Основы физики гибридных наноструктур
advertisement

А.В. Федоров, А.В. Баранов, А.О.Орлова, В.Г.Маслов
Основы физики
гибридных
наноструктур
Учебное пособие
Санкт-Петербург
2014
«САНКТ-ПЕТЕРБУРГСКИЙ НАЦИОНАЛЬНЫЙ ИССЛЕДОВАТЕЛЬСКИЙ
УНИВЕРСИТЕТ ИНФОРМАЦИОННЫХ ТЕХНОЛОГИЙ, МЕХАНИК И
ОПТИКИ»
А.В.Федоров, А.В.Баранов, А.О.Орлова, В.Г.Маслов
Основы физики
гибридных
наноструктур
2
СанктПетербург
2014
Федоров А.В., Баранов А.В., Орлова А.О., МасловВ.Г. Основы
физики гибридных наноструктур.
Учебное пособие. - СПб: СПб НИУ ИТМО, 2014. – 122 с.
Рассматриваются основы формирования и физические свойства
гибридных наноструктур, состоящих из полупроводниковых
нанокристаллов и органических молекул разного типа. Особое
внимание уделяется оптическим методам исследования этих
наноструктур, а также возможным применениям гибридных
наноструктур в устройствах фотоники и оптоинформатики.
Учебноепособие предназначено для обучения магистров по
направлению 200700 Фотоника и оптоинформатика для изучения
дисциплины «Основы физики гибридных наноструктур». Материал
может быть рекомендован для студентов старших курсов физикотехнических специальностей, а также при подготовке магистров и
аспирантов, специализирующихся в области применения оптических
методов в нанотехнологиях.
Рекомендовано УМО вузов Российской Федерации по образованию в
области приборостроения и оптотехники для студентов высших
учебных заведений, обучающихся по направлениям подготовки
магистратуры 12.04.03 (200700) «Фотоника и оптоинформатика».
Протокол заседания Президиума УМО № 2 от 09.04.2014.
© Санкт-Петербургский национальный исследовательский университет
информационных технологий, механики и оптики, 2014.
© Федоров А.В., Баранов А.В., Орлова А.О., Маслов В.Г., 2014
3
О ГЛ А В ЛЕ Н И Е
Введение. Гибридные материалы и наноструктуры
6
1. Законы поглощения света; электронно-колебательные
спектры многоатомных молекул
22
1.1. Закон поглощения света (закон Бугера-Ламберта-Бера).
Физические величины, характеризующие поглощение
света атомно-молекулярными системами.
1.2. Аналитические применения спектров поглощения
1.3. Основные случаи нарушения закона Бугера-ЛамбертаБера, “эффект сита”
1.4. Полосы поглощения и колебательная структура
электронных переходов
1.5. Основные физические закономерности, обусловливающие
активность колебаний в электронных спектрах, принцип
Франка-Кондона
2. Основы теории электронных спектров многоатомных
молекул; электронные переходы основных классов
органических соединений
22
24
24
27
28
32
32
2.1. Основные характеристики электронных состояний:
мультиплетность, дипольный момент перехода, сила
осциллятора
2.2. Классификация электронных переходов плоских систем: , 30
и n-электроны,
*иn
* переходы
40
2.3. Понятие о хромофорных группировках
42
2.4. Этиленовый хромофор, линейные полиены
46
2.5. . Теория электронных спектров ароматических
углеводородов, четырехорбитальная модель, бензольный
хромофор
54
2.6. Соединения с неуглеродными атомами и основные
свойства n
*-переходов
61
2.7. Примеры нециклических соединений: полифенилы,
красители
3. Межмолекулярные взаимодействия и их проявления в
электронно-колебательных спектрах
65
3.1. Универсальные взаимодействия: динамический, статический 65
4
и дисперсионный вклады
3.2. Влияние водородной связи на n
* переходы
3.3. Слабые комплексы с переносом заряда, основные свойства
состояний переноса заряда
3.4. Электронные состояния димеров: давыдовское
расщепление, экситонно-резонансные и зарядоворезонансные состояния
4. Молекулярная люминесценции и безызлучательный перенос
энергии
4.1. Основные фотофизические процессы в молекулах:
флуоресценция, фосфоресценция, внутренняя и
интеркомбинационная конверсии. Закономерности
молекулярной люминесценции
4.2. Спектры люминесценции и спектры возбуждения.
Основные правила их изменения
4.3. Квантовый выход люминесценции и методы его измерения
4.4. Флуоресценция и фосфоресценция при наличии n
*
переходов
4.5. Статическое и динамическое тушение люминесценции
4.6. Безызлучательный перенос энергии. Основные
закономерности индуктивно-резонансного переноса
энергии
4.7. Обменно-резонансный перенос энергии,
фотодинамический эффект и основные свойства
синглетного кислорода
4.8. Основные представления теории безызлучательных
переходов в многоатомных молекулах
5. Поглощение и люминесценция полупроводниковых
квантовых точек с позиций молекулярной спектроскопии
5.1. Происхождение и природа электронных состояний
квантовых точек. Понятие о конфайнменте
5.2. Спектры поглощения и люминесценции квантовых точек.
Размерная зависимость спектральных свойств
5.3. Особенности фотофизических процессов в квантовых
точках. Выполнимость основных закономерностей
поглощения и люминесценции
6. Основы колебательной инфракрасной спектроскопии
5
66
66
66
69
69
71
73
74
78
82
84
86
87
87
87
89
90
6.1. Колебательные переходы многоатомных молекул.
Молекула как механическая система с упругими связями
6.2. Групповые характеристические частоты. Классификация
колебаний: валентные и деформационные
6.3. Основные особенности ИК полос поглощения
6.4. Основные характеристические колебания: валентные OHколебания и водородная связь, валентные CO-колебания,
NH-колебания, NO-колебания
6.5. Особенности измерения поглощения в ИК области,
основные способы приготовления образцов
7. Фотофизические свойства комплексов полупроводниковых
квантовых точек CdSe/ZnS с молекулами порфирина в водных
средах
90
Список литературы
115
6
91
92
93
96
97
Введение
Гибридные материалы и наноструктуры
Под
гибридными материалами и наноструктурами понимают
материалы и наноразмерные структуры, содержащие существенно
разнородные компоненты. Под разнородными материалами понимают,
например, неорганические компоненты, в частности, полупроводниковые
квантовые точки, в сочетании с органическими компонентами, например
молекулами красителей, полимеров и т.п.
В данном введении будут рассмотрены следующие вопросы.
1.
Технологии синтеза наночастиц для использования в качестве
«строительных»
блоков
при
создании
различных
наноструктурированных материалов.
2.
Гибридные материалы на основе проводящих полимеров и
наночастиц для производства солнечных батарей.
3.
Мультифункциональные системы наночастиц для доставки
лекарств и диагностики.
4.
Наноуглеродные материалы для микроэлектроники и фотоники.
5.
Функционализированные нанопористые мембраны для очистки
питьевых, бытовых и сточных вод.
В1. Технологии синтеза наночастиц для использования в качестве
«строительных»
блоков
при
создании
различных
наноструктурированных материалов
Наночастица представляет собой объект, характерные размеры
которого хотя бы в одном измерении не превышают нескольких
десятков нанометров. Малость размеров определяет наличие размерных
квантово-механических эффектов, что приводит к появлению новых по
сравнению с объемным материалом свойств: повышенной твердости,
пластичности, магнетизма, селективного поглощения и эмиссии,
усиления каталитических процессов, более эффективного электронного
и оптического отклика. Этими свойствами можно управлять, изменяя
размеры, форму и морфологию частиц.
В настоящее время идет активное формирование библиотеки
наноразмерных строительных блоков - наночастиц. Для изготовления
нанокристаллов используют широкий диапазон материалов (металлы,
полупроводники, бескислородная и кислородсодержащая керамика,
углерод, силикаты). Также существует большой набор возможных форм
наночастиц (сферы, кубы, диски, стержни («родсы»), проволоки,
дендриты, тетраподы). Такое многообразие необходимо для
конструирования из этих строительных блоков новых материалов с
заранее заданными свойствами.
7
В зависимости от материала и формы наночастиц существует
масса методов их синтеза и модификаций: синтез может осуществляться
в жидкой, газообразной или твердой фазе, могут использоваться методы
химического или физического осаждения, а также другие физические
химические процессы, а также их комбинации. Ключевыми барьерами
на пути внедрения всех этих методов в коммерческое производство
являются высокая стоимость и токсичность. Кроме того имеются и
специфические препятствия. Например, в случае самого популярного
метода – коллоидного синтеза – существует проблема последующей
стабилизации синтезированных частиц, которая решается путем
пассивации их поверхности поверхностно-активными веществами.
Синтезированные наночастицы могут быть рассмотрены как
строительные блоки для последующего конструирования объемного
материала. Все существующие подходы к созданию материалов,
элементы которых имеют нанометровые масштабы, можно условно
разделить на два основных класса: подход «снизу-вверх» (bottom-up) и
подход «сверху-вниз» (top-down), которые схематически показаны на
рис. В1.
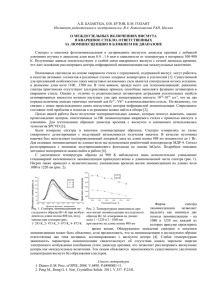
Рис. В1. Основные подходы к получению наноструктурированного
материала: слева – подход «снизу-вверх», справа – подход «сверхувниз» [1].
Подход «сверху-вниз» предполагает переход от объемного
материала к наноструктурированному путем уменьшения размеров за
счет дробления или фотолитографии с последующим травлением.
Обычно для этого используются различные шаблоны-темплейты.
Соответственно, в случае использования подхода «снизу-вверх»
объемный материал формируется путем самосборки из отдельных
наноразмерных блоков. При этом используются различные проявления
химического и физического взаимодействия нанокристаллов. Именно
8
этот подход получил наибольшее распространение ввиду его
перспективности в области создания целых классов новых материалов,
свойствами которых можно будет управлять.
Среди массы продуктов, появление которых на рынке может стать
результатом развития нанотехнологий, как наиболее приоритетные,
можно выделить следующие:
Нанобиоволокна, открывающие
реконструктивной медицины.
новые
возможности
для
Пористые материалы для применения в самых различных областях
от медицины до промышленной очистки и фильтрации.
Легкие,
прочные
материалы
аэрокосмической промышленности.
для
автомобильной
и
Керамические нанокомпозиты для промышленных и медицинских
применений.
Материалы нанофотоники и оптоэлектроники с улучшенными
эксплуатационными
и
потребительскими
свойствами
для
телекоммуникационных приложений.
Новые потребительские свойства, которые могут быть обеспечены
такими материалами, включают в себя следующие:
предельно точно контролируемые оптические, электрические,
химические и механические свойства за счет изменения таких
параметров наночастиц, как размеры, форма, морфология;
мультифункциональность – объединение различных свойств в
одном материале за счет использования технологий контролируемой
сборки наноразмерных строительных блоков;
высокая химическая чистота и качество,
спецификой процесса синтеза наноматериалов.
обусловленные
Появление
коммерчески
эффективных
структурированных на наноуровне, приведет к:
материалов,
формированию новых принципов конструирования устройств,
позволяющих создавать материал под конкретную задачу;
9
существенному расширению существующей базы материалов,
появлению целых классов принципиально новых материалов;
улучшению потребительских свойств уже существующих
материалов (например, суперлегкие материалы для аэрокосмического
приборостроения).
Технологии синтеза частиц нанометрового масштаба в
достаточной степени развиты. Но все они, как правило, не выходят за
рамки лабораторий по причине высокой себестоимости процесса
синтеза. Постоянный поиск более дешевых решений, которые позволят
лучше контролировать размеры и форму синтезируемых наночастиц,
продолжается. Также есть масса научных публикаций и патентов,
посвященных получению материалов путем самосборки наночастиц.
Однако все эти материалы являются лабораторными образцами с
трудновоспроизводимыми параметрами.
В качестве основных и приоритетных областей применения новых
наноматериалов можно назвать:
Энергетический сектор. Здесь новые материалы помогут
оптимизировать энергозатраты и повысить энергоэффективность с
помощью использования возобновляемых источников энергии и
улучшения термоизоляции.
«Зеленые» технологии. Использование материалов на основе
пространственно изолированных наночастиц («мембранных»
материалов) позволит создать технологии очистки воды и воздуха.
Автомобильное и аэрокосмическое производство. Здесь
ключевыми параметрами, которые можно будет улучшить с помощью
использования нанотехнологий, являются вес, прочность и
износостойкость материала.
Биомедицинский сектор. Наноструктурированные объекты
должны лечь в основу новых систем терапии и диагностики
персональной медицины.
10
В2. Гибридные материалы на основе проводящих полимеров и
наночастиц для производства солнечных батарей
Гибридные материалы на основе проводящих полимеров и
полупроводниковых наночастиц рассматриваются в настоящее время в
качестве перспективных для создания функциональных элементов
солнечных батарей (устройств, преобразующего энергию фотонов
солнечного света в электрическую энергию). Технологии синтеза
полупроводниковых наночастиц и технологии создания проводящих
полимеров развивались независимо вплоть до появления в начале 1990-х
годов первых публикаций, посвященных получению гибридных систем
из неорганических наночастиц и проводящих полимеров. Впоследствии,
это привело к созданию целого класса гибридных материалов,
нашедших применение в солнечных батареях, электронике,
электрохромных
и
фотохромных
компонентах,
сенсорах
и
хемирезистивных датчиках, более известных как «искусственные носы».
Гибридные материалы, о которых идет речь, представляют собой
композит из неорганических наночастиц, диспергированных в
органическую матрицу из проводящего полимера. В этой системе
каждый компонент сохраняет свою структуру, и, вместе с тем,
межфазовое взаимодействие, происходящее между компонентами в
нанометровом масштабе, приводит к возникновению оптических и
электрических свойств композита, отсутствующих у отдельных
компонент. Существующие подходы к формированию гибридных
материалов на основе неорганических наночастиц и проводящих
полимеров значительно разнятся в зависимости от предполагаемой
области
применения
материала
и
конкретной
пары
наночастица/проводящий полимер. Эти подходы могут быть
классифицированы и разделены на несколько основных групп.
Смешивание исходных компонентов. Гибридные материалы
могут быть получены смешиванием немодифицированных компонент:
коллоидного раствора наночастиц и проводящего полимера. Этот
процесс приводит к фазовому разделению на микрометровом уровне,
что существенно снижает качество получаемого материала. Причины
фазового разделения следующие:
а) наночастицы, исходно покрытые молекулами лигандов, при удалении
растворителя формируют плотноупакованные агломераты;
б) отвердение сопряженного полимера приводит к образованию
полукристаллической структуры, в которой кристаллизовавшиеся
области отделены от аморфных.
11
Для преодоления этих проблем в данном подходе используются
чисто физические методы, такие как обработка ультразвуком и
использование внешнего электрического поля. Этот подход является
наименее эффективным из используемых в настоящее время. Более
эффективен подход, использующий химические методы, и в частности:
Покрытие
поверхности
наночастиц
сопряженными
макромолекулами, содержащими якорные группы. Наиболее
универсальным методом создания гибридных материалов на основе
сопряженных полимеров и наночастиц является функционализация
сопряженных макромолекул полимера якорными группами, способными
связываться с поверхностью наночастиц. Существует масса вариаций
данного метода, различия между которыми обусловлены структурой
лиганда. Схематически различные пути приготовления гибридных
материалов с использованием сопряженных молекул, содержащих
якорные группы представлены на рисунке В2.
Рис. В2. Схематическое представление различных стратегий
приготовления гибридных материалов с использованием сопряженных
молекул, содержащих якорные группы [2].
Следует
отметить,
что
сохранение
макромолекулярной
регулярности полимера является основным требованием в данном
подходе. В этом смысле использование олигомеров в качестве лигандов
является
предпочтительным,
поскольку
позволяет
сохранить
регулярную структуру в наибольшей степени. Степень упорядоченности
получаемых структур является критическим фактором для всех областей
применения данных гибридных материалов поскольку ухудшение
12
регулярности материала приводит к значительному снижению
мобильности носителей заряда и ухудшению других электрических
параметров материала.
Ковалентное связывание сопряженных макромолекул и
наночастиц, покрытых функционализированными лигандами.
Ковалентное связывание макромолекул сопряженного полимера с
поверхностью
наночастицы
осуществляется
с
помощью
бифункциональных
линковщиков.
Такие
лиганды-линковщики
способны образовывать связи как с молекулой полимера, так и с
поверхностью наночастицы. Здесь, также как и в предыдущем методе,
существует масса вариаций, основные из которых показаны на рисунке
В3.
Рис. В3. Схематическое представление
приготовления
гибридных
материалов
бифункциональных лигандов [2].
различных стратегий
с
использованием
В общем случае можно выделить следующие этапы формирования
гибридного материала:
а) исходные лиганды наночастиц заменяются на бифункциональные
лиганды, описанные выше;
б) выполняется реакция ковалентного связывания макромолекул
полимера и функционализированных наночастиц.
13
Здесь, опять же, ключевым параметром является сохранение
регулярной структуры синтезируемого материала.
Приготовление гибридных материалов через нековалентное
взаимодействие. Как уже отмечалось выше, основным препятствием
при создании гибридных материалов на основе сопряженных полимеров
и наночастиц является фазовое разделение. Однако этот феномен также
может быть использован для синтеза регулярной структуры. В основе
данного подхода лежит специфическое взаимодействие между
поверхностными лигандами наночастиц и молекулами полимера –
молекулярное
распознавание.
Схематически
этот
подход
иллюстрируется на рисунке В3. От обычного образования связи между
молекулами этот процесс отличается селективностью. Молекулярное
распознавание основывается на наличии у молекулы-рецептора участка,
обладающего высоким сродством с молекулой-лигандом. Для этого
рецептор и лиганд должны структурно и энергетически соответствовать
друг другу. Основная сложность при использовании данного метода
заключается в подборе соответствующих пар лиганд/полимер. Именно
поэтому он не получил значительного распространения.
Выращивание нанокристаллов непосредственно в полимерной
матрице. Этот подход существенно отличается от изложенных выше и
представляет собой альтернативный путь преодоления проблемы
фазового разделения. В основе его лежит подбор общего растворителя
для наночастиц и сопряженного полимера. В ходе синтеза гибридного
материала прекурсор, используемый для последующего роста
наночастиц, растворяется в заранее приготовленном растворе полимера.
После чего осуществляется реакция синтеза наночастиц, причем рост
кристаллов происходит непосредственно в полимерной матрице, а
макромолекулярные цепи используются для стабилизации и пассивации
растущих наночастиц.
14
В3. Мультифункциональные системы наночастиц для доставки
лекарств и диагностики
Нанотехнология и наномедицина в частности имеют дело с
объектами нанометрового масштаба. Все процессы в клеточном
организме также происходят в нанометровом или в субнанометровом
масштабе. Это позволяет создать объекты, достаточно маленькие, чтобы
взаимодействовать с клетками напрямую.
Живая клетка может быть рассмотрена как многофункциональный
объект, состоящий из набора функциональных элементов. Воздействуя
на каждый из этих элементов специфическим образом, можно, в
конечном счете, влиять на состояние организма в целом. Именно на этом
принципе основаны все мультифункциональные системы наночастиц
или «лаборатории на чипе».
Рис. В4. Мультифункциональная наночастица. Как правило, включает в
себя специфические метки (обычно антитела или пептиды), контрастные
агенты для формирования изображений (флюоресцирующие или
магнитные наночастицы), якорные агенты для фиксации на поверхности
клетки, молекулы биосовместимого полимера для стабилизации,
биоактивные агенты, запускающие специфические функции клетки и
молекулы лекарственного средства [3].
Такая частица представляет собой гибридный объект, состоящий
из органических и неорганических элементов, помещаемый
непосредственно в организм (см. рис. В4). По кровеносной или
лимфатической системе она доставляется непосредственно к нужным
клеткам и с помощью якорных агентов закрепляется на их поверхности.
Таким образом, формируется био-нано-интерфейс, с помощью которого
происходит
взаимодействие
мультифункциональной
системы
наночастиц и живой клетки. В ходе этого процесса задействуются
15
естественные механизмы: каждая клетка имеет специальные каналы, с
помощью которых она обменивается веществами с окружающей средой.
Через эти же каналы наносистема взаимодействует с клеткой.
Подходы к созданию мультифункциональных систем наночастиц
очень сильно разнятся в зависимости от функций конечного объекта.
Посредством связывания различных молекул и наночастиц можно
комбинировать необходимые функции. Однако, на пути создания таких
по-настоящему мультифункциональных наносистем стоят препятствия,
прежде всего, химического свойства. Чтобы получить стабильный
объект, необходимо использовать ковалентное связывание. Однако в
большинстве случаев этого очень трудно добиться. В созданных на
сегодняшний день бифункциональных наносистем функциональность,
как правило, достигается при помощи использования нековалентных
взаимодействий, таких как электростатическое или специфические
взаимодействия биомолекул.
Основным результатом развития бионанотехнологий станет
создание так называемой «лаборатории на чипе», функции которой
будут существенно модифицироваться в зависимости от данной
конкретной задачи:
Система доставки лекарств. Молекулы лекарственного средства
будут связываться с системой наночастиц и в таком виде доставляться
по основным транспортным артериям организма (кровеносной и
лимфатической системам) к нужным клеткам.
Биосенсор. Молекулы-метки
доставляться к нужным клеткам.
аналогичным
образом
будут
Биоактивный агент. Такие молекулы, взаимодействуя с нужной
клеткой, будут запускать специфические процессы в организме.
Контрастный агент. Введение таких частиц позволит улучшить
качество получаемых изображений. Использование таких систем в
совокупности приведет к объединению в один единый процесс терапии
таких медицинских процедур как детектирование источника
заболевания, соответствующее изменение функций организма, лечение и
передача информации о процессах, происходящих в организме.
16
В4. Наноуглеродные материалы для микроэлектроники и фотоники
Углеродные наноструктуры могут быть определены как
углеродные материалы, характерные размеры и структура которых
находятся в нанометровом масштабе. Углеродные материалы
представляют весь спектр размерностей от нульмерных (фуллерены),
включая одномерные (углеродные нанотрубки), двумерные (графен), и
заканчивая трехмерными структурами (нанокристаллические алмазные
пленки, фуллериты). Такое многообразие аллотропов возможно
благодаря
удивительной
способности
углерода
менять
гибридизациюсвоих электронных орбиталей от sp2 до sp3.
История наноуглерода начинается с открытия в 1985 году одного
из его аллотропов – фуллерена (Крото, Крел и Смолли). В 1991 году
были изготовлены и охарактеризованы углеродные нанотрубки
(Иидзима). Хотя эти объекты были известны и ранее, но только с
появлением
соответствующих
средств
ведения
эксперимента
углеродные нанотрубки заняли свое место в ряду наноструктур. После
этого в 2004 был найден метод получения графена (Гейм и Новоселов).
Наиболее перспективными для практического применения из всего ряда
аллотропов углерода представляются углеродные нанотрубки и графен
(см. рис. В5).
Рис. В5. Аллотропы углерода [4].
Графен представляет собой двумерную плоскость из атомов
углерода толщиной в один атом. Это материал, по сути, представляет
одну плоскость объемного графита. Графен обладает целым рядом
уникальных свойств. Например, его механические (низкий вес, высокая
жесткость) и термические (теплопроводность) свойства делают графен
перспективным материалом для использования в качестве различных
покрытий. Также графен обладает удивительно интенсивным для
одноатомного слоя поглощением. Но благодаря эффекту насыщения
графеновые плоскости в определенных условиях становятся оптически
прозрачными. Эти свойства в совокупности обеспечивают возможность
использования графена сразу в нескольких классах электрооптических
устройств. Кроме того, электрические свойства графена также в высшей
степени примечательны. Это материал обладает высокой подвижностью
17
носителей зарядов (самой высокой из известных к настоящему времени
материалов), что открывает возможности для его использования в
качестве наноразмерного транзистора – основного функционального
элемента многих электронных устройств.
Основными способами производства графена являются:
Микромеханическое отщепление
одноатомных плоскостей от
высокоориентированного пиролитического графита (HOPG). Это
исторически первый метод получения графена. С его использованием до
сегодняшнего дня получают образцы самого высокого качества. Однако,
этот способ не может быть внедрен в промышленное производство.
Эпитаксиальный рост на подложках из карбида кремния. Этот метод
позволяет получать высококачественные образцы, свойства которых
находятся в сильной зависимости от подложки. Такие образцы являются
наиболее
подходящими
для
использования
традиционных
литографических методов производства.
Углеродные нанотрубки. Углеродная нанотрубка представляет
собой полую цилиндрическую наноструктуру из одной или нескольких
гексагональных графитовых плоскостей, по крайней мере с одной
стороны ограниченных полусферой. Эти структуры могут быть очень
протяженными, соотношение длины к диаметру может достигать
значения 132000000:1, что значительно превышает аналогичные
значения для одномерных наноструктур из других материалов.
Углеродным нанотрубкам присущи экстраординарно высокие значения
упругости, жесткости и теплопроводности. Это позволяет использовать
нанотрубки в качестве нановолокон для модификации свойств
традиционных материалов.
Электронные свойства углеродных нанотрубок могут в
значительной степени варьироваться: в зависимости от направления, в
котором была свернута графитовая плоскость, нанотрубка может
демонстрировать
диэлектрические,
полупроводниковые
и
металлические свойства. При этом металлические и полупроводниковые
модификации
обладают
высокой
подвижностью
заряда
и
сверхпроводимостью. В случае, когда материал нанотрубки
соответствует полупроводнику, эта наноструктура может быть
использована в качестве функционального элемента в большом
количестве полупроводниковых устройств.
Основные методы получения углеродных нанотрубок:
Высокотемпературное
электродуговое
испарение
графитовых
стержней. Это исторически первый метод получения нанотрубок. Этот
метод энергозатратен, материалоёмок и дает малый выход конечного
продукта. Однако он же является и самым универсальным: с его
помощью можно получать нанотрубки всех известных типов.
Метод
каталитического
пиролиза
углеводородов
(ChemicalVaporDeposition, CVD). Это первый низкотемпературный
способ получения нанотрубок. При использовании наночастиц металлов
18
в качестве зародышей роста выход конечного продукта увеличивается
до 70-90%.
Существует масса потенциальных применений гибридных
материалов на основе углерода. Все они так или иначе связаны с
уникальными оптическими, электрическими и механическими
свойствами углерода.
Полевые транзисторы. Углеродные нанотрубки и графен могут быть
использованы в качестве функционального элемента полевого
транзистора. Основным технологическим решением здесь является
создание эффективного затвора, так как в углеродных материалах очень
велик ток утечки.
Гибкие дисплеи. Благодаря своей оптической прозрачности графен
может быть использован при проектировании гибких дисплеев с
использованием гибридных материалов на основе графен-полимерных
композитов.
Сенсоры. Углеродные нанотрубки могут применяться в качестве
высокоэффективных сенсоров. Применение их в медицине ограничено
токсичностью.
Суперконденсаторы и батареи. В устройствах хранения энергии
используются высокая проводимость углеродных нанотрубок и их
большая площадь поверхности. Это обеспечивает высокую
эффективность такого рода устройств.
Солнечные батареи. Углеродные нанотрубки используются в качестве
высокоэффективного заменителя традиционного токопроводящего
покрытия ITO.
Светодиоды.
Фотолюминесцентные
свойства
одностеночных
углеродных нанотрубок находят применение при проектировании
светодиодов.
В5. Функционализированные нанопористые мембраны для очистки
питьевых, бытовых и сточных вод
В настоящий момент в водоочистке используются 4 типа мембран:
для микрофильтрации, ультрафильтрации, нанофильтрации и обратного
осмоса. Для нанофильтрации используются наноструктурированные
мембраны (nanostructured membrane (NSM)). Здесь важно понимать
терминологию: термин «нано» используется в водоочистке с 80-х годов
ХХ века. Однако он не соответствует термину «нано», используемому в
нанотехнологии.
Он
соотносится
только
с
размерами
отфильтровываемых частиц. Размеры эти находятся в диапазоне
нескольких нанометров. Из-за возникшего несоответствия новое
поколение мембран, функционализированных наночастицами, получило
название «усовершенствованные мембраны» (nanoenhancedmembranes).
Существует
большое
количество
разработок
мембран,
модифицированных различными типами наночастиц. Однако, эксперты
выделяют три наиболее перспективных направления:
19
мембраны, функционализированные TiO2;
мембраны, функционализированные серебряными наночастицами;
мембраны, функционализированные углеродными нанотрубками.
Полимерные мембраны. Для получения мембраны на полимерной
подложке, функционализированный полиамидный слой наносится на
слой пористого полисульфона. Есть несколько способов получения
наноструктурированного композита на поверхности полисульфона:
наночастицы наносятся непосредственно на поверхность полиамида,
либо синтезируются непосредственно с использованием полиамида в
качестве прекурсора. В этой технологии применяются серебряные
наночастицы, цеолиты и арсенид натрия. Попытки использования в
качестве функционализирующего агента TiO2 успеха не принесли, так
как вследствие фотоактивности TiO2 полимерная мембрана разрушается.
Полимерные
мембраны,
функционализированные
серебряными
наночастицами обладают выраженным бактерицидным эффектом и
являются
приоритетным
направлением,
несмотря
на
ряд
технологических сложностей.
Металлические и керамические мембраны. Мембраны
получаются путем осаждения на подложку из металла или керамики
слоя наночастиц, которые затем отжигаются при высоких температурах
для получения прочного связывания. Наиболее перспективными в этом
классе являются мембраны с TiO2 покрытием. Такие мембраны
сохраняют устойчивость к образованию биопленок. Это позволяет
повысить эффективность водоочистки и снизить частоту замены
мембран.
Мембраны с углеродными нанотрубками. В отдельный класс
могут быть выделены мембраны с интегрированными в их поры
углеродными нанотрубками. Процесс производства такой мембраны
проходит в два этапа. Сначала углеродные нанотрубки упорядочивают в
вертикальном направлении. После этого зазоры между наночастицами
заполняются нитридом кремния. Потенциально такая технология может
обеспечить беспрецедентное качество очистки воды, совмещенное с
высокой эффективностью и низкой стоимостью. Однако, поскольку
направление это только начинает развиваться, есть целый ряд
нерешенных проблем. В частности, имеется ряд исследований,
содержащих данные о токсичности нанотрубок для живых организмов.
Место оптических методов в физике наноструктур
Оптические методы традиционно, занимают значительное место
как при исследовании, так и при производстве различных материалов, в
20
силу их относительной простоты и высокой информативности. Не
являются исключением также исключением также и наноматериалы,
включая гибридные структуры. Данный курс охватывает основы
оптической молекулярной спектроскопии в УФ и видимой областях
спектра и основы ее применения для исследования сложных молекул,
включая сложные многомолекулярные объекты.В качестве объектов
рассматриваются молекулы и квантовые точки. Рассматриваются
переходы как вверх, так и вниз, т.е. спектры поглощения и испускания.
Спектры испускания – это спектры люминесценции. Люминесценция и
ее закономерности также рассматриваются в полном объеме.Следует
заметить, что именно в применении к гибридным наноообъектам методы
спектроскопии являются незаменимыми, т.к. дают принципиально
важную информацию о взаимодействии молекул между собой или с
объектами другой природы.
Рассмотрим
подробнее
задачи,
решаемые
оптической
спектроскопией в УФ, видимой и ИК областях спектра. Этих задач,
вообще говоря, 5:
(а) задачи количественного анализа;
(б) задачи качественного анализа, или идентификации (как
известных, так и неизвестных веществ);
(в) структура вещества по спектру (структурный анализ);
(г) источник информации о фотофизических характеристиках
соединения;
(д) источник информации о межмолекулярных взаимодействиях
(молекула через свой спектр дает информацию о своем
окружении).
Рассмотрим, как эти задачи решаются теми тремя видами
спектров, которые мы будем рассматривать.
Спектроскопия поглощения в области электронных переходов.
(а) – да, до 10-5М,
(б) – да, но широкие полосы, поэтому ограниченные возможности.
(в) – кое-какие выводы иногда можно сделать, но это не самый
лучший подход к решению таких задач.
(г) - ограниченные результаты можно получить, т.е. это только
переходы вверх.
(д) – электронные спектры очень чувствительны к межмолекулярным
взаимодействиям, причем нередко они хорошо интерпретируются.
Люминесценция.
21
(а) – да, до 10-7М,
(б) да, возможности шире, чем у спектров поглощения, т.к. есть еще
одна степень свободы - длина волны возбуждения.
(в) как и в случае спектров поглощения.
(г) – да, возможности шире, чем в случае спектров поглощения, т.к.
переходы вниз.
(д) как и в случае спектров поглощения, но может быть
дополнительная информация. Недостатки метода: не все вещества
люминесцируют, труднее аппаратурно.
ИК спектроскопия.
(а) да, но выше порог обнаружимости, т.к. поглощение слабее.
(б) да, лучше, чем электронные спектры, т.к. спектры более
характеристичны.
(в) это главное применение.
(г) практически нет.
(д) очень ограниченные возможности (напр., водородная связь).
Достоинства метода: узкие полосы, большая информативность.
Недостатки: (i) малая поглощательная способность, необходимость
иметь большие количества вещества; (ii) невозможность работы с
растворами, необходимость работы в безводных средах; (iii) трудность
приготовления образцов.
22
1. Законы поглощения света; электронно-колебательные спектры
многоатомных молекул
1.1. Закон поглощения света (закон Бугера-Ламберта-Бера).
Физические величины, характеризующие поглощение света атомномолекулярными системами
В подавляющем большинстве случаев воспринимаемый нами цвет
окрашенных предметов есть результат избирательного поглощения
веществом определенных цветов в непрерывном спектре. Спектр
поглощения и есть объективная характеристика поглощательной
способности вещества по отношению к свету различных длин волн.
Выведем закон поглощения света (рис.1.1).
Рис.1.1. К выводу закона поглощения света
Полагаем: площадь объекта = 1 см2, а длина оптического
пути=dx.Интенсивность I(энергетическая), или плотность потока
мощности, это энергетическая величина, измеряемая в Вт/см2 (или
эрг/см2 с),безотносительно к длинам волн. (Поэтому это не то же самое,
что световой поток.)
Основное допущение здесь состоит в том, что относительное
ослабление потока мощности зависит только от числа молекул, но не
зависит от величины этого потока:
dI
I
kndx , где k – молекулярный
коэффициент поглощения [см2], n – концентрация (в см-3).
Закон Бугера-Ламберта: каждый тонкий слой внутри однородной
окрашенной среды поглощает определенную долю входящего в него
потока мощности. Закон Бэра: Поглощение тонким слоем
пропорционально числу окрашенных молекул, в нем содержащихся, т.е.
их концентрации.
23
Другая формулировка закона поглощения света (закона БугераЛамберта-Бера), эквивалентная приведенной выше дифференциальной
формулировке:
(1.1)
I l I 0 e knl
Некоторые часто встречающиеся величины.
1) Пропускание:
Il
T
I0
2) Оптическая плотность:
I
1
D lg 0 lg
Il
T
Другие, более распространенные формулировки закона БугераЛамберта-Бера:
I l I 010 D
Il
10 cl
D cl ,
I0
где c – концентрация в молях на литр.
экстинкции.
lg e knl
cl , lg e
n
c
0.43 ,
0.43 6.06 10 20 k , k
3.8 10
21
(1.2)
(1.3)
- молярный коэффициент
NA
10 3
6.06 10 20
(1.4)
(1.5)
1000
, где M – молекулярный
M
вес. Тогда концентрация – должна быть не в моль/л, а в г/см3.
Размерность: [M-1см-1].(M обозначает «моль/л»).
Спектр поглощения - это зависимость молярного коэффициента
экстинкции от длины волны: ( ) (рис.1.2):
Иногда применяют величину
уд .
24
Рис.1.2. Типичная полоса поглощения
Это типичный спектр. Полоса поглощения характеризуется
положением максимума поглощения и полушириной полосы. Нужно
сделать три оговорки. (1) Так изображают спектры, когда имеется
чистое вещество, для которого можно приготовить раствор известной
концентрации. Однако, в большинстве случаев концентрация не
известна. Тогда вместо строится зависимость D от . (2) В справочной
литературе часто вместо приводят lg . (3) Вместо может быть ~ в см1
10 7
1 ~
. [см 1 ]
, в соответствии с общей формулой: ~
.
[нм ]
1.2. Аналитические применения спектров поглощения
Спектры
поглощения
с
успехом
используются
для
количественного и качественного анализа исследуемых структур.
Применения для количественного анализа, т.е. определение
концентраций известного вещества, основано на использовании
соотношения (1.3). Чувствительность метода обычно находится в
пределах 10-7М. Существенным недостатком метода является
невозможность
регистрации
спектров
поглощения
объектов,
расположенных на непрозрачных подложках или внедренных в
непрозрачные среды.
1.3. Основные случаи нарушения закона Бугера-Ламберта-Бера,
“эффект сита”
(1) Неоднородные среды и рассеивающие среды. Это, вообще говоря,
не одно и то же. В общем случае имеем три составляющих
светового потока, проходящего сквозь образец: (a) поток,
25
проходящий сквозь образец без изменения направления, НЕ
прошедший сквозь частицу; (б) поток, прошедший сквозь образец,
прошедший сквозь частицу, но не изменивший направления; (в)
поток, изменивший направление из-за многократных отражений от
частиц.
Рис.1.3. Три составляющие светового потока, проходящего через
пространственно-неоднородный рассеивающий образец.
Одним из характерных проявлений неоднородности среды
является “эффект сита”: при малых плотностях поглощение
уменьшается в определенное число раз, а при больших – происходит
запределивание измеряемой оптической плотности на определенной
величине (рис.1.3).
26
Рис.1.4. Искажение спектра поглощения вследствие «эффекта сита».
Формулы для учета “эффекта сита”.
I l( набл.) I 0
I0
(1.6)
Dcorr.
lg(10
10
D
cl
10
a
D
)
I0
I0
10
Il
I0
D
D
Dcorr .
lg
Il
I0
a 10
a
cl
10
Dcorr.
(1.7)
Здесь I 0 - составляющая интенсивности, проходящая сквозь образец
без поглощения, a – относительная доля этой интенсивности, смысл
остальных величин ясен из приведенных формул.
(2) Применение интегрирующей сферы.
В случае применения интегрирующей сферы проходящий (и,
следовательно, измеряемый) свет содержит в себе значительную
часть составляющей 3 (рис.1.3). Эффект сита при этом не
устраняется.
(3) Учет рассеяния как дополнительного ослабляющего фактора.
Пусть ослабление эквивалентно поглощению с оптической
I
плотности b( ), т.е.: l 10 ecl b ( ) . Вклад рассеяния можно легко
I0
учесть, если в качестве образца сравнения взять образец с таким
27
же рассеянием, но без поглощения: I 0 10 b( ) . Тогда имеем:
Il
10 cl .
I0
(4) Диффузное отражение. Разбавитель.
Это метод измерения спектров поглощения на основе измерения
не прошедшего света, а диффузно-отраженного света. Образец
должен быть в виде непрозрачного порошка. Метод основан на
разбавлении измеряемого порошка разбавителем – «белым»
порошком, не обладающим селективным поглощением в
исследуемом спектральном диапазоне. Измеряется величина:
(r)
I смеси
R
. В этом случае может быть применена формула Кубелки(r)
I разб
.
Мунка:
(1 R)2
2R
2.3 c
,
s
(1.8.)
где s – константа рассеяния окрашенного вещества – доля света,
рассеянного его (вещества) слоем единичной толщины – как
предполагается, не зависит от .
Логарифмическая формулировка:
lg
(1 R) 2
2R
lg
lg c const
(1.9)
(5) Агрегация.
Приводит к тому, что концентрационная зависимость оптической
плотности становится отличной от (1.3).
(6) Зависимость от I.
Молекулярный коэффициент поглощения (k) по своему
физическому смыслу есть не что иное, как молекулярное сечение
поглощения. Реально имеем переходы как прямые (с поглощением
кванта света), так и обратные:
h
A
A*
A*
A
Если интенсивность такова, что число переходов вверх за единицу
времени соизмеримо с константой скорости обратных переходов,
то будет несоблюдение закона Бугера-Ламберта-Бера, т.к. реально
в растворе имеется какой-то процент молекул в возбужденном
28
состоянии,
которое
характеризуется
коэффициента экстинкции.
другим
значением
1.4. Полосы поглощения и колебательная структура электронных
переходов
Основные фотофизические процессы: поглощение и испускание
света:
A h
a
A*
A h
A*
(1.10)
f
Электронные переходы всегда сопровождаются изменением
других степеней свободы колебательных, вращательных. Времена
колебательных релаксаций составляют 10-12-10-13с. Что означает
фактически (физически) полоса поглощения? Ее можно рассматривать
как зависимость вероятности перехода от величины колебательного
кванта (сверх энергии чисто электронного перехода). Почему имеет
место практически сплошное поглощение, т.е. нет дискретных линий,
как в спектрах атомов? Это тоже связано с наличием колебательных
степеней свободы.
Рис.1.5. Схема энергетических уровней молекулы, поясняющая
происхождение колебательной структуры в электронных спектрах.
29
1.5. Основные физические закономерности, обусловливающие
активность колебаний в электронных спектрах, принцип ФранкаКондона
Будем рассматривать электронно-колебательные состояния. Как
выглядит электронно-колебательный спектр в благоприятном случае,
т.е. когда колебательная структура разрешена (см. рис.1.6).
Рис.1.6. Схема спектра поглощения, содержащего три электронных
перехода с разрешенной колебательной структурой.
В случае нескольких электронных переходов критерии выделения
групп полос, соответствующих разным электронным переходам, состоят
в следующем: (1) относительная обособленность групп полос, (2)
наличие закономерности распределения интенсивностей (огибающая с
одним максимумом) внутри группы.
Наиболее часто колебательная структура электронного перехода
образует
прогрессию,
т.е.
приблизительно
эквидистантную
последовательность полос 0-0, 0-1, 0-2 и т.д. В некоторых случаях видна
так называемая «горячая» полоса 1-0 Она может быть выявлена по
температурной зависимости, а также в силу того, что она обычно
существенно «выбивается из закономерности» распределения
интенсивностей внутри группы по сравнению с другими
колебательными полосами.
Что можно сказать о колебательных подуровнях?
1) Вообще, для основных частот молекулярных колебаний имеем:
~ 3400 см 1 . Но для колебаний, проявляющихся в электроннокол.
колебательных спектрах, обычно соблюдается: 100 см 1 кол. ~ 1600 см 1 .
30
Если кажется, что это первый колебательный подуровень, но он отстоит
от 0-0-перехода существенно более, чем на 1600 см-1, то это, скорее
всего, на самом деле, другой электронный переход.2) Переходы 1-1, 2-2
и т.п. налагаются друг на друга. В случае неточного равенства частот
колебаний в основном и возбужденном состояниях образуются так
называемые “секвенции”. Но они бывают видны относительно редко.
Рис.1.7. Схема, поясняющая, какие
проявляются в электронных спектрах.
колебательные
переходы
3) Какие колебания активны в электронных спектрах? Это не трудно
понять из картины адиабатических кривых (рис.1.8).
31
Рис.1.8. Схема, поясняющая франк-кондоновский механизм активизации
колебаний в электронном спектре.
Принцип Франка-Кондона утверждает, что электронный переход
совершается при неподвижных ядрах. Но это не очень строгая
формулировка. Рассмотрим двухатомную молекулу. В основном
состоянии колебательные состояния не заселены. Если кривые не
смещены, то есть только 0-0 переход (в силу правил отбора и
ортогональности волновых функций). Если кривые смещены, то
подуровни появляются тем более, чем более смещены кривые. Кроме
смещения может быть еще изменение силовой постоянной. Но это
обычно не столь существенно. Это означает, что проявляются в спектрах
только такие колебания, для которых меняется положение равновесия.
Для сложных молекул ситуация вполне аналогична рассмотренной.
Таких колебаний обычно немного: доминируют 1-3 колебания, но не
более. Нетрудно сообразить, что если симметрия молекулы – одна и та
же в основном и возбужденном состояниях, то по данному механизму
могут активизироваться только полносимметричные колебания.
32
Рис.1.9. Типичная полоса поглощения с разрешенной колебательной
структурой при наличии прогрессии.
При наличии прогрессии имеем:
. Из-за ангармонизма, как
кол.
правило, при продвижении в сторону высоких частот расстояния между
частотами, составляющими прогрессию, уменьшается. Валентные
колебания с участием атомов водорода, как правило, не проявляются в
электронных спектрах.
Если наблюдается структура в спектре поглощения, то надо
сначала выяснить, есть ли это колебательная структура. Это могут быть
и разные электронные переходы.
Изложенный
механизм
активизации
полносимметричных
колебаний, называют франк-кондоновским механизмом. Однако,
существует
специфический
механизм
активизации
неполносимметричных колебаний в спектрах, это так называемый
герцберг-теллеровский механизм. Он работает тогда, когда в спектре
сосуществуют слабые (запрещенные по симметрии) переходы и
переходы, имеющие высокую интенсивность.
33
2. Основы теории электронных спектров многоатомных молекул;
электронные переходы основных классов органических соединений
2.1.Основные характеристики электронных состояний:
мультиплетность, дипольный момент перехода, сила осциллятора
Уравнение Шредингера для многоэлектронных систем. Система,
содержащая n электронов и N ядер, в квантовой механике описывается
волновой функцией:
.
(
r
,
r
,...,
r
,
R
,
R
,...,
R
en
en 1 2
n
1
2
N)
Если мы интересуемся стационарными состояниями, то эта функция
должна быть решением уравнения Шредингера:
(2.1)
H en E en ,
где H Te Tn V , V Vee Ven Vnn ,
Существенно, что решениями уравнения Шредингера является
набор собственных функций и собственных значений:
E0, E1, E2, …; Ψ0, Ψ1, Ψ2, ….
Если это изобразить на диаграмме (схеме уровней), то это будет
выглядеть следующим образом (рис.2.1):
Рис.2.1. Схема электронных энергетических уровней многоэлектронной
атомно-молекулярной системы.
Приближение
Борна-Оппенгеймера
(адиабатическое
приближение). Полная волновая функция в этом приближении ищется в
виде:
en
e
( ri , R )
n
(R )
(2.2)
где в первом сомножителе зависимость от ядерных координат – только
как от параметра, входящего в уравнение, решением которого является
эта функция. Следующая задача – получить уравнения, решениями
34
которых должны являться первый и второй сомножители. Это и было
сделано Борном и Оппенгеймером.
Первое уравнение имеет следующий вид:
Te Ven ( R ) Vee
e
Ee ( R )
(2.3)
e
Решением является собственные векторы e ( ri , R ) , имеющие слабую
зависимость от ядерных координат и собственные значения Ee Ee (R )
Имеющие тоже такую же параметрическую зависимость.
Второе уравнение имеет следующий вид:
Tn Vnn
Ee ( R )
n
En
(2.4)
n
Здесь существенно, что Ee (R ) входит слагаемым как добавка к
потенциалу. Если бы этой добавки не было, то потенциал был бы только
отталкивательным. Только благодаря этой добавке могут существовать
связанные состояния. Например, в случае двухатомной молекулы имеем
(рис.2.2):
Рис.2.2. Адиабатические кривые в случае двухатомной молекулы.
Молекулярные орбитали и одноэлектронное приближение.
Если у нас имеется система из n электронов (n=2m), то в
одноэлектронном приближении такая система описывается волновой
функцией:
35
1
(1) 1 (1) 2 (1) 2 (1)........
m
(1)
m
(1)
1 1 ( 2) 1 ( 2) 2 ( 2) 2 ( 2)...... m ( 2) m ( 2)
,
0
n! ....................................................
1 ( n ) 1 ( n ) 2 ( n ) 2 ( n )...... m ( n ) m ( n )
n
где m
и:
2
k (i )
k ( ri ) (i )
.
(i )
( ri ) (i )
k
k
(2.5)
(2.6)
Функцией (2.5) описывается состояние, в котором заполнены
низшие m одноэлектронных состояний, а все остальные остаются
незаполненными. Сокращенно эта функция обозначается так:
0
Функции
1
Fi
-
m
i
(
2
1
2
2
...
2
m
).
есть не что иное, как решения уравнения Хартри-Фока:
i
,
(2.7)
где i
( ri ) , индекс у Fi означает, что оператор действует на функцию
от координат i-го электрона.
Это означает, что для построения функции (2.6) для данной системы
решается задача на собственные функции и собственные значения
(вообще говоря, итерационно, т.к. Fi зависит от функций 1 … m ) и
n
берутся m
низших по энергии состояний.
2
Результатом
решения
является
определенное
число
одноэлектронных состояний (молекулярных орбиталей) с их энергиями,
Рис.2.3. Схема молекулярных
многоэлектронной системы.
орбиталей
36
основного
состояния
из которых могут быть построены детерминанты: как для основного
состояния, так и для различных возбужденных состояний системы:
Рис.2.4. Схема построения однодетерминантных возбужденных
состояний из молекулярных орбиталей основного состояния.
Здесь мы имеем тоже энергетические диаграммы, но для
одноэлектронных (орбитальных) состояний.
Из детерминантных волновых функций можно построить
довольно простые линейные комбинации типа суммы или разности,
которые будут характеризоваться некоторым определенным значением
полного спина (рис.2.5-2.7):
Рис.2.5. Схема построения основного состояния, характеризующегося
полным спином S=0.
Энергии синглетных и триплетных состояний. По энергии
синглетное состояние (т.е. состояние с S=0) всегда оказывается
несколько выше, чем соответствующее ему триплетное состояние (т.е.
состояние с S=1). Рассмотрим еще раз общую схему электронных
состояний многоэлектронной системы (см. рис. 2.8).
Из рис.2.8 ясно, в каком взаимоотношении между собой находятся
эти два типа схем уровней: схемы одноэлектронных уровней и общей
энергетической схемы многоэлектронной системы.
37
На рисунке 2.7. приведена схема построения возбужденных состояний,
характеризующихся значениями полного спина S=1 (Sz=-1 и Sz=1).
Рис.2.7.
Схема
построения
возбужденных
состояний,
характеризующихся значениями полного спина S=1 (Sz=-1 и Sz=1).
38
На рисунке 2.8 показана общая схема энергетических состояний
многоэлектронной системы.
Рис.2.8. Общая схема энергетических состояний многоэлектронной
системы.
Интенсивности электронных переходов. Для характеристики
интенсивности оптических переходов из основного состояния в i-е
возбужденное, определяют дипольный момент перехода:
D0
*
0
i
er i d
(2.8)
В спектроскопии применяется величина, называемая силой осциллятора,
которая, с одной стороны, может быть определена экспериментально из
измеренных спектров поглощения, с другой стороны выражается через
дипольный момент перехода:
f0
i
8 2m
D02 i ,
2
he
(2.9)
39
или в атомных единицах:
f0
2
8
i
D02
i
Вектор D0 i вычисляется по правилу Слэтера-Кондона для
одноэлектронных операторов, согласно которому: если
и
i
k
отличаются какой-то одной МО:
входит в i , а - в k , то имеем:
Dij
e
*
(1)r
(1)dr1
(2.10)
Поляризация перехода. Распишем дипольный момент перехода по
компонентам:
px
*
x
dv
py
*
y
dv
pz
*
z
dv
(2.11)
Зная симметрию молекулы и волновых функций, часто бывает можно
сразу сказать, какие из этих матричных элементов равны 0.
Из экспериментальных данных величина fможет быть определена
следующим образом:
f
3mc 9n02
k ( )d
e 2 ( n02 2) 2
9n02
1.3 10
(n02 2) 2
8
( ~ )d ~ (2.12)
2.2. Классификация электронных переходов плоских систем: ,
n-электроны,
*иn
* переходы
и
Классификация МО.
В
случае
плоских
систем
возможна
классификация
одноэлектронных волновых функций по типам симметрии таких систем
(рис.2.9):
40
Рис. 2.9. Классификация молекулярных орбиталей в случае
плоских систем
Случай плоской молекулы – это случай, когда имеется плоскость
симметрии, в которой находятся все или значительная часть атомов. В
этом случае все базисные АО и, в конечном счете, все МО (т.к. они
составлены из АО) могут быть разделены на 2 группы:
и
в
зависимости от того, симметрична или антисимметрична волновая
функция относительно отражения в плоскости. - это те, которые
симметричны. Они содержат АО: s, px, py.
- это те, которые
антисимметричны. Они содержат АО pz.
В этом случае, поскольку для компонент дипольного момента
перехода имеем выражения (2.11), часто можно сказать, какие из этих
*
матричных элементов равны 0. Так, для перехода
: pz=0, px,
*
*
*
py 0.Для переходов n
,
и
: pz 0. Если есть еще другие
элементы симметрии, то можно получить и более жесткие правила
отбора.
2.3. Понятие о хромофорных группировках
Хромофорные группы
Двойные связи, особенно, сопряженные (чередующиеся), играют
особую роль в электронных спектрах. Именно они обусловливают
переходы в УФ, видимой и ближней ИК областях. Без них, т.е. в случае
насыщенных соединений поглощение имеет место только в области
короче 200 нм. Примеры: вода, этанол, метанол, нормальные
углеводороды. Область 185-200 нм – это короткий УФ. Короче 185 нм –
вакуумный УФ – там своя специфическая спектроскопия –
41
спектроскопия так называемых ридберговских состояний. Мы их
рассматривать не будем.
Хромофоры – хотя и не очень четкое понятие, но весьма часто
используемое в спектроскопии. Это некоторая минимальная часть
молекулы, обусловливающая ее спектральные свойства. Если мы знаем,
что поглощение обусловлено -электронами, то можем заменить
молекулу более простой моделью, имеющей тот же хромофор. При
рассмотрении конкретных соединений мы будем это неоднократно
делать. Хромофор – это часть молекулы, внутри которой есть сопряжение, и которая не сопряжена с другими частями молекулы.
Очевидно, эта часть имеет плоское строение. У молекулы может быть
несколько хромофоров (хромофорных групп, группировок). Пример –
молекула белка, где хромофорные группировки, соответствующие
разным аминокислотным остаткам, изолированыдруг от друга (в том
смысле, что отсутствует общая система сопряжения).
Основные понятия и представления структурной химии. Химические
связи
Как образуется химическая cвязь? Например, на атомеC имеем
следующие атомные орбитали (AO): 1s2 – орбиталь «остова» –
двукратно заполненная, а валентную оболочку составляют АО: 2s, 2px,
2py, 2pz. Химическую связь образует пара электронов A и B : один от
одного атома, один – от другого. Как исключение, в некоторых случаях
могут быть и оба электрона с одного атома (так называемая
координационная связь). Но встает вопрос: что за функции
используются в качестве A и B ? В квантовой химии это выясняется
пост-фактум: то, какая комбинация АО оказывается максимально
вовлеченной в комбинацию с другим центром, выясняется в результате
решения квантов-химической задачи. Существенно, что эта комбинация
в значительной мере определяется локальной геометрией окружения
центра. Это и есть то, что называется гибридизованными АО. Тип
гибридизации (которых оказывается не очень много!) зависит от
окружения (локальной геометрии). Но локальная геометрия, в свою
очередь, определяется способом связывания. Для sp-элементов
различают три предельных случая гибридизации, которые мы сейчас
формально и рассмотрим.
42
1) sp3-гибридизация (тетраэдрическая)
метан: CH4:
2) sp2-гибридизация (треугольная)
3) sp-гибридизация (линейная)
Сопряжение и его критерии. С чего нужно начинать, когда надо
узнать структуру по структурной формуле молекулы? Прежде всего,
выявить связи и неподеленные пары электронов.
А) Сначала формально считаем сопряженными те атомы, которые
связаны чередующимися двойными связями: C=C C=C . Затем
смещаем связь на один атом: =C C=C C= и пытаемся замкнуть
формальную структуру. Если это удается, то оказывается, что локальные
системы связаны между собой, т.е. система - единая. Можно сразу
причислить к сопряженному фрагменту все атомы, которые связаны
между собой чередующимися двойными связями.
Б) Правило ароматичности: правило 4n+2. Замкнутый контур образует
сопряженную систему, если число электронов равно 4n+2. Примеры:
бензол, пиррол, нафталин, антрацен. Контрпримеры: циклооктатетраен,
циклобутадиен – не удовлетворяют данному правилу. У них сопряжение
отсутствует.
В) В качестве конкретных примеров можно рассмотреть: этилен, бензол,
ацетилен. У кетонов >C=Oна атоме кислорода 2 неподеленные пары, т.к.
это требуется условиями гибридизации sp2.
Рассмотрим случай пиррола:
NH
Атом азота поставляет в пи-систему 2 пи-электрона, и благодаря этому
правило ароматичности соблюдается.
43
Фуран:
O
изоэлектронен пирролу, но есть неподеленная пара электронов (только
одна!), которая, однако, находится низко и поэтому не проявляется в
спектре. Гибридизация азота- по-прежнему sp2.
1,4-диоксин:
O
O
содержит 8 электронов и поэтому
ароматичности. Поэтому здесь нет
кислорода – sp3.
Анилин – промежуточная ситуация.
не удовлетворяет правилу
сопряжения. Гибридизация
Ксантен:
H2N
O
NH2
Сопряжение через кислород сильно ослаблено из-за того, что для
контура с кислородом не выполняется правило ароматичности.
2.4. Этиленовый хромофор, линейные полиены.
Этилен
H2C=CH2
Полагаем, что
атома.
Орбитали:
1
(
2
1
*
(
2
A
A
- pz-АО 1-го атома углерода,
B
B
- такая же АО 2-го
)
(2.12)
A
B
)
44
Конфигурации:
{ 2}
0
1
1
{
2
{ *2 }
1
*}
1
(
2
*
* )
(2.13)
Рис.2.10. Схема, поясняющая происхождение электронного спектра
этилена
Рис.2.11. Схематическое изображение электронного спектра этилена
Переход в 2-кратно-возбужденное состояние запрещен. Но учет
конфигурационного взаимодействия (КВ) приводит к инверсии
переходов и вкладу разрешенной компоненты.
45
Бутадиен
Структурная
формула
H2C(A)=C(B)H C(C)H=C(D)H2
Орбитали:
1
( A
4
4
1
( A
3
4
1
( A
2
4
1
( A
1
4
B
C
D
)
B
C
D
)
имеет
следующий
вид:
(2.14)
B
C
D
)
B
C
D
)
Рис.2.12. Схема молекулярных орбиталей молекулы бутадиена
Энергия орбитали тем выше, чем большее число раз функция меняет
знак (чем больше число узловых плоскостей) (рис.2.12).
Конфигурации: ( 2
( 1
( 2
( 1
3)
3)
4)
4)
Моменты переходов между конфигурациями. Для его вычисления
удобно поместить начало координат в центр молекулы. В этом случае
легко получить:
T2 3 2 RA 2 RB r
T2
4
0
T1
3
2 RA
T1
4
0
2 RB
(2.15)
r
46
2.13. Схема энергетических уровней молекулы бутадиена
Имеем: E2 (
) E3 (
) . Поэтому учет КВ приводит к полному
перемешиванию данных волновых функций:
2
2
3
1
2
1
2
4
1
(
2
4
3
)
(
1
3
)
(2.16)
(
2
4
)
(
1
3
)
Моменты переходов между состояниями:
T ( S0
S1 )
T ( S0
S2 )
T ( S0
S3 )
r
r
2
r
2
(2.17)
Экспериментально имеем следующий спектр:
Рис.2.14. Экспериментальный спектр молекулы бутадиена.
47
Т.е. теоретическое рассмотрение имеет место качественное
согласие с экспериментом. Здесь мы впервые сталкиваемся с 4орбитальной моделью, т.е. с моделью, в рамках которой оказывается
достаточным рассмотрение только четырех орбиталей: двух из числа
высших заполненных и двух из числа низших вакантных.
2.5. Теория электронных спектров ароматических углеводородов,
четырехорбитальная модель, бензольный хромофор
Правило альтернантности
Ароматический углеводород называется альтернантным, если все его
атомы углерода могут быть разделены на 2 класса так, что атом одного
класса соседствует только с атомом другого класса (см. рис.2.15).
O
X
Рис. 2.15. Схема,
углеводородов.
X
O
O
X
X
O
поясняющая
O
X
X
O
O
X
определение
Основное свойство альтернантных УВ:
расположения заполненных и вакантных МО.
альтернантных
симметрии
взаимного
Рис.2.16. Схема молекулярных орбиталей альтернантного углеводорода
48
Конфигурации:
1
2
n 1
0
1
2
n
2
n 1
( p)
1
n
1
n 1
2
n
2
2
n 1
n
1
n 1
n 1
n 2
(2.18)
Состояния:
1 1
(
2
1 1
(
2
1
( )
1
( )
1
1
1
2
)
2
)
1
1
(2.19)
1
0
1
0
1
( p)
( p)
Рис.2.17. Схема, поясняющая происхождение энергетических уровней
молекулы бутадиена.
Моменты переходов:
В силу альтернантности имеем:
*
0
r 1dv
*
0
r
2
dv
R
Поэтому:
49
D0
D0
0
2
R
2
Таблица 2.1 Обозначения электронных состояний ароматических
углеводородов
Обозначения
Клара
Обозначения 1Lb
Плата
Поляризация X
Поляризация:
и
p
1
La
Y
1
B
X
переходы - по длинной оси, p – по короткой.
50
Рис.2.18. Спектр поглощения нафталина в гексане [5].
Нафталин (рис.2.18):
: 330-340 нм, p: 280-290 нм, : 225 нм.
51
Антрацен (рис.2.19):
: налагается на p: ,p : 380 нм, : 250 нм.
Рис.2.19. Спектр поглощения антрацена в гексане [5].
52
Фенантрен (рис.2.20): : 310-350 нм, p: 270-310 нм, : 250 нм,
1:
210 нм.
Рис.2.20. Спектр поглощения фенантрена в гексане [5].
Общая закономерность спектров АУ - смещение всех полос в
длинноволновую сторону при увеличении размеров системы.
Пери-конденсированные
ароматические
углеводороды
подчиняются тем же закономерностям, что и полиацены.
53
Примеры:
Перилен:
Пирен:
Не все атомы участвуют в переходах. («Внутренние» атомы
углерода не участвуют). Есть тенденция к инверсии альфа и p-состояний
(p может оказаться ниже, чем альфа). У перилена именно это имеет
место.
Бензол как особый случай
Рис.2.21. Схема молекулярных орбиталей молекулы бензола
54
На рисунке 2.22 представлена схема, поясняющая происхождение
энергетических уровней молекулы бензола.
Рис.2.22. Схема, поясняющая происхождение энергетических уровней
молекулы бензола
А на рисунке 2.23 - спектр поглощения бензола (в гексане).
Рис.2.23. Спектр поглощения бензола (в гексане) [5].
55
В одноконфигурационном приближении нижнее синглетное
состояние оказывается четырехкратно вырожденным (рис.2.21,2.22).
При учете КВ происходит снятие вырождения – расщепление на одно
двукратно вырожденное и два невырожденных состояний. Низшими
оказываются два невырожденных состояния, переходы в которые из
основного запрещены по симметрии (рис.2.22). Формально спектр
оказывается очень похожим на спектры других ароматических
углеводородов: имеются кажущиеся аналоги всех трех состояний: , p и
(рис.2.23). Но, на самом деле, реальная аналогия с соответствующими
состояниями других ароматических углеводородов отсутствует. Бензол
следует рассматривать как особый случай.
2.6. Соединения с неуглеродными атомами и основные свойства
n
*-переходов
Когда рассматриваются замещенные производные ароматических
углеводородов и их аналоги с гетероатомами, следует исходить из
ближайших по строению ароматических углеводородов. В чем могут
быть отличия?
1) Простейший случай – когда не меняется число пи-электронов и
нет n-электронов. В этом случае отличия от АУ можно трактовать
как возмущение. Число переходов не меняется
2) Число пи-электронов не меняется, но есть n-электроны. В этом
случае появляются n-пи переходы. Наиболее типичный пример:
аза-замещение: бензол –> пиридин. Этот пример будет рассмотрен
позднее.
3) Если меняется число пи-электронов. Обычный эффект –
батохромное смещение всех полос поглощения при увеличении
числа пи-электронов. Но могут быть и более существенные
изменения.
Рассмотрим случай возмущения на примере бензола. (Надо
помнить, однако, что это особый случай, в силу высокой симметрии
молекулы). У фенола длинноволновая полоса: 1000, =270-280 нм.
Т.е. по сравнению с бензолом имеет место батохромный сдвиг и
заметное увеличение интенсивности (из-за ослабления запрета по
симметрии). Строго говоря, здесь добавляются 2 пи-электрона, но
соответствующие орбитали находятся очень низко, так что можно
считать, что это 1-й случай (возмущение).
Проявление в спектрах n-π-переходов.n-π-переходы легко
выявляются когда это самый длинноволновый переход. Основной
критерий выявления – эффект полярного растворителя. Аналогичным
образом проявляется действие водородной связи или протонирования. В
полярных растоврителях и при образование Н-связи n-π-переходы
56
испытывают сильный гипсохромный сдвиг. При протонировании они
вообще пропадают (рис.2.24):
Рис.2.24. Схема, поясняющая влияние протонирования и образования
водородной связи на n-π-переходы.
Рис.2.25. Спектры поглощения пиридина в толуоле (слева) и в воде
(справа) [5].
57
В спектре пиридина в толуоле (рис.2.25) полоса, соответствующая n-пипереходу, слабая, но структурированная. Это длинноволновая часть
длинноволновой полосы. В воде полоса n-π перехода теряется в альфаполосе.А у пиридиниум-катиона n-π-полоса отсутствует .
Спектр пиразина в толуоле приведен на рис.2.26. В этом случае
полоса, соответствующая n-π-переходу хорошо выражена и содержит
разрешенную колебательную структуру.
Рис.2.26. Спектр поглощения пиразина в толуоле [5].
58
Рис.2.27. Спектр поглощения пиррола (в гексане) [5].
Спектр пиррола изображен на рис.2.27. Здесь n-пи-переходов нет.
Это фактически сильно замытый спектр бензола. Спектр фурана
(рис.2.28) вполне аналогичен спектру пиррола. С точки зрения пиэлектронов пиррол и фуран изоэлектронны. Однако, у фурана, в отличие
от пиррола имеется неподеленная пара электронов. Но эта пара
находится низко и поэтому никак себя не проявляет в электронных
спектрах.
59
Рис.2.28. Спектр поглощения фурана (в циклогексане) [5].
Если мы переходим к более конденсированным системам, то n-пипереходы испытывают не столь сильное батохромное смещение, как пипи.
У хинолина
N
n-π – уже не самая длинноволновая полоса. Низшее возбужденное
состояние: S
.
60
У акридина
N
и феназина
N
N
– тожеn-π – не самая длинноволновая полоса..
Кетоны на примере спектра пара-бензохинона.
O
Структурная формула пара-бензохинона приведена ниже.
O
Спектр поглощения пара-бензохинона приведен на рис.2.29. У этого
соединения n-π-переход выражен исключительно ярко. Из-за того, что
этот переход оказывается в видимой области, именно он обусловливает
окраску пара-бензохинина. Обращает на себя крайне низкая
интенсивность n-π-перехода в данном случае.
Отнесение полос в спектре поглощения пара-бензохинона
(рисунок 2.29) - следующее:
240(π→π*, ε=2·104), 278(π→π*, ε=280), 450 нм (структурированная,
n→π*, ε=10-15).
61
Рис.2.29 Спектр поглощения пара-бензохинона (в гексане) [5].
У нафтахинона:
O
O
Те же полосы поглощения расположены на 246, 330 и 425 (ε=50) нм, т.е.
при
увеличении
размера
системы
n-π-переход
испытывает
гипсохромный сдвиг, а π-π – батохромный. У антрахинона – тенденция
та же. Однако, в обоих последних случаях n-π-переходы остаются
видимы в спектре.
Насыщенные кетоны.
В спектре поглощения ацетона имеем полосы, соответствующие
переходам n->σ* (<200 нм). n->π*(200 нм, ε=277).
62
2.7. Примеры нециклических соединений: полифенилы, красители
Различия между спектрами циклических и нециклических
соединений на примере полифенилов.
Спектры полифенилов характеризуются наличием доминирующей
длинноволновой полосы и отсутствием выраженной колебательной
структуры (рис.2.30).
Рис.2.30. Спектры поглощения полифенилов
Красители на примере незамещенного родамина
В спектре поглощения красителей доминирует длинноволновая
полоса с ε≈105 со слабо выраженной колебательной структурой, как это
показано на рисунке 2.31 для незамещенного родамина в этаноле.
H2N +
O
NH2
COOH
Рис.2.31. Спектр поглощения незамещенного родамина в этаноле.
Справа показана структурная формула красителя.
63
Порфирины и металлопорфирины
Металлопорфирины с непереходными металлами имеют типичный
спектр поглощения, аналогичный спектру, представленному на рис.2.32.
Рис.2.32. Спектр поглощения металлопорфирина с непереходным
металлом. Справа приведена структурная формула металлопорфирина.
Происхождение данного спектра легко объясняется на основе
четырехорбитальной модели. На рисунке 2.33 показана схема
электронных уровней и переходов, поясняющая происхождение спектра
поглощения металлопорфиринов.
Рис.2.33. Схема электронных уровней и переходов, поясняющая
происхождение спектра поглощения металлопорфиринов.
Аналогичным образом объясняется происхождение спектра
поглощения безметального порфирина. Соответствующий спектр и
типичная структурная формула безметального порфирина показаны на
рисунке 2.34.
64
Рис.2.34. Спектр поглощения безметального порфирина
Спектры поглощения ароматических аминокислот и белков
(1) Фенилаланин.
Структурная формула имеет следующий вид.
CH2
H2N CH COOH
Спектр поглощения имеет очевидную аналогию со спектром бензола,
показанным на рис.2.35.
(2) Тирозин.
Структурная формула имеет следующий вид.
HO
CH2
H2N CH COOH
Спектр поглощения аналогичен спектру фенола, который показан на
рисунке 2.35. Тирозин может находиться как в нейтральной, так и в
анионной форме тирозината, отличающейся по спектру от тирозина
батахромным сдвигом.
65
(3) Триптофан.
Структурная формула имеет следующий вид.
NH
CH2
H2N CH COOH
Спектр поглощения триптофана и других ароматических аминокислот
(тирозина и фениланалина), приведенный на рисунке 2.35 явную имеет
аналогию со спектром нафталина. В спектре имеются полосы альфа, «p»
и бета. Триптофан присутствует только в животных белках и, как
правило, отсутствует в растительных.
Рис.2.35. Спектры поглощения ароматических аминокислот
66
3. Межмолекулярные взаимодействия
электронно-колебательных спектрах
и
их
проявления
в
3.1. Универсальные взаимодействия: динамический, статический и
дисперсионный вклады
Универсальные (неспецифические) взаимодействия - это
взаимодействия через макроскопические характеристики: показатель
преломления, диэлектрическую постоянную и т.п. Различают три
вклада, соответствующие трем типам взаимодействия.
1) Динамическое взаимодействие зависит только от показателя
преломления n. Оно имеет место и для бездипольных молекул и
может наблюдаться в неполярных растворителях.
дин.
e2
fe n2 1
,
8 2 mc 0 r 3 2n 2 1
(3.1)
где r – радиус полости (молекулы). Частотный сдвиг всегда только в
длинноволновую сторону и увеличивается с увеличением n.
2) Статический вклад.
2
ст .
2n02 1
D 1
C2
,
2
n0 2
D 1
(3.2)
где C2 ~ P0 P* , D – диэлектрическая постоянная растворителя, n0 –
показатель преломления растворителя.
3) Дисперсионный вклад.
дисп .
n2 1
C 2
,
n 1
(3.3)
*
где C C ( 0
) , α – поляризуемости в основном и возбужденном
*
0 , то
состояниях. Очевидно, если 0
0.
дисп .
Суммарный вклад всех трех механизмов:
дин.
стат .
67
дисп .
3.2. Влияние водородной связи на n
* переходы
К числу микроскопических (специфических) взаимодействий
относится водородная связь. Это взаимодействие рассматривается, когда
обсуждаютсяn-π-переходы в условиях кислотно-основных превращений.
Детальный анализ этих переходов приведен в разделе 2.6.
3.3. Слабые комплексы с переносом заряда, основные свойства
состояний переноса заряда
Донорно-акцепторные взаимодействия, комплексы с переносом
заряда, состояния переноса заряда - это еще один из специфических
типов взаимодействий, имеющий вполне определенные проявления в
электронных спектрах в виде появления новых полос, отсутствующих у
компонентов, не связанных в комплекс. Оно имеет место, когда один
компонент имеет выраженные электронно-донорные свойства, а другой
– электронно-акцепторные. Причины, приводящие к появлению новых
переходов у комплексов, могут быть поняты схемы, приведенной на рис.
3.1 (такой переход показан стрелкой).
Рис.3.1. Схема, поясняющая появление состояний переноса заряда в
комплексах с переносом заряда
68
3.4. Электронные состояния димеров: давыдовское расщепление,
экситонно-резонансные и зарядово-резонансные состояния
Экситонное взаимодействие. Давыдовское расщепление
Предположим, что имеем две одинаковые молекулы A1иA2, находящиеся
в условиях достаточно сильного взаимодействия между собой. В этом
случае основное и возбужденные состояния такого димера описываются
следующим образом:
A1 A2
1
2
A1* A2
1
2
1
2
A1 A2*
( A1 A2 )
0
( A1* A2 )
( A1 A2* )
( A1* A2 )
( A1 A2* )
(3.5)
На рисунке 3.2 приведена схема, уровней, поясняющая появление
экситонно-резонансных состояний димеров
--------------
--------------
-------
-------------
Рис.3.2. Схема, поясняющая
состояний димеров
D
E
cos
2 D2
появление
/ R3
( A)r ( A*)d
0
экситонно-резонансных
3 cos 1 cos
(3.6)
2
где, α0 – угол между моментами переходов молекул 1 и 2, θ1 и θ2 – углы
между дипольными моментами переходов и вектором R, соединяющим
центры молекул.
Влияние геометрии взаимного расположения
Крайними случаями геометрии взаимного расположения
компонентов димеров могут считаться сэндвичеобразная геометрия и
геометрия типа «голова-хвост». Оба этих случая могут рассматриваться
с позиций формулы (3.6) как частные случаи (см. рис.3.3). В первом
69
случае
длинноволновая
компонента
«давыдовского
дублета»
оказывается запрещена, а во втором она, наоборот, имеет максимальную
интенсивность (рис.3.3).
Рис.3.3. Схема, поясняющая влияние взаимного расположения
компонентов димера на распределение интенсивностей между
экситонно-резонансными компонентами в спектре димера.
Зарядово-резонансные состояния.
Это состояния, описываемые следующим образом:
(CR)
1
2
( A1 A2 )
( A1 A2 )
(3.7)
Энергия такого состояния приблизительно выражается так:
E (CR)
I
e2
A
R
(3.8)
В спектрах эти состояния, как правило, не проявляются.
70
4. Молекулярная люминесценции и безызлучательный перенос
энергии
4.1. Основные фотофизические процессы в молекулах:
флуоресценция, фосфоресценция, внутренняя и
интеркомбинационная конверсии. Закономерности молекулярной
люминесценции
Основные
фотофизические
процессы,
происходящие
в
многоатомных молекулах, показаны на рис. 4.1. Эти процессы:
поглощение,
флуоресценция,
внутренняя
конверсия,
инетркомбинационная конверсия, фосфоресценция, синглет-триплетное
поглощение, колебательная релаксация.
Рис.4.1. Схема уровней многоатомной молекулы.
Константы скоростей излучательных и безызлучательных
переходов, показанных на рис.4.1, обозначаются следующим образом:
kf– флуоресценция, kph- фосфоресценция, kTd,kic,- интеркомбинационная
конверсия (интерконверсия), kvr- колебательная релаксация, k**d,k*dвнутренняя конверсия.
Известны следующие определения люминесценции:
(1) общепринятое– как избыток над тепловым излучением;
(2) более простое: люминесценция – это любой спонтанный
излучательный переход из любого возбужденного состояния.
При оптическом возбуждении имеем 2 вида молекулярной
люминесценции: флуоресценция – переходы S1
S0 и фосфоресценция
– переходы T1
S0 .
71
Правило Каша: Излучательные переходы происходят только из
состояний S1 и T1 .
Спектр люминесценции схематически представлен на рис.4.2.
Рис.4.2. Схема взаимного расположения спектров люминесценции и
поглощения.
При изображении спектров люминесценции по оси ординат
откладывается спектральная плотность интенсивности. Спектральная
плотность может быть отнесенная к единичному интервалу, либо длин
волн, либо частот. А сама интенсивность может быть либо
энергетическая, либо квантовая. В результате получается 4 варианта
величин – энергетических характеристик люминесценции, причем все
они реально используются.
Правило Стокса. Первоначальная формулировка: ( h a h f ).
Современная формулировка: максимум в спектре люминесценции
смещен относительно максимума поглощения в сторону длинных волн.
Правило зеркальной симметрии. Первоначальная формулировка:
“Спектральные кривые поглощения и люминесценции в частотном
масштабе являются взаимным зеркальным отражением по отношению к
некоторой линии.” (Лёвшин, 1931г.)
Закон Вавилова. Закон постоянства спектра испускания при
изменении частоты возбуждающего света. При изменении частоты
(длины волны) возбуждения спектр люминесценции изменяется строго
пропорционально, только по интенсивности. На языке констант
скоростей это означает:
kvr
kf
k d**
kd*
kic
(4.1)
k *f *
72
Следствие из закона Вавилова: люминесценция из высоких электронных
состояний невозможна. Закон Вавилова позволяет судить о чистоте
(гомогенности) вещества, т.к. дает экспериментальный критерий,
позволяющий отличить чистое вещество от нечистого.
*
k f kic ).Это правило
Правило Ермолаева-Свешниковой: ( k d
хорошо соблюдается для ароматических соединений и порфиринов. Не
соблюдается для красителей и полиенов.
4.2. Спектры люминесценции и спектры возбуждения
Если поглощения образца невелико, то имеем:
I
I0
cl
10
e
(ln 10 ) ecl
1 (ln 10) cl
(4.2)
Для суммарной (по всему спектру) интенсивности люминесценции
будем иметь:
If
( I0
I)
f
K
K
cI0
f
(4.3)
Или, если интересуемся зависимостью от длины волны, то:
I f ( в,
где
I nf (
f
)
K
f
( в )cI 0 ( в ) I nf (
)
f
)
(4.4)
- нормированный спектр люминесценции.
Спектр возбуждения люминесценции – это зависимость
интенсивности люминесценции (на той или иной длине волны, либо
суммарной по спектру) от длины волны возбуждающего света. При
малых поглощениях спектр возбуждения совпадает со спектром
поглощения.
f
73
Спектрофлуориметры. Оптическая схема
Оптическая схема типичного спектрофлуориметра приведена на рис. 4.3.
Рис. 4.3. Оптическая схема спектрофлуориметра
Отличительной особенностью этого типа спектральных приборов
является наличие двух монохроматоров: одного для возбуждения и
одного для регистрации, каждый из которых может осуществлять
сканирование независимо от другого. Таким образом, могут
регистрироваться как спектры возбуждения люминесценции, так и
спектры люминесценции.
74
4.3. Квантовый выход люминесценции и методы его измерения
Квантовый выход люминесценции это отношение числа квантов,
излученных в виде люминесценции к числу поглощенных квантов.
Экспериментально он может быть определен следующим образом:
f
e
I f De
I e Df
, точнее:
f
e
I f De n 2f
I e D f ne2
(4.5)
где If и Df – соответственно, квантовая интенсивность люминесценции и
оптическая плотность на длине волны возбуждения для исследуемого
вещества, Ie и De – аналогичные величины для эталона, e - квантовый
выход люминесценции эталона, nf и ne– показатели преломления образца
и эталона на длине волны люминесценции. Условия применимости:
малые поглощения D<<~0.1.
Следствие из закона Вавилова и правила Каша: квантовый выход
люминесценции не зависит от длины волны возбуждения (для чистых
веществ!).
Выход в триплет (квантовый выход в триплетное состояние)
определяется следующим выражением:
T
kic
kic
kf
k
*
d
1
(4.6)
Последнее приближенное равенство предполагает выполнение “правила
Ермолаева-Свешниковой”.
75
4.4. Флуоресценция и фосфоресценция при наличии n
* переходов
Схема основных фотофизических процессов в молекулах приведена на
рис.4.4.
Рис.4.4. Схема основных энергетических уровней многоатомной
молекулы (Схема Яблонского)
Основные фотофизические процессы и их константы скоростей.
Основные соотношения между временами затухания люминесценции и
константами скоростей:
Если излучательная константа обозначена какkf, то имеем следующие
соотношения:
r
1
kf
f
r
f
kf
(4.7)
1
k ic
76
k d*
Молекулярная фосфоресценция – это переходы типаS0 ←T1.
Различают фосфоресценцию и замедленную флуоресценцию.Времена в
обоих случаях: 10 1 10 6 с. Спектрально замедленная флуоресценция
совпадает с обычной флуоресценцией, а фосфоресценция смещена в
длинноволновую
сторону.Условия
наблюдения
фосфоресценции:(замороженные)
твердые
растворы.Для
фосфоресценции характерно исключительно сильное тушение
кислородом.В жидких обескислороженных растворах фосфоресценция
тоже может наблюдаться. Процесс, обратный фосфоресценции –это S-Tпоглощение- может наблюдаться в условиях больших длин оптического
пути или (и) насыщения раствора кислородом.
Общие закономерности молекулярной фосфоресценции при
наличии n-пи-переходов.Если низшее возбужденное S-состояние есть
nπ -состояние, то флуоресценция никогда не наблюдается. Основная
причина этого состоит в малости излучательной константы по
сравнению
с
обычными
значениями
константы
скорости
интерконверсии.
Основные эффекты влияния растворителя при наличии nπ и
ππ–состояний.
1-й эффект:
изменение типа фосфоресценции в полярных
растворителей. В качестве иллюстрации на рисунке 4.5 приведена схема,
поясняющая изменение типа фосфоресценции инданона при изменении
растворителя.
O
CH2
CH2
Инданон.
в спирте
в гексане
Рис.4.5. Схема, поясняющая изменение типа фосфоресценции инданона
при изменении растворителя.
77
Этот эффект бывает не всегда. У бензохинона тип фосфоресценции не
меняется и всегда соответствует nπ переходу.
2-й эффект - активация флуоресценция в полярном растворителе. Этот
эффект на примере хинолина проиллюстрирован на рис.4.6.
Хинолин в гептане
Хинолин в спирте
Рис.4.6. Схема, поясняющая эффект активации флуоресценции хинолина
в полярном растворителе
В гептане не обязательно должно быть так, как показано на рисунке 4.6
слева, где флуоресценция хинолина отсутствует. Может быть Sπ→π ниже,
чем Sn→π. Но если Tn→π находится ниже, чем Sπ→π, то флуоресценция
отсутствует, а есть только фосфоресценция. Такова ситуация у
акридина. Таким образом, в основе активизации флуоресценции в
полярных растворителях лежит инверсия уровней Sπ→π и Tn→π .
Люминесценция белков.Люминесцируют все три ароматические
аминокислоты: фенилаланин, тирозин и триптофан. Но люминесценция
фенилаланина практически никогда не наблюдается из-за малого
квантового выхода. Люминесценция тирозина наблюдается только в
отсутствии триптофана и очень слабая. При наличии триптофана
люминесценция
белков
–
это
практически
исключительно
люминесценция триптофана.
нм
Рис.4.7. Пять типов люминесценции триптофана
78
Известно 5 люминесцентных типов триптофана (см.рис.4.7).
Тип A – максимум - 307 нм. Триптофан изоэлектронен нафталину. В
состоянии A флуоресцентный переход – из состояния 1Lb (т.е. α). Это
состояние - низшее возбужденное только в неполярном окружении,
поскольку следующее состояние La (т.е. p) исключительно
чувствительно к полярности окружения. A – это триптофан в
исключительно неполярном окружении, без доступа воды. Этот тип
триптофана бывает очень редко, один из немногих известных случаев –
в составе белка азурина.
Тип S – максимум – 316-317 нм, плечи: 305-307 нм и 320-330 нм. Это
тоже неполярное окружение, но в возбужденном состоянии образуется
комплекс с 1 полярной молекулой. Как правило, данный тип в чистом
виде не встречается. Обычно наблюдается одновременно с типом I.
Важно, что в замороженных растворах комплекс тоже образуется.
Тип I - максимум – 330 нм, полуширина Δ=50 нм. Это триптофан в
полярном, но жестком окружении. В возбужденном состоянии
образуется комплекс 2:1 с полярной молекулой. У типов I-III низшим
синглетным возбужденным состоянием является La (т.е. p), а не 1Lb (α).
Тип II – максимум – 340 нм, полуширина Δ=55 нм. Это триптофан на
внешней поверхности белковой молекулы, находящийся в контакте с
водой.Но именно со связанной водой, а не свободными молекулами
воды.
Тип III – максимум – 350 нм, полуширина Δ=60 нм. Это триптофан на
внешней поверхности белковой молекулы, находящийся в контакте со
свободной водой. Этот тип бывает у белков в их природном состоянии
очень редко. Очень часто он наблюдается у белков в раскрытом
состоянии. Именно этот спектр – у свободного триптофана в водном
растворе.
Тушение люминесценции: типы A, S и I – мало доступны внешним
тушителям. При замораживании у типов I-III спектр смещается в
сторону коротких волн, а у типов A и S – не меняется.
Люминесценция
порфиринов.
Спектры
поглощения
и
люминесценции металло- и безметального порфирина показаны на рис.
4.8.
79
Рис.4.8. Спектры поглощения и люминесценции металлопорфирина
(слева) и безметального порфирина (справа).
Эффект тяжелого атома у порфиринов проявляется в зависимости
от центрального атома металла в следующей последовательности: H2>Be->Mg->Zn->Ca. Специфическая ситуация имеет место в случае
металлов:Cu, VO, Pd. Здесь есть только фосфоресценция (эффект
парамагнитного атома). Порфирины с металлами: Mn, Fe, Co, Ni не
флуоресцируют и не фосфоресцируют во всех валентных состояниях
металла.
4.5. Статическое и динамическое тушение люминесценции
Тушением люминесценции называют любое уменьшение
интенсивности, точнее квантового выхода, при каком-то воздействии на
молекулу. Обратный процесс – возгорание. Среди многочисленных
механизмов тушения люминесценции рассматриваются следующие три.
(1) эффект тяжелого атома
(2) фотохимические реакции
(3) перенос энергии
(1) Эффект тяжелого атома может быть:
(а) внутренний; (б) внешний.
80
Внутренний обусловлен тяжелым атомом, входящим в состав
молекулы.Внешний – атомом, входящим в состав растворителя или
матрицы.
Эффект тяжелого атома обычно проявляется и усиливается при
следующих при следующих заменах:
O S Se
H
Mg
Cl
Br
J
Zn
Li Na K Rb Cs.
Тушение
флуоресценции
по
данному
механизму
обычно
сопровождается возгоранием фосфоресцинции. Но при более сильном
возмущении происходит тушение фосфоресцинции.
(1а) Эффект парамагнитного атома.
Этот эффект проявляется так же, как и эффект тяжелого атома, но
обычно более сильно выражен. Его причина состоит в формальном
снятии спинового запрета перехода для системы в целом. Наиболее
часто встречающиеся примеры: Fe(III) (спин 3/2), Cu(II) (спин ½):
s 1/ 2
[ A( s 1) M ( s
s 1/ 2
[ A( s
1 / 2)]
0) M ( s 1 / 2)]
(3) Фотоперенос электрона определяется следующими процессами:
D*
A
D
A
A*
D
A
D
либо:
Процесс может происходить в результате диффузионных встреч, но
может и при постоянном контакте.
(4) Перенос энергии – это процесс типа:
D*
A
D
A* ,
который будет рассмотрен подробнее ниже.
Общие закономерности тушения люминесценции. Тушение 2-го рода по
Вавилову (динамическое, кинетическое).
81
Это тип тушения характеризуется следующими основными признаками.
(а) Зависимость степени тушения от концентрации тушителя имеет
следующий вид:
c
0
1
1 k q cq
(4.8)
Это формула Штерна-Фольмера.
В другой форме она может быть выражена так:
0
1 kq cq ,
или:
0
k q cq .
Если строить «кривую тушения»(т.е. зависимость интенсивности
люминесценции от концентрации тушителя) в штерн-фольмеровских
координатах (т.е. координатах
и cq ), то Штерн-Фольмеровская
0
зависимость изображается прямой линией (рис.4.9).
Рис.4.9. Схема, поясняющая появление линейной зависимости при
использовании штерн-фольмеровских координат.
(б) Соотношение между длительностью и выходом:
c
c
(4.9)
Штерн-Фольмеровская константа:
82
kq
0
z,
(4.10)
где: (<1) – эффективность (“квантовый выход”) тушения в расчете на 1
диффузионную встречу; z – число диффузионных встреч за единицу
времени, т.е. 0z – число диффузионных встреч за время жизни
возбужденного состояния.
Для жидких растворов Вавиловым в 1928 г. были получены
формулы:
z
2 ( r1 r2 ) 2 kT
3 r1r2
- это число диффузионных встреч за единицу времени (при единичной
концентрации тушителя). Эту величину называют диффузионной
константой (константой диффузии). Другая, более употребительная
форма записи:
kd
2kTN ( rD rA ) 2
30
rD rA
(4.11)
В числителе:
2kTN
2 RT ,
где R=8.31 эрг град-1 моль-1, - коэффициент вязкости растворителя в
сантипуазах.
Формулы выведены в предположении
1 , т.е. повторные
соударения (встречи) несущественны.
Радиусы обычно берут из экспериментов по диффузии. Но иногда
приходится вводить эффективные значения r>>r1,r2.
Фактически
оказывается: ~0.25 0.5, а в расчете на 1 столкновение (которых за 1
встречу происходит несколько сот), ~10-3.
Тушение 1-го рода по Вавилову (статическое) - это тушение в
комплексе:
kr
A Q
A Q
83
kr
[ AQ ]
[ AQ ]
, или: [Q ]
[ A]k r
[ A][Q ]
Полагая, что комплексы не люминесцируют, имеем:
[ A]
1
[ A] [ AQ ] 1 [Q ]k r
0
(4.12)
(4.13)
Если пренебречь той частью молекул тушителя, которые связаны в
комплекс, то это та же Штерн-Фольмеровская зависимость.
Как различить экспериментально статическое тушение и
динамическое. По концентрационной зависимости это не всегда удается,
т.к. зависимости часто оказываются очень близки. Наиболее надежно
эти два типа тушения можно различить по зависимости длительности
люминесценции от выхода люминесценции или от концентрации
тушителя.
При динамическом тушении:
0
1
1 k q cq
При статическом:
0
Часто можно использовать и косвенное различие: данные два типа
тушения имеют противоположные зависимости от температуры. При
повышении температуры статическое тушение уменьшается из-за
диссоциации комплексов, а динамическое, наоборот, увеличивается изза увеличения скорости диффузии.
Тушение неорганическими ионами – всегда динамическое. Причем
эффективность тушения уменьшается в следующем порядке:
J > CNS >Br >Cl >(COO )2 >CH3COO >SO42 >NO3 >F
Возможно также тушение катионами, которые при этом выступают как
акцепторы электрона (т.е. окислители):
Ag+, Cu++, Fe++.
Тушение органическими молекулами может быть как статическим, так и
динамическим.
84
4.6. Безызлучательный перенос энергии. Основные закономерности
индуктивно-резонансного переноса энергии
Перенос энергии - это процесс типа:
A* B
A B* .
Диполь-дипольный индуктивно-резонансный перенос энергии
(Фёрстеровский, FRET) – осуществляется на расстояния до 50-100
ангстрем. Для его количественного описания вводится понятие
критического радиуса (R0) – расстояния на котором константа скорости
переноса энергии равна суммарной скорости всех внутримолекулярных
процессов расселения данного возбужденного состояния. Согласно
теории Фёрстера R0 в случае данного механизма может быть определен
следующим образом:
6
0
R
9000 ln 10 2 q0 D н
ID ( ) A( )
128 5n 4 N
где R0 – в см,
2
4
d ,
– ориентационный фактор:
(4.14)
2
2
при интенсивном
3
броуновском движении, а для твердого замороженного раствора:
2
2
0.84 ; спектр люминесценции I Dн ( ) нормирован таким образом,
3
н
что I D ( )d
1 , - в см-1.
Константа скорости переноса энергии выражается через критический
радиус следующим образом:
dd
k ( R)
1
0D
R0
R
6
(4.15)
По определению для константы скорости переноса энергии имеем:
1
.
k dd ( R0 )
0D
Если имеется очень широкая полоса люминесценции донора, то можно
( )
A
приближенно полагать:
.
4
Условие однонаправленности переноса энергии:
k dd
k vr .
85
Основная модель при создании теории индуктивно-резонансного
переноса энергии – это твердотельные системы со статистическим
распределением донора и акцептора.
Экспериментальные проявления переноса энергии:
(1) тушение люминесценции донора;
(2) возгорание
люминесценции
акцептора,
наличие
сенсибилизированной люминесценции;
(3) спектры возбуждения сенсибилизированной люминесценции
содержат полосы поглощения, характерные для донора;
(4) времена затухания:
удлиняется;
τ донора сокращается, τ акцептора
(5) эффективность процесса зависит от концентрации, но не от
D (в отличие от тривиальной реабсорбции).
Отличительные особенности именно индуктивно-резонансного переноса
энергии:
(1) высокая эффективность тушения донора: кинетический радиус >>
геометрического;
(2) независимость (или очень слабая зависимость) эффективности
процесса от вязкости и(или) температуры.
Особые случаи индуктивно-резонансного переноса энергии
составляют: триплет-синглетный перенос энергии, триплет-триплетная
аннигиляция, концентрационное тушение.
86
4.7. Обменно-резонансный перенос энергии, фотодинамический
эффект и основные свойства синглетного кислорода
Обменно-резонансный перенос энергии описывается формулой
Декстера:
k ex ( R)
2
Y exp
2R
L
I н ( ) н ( )d ,
D
A
(4.16)
где L – средний эффективный Боровский радиус, L и Y – не зависят от R.
Как и в случае индуктивно-резонансного переноса энергии, может
быть введено понятие критического радиуса: k ex ( R0 ) 0 D1 . Тогда
k ex ( R )
1
0D
exp
1
R
R0
2 R0
.
L
,
(4.17)
Основные отличия от случая индуктивно-резонансного переноса
энергии состоят в следующем. (1) В формуле 4.16 отсутствует
квантовый выход люминесценции донора. (2) В формуле 4.16
отсутствуют какие-либо абсолютные характеристики поглощения
акцептора, в частности, коэффициент экстинкции. Это означает, что
этот тип переноса энергии будет иметь место даже если люминесценция
донора, как и поглощение акцептора, практически отсутствуют. Но
переходы, тем не менее, должны иметься, и соответствующие спектры
должны перекрываться, т.к. именно этим обеспечивается соблюдение
закона сохранения энергии.
Модель Перрена: излучают только те молекулы, около которых в
объеме v нет ни одной молекулы акцептора. Зависимость тушения от
4 3
q
R0 .
концентрации в этом случае имеет вид: D exp( vc A ) , где v
3
q0 D
Правило Вигнера – сохранение полного спина – в случае
индуктивно-резонансного переноса энергии должно соблюдаться
отдельно для донора и акцептора. В случае же обменно-резонансного
механизма, из-за того, что имеется более тесное взаимодействие между
донором и акцептором полный спин должен сохраняться для суммарной
системы донор-акцептор, а отделено для донора или акцептора может и
не сохраняться. Примером является триплет-триплетный перенос
энергии, при котором имеет место очевидное нарушение правила
Вигнера отдельно для донора и для акцептора. Тем не менее, этот
процесс имеет место и доказано, что он осуществляется именно по
обменно-резонансному механизму.
Фотоника молекулярного кислорода и фотодинамический
эффект. Кислород представляет собой аномальную молекулу –
87
триплетную в основном состоянии. Низшее возбужденное состояние
молекулы кислорода – синглетное. Его энергия соответствует ближней
ИК области. Время жизни без столкновений, в газовой фазе ~45 мин (!).
Переход кислорода из триплетного состояние в синглетное и обратно –
еще один пример интеркомбинационных переходов, запрет которых
может преодолеваться в ходе обменно-резонансного переноса энергии.
Существует
универсальный
механизм
заселения
(генерации)
синглетного кислорода. Пусть имеется органическая молекула M,
единственным требованием к которой является ненулевой выход в
триплет. На свету осуществляется следующая последовательность
реакций:
h
1
M
1
M*
3
*
M
1
M*
3
3
M*
O2
(4.18)
1
1
M O2
Последняя строчка описывает перенос энергии, осуществляемый по
обменно-резонансному механизму.
С точки зрения химических свойств синглетный кислород –
сильный окислитель. Кроме того, он проявляет склонность к реакциям
присоединения по двойным связям. По своим свойствам он занимает
промежуточное положение между атомарным хлором и молекулярным
хлором. В силу этого, генерация синглетного кислорода приводит к
деструкции среды или каких-либо компонентов системы. В частности,
молекула M тоже может подвергаться деструкции. Данный эффект –
фотоиндуцированная деструкция под действием синглетного кислорода,
генерируемого по механизму (4.18), - получил название
фотодинамического эффекта. Несмотря на быстрое вовлечение в
химические реакции и эффективную дезактивацию при взаимодействии
с молекулами среды, синглетный кислород может обладать остаточной
люминесценцией, наличие которой однозначно свидетельствует о том,
что в данной среде генерируется синглетный кислород.
Тушение синглетного кислорода может осуществляться не только
за счет его вовлечения в химические реакции, но и по чисто
физическому механизму. Так, существуют специфические тушители
синглетного кислорода, наиболее известным из которых является бетакаротин, имеющий триплетное состояние, расположенное ниже уровня
синглетного кислорода. Поэтому бета каротин исключительно
эффективно тушит синглетный кислород по интеркомбинационному
механизму, аналогичному механизму триплет-триплетного переноса
энергии.
88
4.8. Основные представления теории безызлучательных переходов в
многоатомных молекулах
Теория безызлучательных переходов в молекулах была создана
относительно недавно – в конце 60-х годов прошлого века Дж.
Джортнером. В основе его лежит представление о внутренней конверсии
как о процессе, сопровождающемся возбуждением колебаний, в энергию
которых переходит избыток энергии, выделяющийся при переходе в более
низкое электронное состояние. Если вслед за этим происходит достаточно
быстрый процесс колебательных релаксаций, то обеспечивается
необратимый характер внутренней конверсии. В известном смысле можно
рассматривать внутреннюю конверсию как некоторый особый случай
внутримолекулярного переноса энергии с одного электронного состояния
на колебательные подуровни другого, более низкого электронного
состояния.
Очевидно, что в некоторых случаях речь может идти о реальном
переносе энергии, а не об «условном». К числу наиболее важных таких
случаев относятся следующие: тушение люминесценции неорганических
ионов (обычно катионов редкоземельных или переходных металлов)
молекулами лигандов или растворителя (среды), тушение люминесценции
синглетного кислорода молекулами среды и ряд других. Во всех
перечисленных случаях имеет место сильная локализация возбуждения в
относительно небольшом объеме, что, собственно, и позволяет трактовать
процесс тушения люминесценции как перенос энергии. Была доказана
правомерность подобного подхода, подтвержденная, в частности, тем, что
устранение колебаний с участием атомов водорода из окружения
возбуждающих центров (в частности, дейтерозамещение) приводит к
разительному снижению эффекта тушения люминесценции.
89
5. Поглощение и люминесценция полупроводниковых квантовых
точек с позиций молекулярной спектроскопии
5.1. Происхождение и природа электронных состояний квантовых
точек. Понятие о конфайнменте
Квантовые точки (КТ) – полупроводниковые нанокристаллы
размером 2-8 нм. Размер в данном случае определяет так называемый
конфайнмент (т.е. пространственное ограничение) на движение носителей
заряда. В случае КТ имеет место конфайнмент по всем трем
пространственным координатам (рис.5.1).
Рис.5.1. Квантовая точка с оболочкой.
Такой объект моделируется потенциальной ямой, расстояние между
уровнями которой определяется линейными размерами (диаметром) ямы.
5.2. Спектры поглощения и люминесценции квантовых точек.
Размерная зависимость спектральных свойств
Спектры поглощения и люминесценции КТ двух различных
размеров в качестве примера приведены на рисунках 5.2 и 5.3.
Рис.5.2. Спектры поглощения и люминесценции КТ с диаметром ядра 2.5 нм
90
5.3. Спектры поглощения и люминесценции КТ с диаметром ядра 3.3 нм.
Положение длинноволнового максимума поглощения КТ (как и
коэффициент экстинкции) и его размер связаны некоторыми
эмпирическими закономерностями:
Формулы Пенга:
Соотношения,
связывающие
размер
квантовых
точек
полупроводников CdSe, CdTe и CdSс длиной волны максимума их
поглощения:
• CdTe: D = (9.8127 × 10-7)λ3 – (1.7147 × 10-3)λ2 + (1.0064)λ – (194.84)
• CdSe: D = (1.6122 × 10-9)λ4 – (2.6575 × 10-6)λ3 + (1.6242 × 10-3)λ2 –
(0.4277)λ + (41.57)
• CdS: D = (-6.6521 × 10-8)λ3 + (1.9557 × 10-4)λ2 – (9.2352 × 10-2)λ +
(13.29),
где D – размер частиц (в нм), λ - положение максимума длинноволновой
полосы поглощения (в нм).
Коэффициент экстинкции как функция размера (диаметра) КТ.
• CdTe: ε = 10043 (D)2.12
• CdSe: ε = 5857 (D)2.65
• CdS: ε = 21536 (D)2.3,
где ε – коэффициент экстинкции (в М-1 × см-1), D – размер частиц (в нм).
Соблюдение основных закономерностей люминесценции.
1. Правило Стокса – соблюдается. Но стоксов сдвиг может иметь
другую природу, нежели в молекулах, связанную с некоторыми
поверхностными (локальными) уровнями, слабо проявляющимися
в поглощении. На это указывает большая величина стоксова
сдвига, наблюдаемая в ряде случаев.
2. Правило зеркальной симметрии – соблюдается.
3. 2-й закон Вавилова – соблюдается с точностью до неоднородного
уширения, обусловленного дисперсией КТ по размеру.
91
5.3. Особенности фотофизических процессов в квантовых точках.
Выполнимость
основных
закономерностей
поглощения
и
люминесценции
В принципе, фотофизические свойства КТ очень похожи на
свойства
молекул.
Это
касается,
в
частности,
основных
люминесцентных свойств. Однако, есть и особенности, не имеющие
аналогов в фотофизике молекул. У КТ тоже есть триплетные состояния.
Это так называемые «темные состояния». Однако, они имеют ряд
существенных особенностей, отличающих их от аналогичных состояний
молекул.
1. Синглет-триплетное расщепление мало.
2. Отсутствует фосфоресценция.
3. Отсутствие генерации синглетного кислорода.
4. Интеркомбинационная конверсия – не есть существенный канал
для безызлучательной дезактивации возбужденных состояний.
Внутренняя конверсия в КТ. Безызлучательные переходы типа
внутренней конверсии (т.е. между состояниями
одинаковой
мультиплетности) у КТ тоже имеют место. При этом правило Каша –
строго соблюдается. Однако, у КТ имеется ряд особенностей
безызлучательных переходов данного типа:
1. Высокая
эффективность
переходов.
внутризонных
безызлучательных
2. Очень
низкая
эффективность
межзонных
переходов
безызлучательных переходов, в частности, переходов S1 ~~>S0.
Основной механизм таких переходов - с участием промежуточных
состояний, в частности, центров рекомбинации.
Перенос энергии с участием КТ также имеет ряд особенностей по
сравнению с молекулярными системами:
1. Исключительно индуктивно-резонансный механизм (FRET).
2. КТ могут выступать в качестве как донора, так и акцептора
энергии.
3. Исключительно высокая эффективность R0>~ 50 Å.
92
6. Основы колебательной инфракрасной спектроскопии
6.1. Колебательные переходы многоатомных молекул. Молекула как
механическая система с упругими связями
Состояния квантового осциллятора показаны на рис.6.1.
Рис.6.1. Состояния квантового гармонического осциллятора
Энергии этих состояний описываются формулой:
En
n
1
h
2
(6.1)
Правило отбора для дипольных переходов: n
1.
Частоты колебаний многоатомных молекул в общем случаемогут
быть определеныследующим образом, если известна потенциальная
энергия V(ri) как функция всех координат всех атомов:
2
V
;
ri rj
Kij
1
Kij ;
mi m j
Kij
(6.2)
1/ 2
i
i
2
где λi – собственные значения матрицы Kij .
Собственные значения определяют частоты собственных колебаний
системы (нормальные колебания), а собственные векторы – форму этих
колебаний qi.
93
V
1 N
Ki qi2
2i1
E
ni
i
1
h
2
(6.3)
i
Ki
i
i
В случае 2-атомных молекул:
q=r
K
m1m2
m1 m2
Если число атомов – n, то число нормальных колебаний = 3n-6, либо 3n-5
для линейных молекул.
6.2. Групповые характеристические частоты. Классификация
колебаний: валентные и деформационные
Понятие о характеристических частотах.
Характеристическая частота – это частота колебания какой-то пары
атомов (т.е. изменение длины связи) или тройки атомов (т.е. изменение
валентного угла), которые слабо взаимодействует с другими группами
атомов.
Групповые характеристические колебания могут быть:
(1) валентные (в процессе колебания меняется длина связи) и
(2) деформационные (в процессе колебания меняется валентный угол).
Оценка частот колебаний. Наипростейший подход: считать для
одинарной связи: K1=5·105 дин/см, mC=19.8·10-24 г, mH=1.64·10-24 г. Отсюда:
частота должна быть порядка 3040 см-1. Если связь двойная, то K2= 2K1,
если тройная, то K3= 3K1, то есть частоты должны быть соответственно в
два и три раза ниже.
94
Колебания связей X-H. Для 2-атомных молекул имеем следующие
значения частот.
HH(4395см-1) (Запрещено в ИК)
LiH(1406см-1) BeH(2058см-1) BH(2360см-1)
CH(2862см-1) NH(3300см-1)
OH(3735см-1) FH(4138см-1)
NaH(1172см-1) MgH(1496см-1)
AlH(1682см-1) SiH(2080см-1) PH(2380см-1)
SH(2575см-1) ClH(2990см-1)
Дейтерозамещение. Силовая постоянная для связи X-H и X-D не
зависит от того, H или D участвует в этой связи. Меняется только масса.
Для 2-атомной молекулы XH (XD) должно выполняться соотношение:
2 2M
,
2
M
XD
где M –масса атома X. Приближенно можно считать:
XH
XH
2.
XD
6.3. Основные особенности ИК полос поглощения
Интенсивности в ИК спектрах определяются выражением:
P
Di ~
qi
(6.4)
Типичные значения ε: 1-10 (M-1см-1) для углеводородов, до 100 – для
соединений с OH и C=O группами.
95
6.4. Основные характеристические колебания: валентные OHколебания и водородная связь, валентные CO-колебания, NHколебания, NO-колебания
Колебания связей XY. Деформационные колебания.
1.X-Y. Это связи С-С, С-О, C-N. Обычно – в области 700 – 1500 см-1.
Как правило, не характеристичны.
2.X=Y. Это связи: C=C, C=O, C=N, N=O. Обычно – в области 1500-1950
см-1. Очень чувствительны к эффектам сопряжения.
3.X≡Y. Это связи: C≡C, C≡N. Область: 2000-2300 см-1. Почти всегда
характеристичны.
4.Деформационные колебания
С-H: 1700-650 см-1 налагаются на валентные.
N-H: до 1650 см-1
O-H: 900-1400 см-1
X-Y: <500 см-1
Характеристические колебания основных групп соединений
Приведем типичные значения частот характеристических колебаний ряда
углеводородных соединений.
Насыщенные углеводороды.
а) Валентные СH (2750-3000см-1):
2926 см-1–CH2, CH3 асимметричные
2890 см-1– H-C(CH3)3 – трет-бутильные СН
2872 см-1– CH3 симметричные
2853 см-1– CH2 симметричные
б) Деформационные CH (1452-1467, 1370-1397см-1):
1467см-1– CH2 в линейных метиленовых цепях
1460 см-1– CH3 – асимметричные деформационные
1455см-1– CH2 в циклопентане
1452см-1– CH2 в циклогексане
1397см-1 – трет-бутильные CH
1370-1397 см-1– симметричные СH3
Ароматические углеводороды.
CH валентные: 3000-3100см-1 (у алифатических - 2750-3000см-1)
В области 2800-3000 см-1 у ароматических углеводородов есть только
обертоны и составные частоты, а также полосы алкильных заместителей.
96
Гидроксильные соединения.
Частоты воды: асимметричное: 3755см-1, симметричное: 3655см-1,
деформационное: 1595см-1.
OH валентное у спиртов: 3610-3636см-1.
Водородная связь. Пример: OH колебания у спиртов (метанол). В качестве
иллюстрации проявления водородной связи в ИК спектрах гидроксильных
соединений на рисунке 6.2 приведен спектр ИК поглощения метанола.
Типичным проявлением водородной связи является появление
широкой полосы в области на ~150-300 см-1 ниже частоты OH колебания
при отсутствии водородных связей. Ширина этой полосы приблизительно
в 5-10 раз больше, чем ширина полосы валентного изолированного OH
колебания.
Валентные колебания C=O.
Частоты: 1580-1900см-1, обычно – у 1700см-1
R-CO-R
– 1715- 1703см-1
O
Валентное колебание N-H.
Частоты обычно: 3050-3500см-1
N-H+: широкая полоса (как в случае водородной связи) 2800-3000см-1
Свободная NH- связь (без ассоциатов) располагается в области 35003380см-1 и всегда уже, чем OH с водородной связью
Амины: 2 полосы: 3500 см-1(асимм.) и 3400 см-1(симм.). В спектрах
вторичных аминов имеется одна полоса среднего положения.
97
Водородная связь: NH···O=C проявляется аналогично случаю
гидроксильных групп, но в области несколько более низких частот (см.
рис.6.3).
cм-1
Рис.6.3. Проявление водородной связи в ИК спектрах: связь NH···O=C (Nметилацетамид)
Связь C≡N.
У нитрилов 2250 см-1 (ацетонитрил). При сопряжении понижается:
C=C-C≡N: 2225см-1.
У бензонитрила: 2240-2230см-1
Но у HCN: 2089 см-1
Связь N=O.
У нитропарафинов: 1560 см-1(асим.) и 1350 см-1(сим.)
У ароматрических нитросоединений: 1518 см-1(асим.) и 1349 см1
(сим.)
Деформационные неплоские CH-колебания (γCH)
У ароматических соединений: 901-675 см-1.
98
6.5. Особенности измерения поглощения в ИК области, основные
способы приготовления образцов
Особенности аппаратуры и экспериментальной техники в ИК
спектроскопии
1. На образец падает неразложенный ИК свет.
2. Тепловые фотоприемники, характеризующиеся высокой
инерционностью.
3. Особенности оптики: KBr и другие ионные кристаллы как
оптические материалы, отражающая оптика.
Методы приготовления образцов для ИК спектроскопии
А) Сублимация – напыление – пленка на подложке
Б) Испарение раствора – пленка на подложке
В) Пленки без подложек (в основном, полимерные)
Г) Запрессовка в таблетках из KBr.
Д) Натирание – пленка на подложке
Е) Расплав – пленка на подложке
Ж) Растворы в CCl4, CS2, (TiCl4, POCl3) - в кюветах из KBr
З) Жидкости – в кюветах из KBr
И) В вазелиновом масле в виде взвеси.
99
7. Фотофизические свойства комплексов полупроводниковых
квантовых точек CdSe/ZnS с молекулами порфирина в водных
средах [6]
В данном разделе рассматриваются оптические свойства
гибридных
структур
на
конкретном
примере
комплексов
полупроводниковых КТ с молекулами порфиринов [6]. Сегодня
создание таких структур представляет большой интерес, поскольку дает
возможность получения объектов, характеризующихся уникальными
свойствами. Работы по созданию и исследованию свойств комплексов
коллоидных полупроводниковых нанокристаллов, квантовых точек
(КТ), с органическими соединениями в последние два десятилетия
интенсивно ведутся во всем мире.
Как было указано выше, в полупроводниковых квантовых точках
из-за малых размеров нанокристалла (2-10 нм) возникает ограничение
движения носителей заряда, которое приводит к возникновению
квантово-размерного эффекта: дискретности электронных уровней.
Эффект размерного квантования приводит к тому, что спектры
поглощения и люминесценции нанокристаллов зависят не только от
химического состава, но также и от размера частиц. Наряду с квантоворазмерным эффектом квантовые точки характеризуются высоким
квантовым выход люминесценции (до 80%), высокой поглощательной
способностью в широком спектральном диапазоне (~ 105 М-1см-1),
хорошей химической устойчивостью и высокой фотостабильностью.
Уникальные оптические свойства полупроводниковых коллоидных
квантовых точек позволили им найти широкое применение в различных
областях науки, в частности в биомедицинских исследованиях. На
сегодняшний день полупроводниковые квантовые точки используются в
сенсорных системах, для многоцветной визуализации биологических
объектов и в ряде других направлений.
Сегодня достаточно активно развивается направление создания
комплексов квантовых точек с соединениями тетрапиррольного ряда,
которые
могут
использоваться
в
качестве
альтернативных
фотосенсибилизаторов в системах с эффективной генерацией
синглетного кислорода.
Тетрапиррольные соединения являются светочувствительными
органическими веществами. При введении в организм они обладают
способностью селективно накапливаться в зараженных участках ткани.
А под воздействием излучения с длиной волны, соответствующей
полосам
поглощения
вещества,
тетрапиррольные
соединения
генерируют синглетную форму окружающих его молекул кислорода.
Эта активная форма разрушает окружающие клетки ткани. На этом
принципе основан метод лечения онкологических заболеваний –
фотодинамическая терапия рака (ФДТ), в которой тетрапиррольные
соединения выступают в роли фотосенсибилизатора.
100
К сожалению, тетрапиррольные соединения, которые сегодня
применяются в ФДТ, имеют ряд существенных недостатков: длительный
период выведения из организма, необходимость индивидуального
подбора источника возбуждающего излучения. В связи с этим является
актуальным создание комплексов на основе квантовых точек и молекул
фотосенсибилизатора. В комплексах квантовая точка-молекула
фотосенсибилизатора увеличение генерации синглетного кислорода
осуществляется за счет внутрикомплексного переноса энергии
фотовозбуждения от квантовой точки на молекулу. Поскольку
квантовые точки имеют протяженный спектр поглощения, то для
комплексов снимается проблема подбора источника возбуждающего
излучения. Увеличение генерации синглетного кислорода может
позволить снизить терапевтическую дозу фотосенсибилизатора, либо
время облучения пораженных участков ткани.
Как
было
отмечено
выше,
в
большинстве
случаев
внутрикомплексный перенос энергии в системах с участием КТ
происходит по механизму индуктивно-резонансного безызлучательного
переноса энергии (FRET). В качестве донора энергии может выступать
квантовая точка, которая поглощает энергию фотовозбуждения и может
резонансно передавать её акцептору (молекуле фотосенсибилизатора).
Условиями переноса энергии по механизму FRET являются
относительно небольшое расстояние между донором и акцептором (до
10 нм), и наличие перекрытия спектра люминесценции донора со
спектром поглощения акцептора.
В излагаемой работе были исследованы оптические свойства
комплексов водорастворимых полупроводниковых квантовых точек
CdSe/ZnS
с
молекулами
водорастворимого
тетра(pтриметиламинофенил)порфина как в водных растворах, так и в плазме
крови человека. Показано, что в водных растворах для перехода молекул
порфирина, связанных в комплексы с квантовыми точками, в
спектральную форму, которая соответствует равновесному состоянию
системы, необходимо достаточно длительное время (порядка 2-5 часов в
зависимости от концентраций нанокристаллов и молекул порфирина в
смеси). В водных растворах в равновесном состоянии в комплексах
наблюдается эффективный перенос энергии фотовозбуждения от
квантовых точек к молекулам порфирина. В плазме крови обнаружено
взаимодействие молекул тетра(p-триметиламинофенил)порфина с
компонентами плазмы крови. Показано, что при введении из водного
раствора в плазму крови комплексов квантовых точек с молекулами
порфирина наблюдается диссоциация порядка 60% от числа вводимых
комплексов. В оставшихся комплексах наблюдается увеличение
интенсивности люминесценции порфирина, что свидетельствует об
эффективном внутрикомплексном переносе энергии от квантовых точек
на молекулы порфирина в плазме крови человека.
101
В излагаемой работе использовались водорастворимые
отрицательно
заряженные
полупроводниковые
нанокристаллы
(квантовые точки, КТ) CdSe/ZnS, солюбилизированные молекулами
тиогликолевой кислоты, со средним размером ядра 3.0 нм.
Полупроводниковые квантовые точки были получены в результате
высокотемпературного органометаллического синтеза. Для получения
водорастворимых
нанокристаллов
использовалась
стандартная
процедура замещения молекул триоктилфосфин оксида (TOPO),
обеспечивающих
растворимость
нанокристаллов
CdSe/ZnS
в
гидрофобных растворителях, на молекулы тиогликолевой кислоты
(TGA). В результате солюбилизации были получены отрицательно
заряженные водорастворимые КТ. В качестве органической молекулы –
сенсибилизатора
синглетного
кислорода
использовался
водорастворимый безметальный тетра(p-триметиламинофенил)порфин
(ТАФП).
Фотофизические свойства смешанных растворов квантовых точек
и молекул ТАФП в воде и в плазме крови человека были исследованы
методами
стационарной
и
кинетической
люминесцентной
спектроскопии. Для регистрации спектров поглощения растворов
использовался спектрофотометр UV-Probe 3600 (Shimadsu, Япония).
Регистрация спектров люминесцении и возбуждения люминесценции
образцов проводилась на спектрофлуориметрах «Флюорат-Панорама02» («Люмекс», Россия) и CaryEclipse (Varian, Австралия). Для
измерения времен затухания люминесценции образцов использовался
лазерный сканирующий люминесцентный микроскоп MicroTime100
(PicoQuant, Германия).
Результаты и обсуждение. Спектры поглощения и люминесценции
водных растворов (pH=7.0) квантовых точек и молекул порфирина
представлены на рисунке 7.1. Спектр поглощения ТАФП в области Qполос перекрывается со спектром люминесценции нанокристаллов (рис.
1 вставка, кривые 1 и 3 соответственно). Это означает, что в случае
образования комплексов между нанокристаллами и молекулами
порфирина будут выполнены все условия для безызлучательного
резонансного переноса энергии фотовозбуждения от квантовых точек на
молекулы порфирина.
102
оптическая плотность,
интенсивность люминесценции, отн. ед.
3
1
4
0.2
0.1
2
0.0
400
500
600
700
800
длина волны, нм
Рис.7.1. Спектры поглощения и люминесценции водных растворов (pH=7.0)
CdSe/ZnS КТ с диаметром ядра 3.0 нм и молекул тетра(pтриметиламинофенил)порфина: 1,4 – поглощение и люминесценция
порфирина; 2,3 – поглощение и люмнесценция КТ. Возбуждение светом с
длиной волны 405 нм [6].
Образование комплексов CdSe/ZnS КТ с молекулами ТАФП в
водных растворах. Исследование оптических характеристик водных
растворов смеси полупроводниковых квантовых точек CdSe/ZnS и
молекул порфирина показало, что спектры поглощения и
люминесценции молекул ТАФП в смеси с КТ отличаются от
соотвествующих спектров водного раствора свободных молекул. На
рисунке 7.2 приведен спектр поглощения раствора КТ CdSe/ZnS и
молекул порфирина, смешанных в соотношении СКТ:СТАФП равном 1:0.75
(СКТ ~ 10-6 М). Для сравнения на рисунке также приведены спектры
поглощения свободных КТ и молекул ТАФП соответствующих
концентраций.
103
оптическая плотность
1
2
3
4
0.08
0.04
0.4
0.00
500
600
700
0.2
0.0
300
400
500
600
длина волны, нм
Рис.7.2 . Спектр поглощения смешанного водного раствора (pH=7.0)
полупроводниковых квантовых точек CdSe/ZnS и молекул ТАФП: 1 –
спектр поглощения КТ (диаметр ядра 3.0 нм, СКТ 10-6 М); 2 – спектр
поглощения смеси КТ и ТАФП (СКТ:СТАФП равно 1:0.75, СКТ ~10-6 М); 3 –
спектр поглощения молекул ТАФП, связанных в комплекс с КТ, получен
как разность между спектром поглощения смеси (кривая 2) и спектром
поглощения КТ (кривая 1); 4 – спектр поглощения свободного ТАФП
(СТАФП ~ 10-6 М) [6].
Из рисунка 7.2 видно, что по сравнению со спектром водного
раствора молекул ТАФП, в спектре поглощения порфирина в смеси с КТ
происходит заметное уширение и длинноволновое смещение B-полосы
(~ 410 нм), а также наблюдаются сдвиги и изменение соотношения
амплитуд Q-полос в области 500 нм – 650 нм (рис. 7.2, кривые 3 и 4).
Это может являться проявлением того, что при смешивании водных
растворов полупроводниковых квантовых точек и молекул порфирина
происходит образование комплексов КТ/ТАФП. Поскольку квантовые
точки CdSe/ZnS в результате солюбилизации молекулами тиогликолевой
кислоты имеют на поверхности отрицательный заряд, а у молекул
тетра(p-триметиламинофенил)порфина
присутствуют
четыре
положительно заряженные четвертичные аминогруппы, то образование
комплексов
КТ/ТАФП
в
данном
случае
обусловлено
электростатическим
взаимодействием
молекул
солюблизатора
квантовых точек (тиогликолевой кислоты) с молекулами порфирина. Тот
факт, что в спектре поглощения смешанного раствора отсутствуют
полосы, принадлежащие спектру поглощения свободных молекул
104
порфирина (рис.7.2, кривая 4) свидетельствует о том, что все молекулы
ТАФП связаны в комплекс с КТ.
На рисунке 7.3 приведен спектр люминесценции данной смеси КТ
и молекул ТАФП, для сравнения также представлены спектры
люминесценции свободных КТ и молекул ТАФП соответствующих
концентраций.
интенсивность люминесценции, отн. ед.
1
2
3
4
2
1
0
500
550
600
650
700
750
длина волны, нм
Рис.7.3. Спектр люминесценции смешанного водного раствора КТ
CdSe/ZnS с молекулами ТАФП, возбуждение светом с длиной волны
420 нм: 1 – спектр люминесценции свободного ТАФП; 2 – свободные
КТ; 3 - смесь КТ с молекулами ТАФП; 4 – разностный спектр, получен
из зарегистрированного спектра смеси и спектра люминесценции
свободных КТ, кривые 3 и 2 соответственно; для удобства сравнения со
спектром свободного порфирина разностный спектр приведен в
увеличенном масштабе [6].
Видно, что образование комплексов КТ/ТАФП сопровождается
эффективным тушением люминесценции нанокристаллов (~ 80%).
Время жизни люминесценции квантовых точек в смешанном растворе
соответствует времени жизни люминесценции свободных КТ <τКТ> ~11
нс. Это означает, что в смеси с молекулами порфирина наблюдается
люминесценция только свободных квантовых точек, а КТ, связанные с
ТАФП полностью потушены. При этом в спектре люминесценции
ТАФП по сравнению со спектром люминесценции свободного
порфирина наблюдается небольшой сдвиг в длинноволновую сторону
полос с максимумами ~ 645 нм и ~ 700 нм (рис.3., кривые 4 и 1
соответственно) и появление новой полосы в области ~ 610 нм. Следует
отметить, что появление новой полосы с максимумом ~ 610 нм в спектре
люминесценции порфирина ранее наблюдалось при образовании
комплексов CdTe КТ с молекулами ТАФП в щелочном растворе.
105
интенсивность люминесценции, пр.ед.
На рисунке 7.4 приведены спектры возбуждения люминесценции
смешанного раствора КТ и молекул порфирина. В спектре возбуждения
у всех трех полос люминесценции наблюдаются B-полоса (~ 424 нм) и
Q-полоса с максимумом ~ 560 нм, принадлежащие спектру поглощения
ТАФП, связанного с КТ в комплекс. Это свидетельствует о том, что все
полосы люминесценции (610 нм, 660 нм и 715 нм) принадлежат
молекулам ТАФП, связанным с КТ.
1
2
0.02
3
0.01
0.00
400
500
600
длина волны, нм
Рис. 7.4. Спектры возбуждения люминесценции ТАФП, связанного в
комплекс с КТ CdSe/ZnS: 1 – регистрация на длине волны 610 нм; 2 – на
650 нм; 3 – на 710 нм [6].
Интенсивность люминесценции ТАФП в комплексах
с
квантовыми точками CdSe/ZnS, также как и в случае с КТ CdTe,
оказывается в 5 раз меньше интенсивности люминесценции свободных
молекул порфирина (рис.7.3, кривые 1 и 3). Причины наблюдающегося
тушения люминесценции молекул порфирина в комплексах с
квантовыми точками могут заключаться как в изменении излучательной
константы в результате изменения спектральной формы порфирина, так
и в переносе электрона и/или переносе энергии от молекулы на КТ на
локальные уровни, расположенные в запрещенной зоне. Для выяснения
природы наблюдающегося уменьшения интенсивности люминесценции
порфирина необходимо проведение дополнительных исследований.
В ходе исследования оптических свойств комплексов квантовых
точек с молекулами порфирина было установлено, что спектральное
положение и соотношение амплитуд полос в спектрах поглощения и
люминесценции молекул порфирина, связанных в комплекс с КТ,
заметно меняется при изменении концентраций и соотношения
компонент в смешанных растворах. В связи с этим было проведено
106
исследование временной эволюции оптических характеристик
комплексов квантовых точек и молекул порфирина при эквимолярном
соотношении компонент в смеси (C~ 10-6 М).
На рисунке 7.5 приведены зависимости положения и полуширины
B-полосы молекул ТАФП, связанных в комплекс с квантовыми точками
CdSe/ZnS от времени, прошедшего после смешивания компонент.
0.24
2
422
0.23
420
0.22
418
оптическая плотность
положение максимума полосы Соре, нм
1
0.21
416
0
30
60
90
120
150
180
время, мин.
Рис. 7.5. Временная эволюция B-полосы в спектре поглощения ТАФП,
связанного в комплекс с КТ CdSe/ZnS в водном растворе (pH=7.0),
CКТ,ТАФП~ 10-6 М: 1 – положение максимума полосы; 2 – значение
оптической плотности в максимуме полосы [6].
По мере отстаивания водного раствора комплексов КТ/ТАФП в
спектре поглощения порфирина наблюдается плавное смещение Bполосы в длинноволновую область, которое сопровождается
увеличением амплитуды полосы и уменьшением ее ширины. В области
Q-полос
(500–640
нм)
наблюдается
смещение
полос
и
перераспределение их амплитуд. Такие спектральные изменения
свидетельствуют о том, что молекулы порфирина, связанные в комплекс
с квантовыми точками, могут находится, по крайней мере, в двух
спектральных формах. Сразу после образования комплексов КТ/ТАФП
практически все молекулы порфирина в комплексах с КТ находятся в
неустойчивой спектральной форме. В течение 2-5 часов (в зависимости
от концентраций и соотношения компонент в смеси) в комплексах
наблюдается переход всех молекул ТАФП во вторую, устойчивую,
форму. Тот факт, что после двух часов отстаивания водного раствора
комплексов КТ/ТАФП спектральные характеристики молекул
107
порфирина выходят на стационарный уровень (рис.7.5) свидетельствует
о том, что система достигает равновесного состояния.
На рисунке 7.6 приведены спектры поглощения начальной и
конечной спектральных форм молекул порфирина, связанных в
комплекс с квантовыми точками.
1
2
3
4
5
6
отптическая плотность
0.01
0.2
0.1
0.00
500
600
0.0
400
500
600
длина волны, нм
Рис. 7.6. Эволюция спектров поглощения водного раствора (pH=7.0) КТ
CdSe/ZnS и молекул ТАФП в водном растворе, CНК,ТАФП ~ 5·10-7 М: 1 –
спектр поглощения КТ; 2 – спектр поглощения смеси сразу после
добавления молекул ТАФП; 3 – спектр поглощения смеси через 3 часа
после добавления ТАФП к КТ; 4 – спектр поглощения свободного
ТАФП; 5 – спектр поглощения ТАФП, связанного в комплекс с КТ сразу
после смешивания компонент, получен в результате вычитания спектра
КТ из спектра смеси (кривая 1 и кривая 2 соответственно); 6 – спектр
поглощения ТАФП, связанного в комплекс с КТ через 3 часа после
добавления ТАФП к КТ, получен в результате вычитания спектра КТ из
спектра смеси (кривая 1 и кривая 3 соответственно). На вставке в
увеличенном масштабе приведены кривые 4-6 в области Q-полос ТАФП
[6].
В спектре поглощения формы ТАФП, которая образуется сразу
после смешивания квантовых точек с молекулами порфирина, по
сравнению
со
спектром
свободных
молекул
наблюдается
длинноволновый сдвиг B-полосы на 5 нм, а также небольшие сдвиги 1 и
2 полос поглощения и перераспределение амплитуд Q-полос. В спектре
поглощения устойчивой формы молекул порфирина в комплексах с КТ,
которая соответствует равновесному состоянию системы, B-полоса
108
занимает наиболее длинноволновое положение (~424 нм), но ее
полуширина несколько меньше, чем полуширина полосы Соре у формы
ТАФП, образующейся сразу после образования комплексов КТ/ТАФП
(рис.7.6, кривые 6 и 5 соответственно). При этом у данной формы
практически исчезает первая полоса поглощения в области 640 нм и
наблюдается перераспределения амплитуд 3 и 4 полос по сравнению со
спектром поглощения свободного ТАФП (рис.7.6, кривые 6 и 4
соответственно).
Наблюдающиеся различия в спектре поглощения молекул
порфирина, связанных в комплекс с КТ, сразу после образования
комплексов и после установления равновесия, скорее всего, обусловлено
тем, что молекулы порфирина связаны с поверхностью КТ посредством
разного числа аминогрупп. На основании изменений электронного
спектра поглощения молекул порфирина нельзя сделать однозначный
вывод, какая из наблюдаемых нами форм порфирина связана
посредством одной или нескольких аминогрупп.
Логично
предположить, что в первый момент времени после смешивания водного
раствора нанокристаллов с молекулами порфирина большая часть
молекул связываются с КТ через карбоксильные группы тиогликолевой
кислоты посредством одной аминогруппы. По мере отстаивания
раствора с комплексами КТ/ТАФП постепенно все молекулы порфирина
присоединяются к КТ через две аминогруппы, связываясь с двумя
соседними молекулами TGA на поверхности нанокристаллов. Следует
отметить, что скорость перехода молекул порфирина в комплексе с КТ
во вторую спектральную форму коррелирует с молярными
концентрациями КТ и соотношением СКТ:СТАФП в растворе. Так, при
увеличении концентрации КТ и уменьшении среднего числа молекул,
приходящихся в растворе на одну квантовую точку, скорость перехода
молекулы порфирина во вторую спектральную форму возрастает.
Полученные данные свидетельствуют о том, что в комплексах
КТ/ТАФП многообразие спектральных форм ТАФП связано не с
разносортностью молекул порфирина или нанокристаллов, а со
временем, которое необходимо при определенных молярных
концентрациях и соотношениях компонент для перехода всех молекул
ТАФП в стабильную спектральную форму (рис.7.6, кривая 6).
На рисунке 7.7 приведены спектры люминесценции смеси КТ
CdSe/ZnS с молекулами ТАФП сразу и через 3 часа после смешивания
компонент.
109
интенсивность люминесценции, отн.ед.
90
60
1
2
3
4
5
6
10
5
0
600
650
700
750
30
0
550
600
650
длина волны, нм
700
750
Рис. 7.7. Спектры люминесценции смешанного раствора КТ CdSe/ZnS с
молекулами порфирина CНК,ТАФП ~ 5·10-7 М, возбуждение светом с
длиной волны 420 нм: 1 – свободные КТ; 2 – свободные молекулы
ТАФП; 3 – спектр люминесценции смеси сразу после добавления ТАФП
к КТ; 4 – спектр люминесценции смеси через 3 часа после добавления
ТАФП к КТ; 5 – спектр молекул ТАФП, связанных в комплекс с КТ,
сразу после добавления ТАФП к КТ; получен вычитанием с
определенным множителем спектра КТ из спектры смеси (кривые 1 и 3
соответственно); 6 – спектр молекул ТАФП, связанных в комплекс с КТ,
через 3 часа после добавления ТАФП к КТ; получен вычитанием с
определенным множителем спектра КТ из спектры смеси (кривые 1 и 4
соответственно). На вставке в увеличенном масштабе приведены
спектры 5 и 6 [6].
Из рисунка 7.7 видно, что в смешанном растворе наблюдается
эффективное тушение люминесценции КТ (порядка 90%), величина
остаточной люминесценции КТ в ходе эксперимента не меняется
(рис.7.7 кривые 3 и 4). При переходе молекул порфирина, связанных в
комплекс с КТ, в устойчивую форму наблюдается перераспределение
амплитуд полос люминесценции ТАФП.
Хотя спектр поглощения стабильной формы ТАФП, связанной в
комплекс с КТ, в области Q-полос (рис.7.6, кривая 6, область Q-полос на
вставке) согласуется с представлениями о спектре поглощения
металлокомплекса порфирина: характерное коротковолновое смещение
0-0 полосы с 640 нм на 600 нм, а также наличие в системе Q-полос двух
вместо четырех полос поглощения, такая трактовка представляется
маловероятной из-за отсутствия свободных ионов металлов в растворе.
110
Скорее всего появление и увеличение амплитуды полосы
люминесценции 610 нм в спектре ТАФП обусловлено излучательным
переходом с термически заселяемого колебательного уровня первого
возбужденного электронного состояния, т.е. термически активированной
люминесценцией. Окончательный ответ может дать проведение
исследования люминесцентных свойств данного объекта при низкой
температуре.
интенсивность люминесценции.отн. ед.
оптическая плотность
Перенос энергии фотовозбуждения в комплексах КТ/ТАФП. Спектр
люминесценции квантовых точек CdSe/ZnS с диаметром 3.0 нм
перекрывается с тремя из четырех Q-полос в спектре поглощения
ТАФП. Поэтому в комплексах КТ/ТАФП следует ожидать эффективного
переноса энергии фотовозбуждения от КТ на молекулы порфирина по
механизму FRET. Действительно, явные признаки FRET наблюдаются в
спектре возбуждения люминесценции устойчивой формы ТАФП,
связанного в комплекс с квантовыми точками, представленном на
рисунке 7.8 вместе со спектром возбуждения люминесценции КТ и
спектром поглощения данной формы ТАФП.
1
2
3
0,010
4
0,005
0,000
400
500
600
длина волны, нм
Рис. 7.8. Спектр возбуждения люминесценции ТАФП, связанного в
комплекс с CdSe/ZnS КТ: 1 – спектр ТАФП, регистрация на длине волны
660 нм; 2 – спектр возбуждения люминесценци свободных КТ,
регистрация на длине волны 660 нм; 3 – спектр возбуждения
люминесценции свободных КТ, регистрация на длине волны 600 нм; 4 –
спектр поглощения ТАФП, связанного в комплекс с КТ [6].
111
Из рисунка 7.8 видно, что в спектре возбуждения люминесценции
устойчивой формы ТАФП, связанной в комплекс с КТ, присутствуют
полосы ~ 365 нм, ~ 475 нм и ~ 550 нм, которых нет в спектре
поглощения данной формы ТАФП, но которые наблюдаются в спектре
поглощения квантовых точек. Для сравнения на рисунке приведен
спектр
возбуждения
люминесценции
свободных
КТ,
зарегистрированный на длине волны 660 нм (рис.7.8, кривая 2). Видно,
что полосы ~ 365 нм, ~ 475 нм и ~ 550 нм в спектре возбуждения
люминесценции порфирина не могут быть обусловлены остаточной
люминесценцией квантовых точек в смешанном растворе КТ и ТАФП.
Поэтому наличие полос поглощения квантовых точек в спектре
возбуждения люминесценции устойчивой формы порфирина, связанного
в комплекс с КТ, однозначно свидетельствует об эффективном переносе
энергии фотовозбуждения от КТ на молекулы ТАФП. Данная форма
порфирина появляется только в результате образования комплексов
КТ/ТАФП, и не существует у порфирина, не связанного в комплекс с
квантовыми точками. Это обстоятельство не позволяет получить
информацию о спектре возбуждения люминесценции этой формы
порфирина в отсутствие переноса энергии фотовозбуждения от КТ на
ТАФП и, соответственно, дать количественную оценку эффективности
переноса энергии в данном случае.
Оптические свойства молекул ТАФП в плазме крови человека.
Исследование оптических свойств ТАФП в плазме человеческой крови
показало, что в плазме молекулы порфирина переходят в спектральную
форму, отличную от формы свободных молекул ТАФП в водном
растворе. На рисунке 7.9а приведен спектр поглощения молекул ТАФП
в плазме человеческой крови, для сравнения на рисунке 7.9б приведены
также спектры поглощения порфирина в водном растворе и
неустойчивая форма порфирина, связанного в комплекс с КТ в водном
растворе.
112
а)
б)
1
2
3
0,10
0,004
1.0
оптическая плотность
оптическая плотность
1
2
3
0.5
0.02
0,002
0,05
0,000
500
550
600
650
0.01
0,00
0.0
400
500
600
700
800
400
500
600
длина волны, нм
длина волны, нм
0.00
500
600
-6
Рис.7.9. (а) Спектр поглощения ТАФП (C~ 10 М) в плазме
человеческой крови: 1 – поглощение плазмы; 2 – плазма крови с ТАФП;
3 –спектр ТАФП, полученный в результате вычитания спектра 1 из
спектра 2. (б) Спектры поглощения молекул ТАФП: 1 – водный раствор
(pH=7.0); 2 – неустойчивая форма ТАФП, связанного с КТ, в водном
растворе; 3 – в плазме человеческой крови. Вставка: область Q-полос в
увеличенном масштабе [6].
Анализ спектра поглощения порфирина в плазме крови показал,
что положение B-полосы в спектре поглощения порфирина в плазме
крови совпадает с положением данной полосы в спектре неустойчивой
формы порфирина, связанного в комплекс с КТ в водном растворе,
которая образуется сразу после смешивания растворов квантовых точек
и молекул порфирина. Однако положение и соотношение амплитуд Qполос отличается как от спектра свободного, так и связанного с
нанокристаллами порфирина в водном растворе.
Изменение спектральной формы ТАФП при введении в плазму
крови может быть обусловлено образованием комплексов молекул
порфирина с анионами, присутствующими в плазме крови (хлор,
бикарбонат, фосфаты, сульфаты). Образование комплексов молекул
порфирина с компонентами плазмы крови возможно в результате
электростатического взаимодействия разноименно заряженных частиц:
положительно заряженных молекул порфирина с анионами плазмы
крови.
На рисунке 7.10 приведены спектры люминесценции молекул
ТАФП в плазме крови, в водном растворе и в комплексах КТ/ТАФП в
водном растворе сразу после образования комплексов. Уменьшение
интенсивности люминесценции порфирина в плазме крови по
сравнению с люминесценцией свободного ТАФП в водном растворе
(рис.7.10, кривые 3 и 1 соответственно) связано с существенным
поглощением и рассеянием света плазмой крови на длине волны
113
700
интенсивность люминесценции, отн. ед.
возбуждения люминесценции образца (Рис. 7.9а). Нормированные
спектры люминесценции порфирина, приведенные на вставке Рис. 7.10,
ясно показывают, что в спектре порфирина, связанного с компонентами
плазмы, отсутствует полоса 610 нм, а положение оставшихся двух полос
совпадает с их положением в спектре порфирина, связанного в комплекс
с КТ, однако полосы в плазме крови имеют другое соотношение
амплитуд.
90
1
2
3
60
600
700
30
0
600
650
700
750
длина волны, нм
Рис.7.10. Спектры люминесценции ТАФП (C~ 10-6 М), возбуждение
светом с длиной волны 405 нм: 1 – водный раствор; 2 – ТАФП,
связанный с КТ, в водном растворе сразу после смешивания компонент;
3 – в плазме человеческой крови. Вставка: Нормированные спектры
люминесценции ТАФП [6].
Образование комплексов CdSe/ZnS КТ с молекулами ТАФП в
плазме крови человека. Последовательное введение в плазму крови
отдельных компонент: молекул ТАФП и полупроводниковых
нанокристаллов CdSe/ZnS за время проведения эксперимента не
приводит к образованию комплексов КТ/ТАФП. Об этом
свидетельствуют отсутствие тушения люминесценции КТ и
неизменность спектров поглощения и люминесценции молекул ТАФП.
Данный факт вполне объясним, поскольку на скорость образования и
константы устойчивости комплексов КТ/ТАФП существенное влияние
оказывает вязкость растворителя и наличие конкурирующего процесса
образования комплексов молекул ТАФП с компонентами плазмы крови.
Кроме того, на соотношение скоростей образования комплексов
заметное влияние могут оказывать противоионы как тиогликолевой
кислоты, так и молекул порфирина.
114
Для исследования свойств комплексов квантовых точек с
молекулами
порфирина
в
плазму
крови
вводился
высококонцентрированный водный раствор комплексов КТ/ТАФП: 10
мкл водного раствора комплексов (CНК,ТАФП ~ 5·10-4 М) на 10 мл плазмы
крови. На рисунке 7.11 приведены спектры поглощения комплексов
КТ/ТАФП, введенных из водного раствора в плазму крови сразу и
спустя три часа после введения. Видно, что спектр поглощения
комплексов не меняется за время проведения эксперимента. Поскольку в
плазму крови раствор комплексов КТ/ТАФП вводился сразу после
приготовления, то молекулы порфирина, связанные с КТ, имеют
спектральную форму, близкую к форме порфирина, возникающей сразу
после образования комплексов КТ/ТАФП. В тоже время, спектр
поглощения молекул порфирина, связанного с компонентами плазмы,
имеет мало отличий от спектра неустойчивой формы порфирина в
комплексах с КТ. В этом случае по спектру поглощения порфирина
нельзя судить о том, произошла или нет диссоциация комплексов
КТ/ТАФП при введении их из водного раствора в плазму крови.
1
2
3
оптическая плотность
0,3
1,0
4
5
6
0,2
0,1
0,5
0,0
400
0,0
400
500
600
500
600
700
длина волны, нм
Рис.7.11. Спектры поглощения комплексов КТ/ТАФП, соотношение
СКТ:СТАФП равно 1:1, CКТ,ТАФП ~ 5·10-7 М, введенных в плазму крови: 1 –
спектр плазмы крови; 2 –спектр комплексов в плазме крови сразу после
введения; 3 –спектр комплексов в плазме крови через три часа после
введения. На вставке: 4 – спектр поглощения комплексов КТ/ТАФП,
получен в результате вычитания спектра поглощения плазмы из спектра
комплексов сразу после введения их в плазму; 5 – спектр поглощения
комплексов КТ/ТАФП, получен в результате вычитания спектра
поглощения плазмы из спектра комплексов через 3 часа после введения их
в пазму (кривая 1 и 3 соответственно); 6 – спектр поглощения ТАФП в
плазме крови (см. рис.7.9) [6].
115
интенсивность люминесценции, отн. ед.
В спектре поглощения комплексов отчетливо виден вклад
поглощения КТ. Следует отметить, что при введении КТ CdSe/ZnS/TGA
в плазму крови оптические характеристики (спектры поглощения и
спектры люминесценции) нанокристаллов не меняются. Однако при
отстаивании раствора КТ в плазме крови происходит увеличение
квантового выхода люминесценции КТ, в зависимости от типа
(CdSe/ZnS или CdTe) и размеров КТ, от 1.5 до 2 раз по сравнению с
квантовым выходом люминесценции КТ в водном растворе. Такое
увеличение квантового выхода люминесценции КТ без изменения
спектров поглощения и люминесценции может быть обусловлено
пассивацией поверхности нанокристаллов водорастворимыми белками
плазмы.
На рисунке 7.12 приведены спектры люминесценции комплексов
КТ/ТАФП (соотношение СКТ:СТАФП равно 1:1, CКТ,ТАФП ~ 5·10-7 М),
введенных в плазму крови из водного раствора. Видно, что в плазме
крови тушение люминесценции КТ в присутствии молекул порфирина
составило ~ 58%, что заметно меньше, чем для аналогичной смеси КТ и
ТАФП в водных растворах (~ 90%).
1
2
3
4
5
6
90
15
0
60
600
700
30
0
550
600
650
700
750
длина волны, нм
Рис.7.12. Спектры люминесценции комплексов КТ/ТАФП, введенных в
плазму крови из водного раствора, возбуждение светом с длиной волны
405 нм: 1 – КТ в плазме крови; 2 – ТАФП в плазме крови; 3,4 –
комплексы КТ/ТАФП сразу и через 3 часа после введения в плазму; На
вставке: 5,6 – спектры люминесценции ТАФП, связанного в комплекс с
КТ, сразу и через 3 часа после введения в плазму; получены в результате
вычитания спектра свободных КТ с определенным множителем из
соответствующих зарегистрированных спектров (кривая 1 и кривые 3 и
4 соответственно); для сравнения на вставке также приведен спектр
ТАФП в плазме (кривая 2) [6].
116
Наличие достаточно высокого уровня остаточной люминесценции
КТ означает, что сразу после введения водного раствора комплексов
КТ/ТАФП в плазму крови в результате взаимодействия молекул ТАФП с
компонентами плазмы происходит диссоциация порядка 40% от числа
комплексов КТ/ТАФП, существующих при данных молярных
концентрациях компонент в водном растворе. Полагая, что при
соотношении компонент СКТ:СТАФП равном 1:1 в комплексах на каждый
нанокристалл приходится в среднем одна молекула ТАФП, то
диссоциация 40% комплексов приводит к тому, что в плазме крови
оказываются связаны с КТ порядка 60 % молекул порфирина. Из
рисунка 7.12 видно, что в спектре люминесценции ТАФП появляется
полоса с максимумом 610 нм, которая отсутствовала у молекул ТАФП,
связанных с компонентами плазмы, и относится к форме порфирина,
появляющейся сразу после образования комплексов КТ/ТАФП в водном
растворе (см. рис.7.10, кривая 2). Помимо этого наблюдается увеличение
интенсивности люминесценции в полосах на 650 нм и 715 нм (рис.7.12,
вставка, кривые 2 и 5 соответственно). Поскольку спектр поглощения
молекул порфирина остается неизменным появление полосы на 610 нм и
30% увеличение интенсивности полос 650 нм и 715 нм свидетельствует
об
эффективном
внутрикомплексном
переносе
энергии
фотовозбуждения от КТ на молекулы порфирина.
Через три часа после введения комплексов КТ/ТАФП в плазму в
спектре люминесценции ТАФП наблюдается увеличение интенсивности
люминесценции в полосе 610 нм и незначительное уменьшение
люминесценции в полосах 650 нм и 715 нм (рис.7.12, вставка, кривая 6).
Следует отметить, что за время проведения эксперимента было
обнаружено также увеличение интенсивности люминесценции КТ
(рис.7.12, кривые 3 и 4). При этом среднее время жизни люминесценции
данных КТ в плазме крови увеличилось с <τКТ> ~11 нс до <τКТ> ~15 нс.
Это свидетельствует о том, что увеличение интенсивности
люминесценции квантовых точек, скорее всего, обусловлено не
диссоциацией комплексов КТ/ТАФП, а увеличением квантового выхода
люминесценции КТ, которое неоднократно наблюдалось нами при
введении КТ из водного раствора в плазму крови. Увеличение
квантового выхода люминесценции нанокристаллов в плазме крови
должно приводить к увеличению эффективности переноса энергии по
механизму FRET от КТ на молекулы порфирина. Об этом
свидетельствует увеличение интенсивности полосы люминесценции
порфирина с максимумом на 610 нм, которая принадлежит молекулам
порфирина, связанным в комплекс с КТ (рис.7.12, вставка).
Таким образом, исследование условий образования и
фотофизических свойств комплексов полупроводниковых квантовых
точек CdSe/ZnS с молекулами тетра(p-триметиламинофенил)порфина
показало, что в водных растворах образование комплексов КТ/ТАФП
происходит в результате электростатического взаимодействия молекул
117
солюбилизатора КТ тиогликолевой кислоты и положительно
заряженных молекул ТАФП. В равновесном состоянии в водных
растворах наблюдается эффективный внутрикомплексный перенос
энергии фотовозбуждения от КТ на молекулы ТАФП. Установлено, что
молекулы порфирина взаимодействуют с компонентами плазмы крови
человека. Данное взаимодействие конкурирует с процессом образования
комплексов
КТ/ТАФП
в
плазме
крови.
При
введении
концентрированного водного раствора комплексов КТ/ТАФП в плазму
крови в данной среде достаточно быстро устанавливается равновесие
между комплексами молекул порфирина с КТ и с компонентами плазмы
крови. В комплексах КТ/ТАФП в плазме крови наблюдается
эффективный перенос энергии фотовозбуждения от квантовых точек на
молекулы порфирина. Это приводит к появлению полосы с максимумом
на 610 нм в спектре люминесценции ТАФП и к увеличению
интенсивности в полосах люминесценции ТАФП, которые являются
общими для спектра люминесценции ТАФП в плазме крови и ТАПФ,
связанного в комплекс с КТ.
Цитируемая литература:
1. Synthesis and Assembly. Режим доступа:
http://www.wtec.org/loyola/nano/02_01.htm, свободный.
2. P. Reiss, E. Couderc, J. De Girolamo, A. Pron. Nanoscale, V. 3, P. 446–
489 (2011).
3. Sanvicens N., Marco M. P. // Trends in Biotechnology, V.26, №8, P. 425433 (2008)
4. Mauter M. S., Elimelech M. // Environmental Science & Technology, V.
42, № 16 (2008)
5. Э.Штерн, К. Тиммонс Электронная абсорбционная спектроскопия в
органической химии: пер. с англ. М. : Мир, 1974 .— 295 с.
6. А.О. Орлова, М.С. Губанова, В.Г. Маслов, А.В. Баранов, А.В.
Федоров, М.В. Артемьев, Теор. эксп. химия, 48 (1), 56–63 (2012).
Список рекомендуемой литературы
Основная литература:
1. С.Ю.Вязьмин, Д.С.Рябухин, А.В.Васильев. Электронная
спектроскопия органических соединений. Учебное пособие. СПб,
СПбГЛТА, 2011. 43с. Режим доступа:
http://ftacademy.ru/UserFiles/Vasilyev-PosobUV.pdf, свободный.
2. Пентин Ю.А., Курамшина Г.М. Основы молекулярной
спектроскопии. М., Изд-во: Мир, БИНОМ. Лаборатория знаний, 2008.
398c.
3. А.В. Федоров, А.В. Баранов. Оптика квантовых точек. В кн.: Оптика
наноструктур. Под ред. А.В. Федорова: – СПб. Недра, 2005. с. 181.
118
4. Дж.Д. Смолл. Нефотохимическое выжигание стабильных провалов и
дефазировка примесных электронных переходов в органических
стеклах. В кн. Спектроскопия и динамика возбуждений в
конденсированных молекулярных системах. Под ред. В.М.
Аграновича и Р.М. Хохштрассера. – М.: Наука, 1987. С. 316-340.
5. М. Кардона. Резонансные явления. В кн.: Рассеяние света в твердых
телах. Вып.II: Пер. с англ./Под ред. М. Кардоны и Г. Гюнтеродта. –
М.: Мир. 1984, с. 35.
6. Г.Е. Скворцов, В.А. Панов, Н.И. Поляков, Л.А. Федин. Микроскопы. Л.: Машиностроение, 1969. –511 с.
7. S. Nathan. LSCM, Режим доступа:
http://www.olympusfluoview.com/theory/LSCMIntro.pdf, свободный.
8. J. Shah. Ultrafast Spectroscopy of Semiconductors and Semiconductor
Nanostructures. Springer Series in Solid-State Science, 115. Springer,
New York, 1996, – 372 p.
Дополнительная литература:
1. Ю.А.Пентин, Л.В.Вилков. Физические методы исследования в химии
/М.: Мир, 2003. 683 с.
2. Э.Штерн, К. Тиммонс Электронная абсорбционная спектроскопия в
органической химии: пер. с англ. М. : Мир, 1974 .— 295 с.
3. Применение спектроскопии в химии : пер. с англ. / А. Дункан, В.
Горди, Н. Джонс [и др.] ; под ред. В. Веста .— Москва : Изд-во
иностранной литературы, 1959 .— 659 с.
4. А.Н. Теренин. Фотоника молекул красителей и родственных
органических соединений. «Наука», Л., 1967, 616 с.
5. Р.Н. Нурмухаметов. Поглощение и люминесценция ароматических
соединений. «Химия», М., 1971, 216с.
6. В.Л. Ермолаев, Е.Н. Бодунов, Е.Б. Свешникова, Т.А. Шахвердов.
Безызлучательный перенос энергии электронного возбуждения.
«Наука», Л., 1977, 311с..
7. Ельяшевич, М.А. Атомная и молекулярная спектроскопия /
Ельяшевич М.А. — 2-е изд .— Москва : Эдиториал УРСС, 2001 .—
894 с. : ил .— Библиогр.: с.857-873. — ISBN 5-8360-0177-4.
8. Кизель, В. А. Практическая молекулярная спектроскопия :
Учеб.пособие для вузов / Москва : Изд-во МФТИ, 1998 .— 254с
9. К.Бенуэлл. Основы молекулярной спектроскопии, М, Мир, 1985,
384с.
10. Бахшиев Н. Г. Введение в молекулярную спектроскопию. Изд-во
ЛГУ, 1987 год. 215 с.
11. В.В. Лебедева. Экспериментальная оптика. 2005 год. 289 стр.
119
12. О.В. Свердлова, «Электронные спектры в органической химии», Л:
Химия, 1985.
13. Браун, Д. Спектроскопия органических веществ .— М. : Мир, 1992
.— 300с.
14. В.М. Пешкова, М.И. Громова. Методы абсорбционной
спектроскопии в аналитической химии / Москва : Высшая школа,
1976 .— 280 с.
15. Г. В. Майер, В. И. Данилова. Квантовая химия, строение и фотоника
молекул / Томск : Изд-во Том.ун-та, 1984 .— 218 с.
16. Наканиси, К. Инфракрасные спектры и строение органических
соединений : Практическое руководство / Москва : Мир, 1965 . 216 с.
17. Мальцев, А. А. Молекулярная спектроскопия : (теория, практические
работы, задачи) : учебное пособие для вузов /М. : Изд-во
Московского ун-та, 1980 .— 271 с.
18. Л. В. Левшин, А. М. Салецкий. Оптические методы исследования
молекулярных систем. Часть 1. Молекулярная спектроскопия. Изд-во
МГУ, 1984, 320с.
19. Электронно-библиотечная система. Издательство «Лань»
[Электронный ресурс] Фриш С.Э. Оптические спектры атомов. –
Лань, 2010. – Режим доступа:
http://e.lanbook.com/books/element.php?pl1_cid=25&pl1_id=625,
свободный.
20. http://techresearch.intel.com/index.aspx.
21. D. Bimberg, M. Grundmann, N.N. Ledentsov. Quantum Dot
Heterostructures. (John Wiley & Sons, Chichester, 1999).
22. Semiconductor Quantum Dots: Physics, Spectroscopy and
Applications. Edd. by Y. Masumoto and T. Takagahara. (SpringerVerlag, Berlin, 2002).
23. С.В. Гапоненко, Н.Н. Розанов, Е.Л. Ивченко, А.В. Федоров, А.В.
Баранов, А.М. Бонч-Бруевич, Т.А. Вартанян, С.Г. Пржибельский.
Оптика наноструктур. Под ред. А.В. Федорова: СПб. Недра, 2005
г. – 326 с.
24. http://ru.wikipedia.org/
25. Е. Биргер. Фуллерены в роли полупроводников. NanoWeek, 8–14
сентября 2008 г., No. 34.
26. Springer Handbook of Nanotechnology. Edd. by Bharat Bhushan.
(Springer-Verlag, Berlin, 2007).
27. Нанотехнологии. Состояние, направление и тенденции развития.
ОНЭКСИМ группа. http://onexim-group.livejournal.com/4189.html.
28. Zheng Cui. Nanofabrication. (Springer, NY, 2008).
29. http://world-of-photonics.net/link/de/19632839.
30. http://www.jeol.com/PRODUCTS/SemiconductorEquipment/
ElectronBeamLithography/JBX5500FS/tabid/481/Default.aspx.
120
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
http://www.raith.com/?xml=solutions%7CLithography+%26+
nanoengineering.
Молекулярно-лучевая эпитаксия и гетероструктуры. Под ред. Л.
Ченга и К. Плога. Мир. Москва. 1989.
Nanotechnology and Nanoelectronics. Edd. By W. R. Fahrner.
(Springer, NY, 2005).
А.Я. Шик, Л.Г. Бакуева, С.Ф. Мусихин, С.А. Рыков. Физика низкоразмерных систем. Наука. СПб. 2001.
S.V. Gaponenko. Optical Properties of Semiconductor Nanocrystals.
Cambridge University Press. Cambridge. 1998.
C.N.R. Rao, P.J. Thomas, G.U. Kulkarni, Nanocrystals: Synthesis,
Properties and Applications, Springer, NY, 2007).
HRTEM. Режимдоступа: http://www.tf.uni-kiel.de/matwis/amat/
def_en/kap_6/backbone/r6_3_4.html, свободный.
R.V. Lapshin, Nanotechnology,15 (9), 1135-1151 (2004).
R.V. Lapshin, Measurement Science and Technology18 (3): 907-927
(2007).
G. Schitter, M.J. Rost, Materials Today (special issue), 40-48 (2008).
R.V. Lapshin, O.V. Obyedkov, Review of Scientific Instruments,64
(10), 2883-2887 (1993).
R.V. Lapshin, Review of Scientific Instruments,66 (9), 4718-4730
(1995).
Y. Sugimoto et al., Nature, 446, 66 (2007).
Гигантское комбинационное рассеяние: Пер. с англ./Под ред. Р.
Ченга, Т. Фуртака.-М.: Мир, 1984. – 408 с.
S. Raymond, S. Fafard, P.J. Poole, A. Wojs, P. Hawrylak, S.
Charbonneau, R. Leon, D. Leonard, P.M. Petroff, J.L. Merz.Statefilling and time-resolved photoluminescence of excited states in
InGaAs/GaAs self-assembled quantum dots. Phys. Rev. B. 1996. 54. P.
11548-11557.
А.В. Федоров, С.Ю. Кручинин. Акустические фононы в системе
«квантовые точки – матрица»: спектроскопия выжигания
провалов. Опт. испектр. 2004. Т. 97. С. 394–402.
M. Nirmal, C. B. Murray, and M. G. Bawendi. Fluorescence-line
narrowing in CdSe quantum dots: Surface localization of the
photogenerated exciton.Phys. Rev. B. 1994. V. 50. P. 2293-3000.
П. Хокс. Электронная оптика и электронная микроскопия.-М.:
Мир. 1974. 318 с.
Р. Хейденрайх. Основы просвечивающей электронной
микроскопии. – М.: Мир. 1966. 471 с.
Г. Томас, М.Дж. Гориндж. Просвечивающая электронная
микроскопия. –М.: Наука. 1983. 316 с.
Г. Шиммель. Методика электронной микроскопии. – М.: Мир.
1972. 299 с.
121
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
Дж. Спенс. Экспериментальная электронная микроскопия
высокого разрешения. – М.: Наука. 1986. 320 с.
СЗМ. Режим доступа:
http://en.wikipedia.org/wiki/Scanning_probe_microscopy, свободный.
СТМ. Режим доступа:
http://en.wikipedia.org/wiki/Scanning_tunneling_microscope,
свободный.
Ch.Bai. Scanning Tunneling Microscopy and its Application. Berlin:
Springer-Verlag. 1995, – 331 p.
Р.З. Бахтизин. Туннельная микроскопия. Соросовский
образовательный журнал, № 11. 2000 г. Режим доступа:
http://nature.web.ru/db/msg.html?mid=1182775&s, свободный.
R. Schaefer. Design and construction of a scanning tunneling
microscope. CarnegieMellon, 1989. Режим доступа:
http://www.ece.cmu.edu/research/publications/1989/CMU-ECE-1989028.pdf, свободный.
СТМ. Режим доступа:
http://www.all4medicine.ru/?Skaniruyushii_tunnelmznyi_mikroskop,
свободный.
СЗМ методики. Режим доступа: http://ntmdt.ru/SPMTechniques/index.html, свободный.
И.В. Яминский. Зондовая микроскопия. Режим доступа:
http://www.nanoscopy.org/E_Book.html, свободный.
http://www.nanoscopy.org/tutorial/Afm/afm.htm, свободный.
AFM. Режим доступа:
http://www.pacificnano.com/files/img_13_1068824016_0.pdf,
свободный.
К. Кузнецов. IBM напутиктитулу Nanoconstruction Company.
Компьютерное обозрение, 11(628). Режим доступа: http://koonline.com.ua/node/35024, свободный.
D. Bera, L. Qian, T.-K. Tseng and P. H. Holloway, Materials3, 22602345 (2010)
Ch. Ingrosso, A Panniello, R. Comparelli, M. L. Curri and M. Striccoli,
Materials 3, 1316-1352 (2010)
V.Maslov , A.Orlova , A.Baranov. Chapter 7.Combination Therapy:
Complexing of QDs with Tetrapyrrols and Other Dyes. In
Photosensitizers in Medicine, Environment, and Security. T. Nyokong ,
V. Ahsen: Eds, 2012, Springer, Dordrecht, Heidelberg, London, New
York, 658p. pp.355-393.
M. F. Frasco and N. Chaniotakis, Sensors 9, 7266-7286 (2009)
E.I. Zenkevich, Th. Blaudeck, M. Heidernätsch, F. Cichos, C. von
Borczyskowski, Theoretical and Experimental Chemistry, 45, 23-34
(2009)
J. Britton, E. Antunes, T. Nyokong, J Photochem Photobiol A: Chem,
210, 1-7 (2010)
122
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
S. Dayal, N. Kopidakis, D. C. Olson, D. S. Ginley, and Garry Rumbles,
Nano Lett., 10(1), 239–242 (2010).
А.О. Орлова, В.Г. Маслов, Ю.А. Топорова и др, Российские
нанотехнологии, 4 (11-12), 16-21, (2009).
A. V. Baranov, A. O. Orlova, V. G. Maslov et al., J. Appl. Phys., 108,
074306 (1-5) (2010)
А.В.Федоров, А.В. Баранов, Оптика квантовых точек. В кн.
Оптика наноструктур. /Под ред. Федорова А.В. - СПб.: Недра,
2005.
А.В. Федоров, И.Д. Рухленко, А.В. Баранов, С.Ю. Кручинин.
Оптические свойства полупроводниковых квантовых точек. СПб.:
Наука. 2011. 188 с.
Ch. H. Vannoy, A. J. Tavares, M. Omair Noor, U. Uddayasankar and
U. J. Krull, Sensors, 11(10), 9732-9763 (2011).
X. Michalet et al., Science 307, 538 (2005)
R. Freeman, B. Willner, and I. Willner, J. Phys. Chem. Lett., 2 (20),
2667–2677, 2011.
P.O. Anikeeva, J.E. Halpert, M.G. Bawendi, and V. Bulovi, Nano
Letters, 7 [8], 2196-2200 (2007).
J. Chen, W. Lei, C. Et al. Phys. Chem. Chem. Phys., 13, 13182-13184
(2011).
S. D’Souza et al., Journal of Photochemistry and Photobiology A:
Chemistry, 220, 11–19 (2011).
Z.-D. Qi et al., J. Mater. Chem., 21, 2455-2458 (2011).
E. I. Zenkevich et al., J. Phys. Chem. C, 115, 21535–21545 (2011).
А.О. Орлова, В.Г. Маслов, И.Е. Скалецкая, А.В. Баранов, Опт. и
спектр., 101 (4), 616-623 (2006).
А.О.Орлова, В.Г. Маслов, А.В.Баранов, I.Gounko, S.Byrne, Опт. и
спектр., 105 (5), 794-800 (2008).
А.О. Орлова, В.Г. Маслов, А.А. Степанов, А.В. Баранов, I. Gounko,
Опт. и спектр., 105 (8), 889 (2008).
А.О. Орлова, М.С. Губанова, В.Г. Маслов, Г.Н. Виноградова, А.В.
Баранов, А.В. Федоров, I. Gounko, Опт. и спектр., 108 (6), 975–982
(2010).
А. Ф. Миронов, Соровский образовательный журнал, 8, 32-40
(1996).
Ермолаев В.Л., Бодунов Е.Н., Свешникова Е.Б., Шахвердов Т.А.,
Безызлучательный перенос энергии электронного возбуждения. Л.:
«Наука», 1977.
D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase, and H. Weller,
Nano Letters, 1 (4), 207–211 (2001).
А.О. Орлова, М.С. Губанова, В.Г. Маслов, А.В. Баранов, А.В.
Федоров, М.В. Артемьев, Теор. эксп. Химия, 48 (1), 56–63 (2012).
E.Z. Chong, D.R. Matthews, H.D. Summers et al. J Biomed Biotechnol,
2007, Article ID 54169, 7 pages, doi:10.1155/2007/54169 (2007)
123
В 2009 году Университет стал победителем многоэтапного конкурса, в
результате которого определены 12 ведущих университетов России,
которым присвоена категория «Национальный исследовательский
университет». Министерством образования и науки Российской
Федерации была утверждена программа его развития на 2009-2018 годы.
В 2011 году Университет получил наименование «Санкт-Петербургский
национальный исследовательский университет информационных
технологий, механики и оптики (Университет ИТМО).
КАФЕДРА ОПТИЧЕСКОЙ ФИЗИКИ И СОВРЕМЕННОГО
ЕСТЕСТВОЗНАНИЯ
Кафедра, основанная в 2002 году в составе факультета «Фотоники
и оптоинформатики» НИУ ИТМО, готовит высококвалифицированных
исследователей, способных проводить научные и технологические
работы мирового уровня по исследованию и разработке новых
наноматериалов и наноструктур, а также нано- и микросистем на их
основе. Кафедрой руководит доктор физ.-мат. наук, профессор А.В.
Федоров, известный специалист в области физики наноструктур.
Кафедра ведет подготовку бакалавров и магистров по направлению
«200700 Фотоника и оптоинформатика» по специальностям «Оптика
наноструктур» и «Физика наноструктур», а также аспирантов по
специальности «Оптика».
В настоящее время на кафедре преподают: профессора:
А.В. Баранов, Т.А. Вартанян, Е.А. Коншина, В.Г. Маслов Е.Ю. Перлин,
А.В. Федоров, доценты: Л.П. Амосова, Г.Н. Виноградова, В.Л. Комолов,
М.Ю. Леонов, А.О. Орлова, П.С. Парфенов, А.А. Старовойтов, Е.В.
Ушакова.
124
Федоров Анатолий Валентинович
Баранов Александр Васильевич
Орлова Анна Олеговна
Маслов Владимир григорьевич
ОСНОВЫ ФИЗИКИ ГИБРИДНЫХ НАНОСТРУКТУР
Учебное пособие
В авторской редакции
Редакционно-издательский отдел НИУ ИТМО
Заведующая РИО
Лицензия ИД №00408 от 05.11.99.
Подписано в печать 27.06.2014
Заказ № 3150
Тираж 100 экз.
Отпечатано на ризографе.
125
Н.Ф. Гусарова
Редакционно-издательский отдел
Санкт-Петербургского национального
исследовательского университета
информационных технологий, механики
и оптики
197101, Санкт-Петербург, Кронверкский пр., 49
126