончился, превысив отведенное на оптимизацию количество циклов.
В связи с вышесказанным, нами было решено не приводить данные по
геометрическим параметрам и зарядам на атомах для АРДМНА, рассчитанные в
рамках негибридных DFT-методов.
В целом следует отметить, что значения геометрических параметров
АРДМНА (Рисунок 2.3.1(б)), полученные в данной работе, находятся в хорошем
согласии
между
собой
(Таблица
2.3.4).
Исключение
составляют
только
перечисленные выше негибридные DFT методы, использовать которые для изучения
геометрии АРДМНА нецелесообразно.
Таблица 2.3.4. Расчетные значения длин связей и валентных углов анион-радикала
диметинитрамина (длины связей в Å, углы в градусах)
Методы
N1-N10 N1-C6 N10-O11 N10-O12 C2-H3 H3C2N1 C6N1C2 C2N1N10 N1N10O11 O11N10O12
HF
1.432 1.443
1.268
1.268
1.077
109.8
111.7
110.6
113.6
122.5
MP2
1.542 1.449
1.278
1.278
1.089
109.0
110.0
107.0
111.1
122.1
B3LYP
1.559 1.447
1.283
1.283
1.088
108.8
111.8
108.6
111.5
122.2
BHandHLYP 1.465 1.439
1.274
1.274
1.079
109.2
111.7
109.9
112.7
122.3
wB97XD
1.500 1.444
1.280
1.280
1.088
109.1
111.2
109.1
112.1
122.1
mPW2PLYP
1.524 1.447
1.284
1.284
1.086
109.0
111.1
108.4
111.7
122.1
B2PLYP
1.551 1.448
1.285
1.285
1.087
108.9
111.1
108.0
111.3
122.1
M06
1.529 1.438
1.271
1.271
1.090
109.4
110.7
108.1
111.8
122.4
По данным квантово-химических методов присоединение электрона к ДМНА
приводит к увеличению длины связи N1-N10 в среднем на 0.143Å, а также
уменьшению валентных углов C2-N1-C6, C2-N1-N10 и O11-N10-O12 в среднем на 8.4, 6.6
72
и 3.5°. Наряду с увеличением длины связи N-N в АРДМНА по сравнению с
молекулой наблюдается изменение торсионного угла N10-N1-C2-C4 от -55.0° до 175°.
Анализ зарядов, приведенных в таблице 2.3.5, указывает на то, что
присоединение электрона приводит к значительному увеличению абсолютного
значения отрицательного заряда на атомах азота N1 и атомах кислорода O11 и O12.
Таблица 2.3.5. Значения малликеновских зарядов на атомах анион-радикала
динитрамина (е. э.)
N1
Методы
C2
H3
H4
H5
C6
H7
H8
H9
N10
O11
O12
HF
-0.605 -0.059 0.187 0.125 0.135 -0.059 0.125 0.187 0.135 0.654 -0.912 -0.912
MP2
-0.671 -0.069 0.192 0.122 0.138 -0.069 0.122 0.192 0.138 0.648 -0.871 -0.871
B3LYP
-0.374 -0.182 0.166 0.105 0.101 -0.182 0.105 0.166 0.101 0.236 -0.621 -0.621
BHandHLYP
-0.439 -0.149 0.182 0.119 0.120 -0.150 0.119 0.182 0.120 0.393 -0.748 -0.748
wB97XD
-0.429 -0.213 0.187 0.125 0.128 -0.213 0.124 0.187 0.128 0.317 -0.670 -0.670
mPW2PLYP
-0.490 -0.142 0.182 0.117 0.123 -0.142 0.117 0.182 0.123 0.397 -0.734 -0.734
B2PLYP
-0.513 -0.131 0.179 0.114 0.119 -0.131 0.114 0.179 0.119 0.406 -0.728 -0.728
M06
-0.427 -0.269 0.182 0.122 0.137 -0.268 0.122 0.181 0.138 0.409 -0.664 -0.665
Как и в случае нитрометана, для молекулы ДМНА энергии вертикального
сродства к электрону EAv, рассчитанные по уравнению (2.1.1), являются
положительными величинами (Таблица 2.3.2).
Величина энергии адиабатического сродства к электрону (EAad) по данным
неэмпирических методов HF и MP2 к электрону больше нуля. В отличие от оценок
неэмпирических методов, значения EAad, вычисленные на основе данных,
полученных
в
рамках
методов
теории
функционала
плотности,
имеют
отрицательный знак. Аналогичные тенденции наблюдались и для НМ.
В целом следует отметить, что расчетные значения EAad в ДМНА
существенно ниже, чем в НМ. К сожалению, для ДМНА в литературе отсутствуют
экспериментальные оценки энергий вертикального и адиабатического сродства к
электрону.
73
Экспериментальное значение адиабатического потенциала ионизации IPad
ДМНА на 1.55 эВ ниже экспериментальной оценки IPad НМ (Таблица 2.3.2).
Наиболее близкое к эксперименту значение адиабатического потенциала
ионизации (IPad) ДМНА, передают методы BHandHLYP, wB97XD и M06 (Таблица
2.3.2). Расчетные значения, полученные в рамках этих методов, всего на 0.01 эВ
отличаются от экспериментального значения. В целом наихудшее согласие с
экспериментом было получено при использовании метода HF, который по данным
расчета занижает значение IPad на 0.86 эВ.
Прежде чем перейти к обсуждению расчетных значений геометрических
параметров катион-радикала ДМНА (КРДМНА), следует отметить, что оценки,
полученные в рамках гибридных DFT методов OLYP, PBEPBE и PW91PW91 в
отличие от АРДМНА находятся в хорошем согласии с результатами других методов.
Геометрическая
структура
КРДМНА
представлена
на
рисунке
2.3.1(в),
геометрические параметры приведены в таблице 2.3.6.
Таблица 2.3.6. Расчетные значения геометрических параметров в катион-радикале
диметинитрамина (длины связей в Å, углы в градусах)
N1-N10
N1-C6 N10-O11 C2-H3 H3C2N1
HF
1.467
1.462
1.160
1.080
109.1
125.2
115.7
117.4
132.9
MP2
1.580
1.419
1.190
1.098
109.0
125.8
115.8
113.1
135.3
PBEPBE
1.755
1.412
1.183
1.099
110.4
127.4
116.3
111.0
138.1
PW91PW91
1.753
1.412
1.182
1.097
110.3
127.5
116.2
111.0
138.0
OLYP
1.786
1.417
1.177
1.094
110.8
126.3
116.9
110.7
138.5
B3LYP
1.685
1.421
1.175
1.089
110.4
126.2
116.9
111.6
136.8
BHandHLYP
1.533
1.426
1.168
1.089
109.0
125.2
116.0
113.5
133.9
wB97XD
1.578
1.419
1.176
1.096
109.6
125.8
115.9
113.1
134.8
mPW2PLYP
1.643
1.423
1.179
1.087
110.2
125.8
117.3
111.8
136.3
B2PLYP
1.680
1.422
1.181
1.089
110.2
126.0
117.0
111.5
137.0
M06
1.640
1.413
1.169
1.090
110.6
126.4
118.0
111.0
136.9
Методы
C6N1C2 C2N1N10 N1N10O11 O11N10O12
74
Расчетные значения длин связей и валентных углов, полученные с
использованием различных квантово-химических методов находятся в хорошем
согласии. По данным расчета в КРДМНА по сравнению с молекулой ДМНА
наблюдается увеличение длины связи N1-N10 на 0.270 Å. Наибольшее увеличение
данной связи (на 0.389 Å) было получено при использовании метода OLYP.
Все используемые в настоящей работе расчетные методы предсказывают
уменьшение валентного угла N1-N10-O11 в КРДМНА по сравнению с ДМНА в
среднем на 4.6°. При этом значения углов C2-N1-N10 и O11-N10-O12 увеличиваются на
6.5 и 10.4° соответственно.
Необходимо отметить, что данные по зарядам в случае КРДМНА,
полученные в рамках различных квантово-химических методов, достаточно
противоречивы (Таблица 2.3.7).
Таблица 2.3.7. Значения малликеновских зарядов на атомах в катион-радикале
диметилнитрамине (е. э.)
Методы
N1
C2
H3
H4
H5
C6
H7
H8
H9
N10
O11
O12
HF
-0.280 -0.107 0.265 0.239 0.272 -0.107 0.239 0.272 0.265 1.082 -0.571 -0.571
MP2
-0.416 -0.080 0.271 0.244 0.271 -0.146 0.273 0.269 0.273 1.000 -0.491 -0.469
PBEPBE
-0.422 -0.100 0.228 0.245 0.244 -0.100 0.244 0.228 0.245 0.775 -0.293 -0.293
PW91PW91
-0.418 -0.097 0.226 0.243 0.243 -0.097 0.243 0.227 0.243 0.772 -0.293 -0.293
OLYP
-0.583 0.058 0.198 0.210 0.216 0.057 0.216 0.198 0.210 1.014 -0.397 -0.397
B3LYP
-0.349 -0.129 0.237 0.241 0.251 -0.130 0.251 0.237 0.242 0.750 -0.300 -0.300
BHandHLYP -0.143 -0.119 0.265 0.236 0.278 -0.119 0.236 0.265 0.278 0.705 -0.441 -0.441
wB97XD
-0.275 -0.161 0.268 0.234 0.272 -0.214 0.271 0.256 0.269 0.746 -0.342 -0.322
mPW2PLYP -0.383 -0.133 0.252 0.261 0.254 -0.126 0.250 0.253 0.261 0.861 -0.373 -0.376
B2PLYP
-0.390 -0.134 0.250 0.252 0.260 -0.134 0.260 0.250 0.252 0.847 -0.356 -0.356
M06
-0.185 -0.281 0.251 0.264 0.264 -0.218 0.235 0.263 0.265 0.657 -0.247 -0.269
В отличие от АРДМНА, для которого все используемые методы
предсказывают увеличение абсолютной величины отрицательного и уменьшение
75
положительного зарядов по сравнению с молекулой ДМНА, в случае КРДМНА
подобных тенденций не прослеживается. Наиболее достоверными можно считать
значения зарядов, полученные в рамках метода BHandHLYP. Так по данным этого
метода в КРДМНА наблюдается значительное снижение величины отрицательного
заряда на атоме азота N1 и атомах кислорода нитрогруппы, при незначительном
уменьшении абсолютной величины отрицательного заряда на атомах углерода и
увеличении положительного заряда на атомах водорода.
Все используемые в данной работе методы теории функционала плотности,
за исключением BHandHLYP и wB97XD предсказывают увеличение абсолютного
значения отрицательного заряда на атоме азота N1 в КРДМНА по сравнению с
молекулой ДМНА. Отметим также, что по данным методов B3LYP, wB97XD,
B2PLYP и mPW2PLYP в КРДМНА абсолютное значение отрицательного заряда на
атоме углерода С6 больше по сравнению с молекулой ДМНА.
Данные по зарядам, рассчитанные с использованием неэмпирических
методов HF и MP2, указывают на то, что удаление одного электрона из молекулы
ДМНА ведет к уменьшению абсолютного значения отрицательного заряда на атоме
азота N1, что находится в согласии с оценками методов BHandHLYP и wB97XD. В
КРДМНА по сравнению с молекулой по данным методов HF и MP2 наблюдается и
уменьшение абсолютных значений отрицательного заряда на атомах углерода С2 и
С6, что согласуется с результатами дважды гибридных DFT методов.
2.4 Этилнитрат
Этилнитрат (ЭН) можно рассматривать как модельное соединение для
изучения реакций ТЭН. ТЭН широко применяется при проведении взрывных работ в
горнодобывающей промышленности. Также ТЭН используется в медицине при
профилактике стенокардии и ишемической болезни мышц, как и нитроглицерин
относится к категории веществ, называемых вазодилататорами.
76
Геометрическая структура этилнитрата представлена на рисунке 2.4.1(а).
Геометрические параметры этой структуры приведены в таблице 2.4.1.
Рисунок 2.4.1. Геометрические структуры: а) молекулы этилнитрата, б) анионрадикала этилнитрата по данным методов B3LYP, PBEPBE, PW91PW91, wB97XD,
MPW2PLYP, B2PLYP и M06, в) анион-радикала этилнитрата по данным методов HF,
MP2; г) катион-радикала этилнитрата по данным методов HF, MP2, BHandHLYP,
wB97XD, MPW2PLYP и B2PLYP; д) катион-радикала этилнитрата по данным
методов B3LYP, PBEPBE, PW91PW91, OLYP и M06.
Таблица
2.4.1.
Расчетные
и
экспериментальные
значения
геометрических
параметров этилнитрата (длины связей в Å, углы в градусах)
Методы
1
C1-C5 C5-O8 O8-N9 N9-O10 N9-O11 C1C5O8 C5O8N9 O8N9O10 O8N9O11 O10N9O11
2
3
4
5
6
7
8
9
10
11
HF
1.510 1.434 1.324 1.179 1.170
105.8
116.6
116.0
118.0
127.9
MP2
1.515 1.447 1.449 1.213 1.204
105.9
113.2
117.0
112.4
129.9
77
Продолжение таблицы 2.4.1
1
2
3
4
5
6
7
8
9
10
11
PBEPBE
1.516 1.448 1.449 1.213 1.205
105.9
113.3
117.0
112.3
130.6
PW91PW91
1.515 1.447 1.449 1.213 1.204
105.9
113.2
117.0
112.4
130.6
OLYP
1.519 1.446 1.445 1.209 1.202
106.3
114.3
117.4
112.1
130.4
B3LYP
1.514 1.449 1.408 1.206 1.197
105.9
114.5
117.4
112.9
129.6
BHandHLYP 1.504 1.435 1.356 1.191 1.182
105.8
115.3
117.7
113.6
128.71
wB97XD
1.510 1.438 1.381 1.200 1.192
105.9
114.6
117.6
113.2
129.2
MPW2PLYP
1.510 1.444 1.397 1.206 1.198
105.5
114.1
117.4
113.0
129.5
B2PLYP
1.511 1.446 1.408 1.210 1.202
105.5
113.9
117.4
112.9
129.7
M06
1.501 1.434 1.388 1.196 1.188
105.7
114.6
117.3
112.9
129.8
Эксп. [212]
1.528 1.430 1.407 1.215 1.215
109.47
111.0
118.0
111.0
----
Сравнение
с
имеющимися
экспериментальными
данными
расчетных
значений геометрических параметров (Таблица 2.4.1) указывает на хорошее их
согласие между собой.
Как и в случае НМ и ДМНА, расчетные оценки дипольных моментов ЭН
(таблица 2.4.2) также удовлетворительно согласуются с экспериментом.
Анализ зарядов на атомах (Таблица 2.4.3) указывает на то, что отрицательный
заряд сосредоточен на атоме углерода С1 и атомах кислорода. Наибольший по
величине отрицательный заряд по данным расчета находится на атоме азота.
В равновесной структуре анион-радикала ЭН (АРЭН) (рисунок 2.4.1(б)) по
данным методов теории функционала плотности имеет место значительное
увеличение длины связи O8-N9 по сравнению с молекулой, в среднем на 0.805Å
(Таблицы 2.4.1 и 2.4.4).
Следует также отметить, для АРЭН не удалось найти равновесную
конфигурацию методом OLYP, как и для АРДМНА. В отличие от АРДМНА данные
по геометрии молекулы, рассчитанные в рамках негибридных DFT методов PBEPBE
и PW91PW91, находятся в хорошем согласии с оценками других DFT методов. По
данным метода BHandHLYP длина связи O8-N9 составляет 4.472Å. По оценкам
78
неэмпирических методов MP2 и HF, эта величина в АРЭН (Рисунок 2.3.1(в), Таблица
2.4.4) всего на 0.096 и 0.157 Å соответственно, больше, чем в молекуле ЭН. Методы
PBEPBE, PW91PW91, B3LYP и wB97XD предсказывают уменьшение валентного
угла C5-O8-N9 в АРЭН по сравнению с ЭН на 10.5, 10.1, 13.6 и 13.7° соответственно.
По данным неэмпирических и дважды гибридных DFT методов соответствующее
уменьшение
не
превышает
6.5°.
Все
используемые
расчетные
методы
предсказывают уменьшение угла O10-N9-O11 в АРЭН по сравнению с ЭН в среднем
на 5.4°.
Таблица 2.4.2. Расчетные и экспериментальные значения дипольного момента (D, Д),
вертикальной энергии сродства к электрону (EAv, эВ), адиабатической энергии
сродства к электрону (EAad, эВ), вертикального потенциала ионизации (IPv, эВ),
адиабатического потенциала ионизации (IPad, эВ) и вертикального потенциала
ионизации, рассчитанного по теореме Купманса (IPK, эВ) этилнитрата
Методы
D
EAv
EAad
IPv
IPad
IPK
HF
4.10
1.26
0.00
11.41
10.71
13.05
MP2
3.87
1.06
-0.21
11.92
11.27
13.07
PBEPBE
3.26
0.36
-1.18
10.84
9.92
7.46
PW91PW91
3.27
0.30
-1.25
10.92
10.00
7.54
OLYP
3.26
1.05
-
10.72
9.71
7.31
B3LYP
3.57
0.42
-1.32
11.50
10.34
8.91
BHandHLYP
3.83
0.66
-
11.89
10.93
10.89
wB97XD
3.64
0.69
-0.95
11.74
11.12
11.06
MPW2PLYP
3.73
0.45
-0.91
11.65
10.63
11.00
B2PLYP
3.69
0.44
-0.92
11.59
10.98
10.85
M06
3.49
1.23
-1.06
11.66
10.44
9.28
3.39 [212]
-
-
Эксперимент
11.22 [212]
Анализ малликеновских зарядов на атомах в АРЭН (Таблица 2.4.5) показывает
снижение положительного заряда на атомах водорода и уменьшение абсолютной
величины отрицательного заряда на атоме углерода С1, а также увеличение
79
абсолютной величины отрицательного заряда на атоме кислорода O8 и значительное
уменьшение положительного заряда на атоме азота N9 по сравнению с молекулой
ЭН (Таблица 2.4.3).
Таблица 2.4.3. Значения малликеновских зарядов на атомах этилнитрата (е. э.)
Методы
C1
H2
H3
H4
C5
H6
H7
O8
N9
O10
O11
HF
-0.598
0.197 0.189 0.197 0.218
0.185
0.185
-0.672
1.535
-0.709
-0.726
MP2
-0.604
0.198 0.188 0.198 0.232
0.184
0.184
-0.645
1.292
-0.606
-0.621
PBEPBE
-0.575
0.174 0.170 0.174 0.179
0.158
0.158
-0.453
0.884
-0.418
-0.450
PW91PW91
-0.574
0.174 0.169 0.174 0.172
0.159
0.159
-0.435
0.842
-0.402
-0.438
OLYP
-0.496
0.140 0.137 0.139 0.343
0.113
0.113
-0.519
1.106
-0.514
-0.563
B3LYP
-0.562
0.176 0.173 0.176 0.168
0.168
0.168
-0.471
0.910
-0.443
-0.462
BHandHLYP
-0.584
0.189 0.184 0.189 0.163
0.182
0.182
-0.536
1.150
-0.550
-0.571
wB97XD
-0.582
0.188 0.182 0.188 0.181
0.180
0.180
-0.544
1.078
-0.516
-0.535
MPW2PLYP
-0.610
0.196 0.189 0.196 0.102
0.186
0.186
-0.469
0.945
-0.454
-0.466
B2PLYP
-0.583
0.187 0.181 0.187 0.186
0.178
0.178
-0.543
1.056
-0.505
-0.522
M06
-0.612
0.196 0.188 0.196 0.040
0.204
0.204
-0.450
0.835
-0.406
-0.393
Таблица 2.4.4. Расчетные значения геометрических параметров анион-радикала
этилнитрата (длины связей в Å, углы в градусах)
Методы
C1-C5 C5-O8 O8-N9 N9-O10 N9-O11 C1C5O8 C5O8N9 O8N9O10 O8N9O11 O10N9O11
HF
1.518 1.387 1.420 1.260 1.245
107.7
112.4
113.1
110.1
124.1
MP2
1.521 1.391 1.606 1.254 1.245
108.2
109.8
110.8
106.7
125.6
PBEPBE
1.546 1.360 2.329 1.239 1.233
114.0
102.8
111.0
115.1
123.5
PW91PW91
1.545 1.360 2.298 1.239 1.232
113.9
103.1
111.0
114.3
123.6
B3LYP
1.539 1.362 2.327 1.229 1.224
114.2
100.9
113.0
117.0
122.7
wB97XD
1.533 1.360 2.241 1.223 1.216
113.8
100.9
114.5
116.4
122.6
MPW2PLYP 1.536 1.361 2.059 1.222 1.215
112.8
107.6
111.9
109.5
125.4
B2PLYP
1.539 1.362 2.066 1.225 1.219
112.8
108.1
111.6
109.1
125.7
M06
1.524 1.347 2.345 1.221 1.215
114.0
97.5
112.3
117.6
122.6
80
Таблица 2.4.5. Значения малликеновских зарядов на атомах анион-радикала
этилнитрата (е. э.)
Методы
C1
H2
H3
H4
C5
H6
H7
O8
N9
O10
O11
HF
-0.559
0.163 0.140 0.163
0.244
0.153 0.119
-0.672 0.958
-0.876 -0.834
MP2
-0.566
0.161 0.133 0.157
0.256
0.151 0.102
-0.748 0.886
-0.793 -0.740
PBEPBE
-0.467
0.160 0.110 0.134
-0.024
0.152 0.069
-0.597 0.157
-0.336 -0.359
PW91PW91
-0.476
0.166 0.115 0.139
-0.069
0.162 0.077
-0.570 0.070
-0.292 -0.322
B3LYP
-0.443
0.128 0.110 0.154
0.008
0.079 0.158
-0.622 0.229
-0.424 -0.378
WB97XD
-0.502
0.186 0.127 0.144
-0.012
0.159 0.085
-0.600 0.239
-0.408 -0.419
MPW2PLYP -0.509
0.139 0.121 0.164
0.106
0.078 0.164
-0.647 0.370
-0.487 -0.498
B2PLYP
-0.512
0.141 0.123 0.169
0.093
0.083 0.170
-0.639 0.349
-0.480 -0.497
M06
-0.460 0.147 0.133 0.180
-0.132 0.108 0.169
-0.576 0.126
-0.377 -0.316
Использованные
нами
методы
теории
функционала
плотности,
за
исключением MPW2PLYP и B2PLYP, предсказывают наличие отрицательного
заряда на атоме углерода С5 АРЭН.
В
отличие
от
DFT
методов
неэмпирические
методы
HF
и
MP2
предсказывают увеличение величины положительного заряда на атоме углерода С5 в
АРЭН по сравнению с молекулой ЭН.
Интересным является то, что в отличие от неэмпирических методов HF и
MP2, методы B3LYP, PBEPBE, PW91PW91, wB97XD, и B2PLYP предсказывают
уменьшение абсолютного значения отрицательного заряда на атомах кислорода О10
и О11 в АРЭН по сравнению с ЭН. Оценки этих DFT методов указывают на
увеличение абсолютного значения отрицательного заряда на атомах кислорода О10 и
О11. Данное обстоятельство можно объяснить большим значением длины связи О8N9 в АРЭН, предсказываемое методами теории функционала плотности.
Как и в случае НМ и ДМНА, значения вертикального сродства EAv (Таблица
2.4.2), рассчитанные по уравнению (2.1.1) больше нуля. Энергия адиабатического
сродства к электрону (EAad) вычисленная в рамках метода HF равна нулю. Интересно
отметить, что значение EAad, рассчитанное методом MP2, меньше нуля, тогда как для
81
методов оценки EAad, полученные в рамках данного метода для НМ и ДМНА,
больше нуля. Тем не менее, следует отметить, что метод MP2 по сравнению с
другими методами дает существенно более высокое значение EAad.
Как видно из рисунка 2.4.1 (г) и (д) и данных таблицы 2.4.6, расчетные
значения основных геометрических параметров катион-радикала этилнитрата
(КРЭН) существенно зависят от используемого квантово-химического метода.
Таблица 2.4.6. Расчетные значения геометрических параметров катион-радикала
этилнитрата (длины связей в Å, углы в градусах)
Методы
C1-C5 C5-O8 O8-N9 N9-O10 N9-O11 C1C5O8 C5O8N9 O8N9O10 O8N9O11 O10N9O11
HF
1.497 1.588 1.220
1.203
1.186
104.0
119.3
125.9
123.4
110.7
MP2
1.496 1.537 1.276
1.237
1.222
104.1
114.4
125.3
122.6
112.2
PBEPBE
1.560 1.335 1.971
1.164
1.157
113.6
114.7
107.9
103.4
148.7
PW91PW91
1.577 1.333 1.965
1.163
1.157
112.4
114.6
107.9
103.5
148.6
OLYP
1.561 1.336 2.047
1.158
1.153
113.7
118.0
108.0
101.9
150.1
B3LYP
1.613 1.333 1.981
1.146
1.140
107.8
116.4
106.8
102.1
151.1
BHandHLYP 1.489 1.550 1.254
1.152
1.308
104.8
116.4
129.4
111.1
119.5
wB97XD
1.493 1.558 1.263
1.175
1.286
104.9
116.0
129.1
114.2
116.6
MPW2PLYP 1.491 1.569 1.271
1.175
1.314
104.4
115.4
129.7
111.8
118.5
B2PLYP
1.496 1.536 1.283
1.156
1.389
105.7
114.6
128.3
108.8
122.9
M06
1.530 1.336 2.107
1.128
1.124
113.5
118.6
103.1
98.1
158.8
По данным неэмпирических методов HF и MP2, а также методов теории
функционала плотности BHandHLYP, wB97XD, MPW2PLYP и B2PLYP, увеличение
длины связи O8-N9 в КРЭН незначительно по сравнению с молекулой ЭН и
составляет всего 0.1 Å. В свою очередь, значение длины связи O8-N9, рассчитанное в
рамках негибридных методов теории функционала плотности PBEPBE, PW91PW91,
OLYP и гибридных методов B3LYP и M06, намного больше, чем в молекуле ЭН
(Рисунок 2.4.1(д), Таблица 2.4.6). Помимо удлинения связи O8-N9 в КРЭН
происходит поворот нитрогруппы относительно связи O-NO2. Так по данным
методов B3LYP, PBEPBE, PW91PW91 и OLYP значение двугранного угла С1-С5-О8-
82
N9 составляет -110.7, 140.5, 133.3 и -140.7° соответственно. По данным этих методов
в КРЭН по сравнению с молекулой ЭН происходит увеличение валентного угла О10N9-О11 в среднем на 19.3°.
Неэмпирические методы HF и MP2, дважды гибридные DFT методы, а также
методы BHandHLYP и wB97XD предсказывают уменьшение величины угла О10-N9О11 в КРЭН по сравнению с молекулой ЭН в среднем на 12.4°. Уменьшение длины
связи О8-N9 – в среднем на 0.125Å.
Данные по зарядам, приведенные в таблице 2.4.7, свидетельствуют об
уменьшении в КРЭН абсолютной величины отрицательного заряда на атомах
кислорода и углерода С1, а также о значительном увеличении положительного
заряда на атоме азота при незначительном увеличении положительного заряда на
атомах водорода.
Таблица 2.4.7. Значения малликеновских зарядов на атомах КРЭН (е. э.)
Методы
C1
H2
H3
H4
C5
H6
H7
O8
N9
O10
O11
HF
-0.579 0.243 0.247 0.243
0.107
0.250 0.250 -0.579 1.627 -0.395 -0.414
MP2
-0.596 0.241 0.243 0.241
0.119
0.245 0.244 -0.491 1.389 -0.311 -0.325
PBEPBE
-0.497 0.221 0.224 0.228
0.117
0.269 0.227 -0.293 0.943 -0.232 -0.207
PW91PW91
-0.483 0.224 0.228 0.227
0.102
0.267 0.224 -0.285 0.940 -0.231 -0.212
OLYP
-0.417 0.189 0.188 0.183
0.273
0.179 0.230 -0.364 1.190 -0.343 -0.308
B3LYP
-0.491 0.225 0.242 0.233
0.062
0.214 0.246 -0.282 0.986 -0.228 -0.207
BHandHLYP -0.583 0.231 0.237 0.231
0.100
0.242 0.242 -0.443 1.218 -0.380 -0.094
wB97XD
-0.606 0.238 0.244 0.238
0.039
0.246 0.246 -0.333 1.066 -0.287 -0.092
MPW2PLYP
-0.582 0.227 0.232 0.227
0.118
0.235 0.235 -0.389 1.019 -0.267 -0.054
B2PLYP
-0.574 0.230 0.236 0.230
0.113
0.240 0.240 -0.447 1.168 -0.352 -0.085
M06
-0.537 0.230 0.228 0.226 -0.079 0.273 0.237 -0.267 1.114 -0.216 -0.209
Оценки зарядов, полученные с использованием метода wB97XD, не
согласуются
с
данными
других
методов.
Метод
wB97XD
предсказывает
незначительное увеличение абсолютного значения отрицательного заряда на атоме
углерода С1 и положительного заряда на атоме азота. Также все методы, за
83
исключением MPW2PLYP, предсказывают уменьшение положительного заряда на
атоме углерода С5 в КРЭН по сравнению с молекулой ЭН.
Расчетные оценки потенциала ионизации IPad, рассчитанные на основе
выражения (2.1.4) хорошо согласуются с экспериментальным значением (Таблица
2.4.2). Наименьшее абсолютное отклонение от эксперимента получено для метода
MP2 (0.05 эВ).
2.5 Основные выводы по Главе 2
Таким образом, на основе представленных в главе 2 результатов, мы можем
сделать вывод, что все используемые в работе квантово-химические методы с
высокой надежностью передают значения длин связей, валентных углов и
дипольных моментов в молекулах нитрометана, диметилнитрамина, этилнитрата и
нитробензола. Данные по геометрии ион-радикалов нитрометана и нитробензола,
полученные в рамках различных квантово-химических методов, также хорошо
согласуются между собой. Однако использование негибридных методов теории
функционала плотности PBEPBE, PW91PW91 и OLYP в случае анион-радикала
диметилнитрамина не позволяет получать надежные значения, поэтому их
применение для дальнейшего изучения является нецелесообразным. Также следует
отметить, что методы B3LYP, PBEPBE, PW91PW91, wB97XD, MPW2PLYP и
B2PLYP существенно завышают значение длины связи O-NO2 в анион-радикале
этилнитрата, тогда как методы B3LYP, PBEPBE, PW91PW91, OLYP, M06 завышают
значение длины связи O-NO2 в катион-радикале этилнитрата.
Наиболее надежные оценки адиабатического сродства к электрону в
молекулах нитрометана и нитробензола были получены при использовании DFT
методов, тогда как данные методов HF и MP2 хуже всего согласовались с
экспериментом. В отличие от значений EAad расчетные значения вертикального
потенциала ионизации нитрометана и нитробензола хорошо согласуются с
имеющимися экспериментальными значениями.
84
Все
используемые
методы
удовлетворительно
передают
значения
вертикального и адиабатического потенциала ионизации. Наиболее надежно
предсказывает значения потенциала ионизации в молекулах нитрометана и
нитробензола, рассчитанные по теореме Купманса, метод функционала плотности
wB97XD. С использованием этого метода установлена корреляция потенциалов
ионизации и энтальпий диссоциации связи C-NO2 для монофункциональных
паразамещенных нитробензола. Обе коррелирующие величины характеризуют
сопряженную систему нитробензолов и чувствительны к её изменению в ряду
изученных
соединений.
Основным
фактором,
влияющим
на
изменение
коррелирующих величин в ряду, является полярное сопряжение донорных
заместителей с сильным акцептором − нитрогруппой.
85
Глава 3. Особенности механизмов распада ион-радикалов
тринитротолуола
3.1 Основные каналы термодеструкции молекул нитротолуолов
Молекулы нитротолуолов, в том числе и соединений выпускаемых в
промышленных масштабах, способны самопроизвольно разлагаться при умеренных
температурах. Поэтому пристальное внимание уделяется изучению кинетики и
механизма термодеструкции данного класса ароматических нитросоединений. В
таблице
3.1.1
приведены
аррениусовские
параметры
термического
распада
некоторых нитроаренов, полученные экспериментально.
Таблица 3.1.1. Аррениусовские параметры термического распада некоторых
ароматических нитросоединений [227]
Eа,
ΔH≠=Eа-RTср,
кДж/моль
кДж/моль
Соединение
Т,°С
Тср,°С
п-нитротолуол
300-350
325
257.3
252.3
16.73
380-450
415
275,7 ± 4,6
270.0 ± 4.6
16.7 ± 0.4
797-907
852
269.0
259.6
14.89
827-977
902
285.3 ± 8.4
275.5 ± 8.4
14.9 ± 0.5
300-350
325
272.0
267.0
17.88
400-470
435
284.5 ± 5.4
278.6 ± 5.4
16.9 ± 0.4
300-350
325
177.8
172.8
10.22
350-420
385
207.1 ± 3
201.6 ± 3
12.4
797-907
852
257.2
247.8
14.81
797-907
852
215.6
206.2
13.08
827-977
902
280.3 ± 9.2
270.5 ± 9.2
16.4 ± 0.6
2,4-динитротолуол
827-977
902
282.0 ± 7.1
272.2 ± 7.1
15.3 ± 0.4
2,4,6-тринитротолуол
320-340
330
196.2
191.2
12.4
м-нитротолуол
о-нитротолуол
lgА
Как показывает таблица 3.1.1, для некоторых из представленных соединений
они имеют значительный разброс.
86
Наряду
с
проведением
экспериментальных
исследований,
широко
осуществляется теоретическое изучение различных механизмов мономолекулярного
распада нитротолуолов с использованием современных квантово-химических
методов. Вместе с тем, в трактовке механизма термического разложения этих
нитроаренов имеются значительные пробелы. Это относится и к выявлению
особенностей конкуренции различных каналов первичного акта реакций газофазного
мономолекулярного распада, не смотря на то, что в последние годы в этой области
имеются новые результаты, заслуживающие внимание специалистов.
Далее мы приводим результаты теоретического изучения [227,228] ряда
основных механизмов первичного акта газофазного мономолекулярного распада для
изомерных нитротолуолов (о-нитротолуол (о-НТ), м-нитротолуол (м-НТ) и пнитротолуол
(п-НТ)),
динитротолуолов
(2,3-динитротолуол
(2,3-ДНТ),
2,4-
динитротоулол (2,4-ДНТ), 2,5-динитротолуол (2,5-ДНТ), 2,6-динитротолуол (2,6ДНТ), 3,4-динитротолуол (3,4-ДНТ), 3,5-динитротолуол (3,5-ДНТ)), а также 2,4,6тринитротолуола
(ТНТ).
Основная
часть
результатов
была
получена
с
использованием метода B3LYP/6-31+G(2df,p). Вместе с тем, проведённое нами
изучение показало, что близкие значения барьеров активации получаются и при
использовании других DFT-методов [227-229].
Для
нитротолуолов,
имеющих
в
орто-положении
к
нитрогруппе
водородсодержащий заместитель, исследовались шесть альтернативных механизмов
первичного акта реакции:
1. радикальный разрыв связи С-N c отрывом группы NO2•:
R1H O
C
N
R
C
O
C
2
R3
C
C
R4
C
R5
R1H
C
R
C
C
2
R3
C
C
C
+
R5
NO2
;
R4
2. радикальный разрыв связи С-С с отрывом группы CH3•:
(3.1.1)
87
R1H O
C
N
R2
C
C
O
3
R
C
C
C
O
R2
C
C
C
N
C 5
C
R
R3
C
R5
O
+ R1H
;
(3.1.2)
R4
R4
3. нитро-нитритная перегруппировка:
R1H O
C
N
R
C
C
O
R1H
O
C
O
R
N
C
C
2
R
3
C
C
C
2
R5
R
C
3
C
C
R4
R
;
(3.1.3)
5
R4
4. образование аци-форм:
R 1H O
C
N
R2
C
C
O
R
3
C
C
C
R
5
R
2
R1 H O
C
N
C
C
O
R
3
C
C
C
;
(3.1.4)
R5
R4
R4
5. образование замещенных 6-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4триене-8-оксидов [36,83]:
R1H O
C
N
R2
C
O
C
R1H
2
R
C
O
C
N
O
C
C 5
C
R
R3
C
C 5
C
R
R3
C
R4
R4
;
(3.1.5)
6. образование замещенных 2-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4триене-8-оксидов [36,83]:
R
2
R 1H O
C
N
C
C
O
R
3
C
C
C
R5
R4
Для
HR 1 O
C
N
R
C
C
O
C
C
R3
C
R5
4
R
2
;
нитротолуолов,
(3.1.6)
имеющих
в
мета-положении
к
нитрогруппе
водородсодержащий заместитель, исследовались пять альтернативных механизмов
первичного акта реакции:
1. радикальный разрыв связи С-N c отрывом группы NO2•:
R
2
R3
R 1H
C
R5
C
C
C
C
C
R
4
N+
O–
R 1H
C
R5
C
C
+
C
C
R3
C
R2
O
NO2
;
R4
2. радикальный разрыв связи С-С с отрывом группы CH3•:
(3.1.7)
88
R
2
R1H
C
R5
C
C
C
R3
C
C
R2
C
R3
O
N+
C
C
C
C
R5
+
N+
R1H
O
;
(3.1.8)
O–
R4
O–
R4
C
3. нитро-нитритная перегруппировка:
R1H
R5
C
R
C
C
R 1H
C
R5
R
C
C
2
2
3
R
C
C
C
+
N
O
C
3
R
C
4.
O
;
N
(3.1.9)
O
R4
O–
R4
C
образование замещенных 5-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-
2,4-триене-8-оксидов:
R1H
R1H
C
R5
R
C
C
2
3
R
C
C
C
N
+
2
R
O
C
C
3C
C
R
O–
R4
R5
C O
C N
;
(3.1.10)
O
R4
5. образование замещенных 3-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4триене-8-оксидов:
R1H
R1H
2
C
R5
R
C
C
3
R
C
R2
C
3C
C +O
N
C
R
R
Для
C
R4
O–
4
C
C
C
O
R5
;
(3.1.11)
N O
нитротолуолов,
имеющих
в
пара-положении
к
нитрогруппе
водородсодержащий заместитель, исследовались пять альтернативных механизмов
первичного акта реакции:
1. радикальный разрыв связи С-N c отрывом группы NO2•:
R1H
R2
R
3
C
C
O
C
C
N
R 1H
C
R4
R
C +
C
C 4
C
R
R3
C
R5
C
C
2
R4
NO 2
;
(3.1.12)
O
2. радикальный разрыв связи С-С с отрывом группы CH3•:
R 1H
R
2
R3
C
C
O
C
C
N
C
R5
C
R2
C
C
R3
C
–
N+
O
R4
C
O
C
R4
+
C
4
R
R1H
;
O
3. нитро-нитритная перегруппировка:
(3.1.13)
89
R1H
R1H
R2
R3
C
C
C
O
2
R
C
R
C
R3
5
C
N
R4
C R5
C
C
C
N
O
C
O
C
R4
;
(3.1.14)
O
4. образование замещенных 4-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4триене-8-оксидов:
R 1H
R
2
R3
C
C
O
C
C
N
R
C
5
C
R4
R 1H
C
R5
C
C
4
C
C R
R3
C
O
N
R2
O
;
(3.1.15)
O
5. образование замещенных 4-метил-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4триене-8-оксидов:
R 1H
R 1H
R2
R3
C
C
O
C
C
N
R5
C
C
O
R4
R2
C
C
C
R5
R3 C C C R4
O
N
O
;
(3.1.16)
Для всех этих процессов были определены энтальпии активации и реакции.
Для реакций радикального распада, как известно, во многих случаях эти величины
совпадают. Энтальпии реакций рассчитывались на основе энтальпий образования
оптимальных конформаций
исходных соединений
и продуктов. Энтальпии
образования оценивались из полных электронных энергий стандартными методами.
При определении барьеров активации, наличие переходных состояний, здесь и далее
при
исследовании
механизмов
реакции,
подтверждалось
наличием
одного
отрицательного собственного значения матрицы вторых производных. Соответствие
найденных переходных состояний изучаемым процессам подтверждалось спусками
по координате реакции из переходных состояний к реагентам и продуктам.
Экспериментальные данные по кинетике термического распада нитротолуолов
в газообразном состоянии получены в основном в конце прошлого века. Их
достаточно трудно объяснить с позиции единого механизма термического распада.
Вместе с тем не вызывает
сомнений, что в орто-нитротолуоле и его
нитропроизводных механизм реакции не связан с гомолитическим разрывом C–NO2.
90
На это указывают, например, более низкие значения предэкспоненциального
множителя
реакции
для
о-нитротолуолов,
чем
наблюдаемые
в
процессах
мономолекулярного распада м- и п-нитроаренов, для которых радикальный
механизм первичного акта реакции не вызывает сомнений (Таблица 3.1.1).
Барьеры активации (реакции) рассмотренных процессов для соединений, в
которых метильная группа находится в орто-, мета-, и пара-положении к
нитрогруппе представлены, соответственно в таблицах 3.1.2-3.1.4.
Таблица 3.1.2. Энтальпии активации (ΔH≠) и энтальпии реакций
(ΔHреак)
возможных
имеющих
вариантов
первичного
водородсодержащий
заместитель
акта
в
нитроаренов,
о-положении
к
R1H O
C
N
R2
C
O
C
C
R3
C
R5
R4
нитрогруппе (кДж/моль) по данным метода B3LYP/6-31+G(2df,p)
Процесс
C
о-НТ
2,3-ДНТ
2,4-ДНТ
2,5-ДНТ
2,6-ДНТ
ТНТ
R1=CH2,
R1=CH2,
R1=CH2,
R1=CH2,
R1=CH2,
R1=CH2,
R2=R3=
R2=R3= R4=H, R2=R3= R5=H, R2=R4= R5=H, R3=R4= R5=H,
R4=R5=H
R5=NO2
ΔH≠ ΔHреак. ΔH≠
R4=NO2
R3=NO2
R2=NO2
R3=R4=H,
R2= R5=NO2
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
(3.1.1)
-
271.9
-
236.2
-
259.9
-
259.9
-
252.4
-
242.7
(3.1.2)
-
403.2
-
415.9
-
409.9
-
409.1
-
398.6
-
405.2
-3.0
225.6
-21.2
236.7
-12.6
228.9
-15.5
234.0
-14.9
231.6
-24.0
(3.1.4) 169.3 154.6 171.4
154.5
164.3
147.1
169.8
149.1
164.9
146.2
159.0
137.5
(3.1.5) 247.2 221.7 232.7
195.8
231.7
213.8
250.6
220.6
221.3
196.5
207.3
187.8
(3.1.6) 240.4 220.9 219.6
194.4
225.8
209.0
244.1
220.0
215.7
200.8
203.4
189.0
(3.1.3) 238.7
Для нитротолуолов, в которых метильная группа находится в орто-положении
к
нитрогруппе,
наиболее
энергетически
выгодным
является
процесс
внутримолекулярного переноса водорода с образованием аци-нитросоединений.
Этот процесс представляет, безусловно, наибольший интерес среди разнообразных
механизмов
первичного
предполагается
акта
практически
термического
во
всех
распада
опубликованных
нитротолуолов.
теоретических
Он
и
91
экспериментальных
работах
(см.
статью
[36]).
Поэтому
более
подробные
исследования указанного механизма представляют отдельный интерес.
Таблица 3.1.3. Энтальпии активации (ΔH≠) и энтальпии реакций (ΔHреак)
возможных
вариантов
водородсодержащий
первичного
заместитель
в
акта
нитроаренов,
м-положении
к
R1H
C
R5
R
C
C
2
имеющих
R3
нитрогруппе
C
C
C
R4
O
N+
O–
Процесс
(кДж/моль) по данным метода B3LYP/6-31+G(2df,p)
м-НТ
2,3-ДНТ
2,5-ДНТ
3,4-ДНТ
3,5-ДНТ
R1=CH2,
R1=CH2,
R1=CH2,
R1=CH2,
R1=CH2,
R2=R3=R4
R2=R3= R4=H,
R3=R4= R5=H,
R2=R3= R5=H,
R2=R4= R5=H,
=R5=H
R5=NO2
R2=NO2
R4=NO2
R3=NO2
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
(3.1.7)
-
284.8
-
246.3
-
272.9
-
239.5
-
273.0
(3.1.8)
-
408.2
-
415.9
-
409.1
-
414.2
-
413.5
(3.1.9)
258.1
8.7
236.8
-16.1
248.4
-3.5
224.6
-19.6
255.8
-0.4
(3.1.10)
256.2
231.2
232.4
199.0
257.9
229.1
232.9
196.2
240.6
221.8
(3.1.11)
261.1
236.4
243.8
209.1
266.2
241.0
233.7
202.3
248.6
227.6
Таблица 3.1.4. Энтальпии активации (ΔH≠) и энтальпии реакций (ΔHреак)
возможных
вариантов
водородсодержащий
первичного
заместитель
акта
в
нитроаренов,
п-положении
к
имеющих
нитрогруппе
R 1H
R
2
R3
C
C
O
(кДж/моль) по данным метода B3LYP/6-31+G(2df,p)
Процесс
C
C
N
R5
C
C
R4
O
п-НТ
2,4-ДНТ
3,4-ДНТ
ТНТ
R1=CH2,
R1=CH2, R2=R3=
R1=CH2, R2=R3=
R1=CH2, R3=R4=H,
R2=R3= R4=R5=H
R4=H, R5=NO2
R5=H, R4=NO2
R2= R5=NO2
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
ΔH≠
ΔHреак.
(3.1.12)
-
288.9
-
277.0
-
243.5
-
267.7
(3.1.13)
-
409.4
-
409.9
-
414.2
-
405.2
(3.1.14)
262.1
11.4
260.3
2.3
229.4
-17.5
257.3
-6.0
(3.1.15)
257.4
238.9
244.8
227.9
227.1
202.1
239.3
225.6
(3.1.16)
257.4
238.9
249.3
236.6
233.4
205.2
239.3
225.6
92
Накопление нитрогрупп слабо влияет на величину барьера активации
рассматриваемой реакции сигматропного сдвига атома водорода в нитротолуолах.
В ряду о-НТ, 2,4-ДНТ, 2,4,6-ТНТ наблюдается небольшое уменьшение барьера
(на 9.7 кДж/моль, B3LYP/6-31+G(2df,p)). Интересно, что практически на такую же
величину (10 кДж/моль), уменьшается энергия активации реакции термического
распада 2,4,6-тринитротолуола (-207 кДж/моль) в газообразном состоянии по
сравнению с энергией активации реакции термического распада орто-нитротолуола
(197 кДж/моль).
Из данных, приведённых в таблице 3.1.2, следует, что барьер активации
процесса образования аци-формы значительно (на 35-40 кДж/моль) ниже энергии
активации реакции термического распада нитротолуолов (Таблицы 3.1.1, 3.1.2).
Следовательно, этот процесс не может быть лимитирующей стадией механизма
мономолекулярного распада нитротолуолов, в молекулах которых метильная группа
находится в орто-положении к нитрогруппе. С другой стороны, реакция
внутримолекулярного переноса водорода является наиболее выгодным вариантом
изомеризации орто-нитротолуолов, поэтому есть серьёзные основания полагать, что
этот процесс является первичным актом механизма термического распада ортонитротолуола и его нитропроизводных, в том числе и ТНТ.
Тенденцию монотонного снижения энтальпии активации реакции (3.1.4), при
увеличении в молекулах количества нитрогрупп, нарушают результаты, полученные
для 2,3- и 2,5-динитротолуолов. Для реакций с участием этих соединений расчёт
предсказывает небольшое увеличение энтальпии активации по сравнению с
аналогичным процессом для орто-нитротолуола. В случае реакции 2,3-ДНТ
наблюдаемые различия можно связать с тем, что нитрогруппа, участвующая в
реакции дополнительно повёрнута относительно плоскости кольца, вследствие
влияния соседней нитрогруппы.
Интересной особенностью процесса (3.1.4) является монотонность изменения
энтальпии активации и энтальпии реакции. Наличие подобной зависимости
93
подтверждается рисунком 3.1.1. Подобные зависимости обычно встречаются в
реакциях
радикального
распада.
В
процессе
нерадикального
распада
они
наблюдаются редко. Поэтому наличие подобной зависимости в шести реакциях
изомеризации нитроаренов представляет значительный интерес. Закономерности
изменения в ряду изученных соединений энтальпии реакции аналогичны тем,
которые были уже рассмотрены для энтальпии активации. Так, например, энтальпии
реакций 2,3- и 2,5-динитротолуолов превышают соответствующие значения для 2,4и 2,6-динитротолуолов, которые практически не отличаются. В то же время,
изменение в ряду изученных соединений энтальпии реакции значительно превышает
аналогичное изменение энтальпии активации (17.1 кДж/моль и 9.7 кДж/моль
соответственно).
174
172
170
ΔH
≠
168
166
164
162
160
158
136
138
140
142
144
146
148
150
152
154
156
ΔH реакц
Рисунок 3.1.1. Корреляция энтальпий активаций (ΔH≠) и энтальпий (ΔHреакц) реакции
образования аци-нитротолуолов (коэф. корреляции 0,94) по данным метода
B3LYP/6-31+G(2df,p)
Ещё более интересными, по нашему мнению, являются результаты,
полученные
при
теоретическом
изучении
механизма
реакции
образования
бициклических интермедиатов (реакции 3.1.5 и 3.1.6). Одной из интересных
особенностей этих реакций является заметное снижение энтальпии активации при
94
накоплении в молекуле количества нитрогрупп. Подобная тенденция приводит к
тому, что энтальпия активации реакций 3.1.5 и 3.1.6 в ТНТ снижается с
аналогичными процессами в o-нитротолуоле соответственно на 39.9 и 37 кДж/моль.
Энтальпии
активации
этих
реакций
только
незначительно
превышают
экспериментальную оценку энтальпии активации реакции термического распада
ТНТ. Наблюдаемые различия (10-15 кДж/моль) находятся в интервале возможных
погрешностей эксперимента и расчёта. Сказанное дает серьёзные основания
предполагать,
что
механизм
распада
ТНТ
с
образованием
циклических
интермедиатов может быть альтернативным каналом термического разложения ТНТ.
При этом следует учитывать, что реакция образования аци-формы не является
лимитирующей стадией, связанного с этим процессом механизма деструкции ТНТ.
Поэтому возможность альтернативного механизма разложения может представлять
принципиальный научный и практический интерес. К вопросу о механизме
деструкции ТНТ мы еще вернёмся при обсуждении результатов, полученных в ходе
исследования термораспада ион-радикалов этого соединения.
3.2 Основные каналы фрагментации анион-радикала
тринитротолуола
Согласно результатам экспериментальных работ [17,165] основными каналами
распада анион-радикала тринитротолуола (АР-ТНТ) являются отрыв радикалов OH●
и NO●. По данным работы [165] масс-спектр отрицательно заряженного иона ТНТ
содержит относительно интенсивную линию m/z 46, указывающую на образование
аниона NO2-. В отличие от работы [165], результаты более позднего исследования
[17] указывают на низкую интенсивность линии m/z 46. Для интерпретации
результатов
эксперимента
принципиальное
значение
имеет
получение
энергетических оценок элементарных стадий процессов разложения анион-радикала
ТНТ с использованием современных квантово-химических методов. Такие оценки
впервые были получены в нашей работе. Исследования каналов фрагментации ион-
95
радикалов ТНТ (Рисунок 3.2.1) проводилось с использованием метода M06 с
набором базисных функций 6-31+G(d,p). Геометрические структуры анион-радикала
ТНТ, интермедиатов и продуктов реакций распада АР-ТНТ приведены на рисунке
3.2.2. Геометрические структуры переходных состояний приведены на рисунке 3.2.3.
188
.7
2.6
13
9.0
34
193.7
H
198.7
NO
2
.8
82
+
24
OH
141.1
OH
OH
+
2
346.4
NO
H
198.7
193.7
2.6
7
{26
O
.8} N
}
2.8 NO
{27
+
+
4 O
8. N
ON
9
8.
12
21
.9}
25
{2
+
6 NO
2.
33
Рисунок 3.2.1 Схема реакции распада АР-ТНТ по методу М06/6-31+G(d,p).
Энтальпии продуктов, интермедиатов и переходных состояний рассчитаны
относительно АР-ТНТ (А1)
96
Рисунок 3.2.2. Равновесные структуры исходного реагента, интермедиатов и
некоторых продуктов, соответствующих реакциям распада анион-радикала ТНТ (А1)
по данным метода М06/6-31+G(d,p). Длины связей в Å
97
Рисунок 3.2.3. Геометрические структуры переходных состояний основных стадий
распада анион-радикала ТНТ по данным метода М06/6-31+G(d,p). Длины связей в Å
По данным нашего расчета энтальпия активации отрыва NO2- в результате
гетеролитической диссоциации связи С-N (реакция А1→А3, Рисунок 3.2.1)
составляет 280.3 кДж/моль, что на 22.6 кДж/моль превышает энтальпию активации
реакции отрыва NO2● (реакция А1→А2, Рисунок 3.2.1).
98
Реакция отрыва OH● по данным расчёта является многостадийной. Нами было
исследовано несколько возможных каналов отщепления гидроксила от АР-ТНТ.
Первые два канала (реакции А1→А7 и А1→А9, Рисунок 3.2.1) протекают через
образование орто-аци-формы АР-ТНТ (реакция А1→А4, Рисунки 3.2.1 и 3.2.2) и
конформационный переход А4→А5 (Рисунок 3.2.1) на первой и второй стадиях
соответственно. Энтальпия активации образования аци-формы анион-радикала ТНТ
составляет 104.6 кДж/моль (Рисунок 3.2.1), тогда как энтальпия реакции данного
элементарного акта (реакция А1→А4 Рисунок 3.2.1) равна 72.4 кДж/моль.
Конформационный переход А4→А5 (Рисунок 3.2.1), включающий вращение
гидроксильного фрагмента аци-изомера А4 (Рисунки 3.2.1 и 3.2.2) относительно
связи N-O, имеет барьер в 10.4 кДж/моль. Аци-форма А5 (Рисунок 3.2.2) лежит на 2.1
кДж/моль (Рисунок 3.2.1) выше по энергии, чем исходный изомер А4.
В дальнейшем появляются два возможных пути отрыва гидроксила. Первый
путь (реакция А5→А9, Рисунок 3.2.1) соответствует безбарьерному отрыву
гидроксила непосредственно от аци-формы А5. Энтальпия реакции в этом случае
составляет 124.2 кДж/моль. Второй канал (реакция А5→А7, Рисунок 3.2.1) отрыва
гидроксильного радикала от аци-изомера А5 имеет одну дополнительную
промежуточную стадию (реакция А5→А6, Рисунок 3.2.1), соответствующую
повороту фрагмента HONO относительно связи C-N на ~180°. Барьер активации
данного процесса составляет 15.5 кДж/моль. Аци-форма А6 (Рисунок 3.2.2) лежит на
9.2 кДж/моль ниже по энергии, чем аци-изомер А5 (Рисунок 3.2.1). Энтальпия
активации отрыва гидроксильного радикала от аци-формы А6 (Рисунок 3.2.1),
рассчитанная в предположении об отсутствия барьера обратной реакции, равна 128.4
кДж/моль.
Нами также был изучен механизм реакции отрыва OH● от пара-аци-формы АРТНТ (реакция А1→А14, Рисунок 3.2.1). Энтальпия активации образования пара-ациформы АР-ТНТ (реакция А1→А12, Рисунок 3.2.1) составляет 132.6 кДж/моль, что
существенно превышает энтальпию активации реакции образования орто-аци-
99
формы (реакция А1→А4, Рисунок 3.2.1). Пара-аци-форма (А12, Рисунок 3.2.2)
лежит на 138.0 кДж/моль выше по энергии, чем исходная молекула АР-ТНТ и на
65.6 кДж/моль выше по сравнению с орто-аци-формой А4 (Рисунок 3.2.1).
Дальнейший отрыв гидроксила от пара-аци-формы А12 протекает по механизму
аналогичному механизму отщепления OH● от орто-аци-формы АР-ТНТ (Рисунок
3.2.1), рассмотренному ранее. На первой стадии происходит поворот гидроксильного
фрагмента относительно связи O-N (реакция А13→А13, Рисунок 3.2.1). Однако в
случае пара-аци-формы данный процесс связан со значительно большими затратами
энергии (60.7 кДж/моль, Рисунок 3.2.1) по сравнению с аналогичным процессом в
орто-аци-форме (10.4 кДж/моль, Рисунок 3.2.1). Аци-изомер АР-ТНТ А13 (Рисунок
3.2.2) лежит на 54.9 кДж/моль выше аци-изомера А12 (Рисунок 3.2.1) Энтальпия
активации отрыва гидроксила от аци-формы А13 составляет 153.5 кДж/моль.
Таким образом, на основе анализа оценок энтальпий активации, а также
величин энтальпий реакций элементарных стадий, мы можем заключить, что
механизм А1→А7 будет наиболее энергетически выгодным каналом отщепления
гидроксильного радикала от АР-ТНТ, в то время как механизм А1→А9 требует
незначительно большей энергии (на 3.7 кДж/моль, Рисунок 3.2.1). Канал отрыва
гидроксильного радикала, протекающий через образование пара-аци-формы анионрадикала ТНТ (А1→А14, Рисунок 3.2.1), скорее всего не будет конкурировать с
двумя указанными выше механизмами из-за существенно более высокого значения
энтальпии активации.
Мы также предприняли попытку изучить механизм отрыва монооксида азота
от АР-ТНТ. Первоначально мы предположили, что отрыв NO● может протекать по
схожему с молекулой ТНТ механизму, т.е. через образование на первой стадии
нитритного изомера АР-ТНТ в результате нитро-нитритной перегруппировки.
Однако проведённые расчеты показали невозможность данного процесса для АРТНТ. Поэтому, мы изучили альтернативный механизм (реакция А1→А22, Рисунок
3.2.1),
включающий
образование
анион-радикала
8-окса-7-аза-3-метил-2,4-
100
динитробицикло[4.2.0]окта-1(8)-2,4-триен-7-оксида А17 (Рисунки 3.2.1 и 3.2.2) на
начальной стадии. Энтальпия активации реакции образования бициклического
изомера А17 (Рисунок 3.2.1) составляет 188.7 кДж/моль. По данным расчёта бицикл
А17 лежит на 159.4 кДж/моль выше по энергии, чем АР-ТНТ. На следующей стадии
происходит разрыв связи N16-О17 (Рисунок 3.2.2) и образование нитрозо изомера А18
(реакция А17→А18, Рисунок 3.2.1) с барьером активации 34.3 кДж/моль и
энтальпией реакции -39.3 кДж/моль. Отрыву NO● предшествует стадия образования
анион-радикала
7-окса-3-метил-6-нитрозо-2,4-динитробицикло[4.1.0]гепта-2,4-
дииена А19 (Реакция А18→А19, Рисунки 3.2.1 и 3.2.2). Энтальпия активации данной
стадии составляет 21.0 кДж/моль, тогда как изомер А19 лежит на 6.6 кДж/моль ниже
по энергии изомера А18. Отрыв NO● от А19 требует дополнительно 104.9 кДж/моль.
Расчет указывает на то, что реакция отрыва NO● (Реакция А19→А22, Рисунок 3.2.1)
является безактивационной. Как видно из рисунка 3.2.1 механизм отрыва NO●
А1→А22, требует приблизительно на 24.6 кДж/моль больше энергии, чем каналы
отрыва OH● А1→А7 и А1→А9. Более высокий барьер активации соответствует
более низкой интенсивности линии m/z 197 по сравнению с линией m/z 210 в массспектре отрицательно заряженного иона ТНТ.
Нами был также изучен альтернативный канал отрыва NO● – реакция
А19→А21 (Рисунок 3.2.1). Данный механизм имеет одну промежуточную стадию
(реакция А19→А20, Рисунок 3.2.1), соответствующую разрыву связи С1-С2 в
изомере А19 (Рисунок 3.2.2) с последующим образованием анион-радикала 4-метил6-нитрозо-3,5-динитрооксепина А20 (Рисунок 3.2.2). Барьер активации данной
стадии составляет всего 15.4 кДж/моль, а энтальпия реакции равна -28.5 кДж/моль.
Отрыв NO● от интермедиата А20 требует достаточно высокой энергии (247.6
кДж/моль), что на 142.7 кДж/моль выше по сравнению реакцией А19→А22 (Рисунок
3.2.1). Таким образом, мы можем заключить, что отрыв NO● от анион-радикала ТНТ
будет протекать по каналу А1→А22 (Рисунок 3.2.1).
101
Согласно работе [165] масс-спектр отрицательно заряженного иона ТНТ имеет
полосу относительно низкой интенсивности c m/z 180. Данная полоса соответствует
отрыву нейтрального фрагмента HONO. В связи с этим мы изучили несколько
возможных каналов образования иона m/z 180 с брутто формулой С7H4N2O4.
Первые два канала соответствуют безактивационному элиминированию HONO
от орто- и пара-аци-форм АР-ТНТ (реакции А5→А11 и А12→А16, Рисунок 3.2.1).
Энтальпия активация данных элементарных стадий составляет соответственно 168.1
и 211.0 кДж/моль.
Нами также было дополнительно изучено три альтернативных канала
образования иона m/z 180. Все три рассмотренных далее канала соответствуют
поэтапному отрыву радикалов OH● и NO● от аци-изомеров А5, А6 и А13 (Рисунок
3.2.1). Каналы отрыва OH● были рассмотрены ранее в этой главе. Как видно из
рисунка 3.2.1 энтальпия активация отрыва NO● от анионов А7, А9 и А15
соответственно составляет 272.8, 267.8 и 225.9 кДж/моль. Таким образом, мы вправе
предположить, что основным каналом образования иона m/z 180 является
многоступенчатая реакция А1→А11 (Рисунок 3.2.1).
3.3 Основные каналы фрагментации катион-радикала
тринитротолуола
Масс-спектр положительно заряженного иона ТНТ, как и в случае с
отрицательно заряженным ионом, имеет линию наивысшей интенсивности m/z 210,
соответствующую отрыву гидроксильного радикала. Прежде чем приступить к
обсуждению
механизма
распада
КР-ТНТ,
необходимо
отметить
некоторые
особенности проведения квантово-химического расчёта геометрической структуры
КР-ТНТ. В процессе оптимизации геометрических параметров КР-ТНТ методом
M06/6-31+G(d,p),
начиная
со
структуры,
соответствующей
равновесной
конфигурации молекулы ТНТ в основном состоянии, происходит перенос атома
водорода от углерода метильного фрагмента к кислороду нитрогруппы, что в свою
102
очередь приводит к аци-форме КР-ТНТ (Б1, Рисунок 3.3.1). Расчетное значение
адиабатического потенциала ионизации этого соединения (10.16 эВ) хорошо
согласуется с экспериментальной оценкой (10.59 эВ [133]).
Рисунок 3.3.1. Равновесные структуры исходной аци-формы (Б1), интермедиатов
некоторых продуктов и переходного состояния, соответствующих реакциям распада
катион-радикала ТНТ по данным метода М06/6-31+G(d,p). Длины связей в Å, углы в
градусах.
Отрыв OH● от аци-формы КР-ТНТ протекает в несколько стадий (реакция
Б1→Б6, Рисунок 3.3.2). На первой стадии происходит поворот гидроксильного
фрагмента (реакция Б1→Б4, Рисунок 3.3.2) относительно связи N-O. Барьер
активации данной реакции, рассчитанный исходя из полных электронных энергий
переходного состояния и реагента, равен 2.5 кДж/моль (Рисунок 3.3.2), тогда как
энтальпия активации данной стадии составляет -1.3 кДж/моль. Аци-изомер Б4
(Рисунки 3.3.1 и 3.3.2) лежит на 45.2 кДж/моль ниже по энергии, чем исходный
103
реагент (Рисунок 3.3.2). На второй стадии (реакция Б4→Б5, Рисунок 3.3.2)
происходит поворот фрагмента HONO в интермедиате Б4 относительно связи C-N.
Барьер активации данной реакции составляет 67.4 кДж/моль, а энтальпия реакции 10.4 кДж/моль. Геометрическая структура аци-изомера Б5 приведена на рисунке
3.3.1. Обратная реакция отрыву OH● от Б5 (Рисунок 3.3.2) является безбарьерной
реакцией. Требует по данным нашего расчета 74.0 кДж/моль.
Рисунок 3.3.2. Схема распада КР-ТНТ по данным метода М06/6-31+G(d,p).
Энтальпии продуктов, интермедиатов и переходных состояний рассчитаны
относительно аци-формы КР-ТНТ (Б1)
Мы также рассмотрели еще несколько альтернативных отрыву OH●
механизмов распада аци-формы КР-ТНТ. Так, например, по данным расчёта отрыв
104
воды от аци-формы Б5 (Рисунок 3.3.2) требует 279.0 кДж/моль, тогда как энтальпия
активации, рассчитанная для процесса элиминирования молекулы азотистой
кислоты (реакция Б5→Б8, Рисунок 3.3.2) составляет 290.3 кДж/моль. Значительно
более высокие значения энтальпии активации, полученные нами для реакции
элиминирования молекул воды и азотистой кислоты от КР-ТНТ, по сравнению с
отрывом гидроксильного радикала, позволяют объяснить существенно более низкую
интенсивность линий с m/z 209 и m/z 180 по сравнению с интенсивностью линии m/z
210.
Еще меньшую интенсивность имеют линии с m/z 181 и m/z 146,
соответствующие реакциям разрыва связи C-N и отрыву NO2● и NO2+. Это также
можно объяснить исходя из более высоких значений энтальпии активации,
рассчитанных для этих процессов (Рисунок 3.3.2).
3.4 Основные выводы по Главе 3
В связи с отсутствием в настоящее время надёжных теоретических оценок
барьеров различных элементарных стадий термического распада o-нитротолуола и
его нитропроизводных, в том числе и 2,4,6-тринитротолуола, нами было проведено
соответствующее изучение с использованием различных методов функционала
плотности. Было установлено, что наиболее энергетически выгодным для всех
изученных соединений является процесс внутримолекулярного переноса водорода от
метильной
группы
к
нитрогруппе
с
образованием
соответствующих
аци-
нитротолуолов. Однако по данным расчёта барьер этой реакции существенно ниже
экспериментальных
значений
энергии
активации
термического
распада
в
газообразном состоянии. Из сопоставления результатов расчёта с экспериментом
следует, реакция образования аци-формы является первичным актом термического
распада o-нитротолуола и его нитропроизводных, в том числе и тротила. Однако
лимитирующие стадии процесса, очевидно, связаны с вторичными реакциями
распада образующихся аци-нитротолуолов.
105
Для тротила обнаружен альтернативный вариант развития реакции, связанный
с образованием бициклического интермедиата. По данным расчёта лимитирующая
стадия этого процесса близка к энергии активации термического распада ТНТ в
газообразном состоянии. Изучение вторичных стадий распада ТНТ по этому
механизму связано со значительными трудностями. В том случае если окажется, что
лимитирующей стадией процесса разложения является первичный акт, то процесс
изомеризации
ТНТ
с
образованием
бицикла
может
наряду
с
реакцией
внутримолекулярного переноса водорода быть одним из альтернативных каналов
термораспада тротила.
С использованием метода теории функционала плотности M06/6-31+G(d,p)
нами были изучены механизмы фрагментации ион-радикалов ТНТ. Как показывают
результаты расчетов, многоступенчатый отрыв гидроксильного радикала является
энергетически наиболее выгодным процессом распада как катион-, так и анионрадикалов ТНТ. В случае катион-радикала, относительно низкий барьер этой
реакции (74.0 кДж/моль, Рисунок 3.3.2) хорошо коррелирует с более высокой
интенсивностью линии m/z 210 в масс-спектре положительно заряженного иона
ТНТ. Для анион-радикала ТНТ в дополнение к отрыву OH● нами был также изучен
канал элиминирования NO●, протекающий через образование бициклического
изомера АР-ТНТ. Расчетное значение энтальпии активации данного канала всего на
~25 кДж/моль выше барьера лимитирующей реакции цепочки отрыва OH●. Таким
образом, мы можем предположить, что эти два процесса идут параллельно, в то
время как отрыв OH● остается кинетически более выгодным процессом, исходя из
более высокой интенсивности линии m/z 210. Тем не менее, следует отметить, что
для более подробного анализа конкуренции данных механизмов необходимо
проведение расчета констант скоростей на основе теории переходного состояния или
теории RRKM.
106
Таким образом, проведённое теоретическое изучение показало, что для ТНТ
распад молекулы и ион-радикалов в основном протекает по аналогичным
механизмам.
107
Глава 4. Особенности механизмов распада ион-радикалов
этилнитрата и тетранитропентаэритрита
4.1 Основные каналы фрагментации катион-радикалов этилнитрата
и тетранитропентаэритрита1,2
Как уже отмечалось раньше в главе 1, масс-спектры ЭН и ТЭН, полученные c
использованием ионизации электронным ударом (electron ionization), имеют две
наиболее интенсивные линии с массовыми числами m/z 46 и m/z 76 [133], которые
отвечают наличию в продуктах распада положительно заряженных частиц NO2+ и
CH2ONO2+ соответственно (Рисунок 4.1.1). Анализ геометрических структур катионрадикалов этилнитрата (КРЭН) (Рисунок 4.1.2(а)) и ТЭН (ТЭН-КР) (Рисунок 4.1.3,
Таблица 4.1.1), расcчитанных с использованием метода M06/6-31+G(d,p), указывает
на значительное увеличение длин связей O-NO2 по сравнению с молекулами.
Следует отметить, что геометрические параметры ион-радикалов ЭН и ТЭН,
полученные в рамках методов функционала плотности M06 и B3LYP, находятся в
хорошем согласии друг с другом, как это уже отмечалось раньше в главе 2, и
практически не зависят от выбранного базисного набора. Поэтому в данной главе
при обсуждении каналов распада данных соединений мы будем преимущественно
использовать данные по геометриям молекул, полученные в рамках метода M06/631+G(d,p).
Исходя из наличия удлиненной связи О-NO2 в молекулах КРЭН и ТЭН-КР и
интенсивной полосы m/z 46 в масс-спектрах этих двух соединений, можно
предположить, что отрыв нитрогруппы будет одним из основных каналов распада.
Энергетические оценки подтверждают это предположение.
1
Исследование каналов распада ион-радикалов тетранитропентаэритрита проводилось при сотрудничестве с научноисследовательской группой Университета Мэриленда (Колледж Парк, США) под руководством профессора M.M.
Kuklja.
2
Научным консультантом по разделу 4.1 работы был доцент кафедры катализа, к.х.н. Р.В. Цышевский.
108
Рисунок 4.1.1 Основные каналы распада катион- и анион-радикала этилнитрата
Рисунок 4.1.2. Равновесные структуры (M06/6-31+G(d,p)): а) катион-радикала
этилнитрата; б) переходного состояния реакции гомолического разрыва связи O-NO2
с образованием катиона CH3CH-OH; в) катиона CH3CH-OH и г) переходного
состояния реакции отрыва СН4 от катиона CH3CH-OH
109
Рисунок 4.1.3. Геометрические структуры: а) ТЭН; б) катион-радикала ТЭН
Таблица 4.1.1. Основные геометрические параметры молекул ТЭН и ТЭН-КР (длины
связи в Å, углы в градусах)
Геометрические параметры
C4-C3
ТЭН
ТЭН-КР
M06
B3LYP
M06
B3LYP
1.531
1.545
1.718
1.545
3
C -O
8
1.434
1.446
1.312
1.446
8
3
1.406
1.430
1.781
1.430
N3-O7
1.204
1.212
1.157
1.212
O -N
3
11
1
4
N -O
1.196
1.204
1.165
1.204
3
108.6
108.6
103.4
108.6
C4-C3-O8
C -C -C
106.0
106.3
111.3
106.3
3
8
3
114.3
114.1
114.5
114.1
8
3
7
116.9
117.0
105.3
117.0
9
3
11
112.5
112.4
110.3
112.4
C -O -N
O -N -O
O -N -O
O7-N3-O11
130.7
130.5
144.4
130.5
1
4
3
8
-63.5
177.9
-49.4
177.9
4
3
8
3
-179.5
-178.0
112.4
-178.0
0.0
-0.4
179.8
-0.4
C -C -C -O
C -C -O -N
C3-O8-N3-O7
110
Действительно, как видно из таблицы 4.1.2 энтальпия активации отрыва NO2+
от КРЭН, рассчитанная в предположении об отсутствии барьера обратной реакции,
по данным метода M06/6-31+G(d,p) составляет 111.7 кДж/моль, что на ~41.8
кДж/моль
меньше,
чем
удовлетворительном
в
согласии
молекуле
с
ЭН.
Этот
оценками,
результат
полученными
находится
методов
в
M06/6-
311+G(3df,p) и B3LYP/6-311+G(3df,p). Метод B3LYP/6-31+G(d,p) дает ниже
значение энтальпии реакции по сравнению с оценками M06/6-31+G(d,p) примерно
на 29.3 кДж/моль.
Таблица 4.1.2. Энтальпии активации (энтальпии реакции) реакций распада катион- и
анион-радикалов этилнитрата (в кДж/моль)
№
1
M06
Реакция
6-31+G(d,p)
C2H5ONO2+●→C2H5O●+NO2+
2а C2H5ONO2
+●
→CH3●+CH2ONO2+
+
2б CH2ONO2 →CH2O+NO2
3a C2H5ONO2
+●
→NO2●+CH3CHOH+
+
3б CH3CHOH →CH4+COH
4
4
5
+
+
B3LYP
6-311+G(3df,p) 6-31+G(d,p) 6-311+G(3df,p)
111.7(111.7)
98.3 (98.3)
82.8 (82.8)
112.5 (112.5)
75.3 (75.3)
61.5 (61.5)
74.9 (74.9)
64.4 (64.4)
98.7 (98.7)
84.1 (84.1)
59.4 (59.4)
90.8 (90.8)
62.3 (-231.4)
65.7 (-218.0)
68.2(-230.1)
69.0 (-225.9)
403.8(330.1)
402.9 (323.4)
412.5(331.4)
411.7(330.5)
-●
●
-
50.6 (50.6)
57.7 (57.7)
45.2 (45.2)
58.6 (58.6)
-●
-
●
127.2(127.2)
110.5 (57.7)
119.7 (45.2)
115.9(115.9)
C2H5ONO2 →C2H5●+ONO2-
135.6 (46.9)
137.7 (44.8)
137.2 (52.3)
141.8 (54.0)
C2H5ONO2 →C2H5O +NO2
C2H5ONO2 →C2H5O +NO2
-●
В случае с ТЭН-КР энтальпия реакции отрыва NO2+ (Рисунок 4.1.4)
приблизительно на 29.3 кДж/моль меньше, чем для КРЭН и составляет по данным
методов M06/6-31+G(d,p) и B3LYP/6-31+G(d,p) 76.6 и 51.9 кДж/моль соответственно
[230].
Вторым основным каналом фрагментации КРЭН и ТЭН-КР (Рисунки 4.1.1 и
4.1.4) является образование положительно заряженного иона CH2ONO2+ (m/z 76),
связанное с разрывом С1-С5 в молекуле КРЭН (Рисунок 4.1.2) и С4-С3 в ТЭН-КР
(Рисунок 4.1.3). Как видно из таблиц 4.1.2 и 4.1.3, данный механизм требует в
111
среднем на 37.7 кДж/моль меньше энергии, чем отрыв NO2+. Так по данным метода
M06/6-311+G(3df,p)
энтальпия
реакции,
рассчитанная
в
предположении
об
отсутствии барьера обратной реакции, составляет 61.5 кДж/моль (Таблица 4.1.2). По
данным методов M06/6-31+G(d,p) и B3LYP/6-31+G(d,p) энтальпия разрыва связи СС в молекуле ТЭН-КР всего 35.6 и 15.5 кДж/моль соответственно (Таблица 4.1.3).
Рисунок 4.1.4. Схема распада ТЭН-КР и ТЭН-АР
Таблица 4.1.3. Энтальпии активации (энтальпии реакции) реакций распада ТЭН-КР и
ТЭН-АР (в кДж/моль)
№
Реакция
M06/6-31+G(d,p)
B3LYP/6-31+G(d,p)
1
C5H8N4O12+● → C5H8N3O10● + NO2+
76.6
51.9
2а
C5H8N4O12+● → C4H6N3O9● + CH2ONO2+
35.6
15.5
2б
CH2ONO2+ → CH2O + NO2+
98.7 (98.7)
59.4 (59.4)
4
C5H8N4O12-● → C5H8N4O12-●(eq) (ТЭН-АРeq)
4.6 (-67.8)
0.8 (-68.2)
C5H8N4O12-●(eq) → C5H8N3O10● + NO2-
157.7 (157.7)
143.9 (143.9)
4
C5H8N4O12-●(eq) → C5H8N3O10- + NO2●
74.1 (74.1)
66.9 (66.9)
5
C5H8N4O12-●(eq) → C5H8N3O9● + ONO2-
133.1(53.6)
136.0 (67.4)
112
Такое существенное различие в энтальпиях реакции разрыва связи С-С,
полученных в рамках DFT методов, для КРЭН и ТЭН-КР объясняется различием
длин связей C-C в этих соединениях. Так, например, как видно из рисунка 4.1.2(а),
длина связи С-С в КРЭН составляет 1.590 Å, тогда как в равновесной структуре
ТЭН-КР длина связи С4-С3 (Рисунок 4.1.3, Таблица 4.1.1) составляет 1.718 Å, что
приблизительно на 0.2 Å больше, чем в молекуле ТЭН.
Таким образом, на основе сравнения энтальпий реакции мы можем
предположить, что разрыв связи С-С и образование катиона CH2ONO2+ будет
доминирующим первичным каналом распада ТЭН-КР. Это согласуется с тем, что
линия с массовым числом m/z 76 имеет относительно более высокую интенсивность,
чем линия m/z 46. В случае КРЭН линия с массовым числом m/z 46 имеет
интенсивность, приблизительно в два раза превышающую интенсивность линии m/z
76, что противоречит нашим энергетическим оценкам. Мы можем предположить,
что, как и в случае с ТЭН-КР, отщепление CH2ONO2+ от КРЭН будет проходить с
большей скоростью, чем разрыв связи O-NO2. тогда как образование NO2+
происходит на второй стадии реакции в результате распада иона m/z 76 на ион m/z
46 (NO2+) и молекулу формальдегида (Рисунок 4.1.1). Энтальпия реакции данного
процесса по оценке методов M06/6-31+G(d,p) и B3LYP/6-31+G(d,p) составляет 98.7 и
59.4 кДж/моль соответственно (таблица 4.1.2). Не стоит исключать данный механизм
и для распада ионизированной молекулы ТЭН-КР.
Кроме двух, рассмотренных выше линий, масс-спектр ЭН имеет третью по
интенсивности линию с массовым числом m/z 29. Можно было бы предположить,
что данный пик соответствует образованию иона CH3CH2+ в результате отрыва
радикального фрагмента ONO2. Однако, присутствие линии m/z 29 в масс-спектрах
метилнитрата и пропилнитрата, указывают на ошибочность данного предположения.
Также необходимо указать на отсутствие данной линии в масс-спектре ТЭН.
По данным нашего теоретического исследования наличие линии m/z 29 в массспектре ЭН соответствует образованию катиона СОН+, как показано на рисунке
113
4.1.1. Ранее аналогичный механизм был рассмотрен на примере катион-радикала
метилнитрита [231]. Катиона СОН+ образуется в результате многоступенчатой
реакции. Первая стадия данной цепочки превращений соответствует переносу атома
водорода H6 от атома углерода С5 к атому кислорода O8 (Рисунок 4.1.2(б)),
сопровождающийся одновременным отрывом нитрогруппы и образованием катиона
CH3СHОН+ (Рисунок 4.1.2(в)). По данным метода M06/6-31+G(d,p) энтальпия
активации данной реакции составляет 62.3 кДж/моль (Таблица 4.1.2), что хорошо
согласуется с оценками других методов. Энтальпия реакции, рассчитанная методом
M06/6-31+G(d,p)
равна
-231.4
кДж/моль.
Образование
иона
CH3СHОН+
соответствует наличию линии m/z 45 в масс-спектре этилнитрата. На второй стадии
происходит распад катиона CH3СHОН+ на молекулу метана и СОН+. Структура
переходного состояния, найденного для этой стадии, приведена на рисунке 4.1.2(г).
Барьер данной реакции по оценкам метода M06/6-31+G(d,p) составляет 403.8
кДж/моль. Следует также отметить, что наши расчеты указывают на отсутствие
данного канала при распаде катион радикала ТЭН.
4.2 Основные каналы фрагментации анион-радикалов этилнитрата и
тетранитропентаэритрита
Как и в случае с катион-радикалом ЭН, в равновесной структуре анионрадикала ЭН (АРЭН) связь O-N (рисунок 4.2.1(а)) значительно увеличена по
сравнению с молекулой ЭН. Энтальпия реакции гетеролитического разрыва связи ONO2 с отщеплением аниона NO2- (Рисунок 4.1.1) по данным метода M06/6-31+G(d,p)
составляет 50.6 кДж/моль (Таблица 4.1.2), что на 77.4 кДж/моль ниже по сравнению
с гомолитическим отрывом NO2●. Отрыв ONO2- от АРЭН (Рисунок 4.1.1) протекает
через переходное состояние, приведенное на рисунке 4.2.1(б). Энтальпия активация
данного процесса по оценкам методов M06/6-31+G(d,p) и B3LYP/6-31+G(d,p)
составляет 135.6 и 137.2 кДж/моль соответственно (таблица 4.1.2). Как показывают
114
результаты расчета, данная реакция является эндотермической, энтальпия реакции
лежит в пределах 44.8-54.0 кДж/моль в зависимости от используемого метода.
Рисунок 4.2.1. Геометрические структуры (M06/6-31+G(d,p)): а) анион-радикала
этилнитрата; б) переходного состояния реакции гомотического отрыва ОNO2
Рисунок 4.2.2 Молекулярные структуры а) ТЭН-АР и б) ТЭН-АРeq
Структура
анион-радикала
ТЭН
(ТЭН-АР),
полученная
в
результате
оптимизации геометрии, начиная со структуры, соответствующей равновесной
конфигурации молекулы ТЭН в основном состоянии, практически не отличается от
115
молекулы ТЭН, как это видно из рисунков 4.1.3(а) и 4.2.2(а) и таблиц 4.1.1 и 4.2.1.
Однако по данным методов M06/6-31+G(d,p) и B3LYP/6-31+G(d,p) структура ТЭНАР, приведенная на рисунке 4.2.2(а) и в таблице 4.2.1, соответствует нестабильной
конфигурации, которая легко изомеризуется в структуру с одной удлиненной связью
O-NO2 (Рисунки 4.1.4 и 4.2.2(б), Таблица 4.2.1). Барьер данной реакции,
рассчитанный методом B3LYP/6-31+G(d,p), составляет всего 0.8 кДж/моль (Таблица
4.1.3). Полученный изомер (ТЭН-АРeq) соответствует равновесной структуре анионрадикала ТЭН и лежит приблизительно на 66.9 кДж/моль ниже по сравнению с ТЭНАР (Таблица 4.1.3). Структура переходного состояния данной реакции приведена на
рисунке 4.2.3, геометрические параметры – в таблице 4.2.1.
Таблица 4.2.1. Основные геометрические параметры молекул ТЭН-АР, ТЭН-АРeq, а
так же переходных состояний (ПС) реакции изомеризации ТЭН-АР и отрыва ONO2.
Геометрические
параметры
ТЭН-АР
ПС: отрыв ONO2-
ПС: образование
eq
ТЭН-АР
ТЭН-АРeq
М06
B3LYP
М06
B3LYP
М06
B3LYP
М06
B3LYP
C4-C2
1.530
1.544
1.513
1.523
1.547
1.547
1.569
1.586
C2-O5
1.429
1.442
1.808
1.838
1.426
1.426
1.343
1.359
O5-N2
1.407
1.432
1.316
1.331
1.497
1.497
2.245
2.241
N2-O4
1.222
1.232
1.249
1.254
1.242
1.242
1.224
1.233
N2-O6
1.216
1.225
1.265
1.270
1.233
1.233
1.215
1.224
C5-C4-C2
108.6
108.5
109.7
109.9
109.2
109.2
112.2
112.4
C4-C2-O5
106.5
106.8
110.3
110.6
107.1
107.1
112.5
112.4
2
5
2
114.1
113.9
112.1
114.0
112.7
112.7
103.5
104.5
5
2
4
117.3
117.4
117.8
118.3
115.8
115.8
109.8
110.9
5
2
6
112.7
112.6
117.2
117.1
111.3
111.3
111.6
112.4
4
2
6
130.1
130.0
123.1
123.6
129.9
129.9
125.4
125.0
5
4
2
5
-64.0
177.4
156.0
152.6
178.1
178.1
173.4
172.3
4
2
5
2
178.1
179.2
71.5
82.4
172.4
172.4
99.3
101.2
2
5
2
4
1.2
0.3
68.7
83.0
17.4
17.4
24.3
22.7
C -O -N
O -N -O
O -N -O
O -N -O
C -C -C -O
C -C -O -N
C -O -N -О
116
Рисунок 4.2.3 Структуры переходных состояний реакций а) изомеризации ТЭН-АР
→ ТЭН-АРeq; б) отрыва ОNO2-.
Дальнейшее изучение механизмов распада молекулы ТЭН-АРeq указывает на
существенное различие по сравнению с распадом АРЭН. Прежде всего, это касается
реакции разрыва связи O-NO2. Как видно из таблицы 4.1.3 гомолитический отрыв
NO2● от ТЭН-АРeq энергетически более выгоден, чем гетеролитический отрыв
аниона NO2-. Разница в энтальпиях реакции составляет примерно 83.7 кДж/моль.
Преобладание
результатам
гомолитической
эксперимента
по
реакции
над
гетеролитической
диссоциативному
противоречит
присоединению
электрона,
указывающим на то, что NO2- вместе с ONO2- является основным продуктом распада
анион-радикала ТЭН. Все наши попытки найти альтернативный более выгодный
канал образования NO2- на данный момент не увенчались успехом. Поэтому
механизм этого процесса остается дискуссионным. Большой размер молекулы
117
делает в настоящее время невозможным использование мощных неэмпирических
квантово-химических методов с базисами большого размера.
В отличие от реакции разрыва связи O-NO2, как это видно из таблиц 4.1.2 и
4.1.3, отрыв ONO2- от ТЭН-АРeq (Рисунок 4.1.4) имеет достаточно близкие значения
энтальпии активации и энтальпии реакции, рассчитанные для аналогичного процесса
распада АРЭН. Так по данным метода M06/6-31+G(d,p) энтальпия активации и
энтальпия реакции отрыва ONO2- в молекуле ТЭН-АРeq составляет 133.1 и 53.6
кДж/моль соответственно (Таблица 4.1.3), тогда как значение барьера активации
отрыва ONO2- от АРЭН, рассчитанное в рамках этого же метода, составляет 135.6
кДж/моль, энтальпия реакции равна 46.9 кДж/моль (Таблица 4.1.2).
Как видно из таблицы 4.1.3 отрыв ONO2- требует несколько меньших затрат
энергии, чем отрыв NO2-. Данный факт хорошо согласуется с результатами
эксперимента, указывающих на то, что ONO2- является основным продуктом распада
анион-радикала ТЭН, тогда как процесс образования NO2- имеет значительно
меньшую интенсивность. Тем не менее, для более точного сравнения этих двух
процессов необходимо проведение анализа кинетики этих реакций, что будет целью
наших дальнейших исследований.
4.3 Основные выводы по Главе 4
Проведённое теоретическое изучение показало, что равновесные структуры
ион-радикалов ЭН и ТЭН имеют удлиненную по сравнению с молекулами связь ONO2. В равновесной структуре катион-радикала ТЭН также увеличивается длина
связи С-С, примыкающая к нитратной группе. Значительное изменение структуры
ион-радикалов нитроэфиров отражается на энергетике и механизмах распада
соединений. Так, например, наиболее выгодным механизмом распада катион- и
анион-радикалов
ЭН
является
гетеролитический
разрыв
связи
O-NO2,
сопровождающийся образованием ионов NO2+ и NO2- соответственно. В случае
катион-радикала ТЭН наиболее выгодным процессом является разрыв связи С-С.
118
Барьер этой реакции по данным расчета составляет 16.7-37.7 кДж/моль (Таблица
4.3.3). Распад катиона CH2ONO2+, образующегося в результате разрыва связи С-С в
ионизированных молекулах ЭН и ТЭН, на катион NO2+ и молекулу формальдегида
по данным нашего расчета также может конкурировать с прямым отщеплением NO2+
от молекулярных ионов ЭН и ТЭН.
Нами, также был предложен механизм многоступенчатой реакции образования
катиона COH+, объясняющий наличие линии m/z 29 с относительно высокой
интенсивностью в масс-спектре ЭН.
Результаты исследования указывают на существенное (~ 209 кДж/моль)
снижение барьера активации отрыва ONO2- от анион-радикалов ЭН и ТЭН по
сравнению с аналогичными реакциями молекул. Установлено также, что в отличие
от молекул, где реакция рекомбинации радикалов, образующихся при разрыве связи
С-ONO2
является
безактивационной,
в
анион-радикалах
имеет
хорошо
локализованное переходное состояние. Показано, что в АРЭН отрыв ONO2- требует
значительно (~ на 84 кДж/моль) больших затрат энергии, чем отрыв NO2-, тогда как
при распаде ТЭН-АР разрыв связи O-NO2 с последующим отщеплением NO2является менее энергетически выгодной реакцией.
119
Глава 5. Особенности механизмов распада ион-радикалов
диметилнитрамина и 1,3,5,7-тетранитро-1,3,5,7тетраазациклооктана (октогена)
5.1 Основные каналы фрагментации катион- и анион-радикалов
диметилнитрамина
Как показывает анализ масс-спектра ДМНА [133], линии наивысшей
интенсивности соответствуют массовым числам m/z 90, m/z 44, m/z 43, m/z 42.
Первая из данной серии линия (m/z 90) соответствует иону КР-ДМНА. Линия с
массовым числом m/z 44 имеет самую низкую интенсивность среди этих четырех
линий и, скорее всего, соответствует образованию катиона C2H6N+ в результате
гомолитического разрыва связи N-N (реакция В1→В2, Рисунок 5.1.1, Таблица 5.1.1).
Энтальпия реакции данного процесса, рассчитанная в предположении об отсутствии
барьера обратной реакции, по данным метода M06/6-31+G(d,p) составляет 136.4
кДж/моль.
Энтальпия реакции гетеролитической диссоциации связи N-N на 100.8
кДж/моль (реакция В1→В3, Рисунок 5.1.1, Таблица 5.1.1) выше энтальпии реакции
гомолитического отрыва нитрогруппы. Это хорошо согласуется с экспериментом,
т.к. линия m/z 46, соответствующая отщеплению NO2+ имеет более низкую,
примерно в 40 раз, по сравнению с линией m/z 44 интенсивность.
Наличие второй по интенсивности линии m/z 44 объясняется наличием среди
продуктов распада КР-ДМНА иона C2H5N●+, образующегося в результате отрыва
азотистой кислоты (реакция В1→В6, Рисунок 5.1.1) Данная реакция включает две
основные стадии. Первая стадия (реакция В1→В4, Рисунок 5.1.1) включает
образование аци-формы КР-ДМНА в результате миграции атома водорода от
углерода к атому кислорода нитрогруппы. Структуры переходного состояния данной
реакции и аци-формы представлены на рисунке 5.1.2. Энтальпия активации данного
120
процесса по данным метода M06/6-31+G(d,p) составляет 98.3 кДж/моль, тогда как
аци-изомер КР-ДМНА лежит на 55.6 кДж/моль ниже исходного реагента.
Рисунок 5.1.1. Схема реакции распада КР-ДМНА по данным метода М06/631+G(d,p). Энтальпии продуктов, интермедиатов и переходных состояний
рассчитаны относительно КР-ДМНА (В1)
Вторая стадия данного процесса соответствует образованию ион-нейтрального
комплекса В5 (Рисунки 5.1.1 и 5.1.2) с удлиненной связью N-N. Энтальпия
активации стадии В4→В5 составляет 23.4 кДж/моль (Рисунок 5.1.1, Таблица 5.1.1).
Комплекс В5 лежит на 1.3 кДж/моль выше по энергии относительно аци-формы В4
(Рисунок 5.1.1, Таблица 5.1.1). Структура переходного состояния для данной
реакции приведена на рисунке 5.1.2.
121
Таблица 5.1.1. Относительные КР ДМНА энтальпии интермедиатов, продуктов и
переходных состояний реакций распада КР ДМНА (кДж/моль)
Структура
M06
6-31+G(d,p)
6-311+G(3df,p)
B1
0.0
0.0
B2
136.4
141.0
B3
237.2
200.4
B4
-55.6
-49.4
ПС(В1-В4)
98.3
98.7
B5
-54.4
-47.3
ПС(В4-В5)
-32.2
-26.4
B6
-233.0
-145.2
ПС(В5-В6)
-10.9
-13.0
B7
38.1
-14.2
B8
0.0
4.6
ПС(В4-В8)
18.0
25.1
B9
246.0
219.7
Третья стадия данной реакции непосредственно включает элиминирование
азотистой кислоты от комплекса В5 и образование иона C2H5N●+. Энтальпия
активации данной реакции составляет 92.5 кДж/моль (Рисунок 5.1.1, Таблица 5.1.1).
Как видно из рисунка 5.1.1 и таблицы 5.1.1 лимитирующей стадией реакции отрыва
HNO2 является образование аци-формы с барьером активации 98.3 кДж/моль, что на
38.1 кДж/моль ниже по сравнению с реакцией отрыва молекулы NO2 (реакция
В1→В2, Рисунок 5.1.1). Столь значительная разница в значениях энтальпий
активации, рассчитанных для двух этих механизмов, объясняет более высокую
интенсивность линии m/z 43.
Линия m/z 42 имеет наивысшую интенсивность. Как показывает наш расчет
[232], выполненный с использованием разных методов теории функционала
плотности, данная линия соответствует образованию катиона C2H4N+ (реакция
122
В1→В6, Рисунок 5.1.1). Данная реакция протекает в три стадии. Первая и вторая
стадии этого механизма включают соответственно образование аци-формы КРДМНА (реакция В1→В4, Рисунок 5.1.1) и образование ион-нейтрального комплекса
В5 (Рисунок 5.1.1), как и в случае реакции элиминирования азотистой кислоты
(реакция В1→А7, Рисунок 5.1.1). На третьей стадии происходит отрыв молекулы
воды и монооксида азота от комплекса В5 с последующим образованием C2H4N+
(реакция В5→В6, Рисунок 5.1.1). Переходное состояние, найденное для данного
процесса, приведено на рисунке 5.1.2.
Рисунок 5.1.2. Равновесные структуры исходного реагента, интермедиатов и
некоторых продуктов реакций распада катион-радикала ДМНА (В1) по данным
метода М06/6-31+G(d,p). Длины связей в Å.
123
Энтальпия активации элементарной стадии В5→В6 (Рисунок 5.1.1) составляет
43.5 кДж/моль, что на 54.8 кДж/моль ниже, чем энтальпия активации (98.3
кДж/моль) образования аци-формы В1→В4 и на 49.0 кДж/моль ниже энтальпии
активации элиминирования азотистой кислоты от комплекса В5 (реакция В5→В7,
Рисунок 5.1.1, Таблица 5.1.1). Более низкий барьер активации реакции В1→В6
(Рисунок 5.1.1, Таблица 5.1.1) по сравнению с другими рассмотренными процессами
объясняет наибольшую интенсивность линии m/z 42. Следует также обратить
внимание на высокую экзотермичность канала фрагментации КР-ДМНА В1→В6
(Рисунок 5.1.1). Так по данным метода M06/6-31+G(d,p) энтальпия данной реакции
составляет -233.0 кДж/моль.
Также интересным является механизм отщепления катиона NO+, образование
которого соответствует наличию линии в масс-спектре ДМНА с массовым числом
m/z 30. Как уже указывалось в главе 1, отщеплению монооксида азота от молекулы
предшествует
стадия
нитро-нитритной
перегруппировки,
которая
в
случае
нитроалканов, нитраминов, нитроэфиров имеет высокий барьер активации для
конкуренции при газофазном мономолекулярном распаде. Однако все наши попытки
изучить процесс нитро-нитритной перегруппировки для КР-ДМНА окончились
неудачей. В работе [123] также было указано на невозможность протекания нитронитритной перегруппировки в случае катион-радикала 1-нитропропана.
По данным наших расчетов существует альтернативный механизм, связанный
с нитро-нитритной перегруппировкой на первой стадии, − механизм отщепления
катиона NO+ от КР-ДМНА. Данный механизм (реакция В1→В9, Рисунок 5.5.1)
включает в себя несколько стадий. Первая стадия В1→В4 (Рисунок 5.1.1), общая с
реакциями В1→В6 и В1→В7, включает образование аци-формы с барьером
активации 98.3 кДж/моль и энтальпией реакции -55.6 кДж/моль. На второй стадии
происходит отрыв радикала OH● (реакция В4→В8, Рисунок 5.1.1) и образование
катиона C2H5N2O+. Образование катиона C2H5N2O+ в свою очередь соответствует
низко-интенсивной линии в масс-спектре ДМНА с массовым числом m/z 73. Данная
124
реакция требует 73.6 кДж/моль. Переходное состояние, рассчитанное для указанной
реакции, приведено на рисунке 5.1.2. Последняя стадия (реакция В8→В9, Рисунок
5.1.1) – отрыв катиона NO+ от катиона C2H5N2O+, обратная реакция которого
является безбарьерной. Энтальпия реакции процесса В8→В9 равна 246.0 кДж/моль.
Обзор литературы не выявил данных по распаду анион-радикала ДМНА.
Поэтому в нашей работе мы ограничились изучением только одной реакции,
отрывом аниона NO2-. Наш выбор объясняется тем, что образование NO2- является
одной из двух доминирующих реакций при распаде анион-радикалов гексогена и
октогена. В связи с этим изучение механизма отрыва NO2- (реакция Г1→Г2, Рисунок
5.1.3) от более простого представителя нитраминов представлялось важным шагом,
предшествующим исследованию каналов фрагментации анион-радикала реального
взрывчатого вещества с более сложной молекулярной структурой – октогена.
Рисунок 5.1.3. Схема реакции распада АР-ДМНА по данным метода М06/631+G(d,p)
Как видно из рисунка 5.1.3 отрыв NO2- от АР-ДМНА (реакция Г1-Г2), требует
всего 3.8 кДж/моль, что значительно (примерно на 43.1 кДж/моль) ниже по
сравнению с энтальпией диссоциации связи N-N в молекуле ДМНА [172,173].
Энтальпия реакции данного процесса равна -49.1 кДж/моль. Геометрические
структуры АР-ДМНА и переходного состояния отрыва NO2-, рассчитанные методом
М06, приведены на рисунке 5.1.4.
125
Рисунок 5.1.4. Геометрические структуры: Г1 – анион-радикал ДМНА; переходного
состояния реакции гомолического отрыва NO2 (Г1→Г2)
5.2 Основные каналы фрагментации катион- и анион-радикалов
октогена
В отличие от АР-ДМНА распад анион-радикала октогена был изучен
экспериментально методом диссоциативного присоединения электрона [190]. Как
уже отмечалось ранее в главе 1, масс-спектр анион-радикала октогена содержит две
наиболее интенсивные линии с массовыми числами m/z 176 и m/z 46, которые в
свою очередь соответствуют анионам C3H6N5O4- и NO2-.
В нашей работе мы изучили три возможных первичные стадии распада анионрадикала октогена. Первая стадия (реакция Д1→Д2, Рисунок 5.2.1), включает отрыв
радикала NO2● в результате гомолитической диссоциации связи одной из четырех
связей N-N. Эта реакция по данным метода M06/6-31+G(d,p) требует 162.8
кДж/моль, что на 99.6 кДж/моль выше по сравнению с реакцией отрыва NO2- в
результате гетеролитического разрыва связи N-N (реакция Д1→Д3, Рисунок 5.2.1).
Третий изученный процесс (реакция Д1→Д5, Рисунок 5.2.1) также соответствует
отрыву фрагмента NO2●. Однако, механизм Д1→Д5 отличается от механизма
Д1→Д2 тем, что отрыв NO2● сопровождается раскрытием тетраазациклооктанового
кольца. Данная реакция требует 24.3 кДж/моль, тогда как энтальпия реакции
126
составляет 15.1 кДж/моль. Структура переходного состояния данной реакции
приведена на рисунке 5.2.2. Однако необходимо отметить, что спуски по координате
реакции, рассчитанные для данного переходного состояния, приводят к структуре
анион-радикала октогена с одной растянутой связью N-N (Д4, Рисунки 5.2.1 и 5.2.2).
Данная структура лежит на 1.7 кДж/моль выше, чем исходная структура анионрадикала октогена (Рисунок 5.2.2). Тем не менее мы не смогли найти переходного
состояния, находящегося на пути между структурами Д1 и Д4. В отличие от
результатов нашей работы, данные работы [190], полученные в рамках метода
MP2/6-311++G(2d,p), указывают на то, что структура Д4 с одной растянутой N-N
связью соответствует наиболее выгодной по энергии конфигурации анион-радикала
октогена.
Рисунок 5.2.1 Механизм первичных стадий распада анион-радикала октогена.
Энтальпии продуктов, интермедиатов и переходных состояний рассчитаны методом
M06/6-31+G(d,p) относительно структуры Д1
Таким образом, мы можем предположить, что основной первичной стадией
распада анион-радикала октогена является механизм Д1→Д5 (Рисунок 5.2.1),
протекающий с отрывом радикала NO2● и образованием аниона C4H8N7O6- (m/z 250)
127
геометрическая структура которого приведена на рисунке 5.2.2. Анион Д5, по
данным наших расчетов, является интермедиатом, при распаде которого образуются
анионы C3H6N5O4- (m/z 176) и NO2- (m/z 46) (Рисунок 5.2.3). Низкая интенсивность
линии m/z 250 также может свидетельствовать в пользу быстрого дальнейшего
распада аниона C4H8N7O6-.
Рисунок 5.2.2. Геометрические структуры исходного реагента, интермедиатов,
продуктов реакций и переходных состояний распада анион-радикала октогена (Д1)
по данным метода М06/6-31+G(d,p). Длины связей в Å
128
Рисунок 5.2.3. Механизм распада аниона С4H8N7O6- (Д5) Энтальпии продуктов,
интермедиатов и переходных состояний рассчитаны методом M06/6-31+G(d,p)
относительно структуры Д5
Мы изучили три возможных реакции распада аниона C4H8N7O6-. Схема распада
приведена на рисунке 5.2.3. Первый механизм (реакция Д5→Д6, Рисунок 5.2.3)
соответствует отрыву молекулы CH2NNO2, образованию иона C3H6N5O4- с массовым
числом m/z 176 (обратная реакция является безбарьерной). Энтальпия реакции
данного процесса равна 137.7 кДж/моль.
Второй изученный процесс соответствует образованию NO2- (реакция Д5→Д8,
Рисунок 5.2.3). Как показывает наш расчет, отрыв NO2- сопровождается циклизацией
молекулярного фрагмента C4H8N6O4 с образования связи N13-N20 (Рисунок 5.2.2).
Переходное состояние для данной реакции приведено на рисунке 5.2.2. Энтальпия
активации по данным метода M06/6-31+G(d,p) составляет 185.8 кДж/моль (Рисунок
5.2.3), тогда как энтальпия реакции данного процесса равна 162.8 кДж/моль. Как
видно из рисунка 5.2.3 энтальпия активации отрыва NO2- (m/z 46) примерно на 48.1
кДж/моль выше по сравнению с реакцией образования иона C3H6N5O4- (m/z 176)
129
(реакция Д5→Д6), что в принципе объясняет более высокую интенсивность второй
линии по сравнению с первой.
Наконец, последний процесс распада C4H8N7O6-, который был рассмотрен в
данной работе, связан с отрывом азотистой кислоты и образованием аниона
C4H7N6O4- (m/z 203) (реакция Д5→Д7, Рисунок 5.2.3). Энтальпия активации данного
процесса составляет 204.2 кДж/моль. Структура переходного состояния приведена
на рисунке 5.2.2.
Распад катион-радикала октогена, как и в случае с анион-радикалом,
представляется сложным процессом, включающим в себя множество различных
элементарных стадий. Как уже отмечалось ранее масс-спектры октогена и гексогена
имеют две ярко выраженные линии с массовыми числами m/z 30 и m/z 46 [133].
Данные линии, по нашему мнению, соответствуют образованию катионов NO+ и
NO2+ соответственно.
Вначале рассмотрим механизм отщепления NO2+ от катион-радикала октогена.
По данным нашего расчета катион NO2+ образуется входе реакции гетеролитической
диссоциации связи N-N (реакция Е1→Е2, Рисунки 5.2.4 и 5.2.5). Энтальпия
активации по данным метода M06/6-31+G(d,p) составляет 156.0 кДж/моль (Рисунок
5.2.4). В тоже время необходимо отметить, что гомолитический отрыв NO2● требует
всего 81.2 кДж/моль (реакция Е1→Е3, Рисунок 5.2.4). Последняя реакция имеет
хорошо локализованное переходное состояние (Рисунок 5.2.6), тогда как энтальпия
реакции данного процесса равна -5.9 кДж/моль.
Как и в случае КР-ДМНА, отрыв NO+ от катион-радикала октогена является
сложным многостадийным процессом (реакция Е1→Е9, Рисунок 5.2.4). Начальная
стадия (реакция Е1→Е4, Рисунок 5.2.4) включает образование аци-формы катионрадикала октогена и требует 43.1 кДж/моль. Аци-форма является более стабильной
по энергии изомером ионизированной молекулы октогена и лежит на 56.5 кДж/моль
ниже исходного катион-радикала. Структура переходного состояния данной реакции
приведена на рисунке 5.2.6. На второй стадии происходит конформационный
130
переход (реакция Е4→Е5, Рисунок 5.2.4), заключающийся в повороте группы OH в
аци-изомере относительно связи N-O. Этот конформационный переход требует 14.2
кДж/моль, тогда как аци-форма Е5 лежит на 106.7 кДж/моль ниже по энергии, чем
исходный катион-радикал и на 50.2 кДж/моль ниже по сравнению с аци-формой Е4
(Рисунок 5.2.4).
Рисунок 5.2.4. Схема распада катион-радикала октогена. Энтальпии продуктов,
интермедиатов и переходных состояний рассчитаны методом M06/6-31+G(d,p)
относительно структуры Е1
Следующая стадия включает отрыв гидроксила от аци-формы Е5 (реакция
Е5→Е6, Рисунок 5.2.4). Геометрическая структура переходного состояния данной
реакции приведена на рисунке 5.2.6. Энтальпия активации процесса Е5→Е6
составляет 50.6 кДж/моль, энтальпия реакции равна 13.4 кДж/моль.
Наконец, отрыв NO+ (обратная реакция является безбарьерной) требует 237.7
кДж/моль. Данная оценка находится в хорошем согласии с оценкой, полученной для
отрыва NO+ в случае распада КР-ДМНА (245.6 кДж/моль).
131
Рисунок 5.2.5. Геометрические структуры исходного реагента, интермедиатов и
продуктов реакций распада катион-радикала октогена (Е1) по данным метода М06/631+G(d,p). Длины связей в Å
Рисунок 5.2.6. Геометрические структуры переходных состояний реакций распада
катион-радикала октогена (Е1) по данным метода М06/6-31+G(d,p).
Длины связей в Å.
132
5.3 Основные выводы по данной Главе 5
С помощью методов теории функционала плотности нами были изучены
основные механизмы распада ион-радикалов ДМНА и октогена.
В случае катион-радикала ДМНА нами были изучены пять основных каналов,
включающих образование катионов NO2+, C2H6N+, C2H5N+, C2H4N+ и NO+.
Присутствие данных катионов в продуктах распада катион-радикала ДМНА
подтверждается наличием линий с соответствующими массовыми числами в массспектре ДМНА. Расчетные значения энтальпий активации хорошо коррелирует с
интенсивностью линий. Так, например, отрыв NO2+ требует наибольшей энергии
(237.2 кДж/моль) по сравнению с остальными рассмотренными процессами,
вследствие чего, линия m/z 46 имеет наименьшую интенсивность. С другой стороны
образование катиона C2H4N+ в результате отрыва монооксида азота и молекулы воды
требует значительно меньшей энергии (98.3 кДж/моль), что объясняет более
высокую интенсивность линии m/z 42.
В отличие от ДМНА, анализ масс-спектра октогена указывает на то, что
отрывы NO2+ и NO+ будут доминировать над другими механизмами при распаде
катион-радикала октогена. Наши расчеты указывают на важную роль, как и в случае
с КР-ДМНА, реакции образования аци-формы катион-радикала октогена при отрыве
NO+. Однако, оценки энтальпии реакции, полученные для отрыва NO2+ от катионрадикала октогена достаточно высоки, что не позволяет рассматривать данный
механизм в роли доминирующего канала распада. Более того мы имеем полное
право предположить существование альтернативного канала отрыва NO2+ от катионрадикала октогена. На данный момент нам не удалось найти более выгодный
механизм данной реакции.
В случае анион-радикала октогена мы изучили два основных канала распада,
связанные с образованием ионов NO2- (m/z 46) и C3H6N5O4- (m/z 46). Наше
теоретическое исследование показало, что образование этих ионов является
133
результатом многостадийных реакций с общей стадией, включающей раскрытие
тетраазациклооктанового кольца в анион-радикале октогена и отрыв NO2●.
Для анион-радикала ДМНА нами была изучена одна реакция, связанная с
отрывом NO2-. Результаты, полученные для реакции отрыва NO2- от анион-радикала
ДМНА, существенно ниже, чем для анион-радикала октогена.
134
Заключение и основные выводы
Представленный в работе материал показывает, что современные квантовохимические методы позволяют успешно решать различные задачи, связанные с
исследованием геометрической и электронной структур нитросоединений, а также
их ион-радикалов, и влиянием молекулярной структуры на механизмы и кинетику
распада. С использованием методов квантовой химии были получены сведения по
структуре катион- и анион-радикалов молекул трех важнейших взрывчатых веществ:
2,4,6-тринитротолуола, октогена и тетранитропентаэритрита, и их модельных
соединений. Результаты расчета позволяют надежно интерпретировать масс-спектры
положительно и отрицательно заряженных ионов нитросоединений. Основные
результаты работы можно кратко сформулировать в виде следующих выводов:
• На
основе
сопоставления
расчетных
и
экспериментальных
значений
геометрических параметров, дипольных моментов, потенциалов ионизации,
сродства к электрону изученных простейших представителей C-, N-, Oнитросоединений показано, что все использованные в работе DFT-методы
хорошо передают указанные характеристики.
• Установлена
корреляционная
зависимость
расчетных
значений
адиабатических потенциалов ионизации и энергий диссоциации связи C-NO2
для пара-замещенных монофункциональных производных нитробензола
Коэффициент корреляции при использовании метода wB97XD/6-31+G(2df,p)
составляет -0.965. Обнаружено, что донорные заместители снижают потенциал
ионизации и увеличивают энергию диссоциации, а акцепторные заместители,
наоборот, увеличивают потенциал ионизации и уменьшают прочность связи CNO2 пара-замещенных монофункциональных производных нитробензола.
• Показано, что для изученных реакций распад ион-радикалов в ряде случаев
протекает по механизмам, отличным от деструкции нейтральных молекул с
существенно более низкими значениями энтальпии активации.
135
• Доказано, что механизм распада октогена и ТЭНа отличен от механизма
распада диметилнитрамина и этилнитрата соответственно. Последние часто
используются в качестве модельных соединений при изучении механизма
распада октогена и ТЭНа.
• Установлено, что процесс внутримолекулярного переноса водорода с
образованием аци-формы является одним из основных каналов распада
молекулы, катион-радикалов и анион-радикалов 2,4,6-тринитротолуола. В то
же время процесс отрыва радикала гидроксила от аци-формы ион-радикалов
является сложным многостадийным процессом.
• Выявлено путем сопоставления расчетных данных по механизму распада
молекулы, анион-радикала и масс-спектра 2,4,6-тринитротолуола наличие
альтернативного
бициклического
канала
деструкции,
интермедиата
связанного
с
образованием
(6S)-2-метил-3,5-динитро-7-окса-8-аза-
бицикло[4.2.0]-окта-1(8)-2,4-триене-8-оксида,
который
в
дальнейшем
распадается по сложному многостадийному механизму. Существенно, что
расчетные значения энтальпии активации образования (6S)-2-метил-3,5динитро-7-окса-8-аза-бицикло[4.2.0]-окта-1(8)-2,4-триене-8-оксида по данным
метода B3LYP/6-31+G(2df,p) (203.4 кДж/моль) близки к экспериментальному
значению энергии активации термического распада 2,4,6-тринитротолуола в
газообразном состоянии.
• Отмечено, что образование аци-формы также предшествует распаду катионрадикалов
диметилнитрамина
и
октогена.
Распад
анион-радикал
диметилнитрамина скорее всего, начинается с диссоциации связи N-NO2.
Фрагментация анион-радикала октогена является более сложным процессом и
включает в себя раскрытие тетраазациклооктанового кольца на ранних стадиях
развития реакции.
136
• Показано, что основными каналами распада катион-радикалов этилнитрата и
ТЭНа являются соответственно разрывы связей O-NO2 и C-C.
• Впервые изучен процесс образования катиона COH+, объясняющий наличие
линии m/z 29 в масс-спектрах простейших алифатических нитратов. Распад
анион-радикалов этилнитрата и ТЭНа протекает по аналогичным каналам и
включает в себя отрыв анионов NO2- и O-NO2-.
137
Список используемой литературы
1.
Alcamí, M. Computational Chemistry: A useful (Sometimes Mondatory) Tool in
Mass Spectrometry Studies / M. Alcamí, O. Mó, M. Yáñez // Mass. Spectrom. Rev.
– 2001. – V. 20. – Is.3. – p. 195-245.
2.
Bhattacharya, A. Nonadiabatic reaction of energetic molecules / A. Bhattacharya,
Y.Q. Guo, E.R. Bernstein // Acc. Chem. Res. – 2010. – V. 43. – N 12. – p.1476–
1485.
3.
Guo, Y.Q. Reaction of carbon monoxide and hydrogen on neutral Nb8 clusters in the
gas phase / Y.Q. Guo, A. Bhattacharya, E.R. Bernstein // J. Chem. Phys. – 2008. –
V. 128. – N 4. – p.034303:1-034303:6.
4.
Yu, Zijun. Decomposition of pentaerythritol tetranitrate [C(CH2ONO2)4] following
electronic excitation / Zijun Yu, E. R. Bernstein // J. Chem. Phys. – 2011. – V. 135.
– p.154305:1-154305:10.
5.
Bhattacharya, A. A comparison of the decomposition of electronically excited nitrocontaining molecules with energetic moieties C-NO2, N-NO2, and O-NO2 / A.
Bhattacharya, Y.Q. Guo, E. R. Bernstein // J. Chem. Phys. – 2012. – V. 136. –
p. 024321:1-024321:9.
6.
Walker, I.C. Spectroscopy and dynamics of nitromethane (CH3NO2) and its anionic
states / I.C. Walker, M.A.D. Fluendy // Int. J. Mass Spectrom. – 2001. – V. 205. – p.
171–182.
7.
Alizadeh, E. Dissociative electron attachment to nitromethane / E. Alizadeh, F.
Ferreira da Silva 1, F. Zappa 2, A. Mauracher, M. Probst, S. Denifl, A. Bacher, T.D.
Mark, P. Lim˜ao-Vieira 1, P. Scheier // Int. J. Mass Spectr. – 2008. – V. 271. – Is. 13. – p. 15–21.
8.
Adamowicz, L. Dipole-Bound Anionic State of Nitromethane. Ab-Initio Coupled
Cluster Study with First-Order Correlation Orbitals / L. Adamowicz // J.Chem. Phys.
– 1989. – V. 91. – p. 7787-7790.
138
9.
Compton, R.N. On the binding of electrons to nitromethane: Dipole and valence
bound anions / R.N. Compton, H. S. Carman, C. Desfrançois, H. Abdoul-Carime,
J.P. Schermann, J.H. Hendricks, S.A. Lyapustina, K.H. Bowen // J. Chem. Phys. –
1996. – V. 105. − N 9. − p. 3472-3478.
10.
Gutsev, G.L. A theoretical study of the valence- and dipole-bound states of the
nitromethane anion / G.L. Gutsev, R.J. Bartlett // J. Chem. Phys. – 1996. – V. 105, –
p. 8785-8792.
11.
Arenas, J.F. Multiconfigurational second-order perturbation study of the
decompositionof the radical anion of nitromethane / J.F. Arenas, J.C. Otero,
D.Peláez, J. Soto, L. Serrano-Andrés // J. Chem. Phys. – 2004. – V. 121. – N 9. – p.
4127-4132.
12.
Sailer, W. Dissociative electron attachment study to nitromethane / W. Sailer, A.
Pelc, S. Matejcik, E. Illenberger, P. Scheier, T.D. Märk // J. Chem. Phys. − 2002. −
V. 117. − p. 7989-7994.
13.
Edtbauer, A. Dissociative electron attachment to pentaerythritol tetranitrate:
Significant fragmentation near 0 eV / A. Edtbauer, P. Sulzer, A. Mauracher, C.
Mitterdorfer, F. Ferreira da Silva, S. Denifl, T.D. Märk, M. Probst, Y. Nunes, P.
Limão-Vieira, P. Scheier // J. Chem. Phys. − 2010. − V. 132. − p. 134305:1134305:8.
14.
Sulzer, P. Identification of Isomers of Nitrotoluene via Free Electron Attachment / P.
Sulzer, A. Mauracher, S. Denifl, F. Zappa, S. Ptasinska, M. Beikircher, A. Bacher,
N. Wendt, A. Aleem, F. Rondino, S. Matejcik, M. Probst, T.D. Märk, P. Scheier //
Anal. Chem. − 2007. − V. 79. − N 17. − p. 6585-6591.
15.
Sulzer, P. Probing dinitrobenzene by low energy electrons / P. Sulzer, A. Mauracher,
S. Denifl, M. Probst, T.D. Märk, P. Scheier, E. Illenberger // Int. J. Mass Spectrom.
− 2007. − V. 266. − p. 138-148.
139
16.
Sulzer, P. Probing royal demolition explosive 1,3,5-trinitro-1,3,5-triazocyclohexane
by low-energy electrons: Strong dissociative electron attachment near 0 eV / P.
Sulzer, A. Mauracher, F. Ferreira da Silva, S. Denifl, T.D. Märk, M. Probst, P.
Limão-Vieira, P. Scheier, // J. Chem. Phys. − 2009. − V. 131. − p. 144304:1144304:7.
17.
Sulzer, P. Probing trinitrotoluene (TNT) by low-energy electrons: Strong
fragmentation following attachment of electrons near 0 eV / P. Sulzer, F. Rondino,
S. Ptasinska, E. Illenberger , T.D. Märk, P. Scheier // Int. J. Mass Spectrom. − 2008.
− V. 272. − Is. 2-3. − p. 149-153.
18.
Aluker, E.D. Laser initiation of energetic materials: selective photo initiation regime
in PETN / E.D. Aluker, A.G. Krechetov, A.Y. Mitrofanov, D.R. Nurmukhametov,
M.M. Kuklja // J. Phys. Chem. − 2011. − V. 115. − N 14. − p. 6893-6901.
19.
Ewing, R.G. A critical review of ion mobility spectrometry for the detection of
explosives and explosive related compounds / R.G. Ewing, D.A. Atkinson, G.A.
Eiceman, G.J. Ewing // Talanta. − 2001. − V. 54. − Is. 3. − p. 515-529.
20.
Walsh, M.E. Determination of nitroaromatic, nitramine, and nitrate ester explosives
in soil by gas chromatography and an electron capture detector / M.E. Walsh //
Talanta. − 2001. − V. 54. − Is. 3. − p. 427-438.
21.
Sharia, O. Surface-accelerated decomposition chemistry of δ-HMX / O. Sharia, R.
Tsyshesvky, M.M. Kuklja // J. Phys. Chem. Lett. − 2013. − V. 4. − N 5. p. 730-734.
22.
Kuklja, M.M. Effect of polar surfaces on decomposition of molecular materials /
M.M. Kuklja, R. Tsyshevsky, O. Sharia // J. Am. Chem. Soc. − 2014. − V. 136. − N
38. − p. 13289-13302.
23.
Kuklja, M.M. Quantum-Chemical Modeling of Energetic Materials: Chemical
Reactions Triggered by Defects Deformations, and Electronic Excitations / M.M.
Kuklja // Adv. Quant. Chem. − 2014. − V. 69. − p. 71-146.
140
24.
Николаева, Е.В. Особенности механизма первичного акта газофазного
мономолекулярного распада C-нитросоединений по результатам квантовохимических расчетов: дисс. канд. хим. наук: 02.00.04 / Николаева Екатерина
Валерьевна. − Казань, 2002. − 203 c.
25.
Цышевский, Р.В. Молекулярная структура и механизмы реакций газофазного
распада некоторых алканов и нитроалканов: дисс. канд. хим. наук: 02.00.04 /
Цышевский Роман Витальевич. – Казань, 2008. – 185 c.
26.
Храпковский, Г.М. Особенности механизма первичного акта газофазного
мономолекулярного распада нитроэтана и его катион-радикала / Г.М.
Храпковский, Д.В. Чачков, Е.В. Николаева, А.Г. Шамов // Вестник Казанского
технологического ун-та. – 2011. – № 20. – p. 55-67.
27.
Tsyshevsky, R.V. Theoretical Study of the Tautomeric Reactions of Dinitromethane
and Its Radical Cation / R.V. Tsyshevsky, G.G. Garifzianova, D.V. Chachkov, A.G.
Shamov, G.M. Khrapkovskii // J. Energ. Mat. – 2009. – V. 4. – Is. 4. – p. 263-295.
28.
Назин, Г.М. Термическое разложение алифатических нитросоединений / Г.М.
Назин, Г.Б. Манелис, Ф.И. Дубовицкий // Успехи химии. – 1968. – Т. 37. – № 8.
– с. 1443-1461.
29.
Назин, Г.М. Предэкспоненциальный фактор газовых мономолекулярных
реакций / Г.М. Назин // Успехи химии. – 1972. – Т. 41. № 9. – с. 1537-1565.
30.
Манелис, Г.Б. Термическое разложение и горение взрывчатых веществ и
порохов / Г.Б. Манелис, Г.М. Назин, Ю.И. Рубцов, В.А. Струнин. – М.: Наука,
1996. – 223 с.
31.
Назин, Г.М. Термическое разложение алифатических нитросоединений / Г.М.
Назин, Г.Б. Манелис // Успехи химии. – 1994. – Т. 63. – № 4. – c. 327-337.
32.
Назин, Г.М. Исследования реакций термического разложения алифатических
нитро-дифтор-аминосоединений: дисс. докт. хим. наук: 02.00.04 / Назин
Геннадий Михайлович. – Черноголовка, 1975. – 212 c.
141
33.
Назин, Г.М. Предэкспоненциальный множитель реакций радикального
разложения нитросоединений / Г.М. Назин, Г.Б. Манелис // Изв. АН СССР.
Сер. хим. – 1972. – № 4. – c. 811-816.
34.
Cottrel, T.H. Thermal decomposition of nitroethane and 1-nitropropane / T.H.
Cottrel, T.E. Crahm, T.J. Reid // Trans. Farad. Soc. – 1951. – V. 47. – p. 1089-1092.
35.
Храпковский, Г.М. Влияние строения молекул на кинетические параметры
мономолекулярного распада С- и О-нитросоединений / Г.М. Храпковский, Г.Н.
Марченко, А.Г. Шамов. Казань: ФЭН, 1997. – 224 с.
36.
Khrapkovskii, G.M. Mechanisms of gas phase decomposition of C-nitro compounds
from quantum chemical data / G.M. Khrapkovskii,A.G. Shamov, E.V. Nikolaeva,
D.V. Chachkov // Rus. Chem. Rev. – 2009. – V. 78. – № 10. – p. 903-943.
37.
Kiselev, V.G. Theoretical Study of the Nitroalkane Thermolysis. 1. Computation of
the Formation Enthalpy of the Nitroalkanes, Their Isomers and Radical Products /
V.G. Kiselev, N.P. Gritsan // J. Phys. Chem. A. – 2008. – V. 112. – N 19. – p. 44584464.
38.
Khrapkovskii, G.M. Estimations of activation and enthalpies of reaction for HONO
elimination from C2-C4 mononitroalkanes: A theoretical study / G.M. Khrapkovskii,
A.G. Shamov, R.V. Tsyshevsky, D.V. Chachkov, D.L. Egorov, I.V. Aristov //
Comput. Theoret. Chem. – 2011. – V. 966. – p. 265-271.
39.
Khrapkovskii, G.M. Formation enthalpies and bond dissociation enthalpies for C1–
C4 mononitroalkanes by composite and DFT/B3LYP methods / G.M. Khrapkovskii,
R.V. Tsyshevsky, D.V. Chachkov, D.L. Egorov, A.G. Shamov // J. Mol. Struct.:
THEOCHEM. – 2010. – V. 958. – Is. 1-3. – p. 1-6.
40.
Туровцев, В.В. Изучение внутреннего вращения в н-мононитроалкильных
радикалах / В.В. Туровцев, М.Ю. Орлов, Р.В. Туровцев, Ю.Д. Орлов // ЖОХ. –
2014. –Т. 84. – № 1. – с. 11-17.
142
41.
Туровцев, В.В. Потенциальные функции внутреннего вращения в гошизомерах н-мононитроалканов / В.В. Туровцев, Ю.Д. Орлов // Журн. физ. хим.
– 2014. – Т. 88. – № 7. – с. 1183-1189.
42.
Туровцев, В.В. Электронное строение н-моноалкильных радикалов / В.В.
Туровцев, Ю.Д. Орлов // Хим. физ. − 2014. − Т. 33. − № 1. − с. 3-9.
43.
Туровцев, В.В. Распределение электронной плотности в н-мононитроалнанах /
В.В. Туровцев, М.Ю. Орлов, Р.В. Туровцев, Ю.Д. Орлов // Журн. физ. хим. −
2012. − Т. 86. − № 7. − с. 1188-1193.
44.
Туровцев,
В.В.
Влияние
свободной
валентности
на
характеристики
внутреннего вращения в н-алькильных радикалах / В.В. Туровцев, Ю.Д. Орлов
// ЖОХ. − 2011. − Т. 81. − № 9. − с. 1458-1464.
45.
Орлов, Ю.Д. Термохимия органических свободных радикалов. / Ю.Д. Орлов,
Ю.А. Лебедев, И.Ш. Сайфуллин. − Москва: Федеральное государственное
бюджетное учреждение науки Институт химии растворов им. Г.А. Крестова
РАН., 2001. − 304 с.
46.
Орлов, Ю.Д. Закономерности связи строение-свойство и база количественных
данных в термохимии органических свободных радикалов: автореф. дис. …
докт. хим. наук: 02.00.04 / Орлов Юрий Дмитриевич. − Тверь, 1996. − 34 с.
47.
Turovtsev, V.V. Determination of thermodynamic parameters of C2–C3 nitroalkanes
using anharmonic oscillator approximation and explicit treatment of internal rotation
/ V.V. Turovtsev, G.M. Khrapkovskii, A.G. Shamov, Yu.D. Orlov, R.V. Tsyshevsky
// Comput. Theoret. Chem. − 2014. − V. 1039. − p. 55-61.
48.
Фаустов, В.И. Расчет методом MINDO/3 переходного состояния реакции
мономолекулярного отщепления HNO2 от нитроэтана / В.И. Фаустов, С.А.
Шевелев, Н.А. Аникин // Изв. АН СССР. Сер. хим. − 1979. − № 11. − с. 28002803.
143
49.
Фаустов, В.И. Квантово-химические расчеты переходного состояния и энергии
молекулярного 1,2-отщепления (1,2-присоединения) HNO2 / В.И. Фаустов,
С.А. Шевелев, Н.А. Аникин // Изв. АН СССР. Сер. хим. − 1980. − № 10. − с.
2200-2005.
50.
Шамов, А.Г. Структура переходного состояния и некоторые особенности
влияния строения молекул на энергию активации реакции газофазного
элиминирования азотистой кислоты из нитроалканов / А.Г. Шамов, Г.А.
Шамов, Г.М. Храпковский // Int. Symposium CARC-96. − Москва, 1996. −
C. 112.
51.
Шамов, А.Г. Теоретическое изучение механизма газофазного элиминирования
азотистой кислоты из нитроалканов / А.Г. Шамов, Д.В. Чачков, Е.В.
Николаева, Г.М. Храпковский // Сб. ст. VIII Всероссийской конференции
"Структура и динамика молекулярных систем". – Йошкар-Ола. − 2001. − Ч. 2.
− с. 186-190.
52.
Шамов, А.Г. Неэмпирическое изучение механизма мономолекулярного
распада нитросоединений / А.Г. Шамов, Г.М. Храпковский, Г.А. Шамов // Сб.
ст. IV Всероссийской конференции "Структура и динамика молекулярных
систем". − Йошкар-Ола-Казань-Москва. − 1998. − Т. 3. − Вып. IV. − c. 183-187.
53.
Храпковский, Г.М. Теоретическая оценка предэкспоненциального множителя
константы скорости элиминирования нитроалканов / Е.А. Ермакова, В.А.
Захаров, А.Г. Шамов, В.А. Тихомиров // Журн. физ. хим. − 1990. − Т. 64. − №
7. − с. 2247-2250.
54.
Denis, P.A. Density functional study of the decomposition pathways of nitroethane
and 2-nitropropane / P.A. Denis, O.N. Ventura, H.T. Le, M.T. Ngyen // Phys. Chem.
Chem. Phys. − 2003. − V. 5. − p. 1730-1738.
55.
Shamov, A.G. Ab initio study of mechanism of gas-phase monomoleculars
destruction of nitroethane / A.G. Shamov, G.M. Khrapkovskii // Coll. art. 30th Int.
144
Annual Conference of ICT "Energetic Materials". − Federal Republic of Germany,
Karlsruhe, 1999. − p. 60:1-60:10.
56.
Дубихин, В.В. Полярный характер переходного состояния в реакциях
элиминирования HNO2 из мононитросоединений / В.В. Дубихин, Г.М. Назин //
Изв. АН СССР. − 1974. − № 6. − c. 1345-1349.
57.
Чачков, Д.В. Влияние молекулярной структуры на особенности конкуренции
различных
механизмов
первичного
акта
газофазного
распада
С-
нитросоединений по результатам квантово-химических расчетов: дисс. канд.
хим. наук: 02.00.04. − Казань, 2005. − 206 c.
58.
Wodtke, A.M. Infrared Multiphoton Dissociation of Three Nitroalkanes / A.M.
Wodtke, E.J. Hintsa, Y.T. Lee // J. Phys. Chem. − 1986. − V. 90. − N 16. − p. 35493558.
59.
Dewar, M.J.S. Thermolysis of Molecules Containing NO2 Groups / M.J.S. Dewar,
J.P. Ritchie, J. Alster // J. Org. Chem. − 1985. − V. 50. − p. 1031-1036.
60.
McKee, M.L. Ab Initio Study of Rearrangements on the CH3NO2 Potential Energy
Surface / M.L. McKee // J. Am. Chem. Soc. − 1986. − V. 108. − N 19. − p. 57845792.
61.
McKee, M.L. MCSCF Study of the Rearrangement of Nitromethane to Methyl
Nitrite / M.L. McKee // J. Phys. Chem. − 1989. − V. 93. − N 21. − p. 7365-7369.
62.
Николаева, Е.В. Строение и механизм мономолекулярного распада C-, N-, Oнитросоединений. Часть IV. Роль нитро-нитритной перегруппировки в
механизме мономолекулярного распада алифатических нитросоединений / Е.В.
Николаева, А.Г. Шамов, Д.В. Чачков, Д.А. Гордеев, Г.М. Храпковский // Хим.
и комп. моделир. Бутлеровские сообщ. − 2000. − Т. 1. − № 3. − с. 15-24. Режим
доступа: [http.://chem.kstu.ru]
63.
Khrapkovskii, G.M. Nitro-nitrite rearrangement and mechanism of gas-phase
monomolecular decomposition of C-nitrocompounds / G.M. Khrapkovskii, E.V.
145
Nikolaeva, D.V. Chachkov, A.G. Shamov // IV Seminar "New trends in research of
energetic materials". − Pardubice, 2001. − p. P1-17-P1-22.
64.
Шамов,
А.Г.
Теоретическое
перегруппировки
нитрометана
изучение
/
А.Г.
механизма
Шамов,
Е.В.
нитро-нитритной
Николаева,
Г.М.
Храпковский // Сб. ст. VII Всероссийской конферен."Структура и динамика
молекулярных систем". − Мoсква. − 2000. − Вып. VII. − с. 447-450.
65.
Шамов,
А.Г.
Теоретическое
перегруппировки
нитрометана
изучение
/
А.Г.
механизма
Шамов,
Е.В.
нитро-нитритной
Николаева,
Г.М.
Храпковский // VII Всерос. конф. «Структура и динамика молекулярных
систем»: тез. докл. − М.-Казань-Йошкар-Ола, 1999. − С. 117.
66.
Николаева,
Е.В.
перегруппировки
Квантово-химическое
нитрометана
/
Е.В.
изучение
Николаева,
нитро-нитритной
А.Г.
Шамов,
Г.М.
Храпковский // VI Междунар. конф. «Математические модели нелинейных
возбуждений, переноса, динамики, управления в конденсированных системах
и других средах»: тез. докл. − М.: Из-во «Станкин», 2000. − С. 84.
67.
Hu, W.-F. Theoretical Study of the CH3NO2 Unimolecular Decomposition Potential
Energy Surface / W.-F. Hu, T.-J. He, D.-M. Chen, F.-Ch. Liu // J. Phys. Chem. A. −
2002. − V. 106. − p. 7294-7303.
68.
Nguyen,
M.T.
Nitromethane-Methyl
Nitrite
Rearrangement:
A
Persistent
Discrepancy between Theory and Experiment / M.T. Nguyen, H.T. Le, B. Hajgató,
T. Veszprémi, M.C. Lin // J. Phys. Chem. A. − 2003. − V. 107. − p. 4286-4290.
69.
Zhan70g, C. Isomers and Isomerization Reactions of Four Nitro Derivatives of
Methane / C. Zhang, X. Wang, M. Zhou // J. Comput. Chem. − 2011. − V. 32. − Is.
8. − p. 1760-1768.
70.
Шамов, А.Г. Строение и механизм мономолекулярного распада C-, N-, Oнитросоединений. Часть V. Механизм образования и деструкции аци-форм Cнитросоединений / А.Г. Шамов, Г.М. Храпковский, Г.А.
Шамов, Е.В.
146
Николаева, Д.В Чачков, // Хим. и комп. модел. Бутлеровские сообщ. − 2002. −
Т. 2. − № 6. − c. 1-10. Режим доступа: [http.://chem.kstu.ru].
71.
Храпковский, Г.М. Теоретическое изучение механизма образования и
мономолекулярного распада аци-нитрометанов / Г.М. Храпковский, А.Г.
Шамов, Г.А. Шамов, В.А. Шляпочников // Изв. АН. Сер. хим. − 2001. − № 6. −
c. 913-917.
72.
Lammertsma, K. Nitro ↔ aci-Nitro Tautomerism / K. Lammertsma, B.V. Prased //
J. Am. Chem. Soc. − 1993. − V. 115. − p. 2348-2351.
73.
Храпковский, Г.М. Об одном возможном механизме термического распада
алифатических нитросоединений в газовой фазе. Теоретическое изучение
деструкции динитрометана / Г.М. Храпковский, А.М. Розин, В.А. Тихомиров,
А.Г. Шамов, Г.Н. Марченко // Докл. АН СССР. − 1988. − Т. 298. − № 4. − p.
921-924.
74.
Khrapkovskii, G.M. A theoretical study of the formation and destruction of the aciform of nitromethane and dinitromethane / G.M. Khrapkovskii, A.G. Shamov, G.A.
Shamov, V.A Slyapochnikov // Mendeleev Commun. − 1997. − № 5. − p. 169-171.
75.
Храпковский, Г.М. Некоторые особенности влияния строения молекул на
энергию активации радикального распада алифатических нитросоединений /
Г.М. Храпковский, А.Г. Шамов, Г.А. Шамов, В.А. Шляпочников // Изв. АН.
Сер. хим. − 1996. − № 10. − c. 2438-2443.
76.
Матвеев, В.Г. Разложение производных нитробензола в газовой фазе по
радикальному механизму / В.Г. Матвеев, В.В. Дубихин, Г.М. Назин // Изв. АН
СССР. Сер. хим. − 1978. − № 4. − c. 783-786.
77.
Храпковский, Г.М. Некоторые особенности влияния строения молекул на
энергию
активации
радикального
газофазного
распада
ароматических
нитросоединений / Г.М. Храпковский, Е.А. Ермакова, Е.А. Рафеев // Изв. АН.
Сер. хим. − 1994. − № 12. − c. 2118-2122.
147
78.
Shao, J. Density functional calculations of bond dissociation energies for removal of
the nitrogen dioxide moiety in some nitroaromatic molecules / J. Shao, X. Cheng, X.
Yang // J. Mol. Struct.: THEOCHEM. – 2005. – V. 755. – Is. 1-3. – p. 127-130.
79.
Shao, J. The C−NO2 bond dissociation energies of some nitroaromatic compounds:
DFT study / J. Shao, X. Cheng, X. Yang // Struct. Chem. – 2006. – V. 17. – Is. 5. –
p. 547-550.
80.
Rice, B.M. Density functional calculations of bond dissociation energies for NO2
scission in some nitroaromatic molecules / B.M. Rice, S. Sahu, F.J. Owens // Journal
of Molecular Structure: THEOCHEM. – 2002. – V. 583. – Is. 1-3. – p. 69-72.
81.
Khrapkovskii, G.M. Theoretical study of substituents effect on C–NO2 bond strength
in monosubstituted nitrobenzenes / G.M. Khrapkovskii, D.D. Sharipov, A.G.
Shamov, D.L. Egorov, D.V. Chachkov, B. Nguyen Van, R.V. Tsyshevsky //
Comput. Theoret. Chem. – 2013. – V. 1017. – p. 7-13.
82.
Храпковский, Г.М. Некоторые особенности влияния строения молекул на
структуру
переходного
состояния
и
энергию
активации
реакций
молекулярного газофазного распада C-нитросоединений / Г.М. Храпковский,
А.Г. Шамов, В.А. Шляпочников // Журн. орг. хим. – 1999. – Т.35. – № 6. –
c. 891-901.
83.
Шамов,
А.Г.
Новый
механизм
термического
распада
ароматических
нитросоединений / А.Г. Шамов, Г.М. Храпковский // Вестник казанского
технологического ун-та. Специальный выпуск. – 2009. – Спец. вып. – с. 37-44.
84.
Матвеев, В.Г. Разложение изомеров нитротолуола в газовой фазе / В.Г.
Матвеев, В.В. Дубихин, Г.М. Назин // Кинетика и катализ. – 1976. – Т. XVII. –
Вып. 2. – с. 280-284.
85.
Чачков, Д.В. Некоторые особенности влияния строения молекул на энергию
активации радикального распада нитробензолов / Д.В. Чачков, Е.В. Николаева,
А.Г. Шамов, Г.М. Храпковский // Сб. ст. VIII Всероссийской конференц.
148
"Структура и динамика молекулярных систем". – Йошкар-Ола. – 2001. – Вып.
VIII. – Ч. 2. – c. 198-201.
86.
Meyerson, S. Organic Ions in the Gas Phase. XVIII. Mass Spectra of Nitroarenes / S.
Meyerson, I. Pusks, E.K. Fields // J. Org. Chem. – 1966. – V. 88. – N 21. – p. 49744980.
87.
Fields, E.K. Mass spectral and thermal reactions of dinitrobenzenes / E.K. Fields, S.
Meyerson // J. Org. Chem. – 1972. – V. 37. – N 24. – p. 3861-3866.
88.
Brill, T.B. Kinetics and Mechanism of Thermal Decomposition of Nitroaromatic
Explosives / T.B. Brill, K.J. James // Chem. Rev. – 1993. – V. 93. – N 8. – p. 26672692.
89.
Матвеев,
В.Г.
Согласованный
механизм
разложения
ароматических
нитросоединений в газовой фазе / В.Г. Матвеев, В.В. Дубихин, Г.М. Назин //
Изв. АН СССР. Сер. хим. – 1978. – № 2. – c. 474-477.
90.
Beynon, J.H. Some Specific Molecular Rearrangements in the Mass Spectra of
Organic Compounds / J.H. Beynon, G.R. Lester, A.E. Williams // J. Phys. Chem. –
1959. – V. 63. – N 11. – p. 1861-1868.
91.
Turner, A.G. Thermal decomposition of TNT / A.G. Turner, I.P. Devis // J. Am.
Chem. Soc. – 1984. – V. 106. – p. 5447-5451.
92.
Храпковский, Г.М. Теоретическое изучение механизма нитро-нитритной
перегруппировки и ее роли в процессах мономолекулярного распада Снитросоединений / Г.М. Храпковский, Е.В. Николаева, Д.В. Чачков, А.Г.
Шамов // ЖОХ. – 2004. – Т. 74. – № 6. с. 983-986.
93.
Glenwinkel-Meyer, T. The isomerization of nitrobenzene to phenilnitrite / T.
Glenwinkel-Meyer, F.F. Crim // J. Mol. Struct. (Theochem). – 1995. – V. 337. – p.
209-224.
94.
Nibbering, N.M.M. The role of mass spectrometric methods in ionic reaction
mechanistic studies / N.M.M. Nibbering // Int. J. Mass Spectrom. – 2000. – V. 200.
– Is. 1-3. – p. 27-42.
149
95.
Хмельницкий, Р.А. Пиролитическая масс-спектрометрия высокомолекулярных
соединений / Р.А. Хмельницкий, И.М. Лукашенко, Е.С. Бродский. – М.:
Химия, 1980, – 280 с.
96.
Вилков, Л.В. Физические методы исследования в химии. Структурные методы
и оптическая спектроскопия. Учеб. для хим. спец. ВУЗов. / Л.В. Вилков, Ю.А.
Пентин. – М.: Высш. шк., 1987. – 367 с.
97.
Мусин, Р.З. Основы масс-спектрометрии: учебно-метод. пособие / Р.З. Мусин,
Н.И. Мовчан, С.М. Горюнов, С.Ф. Мухарлямов. – Казань: КГТУ, 2000. – 64 с.
98.
Дьюар, М. Теория возмущений молекулярных орбиталей в органической
химии / М. Дьюар, Р. Догерти. – М.: Мир, 1977. – 696 с.
99.
Polášek, M. High-energy [C,H3,N,O] cation radicals and molecules / M. Polášek, M.
Sadilek, F. Tureček // Int. J. Mass Spectrom. – 2000. – V. 195/196. – p. 101-114.
100. Ogden, I.K. Сompeting dissociation channels of nitromethane and methyl nitrite ions
and the role of electronic and internal modes of excitation / I.K. Ogden, N. Shaw,
C.J. Danby, I. Powis // Int. J. Mass Spectrom. Ion Process. – 1983. – V. 54. – p. 4153.
101. Gilman, J.P. Competition between Isomerization and fragmentation of gaseous Ions.
II Nitromethane and methylnitrite Ions / J.P. Gilman, T. Hsies, G.G. Meisels // J.
Chem. Phys. – 1983. – V. 78. – N 3. – p. 1174-1179.
102. Gilman, J.P. The unimolecular decomposition rates of energy selected methylnitrite
and deuterated methylnitrite ions / J.P. Gilman, T. Hsies, G.G. Meisels // J. Chem.
Phys. – 1983. – V. 78. – N 6. – p. 3767-3773.
103. Egsgaard, H. Isomerisation of the Nitromethane Radical Cation in the Gas Phase / H.
Egsgaard, L. Carlsen, S. Elbel // Ber. Bungenges. Phys. Chem. – 1986. – V. 90. – N
4. – p. 369-374.
104. Lifshitz, C. Unimolecular Fragmentations of the Nitromethane Cations / C. Lifshitz,
M. Rejwan, I. Levin, T. Peres // Int. J. Mass Spectrom. Ion Process. – 1988. – V. 84.
– p. 271-282.
150
105. Alizadeh, E. Dissociative electron attachment to nitromethane / E. Alizadeh, F.
Ferreira da Silva, F. Zappa, A. Mauracher, M. Probst, S. Denifl, A. Bacher, T.D.
Mark, P. Limao-Vieira, P. Scheier // Int. J. Mass Spectrom. – 2009. – V. 271. – Is. 13. – p. 15-21.
106. Hindawi, S.K. Reactions induced by the γ-hydrogen shift in the molecular ion of 1nitropropane / S.K. Hindawi, R.H. Fockkens, F.A. Pinkse, N.M.M. Nibbering //
J. Mass. Spectrom. – 1986. – V. 21. – Is. 5. – p. 243-250.
107. Nibbering, N.M.M. Mass Spectrometry of Nitro Compounds. Part I. Mass spectra of
α-, β-, and γ-Deuterated 1-Nitropropane / N.M.M. Nibbering, T.J. de Boer, H.J.
Hofman // Rec. Trav. Chim. Pays-Bas. – 1965. – V. 84. – p. 481-487.
108. Nibbering, N.M.M. The McLafferty Rearrangement: A Personal Recollection /
N.M.M. Nibbering // J. Am. Soc. Mass. Spectrom. – 2004. – V.15. – p. 956-958.
109. Nibbering, N.M.M. Four Decades of Joy in Mass Spectrometry / N.M.M. Nibbering
// Mass. Spectrom. Rev. – 2006. – V. 25. – Is. 6. – p. 962-1017.
110. McLafferty, F.W. Spectrometric Analysis Broad Applicability to Chemical Research
/ F.W. McLafferty // Anal. Chem. Mass. – 1956. – V. 28. – p. 306-316.
111. Stone, D.J.M. Mechanistic studies of unimolecular ionic decompositions by
deuterium and heavy-atom isotope effects. Concerted elimination of acetaldehyde
from the benzyl ethyl ether radical cation / D.J.M. Stone, J.H. Bowie, D.J.
Underwood // J. Am. Chem. Soc. – 1983. – V. 105. – N 6. – p. 1688-1689.
112. Allison, C.E. Combined deuterium and oxygen-18 isotope effects in support of a
concerted, synchronous elimination of acetaldehyde from a bis(benzyl ethyl ether)
radical cation / C.E. Allison, M.B. Stringer, J.H. Bowie, P.J. Derrick // J. Am. Chem.
Soc. – 1988. – V. 110. – N 19. – p. 6291-6297.
113. Wesdemiotis, C. Distonic radical ions. Stepwise elimination of acetaldehyde from
ionized benzyl ethyl ether / C. Wesdemiotis, R. Feng, F.W. McLafferty // J. Am.
Chem. Soc. – 1985. – V. 107. – N 3. – p. 715-716.
151
114. Tureček, F. The stepwise nature of the .gamma.-hydrogen rearrangement in
unsaturated ions / F. Tureček, D.E. Drinkwater, F.W. McLafferty // J. Am. Chem.
Soc. – 1990. – V. 112. – N 3. – p. 993-997.
115. Osterhold, T.H. Infrared multiple photon dissociation of butyrophenone cation. A
stepwise McLafferty rearrangement / T.H. Osterhold, J.I. Brauman // J. Am. Chem.
Soc. – 1990. – V. 112. – N 5. – p. 2014-2016.
116. Hudson, C.E. Ab initio and RRKM evidence that the McLafferty rearrangement of
ionized n-butanal is stepwise / C.E. Hudson, L.L. Griffin, D.J. McAdoo // Org.
Mass. Spectrom. – 1989. – V. 24. – Is. 10. – p. 866-870.
117. Dorgio, A.E. Transition structures for hydrogen atom transfers to oxygen.
Comparisons of intermolecular and intramolecular processes, and open- and closedshell systems / A.E. Dorgio, M.A. McCarrick, R.J. Loncharich, K.N. Houk // J. Am.
Chem. Soc. – 1990. – V. 112. – N 21. – p. 7508-7514.
118. Liu, R. Ab initio evidence for the stepwise mechanism of the McLafferty
rearrangement of the butanal radical cation / R. Liu, P. Pulay // J. Comput. Chem. –
1992. – V. 13. – Is. 2. – p. 183-186.
119. Norberg, D. McLafferty Rearrangement of the Radical Cations of Butanal and 3Fluorobutanal: A Theoretical Investigation of the Concerted and Stepwise
Mechanisms / D. Norberg, N. Salhi-Benachenhou // J. Comput. Chem. – 2008. – V.
29. – Is. 3. – p. 392-406.
120. Schroder, D. Dissociation behavior of ionized valeramide: Part II. Theoretical
exploration of the potential-energy surface / D. Schroder, J. Loos, M. Semialjac, T.
Weiske, H. Schwarz, G. Hohne, R. Thissen, O. Dutuit // Int. J. Mass Spectrom. –
2002. – V. 214. –, Is. 1. – p. 129-154.
121. Loos, J. Competitive reactions and diastereoselective C-H bond activation in the
McLafferty rearrangement of photoionized 3-methyl valeramide / J. Loos, D.
Schroder, H. Schwarz, R. Thissen, O. Dutuit // Int. J. Mass Spectrom. – 2005. – V.
240. – Is. 2. – p. 121-137.
152
122. Semialjac, M. Car–Parrinello Molecular Dynamics Study of the Rearrangement of
the Valeramide Radical Cation / M. Semialjac, D. Schröder, H. Schwarz //
Chemistry. – 2003. – V. 22. – Is. 18. – p. 4396-4040.
123. Tsyshevsky, R.V. Fragmentation reactions in the 1-nitropropane radical cation
induced by γ-hydrogen shift: Ab initio study / R.V. Tsyshevsky, G.G. Garifzianova,
A.G. Shamov, G.M. Khrapkovskii // Int. J. Mass Spectrom. – 2014. – V. 369. – p.
36-43.
124. Fields, E.F. Arylation by aromatic nitrocompounds at high temperatures. II.
Nitrobenzene alone and with benzene and benzene - d6 / E.F. Fields, S. Meyerson //
J. Am. Chem. Soc. – 1967. – V. 89. – N 13. – p. 3224-3228.
125. Fields, E.F. Interchange of hydrogen and oxygen atoms in o-nitrotoluenes / E.F.
Fields, S. Meyerson // Tetrahedron Lett. – 1968. – V. 9. – Is. 10. – p. 1201-1203.
126. Fields, E.F. Arylation by aromatic nitrocompounds at high temperatures. V.
Reactions of nitrotoluenes / E.F. Fields, S. Meyerson // J. Org. Chem. – 1968. – V.
33. – N 12. – p. 4487-4493.
127. Meyerson, S. Organic ions in the gas phase. XXVI. Decomposition of 1,3,5trinitrobenzene nuclear electron impact / S. Meyerson, R.W.V. Haer, E.F. Fields // J.
Org. Chem. – 1972. – V. 37. – p. 4114-4119.
128. Cooper, L. Time-of-flight mass spectrometry study of the fragmentation of valence
shell ionized nitrobenzene / L. Cooper, L.G. Shpinkova, E.E. Rennie, D.M.P.
Holland, D.A. Shaw // Int. J. Mass Spectrom. – 2001. – V. 207. – Is. 3. – p. 223-239.
129. Osterheld, T.H. Infrared Multiple-Photon Dissociation of the Nitrobenzene Radical
Cation. A Paradigm for Competitive Reactions / T.H. Osterheld, T. Baer, J.I.
Brauman // J. Am. Chem. Soc. – 1993. – V. 115. – N 14. – p. 6284-6289.
130. Николаева, Е.В. Теоретическое изучение механизма газофазного распада
катион-радикала
нитробензола
/
Е.В.
Николаева,
А.Г
Шамов,
Г.М.
Храпковский // Вестник казанского технологического ун-та. – 2011. – № 20. –
с. 32-39.
153
131. Корсунский, Б.Л. Изучение электронно-ударной фрагментации молекулярных
ионов нитро- и нитрозобензолов / Б.Л. Корсунский, Г.М. Назин, В.Р. Степанов,
А.А. Федотов // Кинетика и катализ. – 1993. – Т. 34. – № 4. – c. 608-611.
132. Корсунский,
Б.Л.
Изучение
первичных
стадий
термического
распада
некоторых нитробензолов методом высоковакуумного пиролиза / Б.Л.
Корсунский, Г.М. Назин, В.Р. Степанов, А.А. Федотов // Кинетика и катализ. –
1993. – Т. 34. – № 5. – c. 775-777.
133. Linstrom, P.J. NIST Chemistry WebBook / P.J. Linstrom, W.G. Mallard, Eds. //
NIST Standard Reference Database Number 69. – National Institute of Standards
and
Technology.
–
Gaithersburg
MD,
20899.
–
Режим
доступа:
http://webbook.nist.gov, (retrieved September 3, 2014).
134. Светлов, Б.С. Термическое разложение нитроэфиров: дис. … докт. хим. наук:
05.17.10 / Светлов Борис Сергеевич. – Москва, 1970. – 260 с.
135. Лурье, Б.А. Кинетические параметры первичной стадии термического
разложения органических нитратов / Б.А. Лурье, Б.С. Светлов // Кинетика и
катализ. – 1994. – Т. 35. – № 2. – c. 165-175.
136. Shamov, A.G. The role of by-radical stages in the thermal decomposition of
aliphatic C-nitrocompounds process / A.G. Shamov, E.V. Nikolaeva, D.V.
Chachkov, G.M. Khrapkovskii // Coll. art. 35th International Annual Conference of
ICT "Energetic Materials: Structure and properties". Federal Republic of Germany,
Karlsruhe. – 2004. – p. 127:1-127:14.
137. Шляпочников, В.А. Колебательные спектры алифатических нитросоединений /
В.А. Шляпочников. – М.: Наука, 1989. – 136 с.
138. Еременко, Л.Т. Нитроцеллюлоза и ее использование в технике /
Л.Т.
Еременко, А.Н. Королев, Л.И. Березина. – Черноголовка, 1981. – 272 с.
139. Шамсутдинов, Т.Ф. Теоретическая оценка энергий диссоциации связей
нитроэфиров β-D-глюкозы / Т.Ф. Шамсутдинов, Д.В. Чачков, Е.В. Николаева,
154
А.Г. Шамов, Г.М. Храпковский // Вестник казанского технологического ун-та.
– 2004. – № 1-2. – c. 24-27.
140. Шамсутдинов, Т.Ф. Влияние величины предэкспоненциального множителя на
конкуренцию различных механизмов газофазного распада c-нитросоединений:
дис. … канд. хим. наук: 02.00.04 / Шамсутдинов Тимур Фаритович. – Казань,
2007. – 170 с.
141. Коваленко,
В.И.
Нитрат
целлюлозы:
молекулярно-структурная
неоднородность / В.И. Коваленко, О.В. Михайлов, Г.М. Храпковский. –
Казань: Фэн, 2003. – 152 с.
142. Gruzdkov, Y.A. Shock Wave Initiation of Pentaerythritol Tetranitrate Single
Crystals: Mechanism of Anisotropic Sensitivity / Y.A. Gruzdkov, Y.M. Gupta // J.
Phys. Chem. A. – 2000. – V. 104. – N 47. – p. 11169-11176.
143. Gruzdkov, Y.A. Vibrational Properties and Structure of Pentaerythritol Tetranitrate /
Y.A. Gruzdkov, Y.M. Gupta // J. Phys. Chem. A. – 2001. – V. 105. – N 25. – p.
6197-6202.
144. Zhang, G. Crystal growth of organic energetic materials: pentaerythritol tetranitrate /
G. Zhang, B.L. Weeks, X. Zhang // Cent. Eur. J. Eng. – 2012. – V. 2. – Is. 3. – p.
336-346.
145. Lindner, V. Explosives and propellants In Kirk-Othmer Encyclopedia of Chemical
Technology, 4th ed.; / V. Lindner, J.I. Kroschwitz, M. Howe-Grant, Eds. – Wiley,
New York, 1993. – V. 10. – pp. 22-60.
146. Ng, W.L. Study of the thermal decomposition of pentaerythritol tetranitrate / W.L.
Ng, J.E. Field, H.M. Hauser // J. Chem. Soc. Perkin Trans. – 1976. – V. 2. – Is. 6. –
p. 637-639.
147. Ng, W.L. Thermal, fracture and laser-induced decomposition of pentaerythritol
tetranitrate / W.L. Ng, J.E. Field, H.M. Hauser // J. Appl. Phys. – 1986. – V. 59. – p.
3945-3952.
155
148. Andreev,
K.K.
Thermal
Decomposition
of
Nitrate
Esters.
II.
Thermal
Decomposition of Pentaerythritol Tetranitrate / K.K. Andreev, B.I. Kaidymov //
Russ. J. Phys. Chem. – 1961. – V. 35. – p. 1324-1330.
149. Chambers, D.M. Perspectives on Pentaerythritol Tetranitrate (PETN) Decomposition
/ D.M. Chambers // UCRL-JC-148956. – 2002. – p. 1-18.
150. Gibbs, T.R. LASL Explosive Property Data / T.R. Gibbs, A. Popolato. – University
of California Press: Berkley, USA, 1980, – p. 131-140.
151. Robertson, A.J.B. The thermal Decomposition of Pentaerythritol Tetranitrate,
Nitroglycerin, Ethylenediamine Dinitrate and Ammonium Nitrate / A.J.B. Robertson
// J. Soc. Chem. Ind. – 1948. – V. 67. – Is. 6. – p. 221-224.
152. Rogers, R.N. Estimating Activation Energies with a Differential Scanning
Calorimeter / R.N. Rogers, Jr. Morris, E.D. On // Anal. Chem. – 1966. – V. 38. – N
3. – p. 412-414.
153. Maycock, J.N. Characterization of thermal and photosublimation of organic
explosives by thermobarogravimetric techniques / J.N. Maycock, V.R. Pai Verneker
// Thermochim. Acta. – 1970. – V. 1. – Is. 2. – p. 191-198.
154. Volltrauer, H.N. Real time low temperature decomposition of explosives - PETN /
H.N. Volltrauer // J. Haz. Mater. – 1982. – V. 5. – Is. 4. – p. 353-357.
155. Oxley, J.C. Characterization and Analysis of Tetranitrate Esters / J.C. Oxley, J.L.
Smith, J.E. Brady IV, A.C. Brown // Propellants Explos. Pyrotech. – 2012. – V. 37.
– Is. 1. – p. 24-39.
156. Tsyshevsky, R.V. Thermal Decomposition Mechanisms of Nitroesters: Ab Initio
Modeling of Pentaerythritol Tetranitrate / R.V. Tsyshevsky, O. Sharia, M.M. Kuklja
// J. Phys. Chem. – 2013. – V. 117. – N 35. – p. 18144-18153.
157. Liu, W.G. Explanation of the Colossal Detonation Sensitivity of Silicon
Pentaerythritol Tetranitrate (Si-PETN) Explosive / W.G. Liu, S.V. Zybin, S.
Dasgupta, T.M. Klapötke, W.A. Goddard III // J. Am. Chem. Soc. – 2009. – V. 131.
– N 22. – p. 7490-7491.
156
158. Brook, A.G. Molecular rearrangements of organosilicon compounds / A.G. Brook //
Acc. Chem. Res. – 1974. – V. 7. – N 3. – p. 77-84.
159. Landerville, A.C. Reactive Molecular Dynamics of Hypervelocity Collisions of
PETN Molecules / A.C. Landerville, I.I. Oleynik, C.T. White // J. Phys. Chem. A. –
2009. – V. 113. – N 44. – p. 12094-12104.
160. Fried, L.E. Design and Synthesis of Energetic Materials / L.E. Fried, M.R. Manaa,
P.F. Pagoria, R.L. Simpson // Annu. Rev. Mater. Res. – 2001. – V. 31. – p. 291-321.
161. Khrapkovskii, G.M. Energy of the O–NO2 bond dissociation and the mechanism of
the gas-phase monomolecular decomposition of aliphatic alcohol nitroesters / G.M.
Khrapkovskii, T.F. Shamsutdinov, D.V. Chachkov, A.G. Shamov // J. Mol. Struct.:
THEOCHEM. – 2004. – V. 686. – Is. 1-3. – p. 185-192.
162. Zhu, L. Temperature Dependence of the Near UV Absorption Spectra and Photolysis
Products of Ethyl Nitrate / L. Zhu, C.F. Ding // Chem. Phys. Lett. – 1997. – V. 265.
– Is. 1-2. – p. 177-184.
163. He, Sh. Photochemical Reactions of Methyl and Ethyl Nitrate: a Dual Role for Alkyl
Nitrates in the Nitrogen Cycle / Sh. He, Zh. Chen, X. Zhang // Environ. Chem. –
2011. – V. 8. – N 6. – p. 529-542.
164. Rebbert, R.E.Primary Processes in the Photolysis of Ethyl Nitrate / R.E. Rebbert // J.
Phys. Chem. – 1963. – V. 67. – N 9. – p. 1923-1925.
165. Boumsellek, S. Negative-Ion Formation in the Explosives RDX, PETN, and TNT By
Using the Reversal Electron Attachment Detection Technique / S. Boumsellek, S.H.
Alajajian, A. Chutjian // J. Am. Soс. Mass Spectrom. – 1992. – V. 3. – Is. 3. – p.
243-247.
166. Дубовицкий, Ф.И. Кинетика термического разложения N-нитросоединений /
Ф.И. Дубовицкий, Б.Л. Корсунский // Успехи химии. – 1981. – Т. 50. – № 10. –
с. 1828-1871.
157
167. Шу, Ю. Механизм термического разложения вторичных нитраминов / Ю. Шу,
Б.Л. Корсунский, Г.М. Назин // Успехи химии. – 2004. Т. 73. – № 3. – c. 321335.
168. Петухова, Т.В. Исследование строение и механизмов термического распада
полуфункциональных N-нитросоединений расчетными методами: автореф.
дис. … канд. хим. наук: 02:00:04 / Петухова Татьяна Виниаминовна. – ЙошкарОла, 2004. – 24 с.
169. Фридман, А.Л. Успехи химии первичных алифатических нитраминов / А.Л.
Фридман, В.П. Ившин, С.С. Новиков // Успехи химии. – 1969. – Т. 38. – № 8. –
с. 1448-1478.
170. Королев, В.Л. Дифференциация молекулярного строения нитросоединений как
основа моделирования процессов их термодеструкции / В.Л. Королев, Т.С.
Пивина, А.А. Поролло, Т.В. Петухова, А.Б. Шереметев, В.П. Ившин // Успехи
химии. – 2009. – Т. 78. – № 10. – с. 1022-1047.
171. Кекин,
Ю.В.
Кинетика
термического
разложения
первичных
алкилнитроаминов в газовой фазе / Ю.В. Кекин, В.Н. Шанько, Р.С. Степанов //
Кинетика и катализ. – 1989. – Т. 30. – Вып. 4. – c. 963-965.
172. Мазилов, E.A. Молекулярная структура и механизм газофазного распада
первичных алифатических N-нитраминов: дис. … канд. хим. наук: 02.00.04 /
Мазилов Елисей Александрович. – Казань, 2009. – 143 c.
173. Мазилов, Е.А. Молекулярная структура и механизмы мономолекулярного
распада первичных N-нитраминов / Е.А. Мазилов, Д.В. Чачков, А.Г. Шамов,
Г.М. Храпковский // ЖОХ. – 2009. – № 3. – c. 425-438.
174. Mazilov, E.A. A theoretical study on the mechanism of gas-phase monomolecular
decomposition of N-methylnitramine / E.A. Mazilov, E.V. Nikolaeva, A.G. Shamov,
G.M. Khrapkovskii // Mendeleev Commun. – 2007. – V. 17. – p. 359-361.
175. Khrapkovskii, G.M. Structure of transient and kinetic parameters of gas phase
reaction of elimination HNO2 C-, N-, O-nitrocompounds / G.M. Khrapkovskii, E.A.
158
Mazilov, D.V. Chachkov, A.G. Shamov, T.F. Shamsutdinov, A.F. Shamsutdinov //
The 10-th of the V.A. Fock Meeting on Quantum and Computational Chemistry.
Kazan, 2006. – P. 51.
176. Korsunskii,
B.L.
Thermochemical
and
kinetic
investigation
of
N,N-
dimethylnitrosoamine / B.L. Korsunskii, V.I. Pepekin, Yu.A. Lebedev, A.Y. Apin //
Russ. Chem. Bull. – 1967. – V. 16. – Is. 3. – p. 509-511.
177. Korsunskii, B.L. Kinetics of the thermal decomposition of N,N-diethylnitroamine
and N-nitropiperidine / B.L. Korsunskii, F.I. Dubovitskii, E.A. Shurygin // Russ.
Chem. Bull. – 1967. – V. 16. – Is. 7. – p. 1405-1407.
178. Johnson, M.A. High-Level ab Initio and Density Functional Theory Evaluation of
Combustion
Reaction
Energetics:
NO2
and
HONO
Elimination
from
Dimethylnitramine / M.A. Johnson, T.N. Truong // J. Phys. Chem. A. – 1999. – V.
103. – N 44. – p. 8840-8846.
179. Cass, R.C. Heats of combustion and molecular structure / R.C. Cass, S.E. Fletcher,
C.T. Mortimer, P.G. Quincey, H.D. Springall // J. Chem. Soc. – 1958. – Is. 0. – p.
958-963.
180. Корсунский, Б.Л. Кинетика термического распада N,N-диметилнитрамина /
Б.Л. Корсунский, Ф.И. Дубовицкий, Г.В. Ситонина // Докл. АН СССР. – 1967.
– Т. 174. – с. 1126-1132.
181. Петухова, Т.В. Исследование строение и механизмов термического распада
полуфукциональных N-нитросоединений расчетными методами: дис. … канд.
хим. наук: 02.00.04 / Петухова Татьяна Виниаминовна. – Йошкар-Ола, 2004. –
136 с.
182. Petukhova, T.V. Polyfunctional N-nitramines structure differentiation as a basis for
simulation of their decomposition mechanism / T.V. Petukhova, V.P. Ivshin, V.L.
Korolev, T.S. Pivina // Proceedings of the 9th Seminar of the New Trends in
Research of Energetic Materials. – Pardubice. – 2006. – p. 260-267.
159
183. Brill, T.B. Thermal Decomposition of Energetic Materials. 66. Kinetic
Compensation Effects in HMX, RDX, and NTO / T.B. Brill, P.E. Gongwer, G.K.
Williams // J. Phys. Chem. – 1994. – V. 98. – N 47. – p. 12242-12247.
184. Chakraborty, D. Mechanism for unimolecular decomposition of HMX (1, 3, 5, 7tetranitro-1, 3, 5, 7-tetrazocine), an ab initio study / D. Chakraborty, R.P. Muller, S.
Dasgupta, W.A. Goddard // J. Phys. Chem. A. – 2001. – V. 105. – N 8. – p. 13021314.
185. Sharia, O. Ab initio kinetics of gas phase decomposition reactions / O. Sharia, M.M.
Kuklja // J. Phys. Chem. A. – 2010. – V. 114. – N 48. – p. 12656-12661.
186. Sharia, O. Modeling thermal decomposition mechanisms in gaseous and crystalline
molecular materials: application to β-HMX / O. Sharia, M.M. Kuklja // J. Phys.
Chem. B. – 2011. – V. 115. – N 44. – p. 12677-12686.
187. Sharia, O. Surface-Enhanced Decomposition Kinetics of Molecular Materials
Illustrated with Cyclotetramethylene-tetranitramine / O. Sharia, M.M. Kuklja // J.
Phys. Chem. C. – 2012. – V. 116. – N 20. – p. 11077-11081.
188. Sharia, O. Rapid materials degradation induced by surfaces and voids: ab initio
modeling of β-octotetramethylene tetranitramine / O. Sharia, M.M. Kuklja // J. Am.
Chem. Soc. – 2012. – V. 134. – p. 11815-11820.
189. Chakraborty, D. The Mechanism for Unimolecular Decomposition of RDX (1,3,5Trinitro-1,3,5-triazine), an ab Initio Study / D. Chakraborty, R.P. Muller, S.
Dasgupta, W.A. Goddard // J. Phys. Chem. A. – 2000. – V. 104. – N 11. – p. 22612272.
190. Postler, J. Dissociative Electron Attachment to the Nitroamine HMX(Octahydro1,3,5,7-Tetranitro-1,3,5,7-Tetrazocine) / J. Postler, M.M. Goulart, C. Matias, A.
Mauracher, F. Ferreira da Silva, P. Scheier, P. Limão-Vieira, S. Denifl // J. Am. Soc.
Mass Spectrom. – 2013. – V. 24. – Is. 5. – p.744-752.
160
191. Florián, J. Nitramine Anion Fragmentation: A Mass Spectrometric and Ab Initio
Study / J. Florián, L. Gao, V. Zhukhovskyy, D.K. Macmillan, D.K. Chiarelli //
J. Am. Soc. Mass Spectrom. – 2007. – V. 18. – Is. 5. – p. 835-841.
192. Sharipov, D.D. Effect of the molecular structure on the strength of the C-NO2 bond
in a series of monofunctional nitrobenzene derivatives / D.D. Sharipov, D.L.
Egorov, D.V. Chachkov, A.G. Shamov, G.M. Khrapkovskii // Rus. J. Gen. Chem.
2011. – V. 81. – № 11. – р. 2273-2287.
193. Garivzianova, G.G. A quantum-chemical study of n-butane and of butane cation
radical / G.G. Garivzianova, R.V. Tsyshevskii., A.G.Shamov, G.M. Khrapkovskii //
Int. J. Quantum. Chem. – 2007. – V. 107. – Is. 13. – р. 2489-2493.
194. Olivella, S. Unimolecular Reactions of Ionized Alkanes: Theoretical Study of the
Potential Energy Surface for CH3 and CH4 Losses from Ionized Butane and
Isobutane / S. Olivella, Al. Sole, D.J. McAdoo, L.L. Griffins // J. Am. Chem. Soc. –
1994. – V.116. – N 24. – р.11078-11088.
195. Tsyshevsky, R.V. Study of Geometry and Electronic Structure of Molecules, CationRadicals, and Anion-Radicals of Nitromethane, Dimethylnitramine, and Ethyl
Nitrate / R.V. Tsyshevsky, B. Nguyen Van, A.G. Shamov, and G.M. Khrapovskii //
Rus. J. Gen. Chem. – 2013. – V. 83. – № 10. – p. 1823-1839.
196. Perdew, J.P. Generalized gradient approximation made simple / J.P. Perdew, K.
Burke, M. Ernzerhof // Phys. Rev. Lett. – 1996. – V. 77. – p. 3865-3868.
197. Perdew, J.P. Atoms, molecules, solids, and surfaces: Applications of the generalized
gradient approximation for exchange and correlation / J.P. Perdew, J.A. Chevary,
S.H. Vosko, K.A. Jackson, M.R. Pederson, D.J. Singh, C.Fiolhais // Phys. Rev. –
1992. – V. 46. – p. 6671-6687.
198. Perdew, J.P. Generalized gradient approximation for the exchange-correlation hole
of a many-electron system / J.P. Perdew, K. Burke, Y. Wang // Phys. Rev. – 1996. –
V. 54. – p. 16533-16539.
161
199. Handy, N.C. Left-right correlation energy / N.C. Handy, A.J. Cohen // Mol. Phys. –
2001. – V. 99. – Is. 5. – p. 403-412.
200. Lee, C. Development of the Colle-Salvetti correlation-energy formula into a
functional of the electron density / C. Lee, W. Yang, R.G. Parr // Phys. Rev. – 1988.
– V. 37. – p. 785-789.
201. Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange /
A.D. Becke // J. Chem. Phys. – 1993. – V. 98. – p. 5648-5652.
202. Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange /
A.D. Becke // J. Chem. Phys. – 1993. – V. 98. – p. 1372-1377.
203. Zhao, Y. The M06 suite of density functionals for main group thermochemistry,
thermochemical kinetics, noncovalent interactions, excited states, and transition
elements: two new functionals and systematic testing of four M06-class functionals
and 12 other functional / Y. Zhao, D.G. Truhlar // Theor. Chem. Acc. – 2008. – V.
120. – Is. 1-3. – p. 215-241.
204. Grimme, S. Semiempirical hybrid density functional with perturbative second-order
correlation / S. Grimme // J. Chem. Phys. – 2006. – V. 124. – p. 034108:1034108:16.
205. Schwabe, T. Towards chemical accuracy for the thermodynamics of large
molecules: new hybrid density functionals including non-local correlation effects /
T. Schwabe, S. Grimme // Phys. Chem. Chem. Phys. – 2006. – V. 8. – Is. 38. – p.
4398-4401.
206. Hohenberg, P. Inhomogeneous Electron Gas / P. Hohenberg, W. Kohn // Phys. Rev.
– 1964. – V. 136. – p. B864-B871.
207. Kohn, W. Self-Consistent Equations Including Exchange and Correlation Effects /
W. Kohn, L.J. Sham // Phys. Rev. – 1965. – V. 140. – p. A1133-A1138.
208. Chai, J.D. Long-range corrected hybrid density functionals with damped atom-atom
dispersion corrections / J.D. Chai, M. Head-Gordon // Phys. Chem. Chem. Phys. –
2008. – V. 10. – p. 6615-6620.
162
209. Chai, J.D. Systematic optimization of long-range corrected hybrid density
functionals / J.D. Chai, M. Head-Gordon // J. Chem. Phys. – 2008. – V. 128. –
084106-1 - 084106-15.
210. Møller, C. Note on an approximation treatment for many-electron systems / C.
Møller, M.S. Plesset // Phys. Rev. – 1934. – V. 46. – p. 0618-0622.
211. Frisch, M. J. Gaussian 09, Revision A.1 / M.J. Frisch, G.W. Trucks, H.B. Schlegel,
G.E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.
A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J.
Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J.
Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A.
Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N.
Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C.
Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J.E.
Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O.
Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K.
Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich,
A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox. –
Gaussian, Inc., Wallingford CT, 2009.
212. Johnson III, R.D. NIST Computational Chemistry Comparison and Benchmark
Database, NIST Standard Reference Database Number 101, Release 15b / Editor:
Russell D. Johnson III. – August 2011,
213. Hellwege, K.H. Landolt-Bornstein: Group II: Atomic and Molecular Physics
Volume 7: Structure Data of Free Polyatomic Molecules / K.H. Hellwege, A.M.
Hellwege (ed.). – Springer-Verlag. Berlin, 1976.
214. Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave
Functions. I / R.S. Mulliken // J. Chem. Phys. – 1955. – V. 23. – p .1833-1840.
215. Lias, S.G. "Ion Energetics Data" in NIST Chemistry WebBook, NIST Standard
Reference Database Number 69, / S.G. Lias, R.D. Levin, S.A. Kafafi, Eds. P.J.
163
Linstrom, W.G. Mallard. – National Institute of Standards and Technology,
Gaithersburg MD, 20899. – Режим доступа: http://webbook.nist.gov (retrieved
March 10, 2013).
216. Rosenstock, H.M. "Ion Energetics Data" in NIST Chemistry WebBook, NIST
Standard Reference Database Number 69, Eds. / H.M. Rosenstock, K. Draxl, B.W.
Steiner, J.T. Herron, Eds. P.J. Linstrom, W.G. Mallard. – National Institute of
Standards and Technology, Gaithersburg MD, 20899. – Режим доступа:
http://webbook.nist.gov (retrieved March 10, 2013).
217. Manelis, G.B. Thermal Decomposition and Combustion of Explosives and
Gunpowders / G.B. Manelis, G.M. Nazin, Yu.I. Rubtsov, V.A. Strunin. – CRC
Press, Moscow, 2003. – 376 p.
218. Luo, Y.R. Handbook of Bond Dissociation Energies in Organic Compounds / Y.R.
Luo. – CRC Press, 2003
219. Sumpter, B.G. Unimolecular reaction dynamics of dimethylnitramine / B.G.
Sumpter, D.L. Thompson // J. Chem. Phys. – 1988. – V. 88. – p. 6889-6897.
220. Nigenda,
E.S.
Thermal
decomposition
of
dimethylnitramine
and
dimethylnitrosamine by pulsed laser pyrolysis / E.S. Nigenda, D.F. McMillen, D.M.
Golden // J. Phys. Chem. – 1989. – V. 93. – N 3. – p. 1124-1130.
221. Flournoy, J.M. Thermal Decomposition of Gaseous Dimethylnitranine / J.M.
Flournoy // J. Chem. Phys. – 1962. – V. 36. – p. 1106-1107.
222. Harris, N.J. Ab Initio Density Functional Calculations of Deuterium Kinetic Isotope
Effects for Decomposition of Dimethylnitramine / N.J. Harris, K. Lammertsma //
J. Phys. Chem. A. – 1997. – V. 101. – N 7. – p. 1370-1373.
223. Oxley,
J.C.
Thermal
decomposition
of
nitramines:
dimethylnitramine,
diisopropylnitramine, and N-nitropiperidine / J.C. Oxley, M. Hiskey, D. Naud, R.
Szekeres // J. Phys. Chem. – 1992. – V. 96. – N 6. p. 2505-2509.
164
224. Velardez, G.F. Theoretical predictions of the initial decomposition steps of
dimethylnitramine / G.F. Velardez, S. Alavi, D.L. Thompson // J. Chem. Phys. –
2005. – V. 123. – p. 074313:1-074313:7.
225. George, M.V. The Steric Conformations of Nitro- and Nitroso-bis-piperazines /
M.V. George, G. F. Wright // J. Am. Chem. Soc. – 1958. – V. 80. – N 5. – p. 12001204.
226. Johnson, M.A. Importance of Polarization in Simulations of Condensed Phased
Energetic Materials / M.A. Johnson, T.N. Truong // J. Phys. Chem. – 1999. – V. 103.
– N 44. – p. 9392-9393.
227. Храпковский, Г.М. Барьеры реакций газофазного мономолекулярного распада
нитротолуолов. 1. Общие закономерности / Г.М. Храпковский, Е.В. Николаева,
Д.Л.
Егоров,
А.Г.
Шамов,
Б.
Нгуен
Ван
//
Вестник
казанского
технологического ун-та. – 2014. – Т. 17. – № 13. – с. 55-61.
228. Нгуен Ван, Б. Геометрическая и электронная структуры молекул, катион- и
анион-радикалов нитробензола по данным квантово-химических расчетов / Б.
Нгуен Ван, Р.В. Цышевский, Г.М. Храпковский // Вестник казанского
технологического ун-та. – 2014. – Т. 17. – № 13. – с. 62-67.
229. Храпковский, Г.М. Барьеры реакций газофазного мономолекулярного распада
нитротолуолов. 2. Влияние молекулярной структуры на барьеры реакций
внутримолекулярного переноса водорода с образованием аци-нитротолуола /
Г.М. Храпковский, Е.В. Николаева, Д.Л. Егоров, А.Г. Шамов, Б. Нгуен Ван //
Вестник казанского технологического ун-та. – 2014. – Т. 17. – № 13. – C. 68-72.
230. Цышевский, Р.В. Теоретическое исследование газофазного распада анион- и
катион-радикалов пентаэритриттетранитата / Р.В. Цышевский, Б. Нгуен Ван,
Г.М. Храпковский // Тез. докл. XXX Всероссийского симпозиума молодых
ученых по химической кинетике. – Москов. обл. – 2012. – С. 80.
231. Schroeder, D. Further Insight in the Surprisingly Complex Unimolecular
Fragmentations of the Methyl Nitrite Radical Cation / D. Schroeder, D. Suelzle, O.
165
Dutuit, T. Baer, H. Schwarz // J. Am. Chem. Soc. – 1994. – V. 116. – N 14. – p.
6395-6400.
232. Нгуен Ван, Б. Исследование механизмов термического распада анион- и
катион-радикалов
диметилнитрамина
по
данным
квантово-химических
расчетов / Б. Нгуен Ван, Р.В. Цышевский, Г.М. Храпковский // Сб. ст.
международной научно-практической конференции. – Уфа, 2014. – c. 34-35.